

Abstract
Mechanical stress induces auto/paracrine ATP release from various cell types, but the mechanisms underlying this release are not well understood. Here we show that the release of ATP induced by hypotonic stress (HTS) in bovine aortic endothelial cells (BAECs) occurs through volume-regulated anion channels (VRAC). Various VRAC inhibitors, such as glibenclamide, verapamil, tamoxifen, and fluoxetine, suppressed the HTS-induced release of ATP, as well as the concomitant Ca2+ oscillations and NO production. They did not, however, affect Ca2+ oscillations and NO production induced by exogenously applied ATP. Extracellular ATP inhibited VRAC currents in a voltage-dependent manner: block was absent at negative potentials and was manifest at positive potentials, but decreased at highly depolarized potentials. This phenomenon could be described with a “permeating blocker model,” in which ATP binds with an affinity of 1.0 ± 0.5 mM at 0 mV to a site at an electrical distance of 0.41 inside the channel. Bound ATP occludes the channel at moderate positive potentials, but permeates into the cytosol at more depolarized potentials. The triphosphate nucleotides UTP, GTP, and CTP, and the adenine nucleotide ADP, exerted a similar voltage-dependent inhibition of VRAC currents at submillimolar concentrations, which could also be described with this model. However, inhibition by ADP was less voltage sensitive, whereas adenosine did not affect VRAC currents, suggesting that the negative charges of the nucleotides are essential for their inhibitory action. The observation that high concentrations of extracellular ADP enhanced the outward component of the VRAC current in low Cl− hypotonic solution and shifted its reversal potential to negative potentials provides more direct evidence for the nucleotide permeability of VRAC. We conclude from these observations that VRAC is a nucleotide-permeable channel, which may serve as a pathway for HTS-induced ATP release in BAEC.
Keywords: ATP release, nitric oxide, hypotonic stress, volume-regulated anion channel, calcium
INTRODUCTION
Mechanical stimuli, such as shear stress (Bodin et al., 1991), mechanical strain (Sauer et al., 2000), and hypotonic stress (HTS)* (Oike et al., 2000), evoke a release of ATP from a variety of cells, including vascular endothelial cells. It is also known that mechanical stress induces the release of the P2 agonist UTP in various cell types (Lazarowski and Harden, 1999). The released nucleotides bind to P2 receptors in an autocrine and/or paracrine manner and regulate cellular functions such as NO production (Kimura et al., 2000), chloride secretion (Grygorczyk and Hanrahan, 1997), and cell volume regulation (Braunstein et al., 2001).
The cystic fibrosis transmembrane conductance regulator (Reisin et al., 1994; Schwiebert et al., 1995; Cantiello et al., 1998) and the mdr-1 gene product P glycoprotein (Abraham et al., 1993; Roman et al., 1997) have been proposed as putative candidates for the mechanically or HTS-induced ATP release pathway, but more recently evidence has been presented that these proteins are modulators of ATP release through an ATP channel (Grygorczyk and Hanrahan, 1997; Braunstein et al., 2001; Roman et al., 2001). Recently, it has been reported that the large conductance anion channel (Sabirov et al., 2001) may be the pathway for HTS-induced ATP release in cultured mammary tumor cells, and that the mechanosensitive ATP release in Xenopus oocyte might be mediated by a membrane trafficking mechanism that is suppressed by brefeldin A and cytochalasin D (Maroto and Hamill, 2001).
The ubiquitously expressed volume-regulated anion channel (VRAC) (Nilius et al., 1996), which has been shown to be permeable for large anions (Strange et al., 1996; Nilius et al., 1997a; Okada, 1997), is an alternative putative pathway for the release of negatively charged nucleotides, including ATP and UTP, all the more so because VRAC currents and the ATP release pathway share a number of common properties: (a) The HTS-induced ATP release in bovine aortic endothelial cells (BAEC) (Koyama et al., 2001) and the activation of VRAC (Voets et al., 1998; Nilius et al., 1999) are both mediated by Rho/Rho-kinase and tyrosine kinase. (b) The HTS-induced ATP release and activation of VRAC are concurrent, i.e., both responses are activated within 1 min after starting hypotonic challenge and reach their maximum after a few minutes (Nilius et al., 1994a; Koyama et al., 2001). (c) Extracellular ATP is a voltage-dependent blocker of VRAC (Ackerman et al., 1994; Jackson and Strange, 1995; Tsumura et al., 1996), which is reminiscent for open pore block, and a number of open pore blockers with dimensions even larger than those of ATP have been shown to permeate through VRAC (Droogmans et al., 1998, 1999).
In this study, we first examined the effects of VRAC inhibitors on the HTS-induced ATP release and the concomitant cellular responses in BAEC. Since the currently available VRAC inhibitors are not selective, we have used four different chemicals with distinct physicochemical properties and structure that have been reported to inhibit VRAC, i.e., tamoxifen (Nilius et al., 1994b), fluoxetine (Maertens et al., 1999), verapamil (Nilius et al., 1994a), and glibenclamide (Yamazaki and Hume, 1997), and found that as well as their blocking action on VRAC currents, they all inhibited HTS-induced ATP-release but not the responses to exogenously applied ATP. In a second series of experiments, we performed a detailed quantitative analysis of the voltage-dependent inhibition of the VRAC current by various nucleotides (ATP, ADP, UTP, CTP, GTP) using a permeating blocker model. This model does not only predict the binding of these nucleotides at an electrical distance of 0.4 inside the channel, but also their permeation across the membrane. Furthermore, we observed that high concentrations of extracellular ADP enhanced the outward component of the VRAC current and shifted its reversal potential to more negative values under conditions where the contribution of Cl− ions to the VRAC current was minimized by reducing intra- and extracellular Cl− concentrations. It is concluded that VRAC is permeable for these nucleotides, and provides a pathway for HTS-induced ATP release in BAEC.
MATERIALS AND METHODS
Cell Culture
Bovine thoracic aorta was obtained from the local slaughterhouse, and endothelial cells were cultured as reported previously (Oike et al., 2000). The cells of the first subculture were used for the present study.
Measurement of Intracellular Ca2+ Concentration
[Ca2+]i was measured at room temperature (20–25°C) with fura-2 fluorescence using an Attofluor digital fluorescence microscopy system (Atto Instruments) as described previously (Koyama et al., 2001).
Measurement of the Intracellular Production of NO
NO was measured with diaminofluorescein-2 (DAF-2), an NO-sensitive fluorescent dye (Kojima et al., 1998), as reported previously (Kimura et al., 2001). Since DAF-2 fluorescence increases almost linearly with NO concentration (Kojima et al., 1998), the fluorescence intensities in each experiment were normalized to a reference image recorded before hypotonic challenge (Kimura et al., 2001).
Measurement of Extracellular ATP Concentration by Luciferase Bioluminescence
The extracellular ATP concentration ([ATP]o) was measured using luciferin–luciferase bioluminescence as described previously (Koyama et al., 2001). ATP concentration in the presence of VRAC inhibitors was calculated from calibration curves in the presence of these inhibitors.
Measurement of Membrane Current
Whole cell membrane current was recorded in the conventional ruptured whole cell configuration (Hamill et al., 1981) with an EPC-9 amplifier (Heka Elekronik GmbH). The pipette solution for examining the effects of VRAC inhibitors and brefeldin A contained (in mM): KCl 40, K-aspartate 100, MgCl2 1, Na2ATP 5, HEPES 10, and EGTA 5 (pH adjusted to 7.3 with KOH). For the effects of 1 mM extracellular nucleotides (Figs. 4 and 5), the pipette solution contained (in mM): CsCl 45, Cs-aspartate 100, MgCl2 1, Na2ATP 5, HEPES 10, BAPTA 5, and CaCl2 1.436 (to give free [Ca2+]i of 30 nM, pH adjusted to 7.3 with CsOH). To examine the contribution of extracellular ADP to the VRAC current (see Fig. 6), we have used a pipette solution with reduced Cl− concentration containing (in mM): Cs-aspartate 145, MgCl2 1, Na2ATP 1, HEPES 10, BAPTA 5, and CaCl2 1.503 (to give free [Ca2+]i of 30 nM, pH adjusted to 7.3 with CsOH). The osmolarity of each solution was adjusted to 300 mOsm with a freezing point depression osmometer (OM-801; Vogel) by adding mannitol. In the experiments with extracellular nucleotides, we pretreated the cells with 1 μM thapsigargin for 30 min to deplete intracellular Ca2+ stores in order to avoid a possible contamination with Ca2+-activated chloride currents (Nilius et al., 1997b) that might be activated by nucleotide-induced Ca2+ release.
Figure 4.

Effects of extracellular ATP on VRAC currents in BAEC. Cells were pretreated with 1 μM thapsigargin for 30 min as described in materials and methods. (A) Reversible inhibition of the HTS (17%) -elicited VRAC current by extracellular ATP (total 1 mM, free 0.15 mM) at 100 mV. Current amplitudes were obtained from voltage ramps from −150 to 150 mV applied every 20s. (B) Current-voltage relations, obtained at the points marked with filled symbols in A. It is obvious that ATP inhibits only the outward component of the current. (C) Current inhibition at positive membrane potentials, calculated from the I-V curves (ii and iii) after correction for the current in isotonic solution (i) and expressed as a percentage of the current in hypotonic solution. Note the prominent increase of inhibition by depolarization and its relief at more pronounced depolarizing potentials. The solid and dashed lines represent the fits of the data points to Eqs. 1 and 2, respectively. Values for Kd(0), δ, and r of the fitted curves were 0.81 mM, 0.42, and 0.29 (solid line) and 1.17 mm, 0.43, and 0.22 (dashed line).
Figure 5.

Representative data showing the effects of adenine nucleotides and triphosphate nucleotides other than ATP on VRAC currents. Total concentration of each nucleotide is 1 mM, the calculated free concentration is given in the figure. (A) Current-voltage relationships in isotonic (i), and in hypotonic solution in the absence (ii) or presence (iii) of the nucleotide. Except for adenosine, all nucleotides affect the outward current but have no virtual effect on inward current. (B) Percent inhibition of VRAC by the various nucleotides. It is obvious that the AMP-block is hardly voltage-dependent. Data of all other nucleotides except for AMP could be fitted to Eq. 2 (solid lines). Values of Kd(0), δ and r for these fits were 0.81 mM, 0.40, and 0.26 for ADP; 4.1 mM, 0.44, and 0.057 for CTP; 0.41 mM, 0.44, and 0.24 for GTP; and 1.1 mM, 0.42, and 0.13 for UTP.
Figure 6.

Effects of extracellular ADP on VRAC currents in low Cl− solution. (A) Current-voltage relationships in isotonic (a) and hypotonic (b) solutions with normal Cl− concentrations, and in low Cl− hypotonic solutions in the absence (c) and presence (d) of 1 mM extracellular ADP. Note the reduced outward current component in the presence of ADP. (B) Current-voltage relationships in low Cl− hypotonic solutions in the absence and presence of extracellular ADP (10 and 20 mM), obtained from the same cell as A. Note the increased outward current and shift in reversal potential with increasing ADP concentration. Similar results were obtained in five other cells.
Drugs and Solutions
The extracellular solution used for Figs. 1, 2, and 3 was a modified Krebs solution (1.5 mM Ca2+ solution) containing (in mM): NaCl 132, KCl 5.9, MgCl2 1.2, CaCl2 1.5, glucose 11.5, HEPES 11.5; pH adjusted to 7.3 with NaOH. The solution was made hypotonic by adding an appropriate amount of distilled water. The isotonic (300 mOsm) and hypotonic (250 mOsm) solutions used for the measurement of 1 mM nucleotide-induced inhibition of VRAC currents (Figs. 4 and 5) had the following ionic composition (in mM): NaCl 75, CsCl 6, CaCl2 1.5, glucose 10, HEPES 10, and 151 and 65 mM mannitol were added to adjust their osmolarity. To examine the effects of extracellular ADP, we have used an extracellular solution of reduced Cl− concentration, which contained (in mM): CsOH 5, MgCl2 1, Ca(OH)2 1.5, glucose 10, HEPES 10, and a solution of the same composition with NaCl 60 as a normal Cl− hypotonic solution. Mannitol was added to both solutions to adjust the osmolarity to 250 mOsm. ADP-containing low Cl− solutions were prepared by replacing the adequate amount of mannitol with 1, 10, or 20 mM NaADP, and adjusting the osmolarity to 250 mOsm.
Figure 1.

VRAC inhibitors on ATP release induced by HTS from BAEC. (A) VRAC activation by HTS (20%) and its reversible inhibition by 100 μM glibenclamide. Current amplitudes at −50 mV were obtained from voltage ramps from −150 to 100 mV applied every 20 s (a). The current-voltage relationships (b) taken at the points marked by the filled symbols in (a) show that glibenclamide inhibits both inward and outward components of the VRAC current. (B) Inhibition of 20% HTS-induced ATP release by various VRAC inhibitors. Fluoxetine (1 μM), glibenclamide (100 μM), tamoxifen (10 μM), and verapamil (10 μM) were present throughout the measurements. Luciferin chemiluminescence was measured for 10 min after starting hypotonic challenge, and was converted into [ATP]o by using standard curves obtained in the presence of each inhibitor. Numbers in parentheses indicate the number of measurements. (**, P < 0.01) (C) Dose–response relationships of glibenclamide (a) and fluoxetine (b) on VRAC current at −50 mV (open symbols) and HTS-induced [ATP]o increase (closed symbols). HTS of 20% was used for both measurements. The solid lines represent the fit of the logistic equation to the [ATP]o data points with IC50 values of 17.1 μM (a) and 0.24 μM (b). Numbers of measurements are 5 for VRAC currents and 6–8 for [ATP]o.
Figure 2.

Effects of VRAC inhibitors on HTS-induced Ca2+ oscillations and NO production in BAEC. (A) HTS (40%) induced Ca2+ oscillations in control cells (a), which were reversibly inhibited by glibenclamide (100 μM) (b). Similar results were obtained in five other cells. Glibenclamide did not affect Ca2+ transients induced by exogenously applied ATP (1 μM) (c). (B) Inhibition by VRAC inhibitors of HTS-induced (a), but not of exogenous ATP (1 μM)-induced (b), NO production, assessed by DAF-2. Numbers in parentheses indicate the number of cells examined. (**, P < 0.01).
Figure 3.

Brefeldin A does not affect HTS (20%)-induced ATP release and VRAC currents in BAEC. Cells were pretreated with 3 μg/ml brefeldin A for 2.5 h before each experiment. (A) Basal and HTS-induced increase in [ATP]o were not different between control and brefeldin A–pretreated cells. n.s., P > 0.05. (B) HTS-induced VRAC currents in control and brefeldin A–treated cells at −50 mV and 100 mV. Differences between are not significant (P > 0.05).
Fluoxetine was purchased from RBI Research Biochemicals International. All other drugs were from Sigma-Aldrich.
Data Analysis
The voltage dependence of current inhibition in the presence of extracellular nucleotides was fitted to Eqs. 1 and 2 using Origin software (OriginLab Corp.).
Data are given as mean ± standard error of the mean. Statistical significance between two groups was determined by using Student's unpaired t test. Probabilities less than 5% (P < 0.05) were regarded as significant.
RESULTS
Effects of VRAC Inhibitors on Hypotonic Stress (HTS)-induced Responses in BAEC
First, we examined the effects of VRAC inhibitors on the HTS-induced current and ATP release. Fig. 1 A, a and b, illustrates the gradual development of an outwardly rectifying current during HTS and its reversible inhibition by 100 μM glibenclamide. The effect of glibenclamide was concentration-dependent, as shown in Fig. 1 C, a. Fluoxetine also inhibited this current in a concentration-dependent manner (Fig. 1 C, b), as did tamoxifen (10 μM) and verapamil (10 μM) (unpublished data).
The time course of activation and the pharmacological properties of this current indicate that it is probably due to activation of the ubiquitously expressed VRAC channels (Nilius et al., 1996). Because it was recorded using a pipette solution containing 5mM EGTA, it is unlikely that it was due to activation of Ca2+-activated Cl− channels by the concomitant HTS-induced Ca2+ release. Moreover, its outward rectification is less prominent than that of Ca2+-activated Cl− channels (Nilius et al., 1997b). Because it is inhibited by tamoxifen, this current is also different from the tamoxifen-activated large conductance Cl− channel current in endothelium (Li et al., 2000). Also, exogenous ATP did not activate a similar current in isotonic solutions, indicating that it was not activated by an autocrine action of ATP released from the cell during the hypotonic challenge.
The current in isotonic solution was insensitive to VRAC inhibitors, and was not suppressed in hypertonic solution (unpublished data), indicating that it is not due to a partial activation of VRAC in isotonic solution.
These VRAC inhibitors also suppressed the HTS-induced release of ATP, assessed from the increase in extracellular ATP concentration ([ATP]o, 9.7 ± 0.5 nM) (Fig. 1 B). Glibenclamide and fluoxetine inhibited this ATP release in a concentration-dependent manner that was similar to their effects on VRAC inhibition (Fig. 1 C). Furthermore, glibenclamide (Fig. 2 A, b), fluoxetine, and verapamil abolished HTS-induced Ca2+ oscillations (Fig. 2 A, a), and the HTS-induced NO production (Fig. 2 B, a) in BAEC, which are both mediated by the released ATP (Oike et al., 2000). Because of its autofluorescence, we could not use tamoxifen in these fluorescent dye assays. These VRAC inhibitors did, however, not affect the responses downstream of ATP release, since they had no effect on Ca2+ transients (Fig. 2 A, c) and NO production (Fig. 2 B, b) induced by exogenously applied ATP (1 μM).
Effects of Brefeldin A on HTS-induced ATP Release and VRAC Current
It has been proposed recently (Maroto and Hamill, 2001) that the HTS-induced ATP release in Xenopus oocytes represents a vesicular transport process sensitive to brefeldin A, an inhibitor of vesicular transport (Klausner et al., 1992). Brefeldin A did not, however, affect HTS-induced ATP release (Fig. 3 A) or VRAC currents (Fig. 3 B), indicating that membrane trafficking does not contribute to HTS-induced ATP release in BAEC.
Voltage-dependent Inhibition of VRAC with Extracellular ATP in BAEC
The close correlation between the effects of VRAC inhibitors on current activation and ATP release in BAEC is compatible with the release of ATP through VRAC channels. The most compelling evidence for permeation of ATP through VRAC would be the demonstration of VRAC currents under conditions where ATP is the only permeant anion. Single-channel and whole-cell ATP currents have been measured through P-glycoprotein (Abraham et al., 1993), the cystic fibrosis transmembrane conductance regulator (Reisin et al., 1994), and through large conductance chloride channels (Sabirov et al., 2001). Unfortunately, all our attempts to record single channel ATP currents from BAEC cells failed so far, whereas application of extracellular ATP at concentrations of 10 mM or higher induced membrane leakiness. We have therefore used an alternative approach to provide more direct evidence for ATP permeation that is based on a quantitative analysis of the voltage-dependent block of VRAC by extracellular ATP (Ackerman et al., 1994; Jackson and Strange, 1995; Tsumura et al., 1996). As shown in Fig. 4, A and B, extracellular ATP (total concentration of 1 mM; free concentration of 0.15 mM) inhibited the outward current through VRAC, but not the inward current. Current inhibition at positive potentials, expressed as the fraction of hypotonic current blocked by extracellular ATP, showed a marked voltage dependency, which is reminiscent of open pore block due to binding of ATP inside the VRAC pore. This inhibition reached a maximum of ∼30–40 mV and declined at more positive potentials (Fig. 4 C).
A similar bell-shaped voltage-dependence of inhibition of endothelial VRAC currents has already been observed with calix[4]arene and suramin (Droogmans et al., 1998), and could be described by a model in which the relief of inhibition at strongly positive potentials was explained by the permeation of this large anion through VRAC. We have used the same “permeating blocker” model, which assumes binding of extracellular ATP to a site at an electrical distance δ inside the channel pore from which it can be released either to the inside or outside, to analyze the voltage-dependence of VRAC inhibition by extracellular ATP, i.e.,
ATPo and ATPi represent extracellular and intracellular ATP, and k01(V), k10(V), k12(V), and k21(V) represent the voltage-dependent transition rate constants between the various states. The fraction of channels occupied by ATP at extra- and intracellular concentrations [ATP]o and [ATP]i and potential V is given by
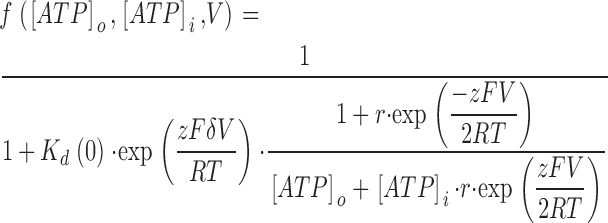 |
where Kd(0) = k10(0)/k01(0) = k12(0)/k21(0) represents the dissociation constant of the ATP–VRAC complex at 0 mV, r = k12(0)/k10(0) = k21(0)/k01(0), z (equal to −4) the valence of ATP. R, T, and F have their usual meaning. The predicted fraction of the current inhibited by 0.15 mM extracellular ATP4− at a potential V is given by
 |
(1) |
The calculated free [ATP]i in the pipette solution is 3.1 mM. This equation was fitted to the experimental data, and shown as the solid line in Fig. 4 C.
From the pooled data from nine cells, we obtained the following values for the parameters: Kd(0) = 1.0 ± 0.5 mM, δ = 0.41 ± 0.03, and r = 0.38 ± 0.08.
We have also fitted the data to a simplified equation (Eq. 2), which does not take into account the binding of intracellular ATP inside the pore at positive potentials (k21(V) ≈ 0) (dashed line in Fig. 4 C).
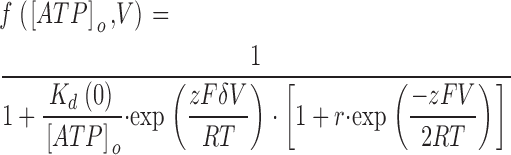 |
(2) |
The values of Kd(0) = 1.0 ± 0.3 mM, δ = 0.42 ± 0.02, r = 0.42 ± 0.16 (n = 9) obtained with Eq. 2 are similar to those obtained with Eq. 1. We have therefore used this simplified equation in subsequent fits (see below).
The value of r = 0.38 indicates that the transition of ATP from the cytoplasm to its binding site in the membrane and vice versa represents the rate limiting step for ATP permeation though the membrane. The probability that ATP bound to its site in the pore will be released to the extracellular side rather than into the cytosol at 0 mV is given by k10(0)/[k10(0)+k12(0)] = 1/(1 + r) = 0.72; i.e. 72% of the total amount of cytosolic ATP entering the pore and binding at its intramembrane site will be released into the extracellular space and contribute to net ATP release at 0 mV. This fraction will be even larger at the resting potential of BAEC cells of about −15 mV, as assessed from the reversal potential of the VRAC current.
Effects of other Nucleotides on VRAC Current in BAEC
The three-dimensional structure of ATP is similar to that of other negatively charged triphosphate nucleotides (UTP, GTP, and CTP) present in the cell. We therefore used the same protocol as in Fig. 4 to examine whether these nucleotides as well as other adenine nucleotides with a reduced number of phosphate residues (ADP, AMP, and adenosine) also permeate through VRAC. We have always used a total concentration of 1 mM for these nucleotides.
All molecules except adenosine inhibited the outward component of the VRAC current (Fig. 5). The block of AMP was weakly voltage-dependent, but all other nucleotides exerted a substantial voltage-dependent block. The current inhibition by all nucleotides except adenosine and AMP could be fitted to Eq. 2, which indicates that they permeate through VRAC (Fig. 5 B). The values of Kd(0), δ, and r for the various nucleotides from the fits to Eqs. 1 and 2 are summarized in Table I.
TABLE I.
The Kd(0), d, and r Values for the Various Nucleotides Calculated from Fits to Eqs. 1 (ATP) and 2 (other Nucleotides)
| ATP | ADP | UTP | GTP | CTP | |
|---|---|---|---|---|---|
| kd (0) (mM) | 1.0 ± 0.5 | 2.3 ± 0.5 | 1.4 ± 0.5 | 0.6 ± 0.3 | 3.1 ± 0.8 |
| δ | 0.41 ± 0.03 | 0.48 ± 0.01 | 0.39 ± 0.04 | 0.41 ± 0.06 | 0.47 ± 0.02 |
| r | 0.38 ± 0.08 | 0.23 ± 0.05 | 0.14 ± 0.05 | 0.70 ± 0.30 | 0.21 ± 0.09 |
| n | 9 | 7 | 5 | 7 | 8 |
ADP Permeation through VRAC
As mentioned above, our failure to record whole-cell ATP currents was related to the fact that elevated concentrations of ATP induced membrane leakiness. Since ADP is likely also permeating VRAC and it appeared from a preliminary experiment that high concentrations of ADP did not cause membrane leakiness, we have tried to measure whole-cell ADP currents to provide more firm evidence for nucleotide permeation through VRAC. To reduce the contribution of Cl− to the VRAC current, we have decreased the extracellular Cl− concentration from 60 to 2 mM and examined under these conditions the effect of adding extracellular ADP (1, 10, or 20 mM) on the outward component of the VRAC current. It was our expectation that the inhibition of the small Cl− component of the VRAC current by extracellular ADP might be overcome at higher ADP concentrations by a current component carried by ADP, which in addition would induce a shift of the reversal potential to more negative values. Fig. 6 shows that this was indeed the case. The VRAC current was drastically lowered at reduced extracellular Cl− concentration, and the outward component was still inhibited by 1 mM ADP (Fig. 6 A). Adding 10 or 20 mM ADP to the low Cl− hypotonic solution significantly enhanced the outward current above the level in ADP-free solution, but had virtually no effect on the inward current (Fig. 6 B). This ADP-induced outward current was suppressed by 100 μM glibenclamide, as was also the case for the VRAC current carried by Cl− (unpublished data). These findings indicate that the effect of extracellular ADP is not due to a nonspecific increase in membrane permeability, and that ADP contributes to the outward current. The concomitant shift of the reversal potential to more negative potentials, as well as the clear dependence of these effects on the ADP concentration further support this contention.
DISCUSSION
We have reported previously that mechanosensitive ATP release induces Ca2+ transients (Oike et al., 2000) and NO production (Kimura et al., 2000) in an auto/paracrine manner in BAEC. We have shown in the present study that glibenclamide, tamoxifen, verapamil, and fluoxetine, in addition to their blocking action on VRAC currents, also suppress HTS-induced ATP release (Fig. 1, B and C) and its downstream responses (Ca2+ transient and NO production, Fig. 2). However, the action of these blockers on the cellular responses downstream of ATP release is indirect, because Ca2+ transients and NO production induced by exogenous ATP are not altered. Moreover, the concentration-dependence of the inhibition of ATP release and VRAC current induced by glibenclamide and fluoxetine is comparable (Fig. 1 C). The close correlation between the effects of these inhibitors with distinct physicochemical and structural properties on VRAC and ATP release therefore suggests a causal relation between both processes. This observation, together with the comparable time courses of VRAC activation and ATP release (Koyama et al., 2001), provide convincing but indirect evidence that the ATP release occurs through VRAC.
VRAC inhibitors are not very specific, and their effects on ATP release appear to be cell specific in the sense they inhibit ATP release in some tissues but are ineffective in others, e.g., NPPB suppressed ATP release in prostate cancer cells (Sauer et al., 2000) and cultured ciliary epithelial cells (Mitchell et al., 1998), whereas tamoxifen was effective in prostate cancer cells (Sauer et al., 2000) but not in ciliary epithelial cells (Mitchell et al., 1998). The effects of these substances on VRAC currents are also quite heterogeneous, and the concentrations for half-maximal inhibition of VRAC currents range, depending on the cell-type, from a few μM to 1 mM (for review see Nilius et al., 1997a). For instance, tamoxifen is a very potent inhibitor in most tissues, but did not significantly inhibit VRAC currents in BC3H1 and C2C12 cells at concentrations up to 100 μM (Voets et al., 1997). These heterogeneous responses therefore seem to point to a family of VRAC channels with different biophysical and pharmacological properties. Hazama et al. (1999) reported a number of experimental findings in a cultured human epithelial cell line that seem to contradict the correlation between VRAC currents and ATP release in this cell type, indicating that ATP release may not be a general property of all VRAC channels. It might therefore be useful to analyze the voltage-dependence of ATP block of VRAC in this cell type, as well as in other cell types, such as intestinal epithelium (Tsumura et al., 1996), glioma cells (Jackson and Strange, 1995), and Xenopus oocytes (Ackerman et al., 1994), in which ATP exerts a voltage-dependent block of VRAC.
Sabirov et al. (2001) reported recently that ATP release in cultured mouse mammary C127i cells occurs through a large conductance anion channel (400pS). It is, however, unlikely that this large conductance anion channel contributes to ATP release in BAEC, because (a) glibenclamide inhibits HTS-induced ATP release (Fig. 1 C) but not the large conductance anion channel (Sabirov et al., 2001), and (b) tamoxifen, which activates a similar large conductance anion channel (368 pS) in porcine aortic endothelial cells (Li et al., 2000), inhibits HTS-induced ATP release (Fig. 1 C).
Our data also exclude a membrane trafficking–mediated ATP release (Maroto and Hamill, 2001) in BAEC, since brefeldin A did not affect ATP release or VRAC currents (Fig. 3). In addition, we have reported previously that disruption of the actin cytoskeleton with cytochalasin B does not inhibit HTS-induced ATP release (Koyama et al., 2001).
The analysis of the ATP-induced, voltage-dependent inhibition of VRAC currents (Fig. 4) provides more direct evidence for permeation of ATP through VRAC. It is unlikely that these effects result from the activation of purinergic receptors, resulting in the activation of protein kinase C and/or Ca2+ mobilization, because these intracellular messengers do not affect VRAC (Nilius et al., 1996). The Kd(0) value of ATP binding of 1.0 mM is within a reasonable range considering the millimolar concentration of intracellular ATP. Also, the value of r (= k12(0)/k10(0) = 0.38) indicates that 72% of cytosolic ATP that binds to the channel permeates through the membrane at 0 mV. The three-dimensional structure of ATP consists of a centered ribose with arms of adenine (9.2 Å) and triphosphate (13.5 Å) residues. Permeation of ATP through the VRAC pore is therefore also compatible with the lower and upper limits of the cross-sectional area of 11 × 12 and 12 × 17 Å2 of the VRAC pore, as derived from the permeation properties of calixarenes in endothelium (Droogmans et al., 1999). From these evidences for ATP permeation through VRAC, we conclude that this channel is the major candidate of the ATP release pathway during hypotonic stimulation in BAEC. This model also predicts that the interaction of intracellular ATP with the channel may account for the outward rectification of VRAC currents.
CTP, GTP, UTP, and ADP also showed a bell-shaped voltage-dependent inhibition of VRAC current, that could be fitted to the permeating blocker model (Fig. 5), suggesting that these nucleotides permeate through VRAC by binding to a common site that is located at ∼40% of the electrical field (Table I). The observation that adenosine does not inhibit VRAC currents indicates that the negatively charged phosphate residues of the nucleotides might be essential for their interaction with the binding site in the VRAC pore. This is also consistent with the weak voltage dependence of the inhibition by AMP. Since these nucleotides, at least ADP and UTP, have been reported to induce Ca2+ transients in endothelium (Viana et al., 1998), the release of nucleotides other than ATP may also contribute to the HTS-induced Ca2+ transients (Fig. 2 A, a) and NO production (Fig. 2 B, a) in BAEC.
Because of the skinning effects of high extracellular ATP concentrations, we were unable to directly demonstrate ATP permeation through VRAC. However, our experimental data with high ADP concentrations provide compelling evidence for the nucleotide permeability of VRAC. Together with the observed close correlation between inhibition of VRAC currents and HTS-induced ATP release and the voltage-dependent inhibition of VRAC currents by ATP and other nucleotides, our data provide unequivocal evidence that VRAC provides a pathway for HTS-induced release of ATP and presumably of other purinergic agonists in BAEC. It might be the subject of future investigations to find out whether VRAC is also the pathway for ATP release induced by other mechanical stimuli, such as shear stress (Bodin et al., 1991) and mechanical strain (Sauer et al., 2000), since it has been reported that shear stress activates a chloride conductance similar to VRAC in vascular endothelial cells (Barakat et al., 1999).
Figure .

Acknowledgments
The authors are grateful for Dr. Bernd Nilius for fruitful discussion.
This study was carried out as a part of “Ground Research Announcement for Space Utilization” promoted by the National Space Development Agency of Japan and Japan Space Forum (M. Oike).
Footnotes
Abbreviations used in this paper: BAEC, bovine aortic endothelial cell; DAF-2, diaminofluorescein-2; HTS, hypotonic stress; VRAC, volume-regulated anion channels.
References
- Abraham, E.H., A.G. Prat, L. Gerweck, T. Seneveratne, R.J. Arceci, R. Kramer, G. Guidotti, and H.F. Cantiello. 1993. The multidrug resistance (mdr1) gene product functions as an ATP channel. Proc. Natl. Acad. Sci. USA. 90:312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman, M.J., K.D. Wickman, and D.E. Clapham. 1994. Hypotonicity activates a native chloride current in Xenopus oocytes. J. Gen. Physiol. 103:153–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat, A.I., E.V. Leaver, P.A. Pappone, and P.F. Davies. 1999. A flow-activated chloride-selective membrane current in vascular endothelial cells. Circ. Res. 85:820–828. [DOI] [PubMed] [Google Scholar]
- Bodin, P., D. Bailey, and G. Burnstock. 1991. Increased flow-induced ATP release from isolated vascular endothelial cells but not smooth muscle cells. Br. J. Pharmacol. 103:1203–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein, G.M., R.M. Roman, J.P. Clancy, B.A. Kudlow, A.L. Taylor, V.G. Shylonsky, B. Jovov, K. Peter, T. Jilling, I.I. Ismailov, et al. 2001. Cystic fibrosis transmembrane conductance regulator facilitates ATP release by stimulating a separate ATP release channel for autocrine control of cell volume regulation. J. Biol. Chem. 276:6621–6630. [DOI] [PubMed] [Google Scholar]
- Cantiello, H.F., G.R. Jackson, Jr., C.F. Grosman, A.G. Prat, S.C. Borkan, Y. Wang, I.L. Reisin, C.R. O'Riordan, and D.A. Ausiello. 1998. Electrodiffusional ATP movement through the cystic fibrosis transmembrane conductance regulator. Am. J. Physiol. 274:C799–C809. [DOI] [PubMed] [Google Scholar]
- Droogmans, G., C. Maertens, J. Prenen, and B. Nilius. 1999. Sulphonic acid derivatives as probes of pore properties of volume-regulated anion channels in endothelial cells. Br. J. Pharmacol. 128:35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droogmans, G., J. Prenen, J. Eggermont, T. Voets, and B. Nilius. 1998. Voltage-dependent block of endothelial volume-regulated anion channels by calix[4]arenes. Am. J. Physiol. 275:C646–C652. [DOI] [PubMed] [Google Scholar]
- Grygorczyk, R., and J.W. Hanrahan. 1997. CFTR-independent ATP release from epithelial cells triggered by mechanical stimuli. Am. J. Physiol. 272:C1058–C1066. [DOI] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Hazama, A., T. Shimizu, Y. Ando-Akatsuka, S. Hayashi, S. Tanaka, E. Maeno, and Y. Okada. 1999. Swelling-induced, CFTR-independent ATP release from a human epithelial cell line: lack of correlation with volume-sensitive Cl− channels. J. Gen. Physiol. 114:525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, P.S., and K. Strange. 1995. Characterization of the voltage-dependent properties of a volume-sensitive anion conductance. J. Gen. Physiol. 105:661–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, C., T. Koyama, M. Oike, and Y. Ito. 2000. Hypotonic stress-induced NO production in endothelium depends on endogenous ATP. Biochem. Biophys. Res. Comm. 274:736–740. [DOI] [PubMed] [Google Scholar]
- Kimura, C., M. Oike, T. Koyama, and Y. Ito. 2001. Impairment of endothelial nitric oxide production by acute glucose overload. Am. J. Physiol. 280:E171–E178. [DOI] [PubMed] [Google Scholar]
- Klausner, R.D., J.G. Donaldson, and J. Lippincott-Schwartz. 1992. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116:1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima, H., N. Nakatsubo, K. Kikuchi, S. Kawahara, Y. Kirino, H. Nagoshi, Y. Hirata, and T. Nagano. 1998. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal. Chem. 70:2446–2453. [DOI] [PubMed] [Google Scholar]
- Koyama, T., M. Oike, and Y. Ito. 2001. Involvement of Rho-kinase and tyrosine kinase in hypotonic stress-induced ATP release in aortic endothelial cells. J. Physiol. 532:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarowski, E.R., and T.K. Harden. 1999. Quantitation of extracellular UTP using a sensitive enzymatic assay. Br. J. Pharmacol. 127:1272–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., Y. Niwa, S. Sakamoto, X. Chen, and Y. Nakaya. 2000. Estrogen modulates a large conductance chloride channel in cultured porcine aortic endothelial cells. J. Cardiovasc. Pharmacol. 35:506–510. [DOI] [PubMed] [Google Scholar]
- Maertens, C., L. Wei, T. Voets, G. Droogmans, and B. Nilius. 1999. Block by fluoxetine of volume-regulated anion channels. Br. J. Pharmacol. 126:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroto, R., and O.P. Hamill. 2001. Brefeldin A block of integrin-dependent mechanosensitive ATP release from Xenopus oocytes reveals a novel mechanism of mechanotransduction. J. Biol. Chem. 276:23867–23872. [DOI] [PubMed] [Google Scholar]
- Mitchell, C.H., D.A. Carre, A.M. McGlinn, R.A. Stone, and M.M. Civan. 1998. A release mechanism for stored ATP in ocular ciliary epithelial cells. Proc. Natl. Acad. Sci. USA. 95:7174–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius, B., M. Oike, I. Zahradnik, and G. Droogmans. 1994. a. Activation of a Cl− current by hypotonic volume increase in human endothelial cells. J. Gen. Physiol. 103:787–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius, B., J. Sehrer, and G. Droogmans. 1994. b. Permeation properties and modulation of volume-activated Cl−-currents in human endothelial cells. Br. J. Pharmacol. 112:1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius, B., J. Eggermont, T. Voets, and G. Droogmans. 1996. Volume-activated Cl− channels. Gen. Pharmacol. 27:1131–1140. [DOI] [PubMed] [Google Scholar]
- Nilius, B., J. Eggermont, T. Voets, G. Buyse, V. Manolopoulos, and G. Droogmans. 1997. a. Properties of volume-regulated anion channels in mammalian cells. Prog. Biophys. Mol. Biol. 68:69–119. [DOI] [PubMed] [Google Scholar]
- Nilius, B., J. Prenen, G. Szucs, L. Wei, F. Tanzi, T. Voets, and G. Droogmans. 1997. b. Calcium-activated chloride channels in bovine pulmonary artery endothelial cells. J. Physiol. 498:381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius, B., T. Voets, J. Prenen, H. Barth, K. Aktories, K. Kaibuchi, G. Droogmans, and J. Eggermont. 1999. Role of Rho and Rho kinase in the activation of volume-regulated anion channels in bovine endothelial cells. J. Physiol. 516:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oike, M., C. Kimura, T. Koyama, M. Yoshikawa, and Y. Ito. 2000. Hypotonic stress-induced dual Ca2+ responses in bovine aortic endothelial cells. Am. J. Physiol. 279:H630–H638. [DOI] [PubMed] [Google Scholar]
- Okada, Y. 1997. Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. Am. J. Physiol. 273:C755–C789. [DOI] [PubMed] [Google Scholar]
- Reisin, I.L., A.G. Prat, E.H. Abraham, J.F. Amara, R.J. Gregory, D.A. Ausiello, and H.F. Cantiello. 1994. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J. Biol. Chem. 269:20584–20591. [PubMed] [Google Scholar]
- Roman, R.M., N. Lomri, G. Braunstein, A.P. Feranchak, L.A. Simeoni, A.K. Davison, E. Mechetner, E.M. Schwiebert, and J.G. Fitz. 2001. Evidence for multidrug resistance-1 P-glycoprotein-dependent regulation of cellular ATP permeability. J. Membr. Biol. 183:165–173. [DOI] [PubMed] [Google Scholar]
- Roman, R.M., Y. Wang, S.D. Lidofsky, A.P. Feranchak, N. Lomri, B.F. Scharschmidt, and J.G. Fitz. 1997. Hepatocellular ATP-binding cassette protein expression enhances ATP release and autocrine regulation of cell volume. J. Biol. Chem. 272:21970–21976. [DOI] [PubMed] [Google Scholar]
- Sabirov, R.Z., A.K. Dutta, and Y. Okada. 2001. Volume-dependent ATP-conductive large-conductance anion channel as a pathway for swelling-induced ATP release. J. Gen. Physiol. 118:251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer, H., J. Hescheler, and M. Wartenberg. 2000. Mechanical strain-induced Ca2+ waves are propagated via ATP release and purinergic receptor activation. Am. J. Physiol. 279:C295–C307. [DOI] [PubMed] [Google Scholar]
- Schwiebert, E.M., M.E. Egan, T.H. Hwang, S.B. Fulmer, S.S. Allen, G.R. Cutting, and W.B. Guggino. 1995. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell. 81:1063–1073. [DOI] [PubMed] [Google Scholar]
- Strange, K., F. Emma, and P.S. Jackson. 1996. Cellular and molecular physiology of volume-sensitive anion channels. Am. J. Physiol. 270:C711–C730. [DOI] [PubMed] [Google Scholar]
- Tsumura, T., S. Oiki, S. Ueda, M. Okuma, and Y. Okada. 1996. Sensitivity of volume-sensitive Cl− conductance in human epithelial cells to extracellular nucleotides. Am. J. Physiol. 271:C1872–C1878. [DOI] [PubMed] [Google Scholar]
- Viana, F., H. de Smedt, G. Droogmans, and B. Nilius. 1998. Calcium signalling through nucleotide receptor P2Y2 in cultured human vascular endothelium. Cell Calcium. 24:117–127. [DOI] [PubMed] [Google Scholar]
- Voets, T., V. Manolopoulos, J. Eggermont, C. Ellory, G. Droogmans, and B. Nilius. 1998. Regulation of a swelling-activated chloride current in bovine endothelium by protein tyrosine phosphorylation and G proteins. J. Physiol. 506:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets, T., L. Wei, P. De Smet, W. Van Driessche, J. Eggermont, G. Droogmans, and B. Nilius. 1997. Downregulation of volume-activated Cl− currents during muscle differentiation. Am. J. Physiol. 272:C667–C674. [DOI] [PubMed] [Google Scholar]
- Yamazaki, J., and J.R. Hume. 1997. Inhibitory effects of glibenclamide on cystic fibrosis transmembrane regulator, swelling-activated, and Ca2+-activated Cl− channels in mammalian cardiac myocytes. Circ. Res. 81:101–109. [DOI] [PubMed] [Google Scholar]