

Abstract
The p16-pRb and p53-MDM2 pathways represent vital cell cycle checkpoints. Recent studies provide evidence that these pathways are directly linked via MDM2-pRb interaction and p53 suppression of the RB1 gene. In the present study we investigated the alterations of this G1 phase protein network using immunohistochemical and molecular methods in a series of 68 non-small-cell lung carcinomas (NSCLCs) and correlated the findings with clinicopathological features and prognosis of the patients. Aberrant expression (Ab) of p16 and pRb was observed in 33 (49%) and 27 (40%) of the carcinomas, respectively. Analysis of the region that encodes for p16 by deletion mapping, a polymerase chain reaction (PCR)-based methylation assay and PCR single-strand conformation polymorphism (SSCP) analysis revealed that deletions and transcriptional silencing by methylation might represent the main mechanisms of CDKN2/p16ink4a inactivation in NSCLCs. The results of deletion mapping also suggest that other tumor suppressor genes may reside at the 9p21–22 region, which encodes for CDKN2/MTS1/p16ink4a, p14ARF, and MTS2/p15ink4b. In addition, microsatellite instability was observed with a frequency of 16% in the 9p21–22 chromosome area. Overexpression (P) of p53 and MDM2 proteins was found in 39 (58%) and 47 (70%) of the cases, respectively. A highly significant association was observed between p53 overexpression and p53 mutations (P = 0.006). Statistical analysis of the expression patterns of the biologically relevant molecules (p16/pRb, p53/MDM2, MDM2/pRb, and p53/pRb) showed coincident overexpression of p53 and MDM2 (P = 0.04) and that abnormal pRb was correlated with elevated levels of MDM2 (P = 0.013) and p53 (P = 0.01), respectively. We suggest that deregulated expression of these molecules may act synergistically. An important finding of the study was that multiple impairments (three and four molecules affected) of the p16/pRb/p53/MDM2 network occurred in a large proportion (43%) of the carcinomas. This finding in addition to the absence of correlation with clinical stage of the tumors suggests that multiple hits of this network may be a relatively early event in the development of a subset of NSCLCs. The relationship between the factors examined in the present study, clinicopathological features, and survival of the patients did not reveal any significant correlations with the exception of smoking, which was associated with microsatellite alterations (loss of heterozygosity and microsatellite instability) at the 9p21–22 locus (P = 0.04) and the immunophenotypes p53(P)/MDM2(P) (P = 0.04) and p16(Ab)/pRb(Ab)/p53(P)/MDM2(P) (P = 0.03), respectively. We suggest that in a subset of NSCLCs, simultaneous deregulation of the members of this network may represent one way of initiating the oncogenic procedure whereas in other NSCLC subgroups alternative pathways may play this role.
Non-small-cell lung cancer (NSCLC) represents one of the most frequent fatal human malignancies. Although tobacco and its derivatives are considered to be leading causes in NSCLC development, their underlying genetic targets are not yet completely clarified. 1 It is not surprising that alterations in the molecular machinery that controls transition from G1 to S phase might represent central events leading to NSCLC generation. In this respect, there is a strong body of evidence that cell-cycle regulators are involved in human cancer development. 2
Two of the most conspicuous molecules that emerge from ongoing studies in this field are the products of the CDKN2/MTS1 gene, which is located on locus 9p21–22. The CDKN2 gene encodes for two proteins: p16 and p14ARF (the human homologue of p19ARF). 3 P16 consists of 156 amino acids (aa) and functions as an inhibitor of cyclin-dependent kinase 4 and 6 (CDK4 and CDK6). 2,4,5 CDK4- and CDK6-mediated phosphorylation of the retinoblastoma protein (pRb) is a critical step in cell cycle progression. The pRb protein (p105RB1) is a 928-aa, 105-kd nuclear phosphoprotein encoded by the RB1 tumor suppressor gene, which has been mapped to chromosomal region 13q14.2 (reviewed in Ref. 6 ). pRb phosphorylation is stimulated by cyclin D1 and inhibited by p16. Therefore, p16 and pRb are suggested to function in a single regulatory pathway of the cell cycle. 2 Unregulated phoshorylation of pRb by CDK4/6 due to either cyclin D1 overexpression or loss of functional p16 could lead to uncontrolled cellular proliferation. 2 The CDKN2/p16ink4a gene product has been found to be nonfunctional in a high percentage of cell lines (75%) and various malignancies, suggesting that it is a candidate tumor suppressor protein. 2,7,8 The most common mechanisms for CDKN2/p16ink4a inactivation appear to be homozygous deletions, mutations, loss of one allele, and inactivation of the other, probably due to hypermethylation. 2,7,8 Several reports have pointed out a difference in the frequency of CDKN2/p16ink4a genetic alterations between cell lines and primary tumors. 8-11 Contamination of the cancerous tissue by the material derived from the surrounding stromal cells and artifactual or cellular adaptation during culture account for this difference. A similar situation has been described for RB1 gene alterations in tumors. 12 Mutational inactivation of the RB1 gene has been detected in a wide spectrum of malignancies, including retinoblastomas, small-cell lung, bladder, pancreatic, and breast carcinomas (reviewed in Ref. 6 ). The problem of evaluating p16 and pRb alterations, avoiding false positive results of the conventional molecular biology methods (polymerase chain reaction (PCR) and Southern blot) due to stromal contamination, can be circumvented by immunohistochemical methods that allow the investigation of protein abnormal expression at the single-cell level.
The data concerning the frequency and mechanisms of CDKN2/p16ink4a gene inactivation in NSCLCs are controversial. 13-26 In reviewing the world literature published so far, we found six reports dealing with aberrant p16 immunoexpression and in situ distribution with percentages ranging from 27% to 67%. 13,20-22,24,25 Although most studies agree with the point that mutations are a rather rare mechanism of CDKN2/p16ink4a inactivation (1% to 10%), 14-19,23,25 there is a great discrepancy regarding CDKN2/p16ink4a gene deletion, with percentages ranging from 0% to 83%. 13,15-17,21-23,25 In addition, to the best of our knowledge, there are only two reports dealing with the methylation status of the CDKN2/p16ink4a gene in NSCLCs. 25,26 On the other hand, inactivation of RB1 gene has been shown in 9 to 38% of NSCLCs. 12,20-22,25-30 Recent, in vitro evidence has shown that RB1 expression is negatively regulated by p53 31 and that MDM2 protein binds to the carboxy terminus of pRb, relieving pRb suppression of E2F transactivating function, thus permitting the cell to enter the S phase. 32 The MDM2 gene is located on chromosome 12q13–14 and can generate various MDM2 protein isoforms. The full-length MDM2 gene product p90 forms an autoregulatory feedback loop with p53 protein that seems to be vital for normal cell proliferation. Overexpression of MDM2 protein(s) has been observed in several human malignancies and associated with unfavorable prognosis (reviewed in Refs. 33 and 34 ). Recently, we reported that MDM2 protein isoforms are overexpressed and coexist with the mutant p53 protein (mt p53) in a subset of lung carcinomas, suggesting that this association may reflect a gain of function phenotype. 35-37
The in vitro evidence mentioned above suggests that the p53-MDM2 duet is linked directly to the p16-pRb pathway (Figure 1) ▶ . Alterations in this network may disrupt normal cell-cycle control. To investigate the involvement of this G1 protein network in non-small-cell lung carcinogenesis we examined, by immunohistochemical and molecular methods, its status in a series of 68 NSCLCs and correlated the findings with clinicopathological features and prognosis of the patients. Furthermore, we examined the state of the chromosomal region 9p21–22 by performing a mapping using a tight cluster of highly polymorphic microsatellite markers because there are indications that, in addition to CDKN2/p16ink4a and MTS2/p15ink4b tumor suppressor genes (TSGs), this region harbors other TSG(s) 38 (Figure 2a) ▶ .
Figure 1.

Schematic representation of the p16/pRb/p53/MDM2 protein network. p16 inhibits cyclin-dependent phosphorylation of pRb. 2 p53 and MDM2 form a feedback loop. 33 MDM2 binds the carboxy terminus of pRb, relieving pRb suppression of E2F transactivation function, thus permitting the cell to enter the S phase. 32 RB1 is negatively regulated by p53. 31 *Recent evidence indicates that the other product of the CDKN2 gene, p14ARF (the human homologue of p19ARF), binds the carboxy terminus of MDM2, regulating its actions. 48,49
Figure 2.

a: Schematic representation of the 9p21–22 region, the polymorphic microsatellite markers used in the study, and the location of the CDKN2 and p15ink4b genes. b: Deletion frequencies observed at the D9S162, IFNA, D9S171, and D9S126 loci.
Materials and Methods
Tissue Samples
Specimens from 68 NSCLCs were obtained in less than 15 minutes after surgery. Two samples of each tumor were taken. One was snap-frozen in liquid nitrogen and stored at −70°C; the other was formalin fixed and paraffin embedded (FFPE). In addition, adjacent normal tissue was included from each specimen examined. The patients had not undergone any chemo- or radiotherapy before surgical resection, thus avoiding up- and down-regulation of p53 and MDM2 proteins, respectively, due to DNA damage. 33 The material comprised 31 squamous cell carcinomas (SCLCs), 32 adenocarcinomas, and 5 undifferentiated large-cell carcinomas. Tumors were classified according to the World Health Organization criteria and TNM system. 39 Thirty carcinomas were stage I (10 T1/N0/M0, 2 T1/N1/M0, and 18 T2/N0/M0), 18 stage II (T2/N1/M0), and 20 stage III (6 T3–4, any N or M, and 14 N2, any T or M). The gender, age, and smoking habits of the patients are presented in Table 1 ▶ .
Table 1.
Summary of Clinicopathological Features, Immunohistochemical Results, and p53 and p16INK4a Gene Analysis
| Clinicopathological features | Immunophenotypes by IHC | |||||||||||
| Sample | Sex | Age (years) | Smoking | Histology | Stage | LN | Patients* follow-up | p16 | pRb | p53 | MDM2 | |
| 19 | M | 70 | YES | AD | I | NO | 22+ | No | No | N | N | |
| 20 | M | 66 | YES | SQ | I | NO | 22+ | No | No | N | N | |
| 22 | M | 70 | YES | AD | I | NO | 22+ | No | No | N | N | |
| 31 | M | 64 | YES | AD | I | NO | 10 | No | No | N | N | |
| 51 | M | 48 | YES | AD | II | YES | 15 | No | No | N | N | |
| 62 | M | 57 | YES | SQ | I | NO | 13 | No | No | N | N | |
| 66 | M | 58 | YES | SQ | II | YES | 3 | No | No | N | N | |
| 30 | M | 75 | YES | UL | I | NO | 20+ | No | No | N | P | |
| 32 | M | 42 | YES | AD | III | YES | 18+ | No | No | N | P | |
| 39 | M | 56 | YES | SQ | III | YES | 9 | No | No | N | P | |
| 40 | M | 54 | YES | UL | I | NO | 18 | No | No | N | P | |
| 43 | M | 60 | NO | AD | II | YES | 18+ | No | No | N | P | |
| 44 | M | 72 | NO | AD | III | YES | 14 | No | No | N | P | |
| 50 | M | 62 | NO | AD | II | YES | 17+ | No | No | N | P | |
| 8 | F | 69 | NO | AD | I | NO | 22+ | No | No | P | N | |
| 13 | M | 64 | YES | SQ | III | YES | 8 | No | No | P | N | |
| 24 | M | 62 | YES | SQ | I | NO | NA | No | No | P | N | |
| 52 | M | 72 | YES | SQ | III | NO | 17+ | No | No | P | N | |
| 67 | M | 71 | NO | AD | I | NO | 10+ | No | No | P | N | |
| 7 | M | 64 | YES | SQ | II | YES | 23+ | No | No | P | P | |
| 10 | F | 68 | YES | UL | II | YES | 9 | No | No | P | P | |
| 16 | M | 63 | YES | AD | I | NO | 7 | No | No | P | P | |
| 18 | M | 73 | YES | SQ | II | YES | 11 | No | No | P | P | |
| 59 | M | 65 | YES | AD | I | NO | 14+ | No | No | P | P | |
| 25 | M | 63 | NO | AD | I | NO | 21+ | No | Ab | N | N | |
| 33 | M | 59 | YES | SQ | I | NO | 14 | No | Ab | N | P | |
| 49 | M | 53 | NO | AD | I | NO | 17+ | No | Ab | N | P | |
| 53 | M | 60 | YES | SQ | III | YES | 15 | No | Ab | P | N | |
| 1 | M | 57 | YES | AD | III | YES | 3 | No | Ab | P | P | |
| 6 | M | 75 | YES | SQ | II | YES | 23+ | No | Ab | P | P | |
| 28 | M | 60 | YES | SQ | I | NO | 21+ | No | Ab | P | P | |
| 48 | M | 73 | YES | SQ | III | NO | 14 | No | Ab | P | P | |
| 55 | M | 52 | NO | AD | II | YES | 10 | No | Ab | P | P | |
| 57 | M | 58 | YES | SQ | III | YES | 8 | No | Ab | P | P | |
| 36 | M | 57 | NO | AD | III | YES | 10 | Ab | No | N | N | |
| 45 | M | 57 | YES | AD | II | YES | 18+ | Ab | No | N | N | |
| 46 | M | 68 | NO | AD | I | NO | 17+ | Ab | No | N | N | |
| 54 | M | 65 | YES | SQ | I | NO | 16+ | Ab | No | N | N | |
| 3 | M | 61 | YES | SQ | I | NO | 25+ | Ab | No | N | P | |
| 21 | M | 70 | YES | SQ | III | YES | 1 | Ab | No | N | P | |
| 37 | M | 71 | NO | AD | II | YES | 12 | Ab | No | N | P | |
| 41 | M | 67 | YES | SQ | I | NO | 18+ | Ab | No | N | P | |
| 38 | M | 53 | YES | SQ | III | YES | 18+ | Ab | No | P | N | |
| 5 | M | 61 | YES | AD | III | YES | 12 | Ab | No | P | P | |
| 11 | M | 70 | YES | SQ | I | NO | 22+ | Ab | No | P | P | |
| 17 | M | 64 | YES | SQ | II | NO | 22+ | Ab | No | P | P | |
| 26 | M | 63 | YES | AD | II | YES | 17 | Ab | No | P | P | |
| 42 | M | 76 | YES | SQ | III | YES | 18+ | Ab | No | P | P | |
| 61 | M | 59 | YES | AD | III | YES | 14+ | Ab | No | P | P | |
| 68 | M | 61 | YES | SQ | II | YES | 10+ | Ab | No | P | P | |
| 23 | M | 79 | YES | AD | II | YES | 18 | Ab | Ab | N | N | |
| 27 | M | 70 | NO | AD | I | YES | 14 | Ab | Ab | N | P | |
| 34 | M | 63 | YES | AD | I | NO | 20+ | Ab | Ab | N | P | |
| 4 | M | 60 | YES | UL | I | NO | 0 | Ab | Ab | P | P | |
| 9 | M | 53 | YES | AD | I | NO | 23+ | Ab | Ab | P | P | |
| 12 | M | 70 | YES | SQ | I | NO | 23+ | Ab | Ab | P | P | |
| 14 | M | 74 | YES | SQ | III | YES | 8 | Ab | Ab | P | P | |
| 15 | M | 69 | YES | SQ | I | NO | 22+ | Ab | Ab | P | P | |
| 29 | M | 61 | YES | UL | III | YES | 0 | Ab | Ab | P | P | |
| 35 | M | 67 | YES | AD | I | YES | 17 | Ab | Ab | P | P | |
| 47 | M | 71 | YES | SQ | I | NO | 17+ | Ab | Ab | P | P | |
| 56 | M | 58 | YES | SQ | II | YES | 11 | Ab | Ab | P | P | |
| 58 | M | 52 | NO | AD | II | YES | 14+ | Ab | Ab | P | P | |
| 60 | M | 64 | YES | AD | III | YES | 14+ | Ab | Ab | P | P | |
| 63 | F | 60 | YES | AD | III | NO | 13+ | Ab | Ab | P | P | |
| 64 | M | 62 | YES | AD | I | NO | 12+ | Ab | Ab | P | P | |
| 65 | M | 56 | YES | SQ | II | YES | 12+ | Ab | Ab | P | P | |
| 2 | M | 80 | YES | SQ | III | YES | 21 | ND | ND | ND | ND |
| Molecular analysis | |||||
|---|---|---|---|---|---|
| p53 gene analysis | p16iNK4a ge ne analysis | ||||
| SSCP analysis | Defective exon | Gene mutation | Amino acid substitution | SSCP analysis | Methylation status |
| − | − | NM | |||
| + | 9 | ND | − | NM | |
| − | − | NM | |||
| − | − | NM | |||
| − | − | NM | |||
| + | 8 | ND | − | NM | |
| − | − | NM | |||
| − | − | NM | |||
| + | 5 | ND | − | NM | |
| − | − | NM | |||
| − | − | NM | |||
| + | 4 | Codon 60, CCA → CCT | P → P | − | NM |
| + | 6 | ND | − | NM | |
| − | − | NM | |||
| + | 7 | Codon 248, CGG → TGG | R → W | − | NM |
| + | 7 | Codon 248, CGG → CTG | R → L | − | NM |
| + | 4 | ND | − | NM | |
| + | 8 | Codon 273, CGT → CAT | R → H | − | NM |
| − | − | NM | |||
| + | 8 | Codon 273, CGT → CTT | R → L | − | NM |
| + | 4 | Codon 93, CTG → ATG | L → M | − | NM |
| − | − | NM | |||
| + | 7 | Codon 230, ACC → AAC | T → N | − | NM |
| + | 5 | Codon 143, GTG → GCC | V → A | − | NM |
| − | − | NM | |||
| − | − | NM | |||
| − | − | NM | |||
| + | 9 | Codon 319, AAG → AAT | K → N | − | NM |
| − | − | NM | |||
| + | 5 | Codon 151, CCC → CAC | P → H | − | NM |
| + | 5 | ND | − | NM | |
| + | 7 | Codon 237, ATG → ATA | M → I | − | NM |
| + | 8 | Codon 275, TGT → TTT | C → F | − | NM |
| + | 5 | Codon 153, CCC → CAC | P → H | − | NM |
| − | − | HM | |||
| − | − | NM | |||
| − | − | HM | |||
| − | − | HM | |||
| − | − | HM | |||
| − | − | HM | |||
| − | − | HM | |||
| + | 4 | Codon 110, CGT → CT | FS | − | HM |
| − | − | NM | |||
| + | 7 | Codon 244, GGC → TGC | G → C | − | NM |
| + | 8 | Codon 280, AGA → ATA | R → I | − | HM |
| − | − | NM | |||
| + | 6 | Codon 196, CGA → CCA | R → P | − | HM |
| + | 5 | Codon 157, GTC → TTC | V → F | − | HM |
| − | − | NM | |||
| − | − | HM | |||
| − | − | NM | |||
| + | 8 | ND | − | HM | |
| − | − | HM | |||
| + | 7 | Codon 249, AGG → AGT | R → S | − | HM |
| + | 7 | Codon 238, TGT → TTT | C → F | − | NM |
| + | 7 | Codon 234, TAC → TGC | Y → C | − | NM |
| + | 7 | Codon 239, AAC → ACC | N → T | − | NM |
| + | 5 | Codon 163, TAC → TGC | Y → C | − | NM |
| + | 4 | Codon 69, GCT → GGT | A → G | − | NM |
| − | − | NM | |||
| − | − | HM | |||
| − | − | HM | |||
| − | − | HM | |||
| − | − | NM | |||
| − | − | NM | |||
| − | − | NM | |||
| − | − | NM | |||
| − | − | HM |
LN, lymph node metastases; IHC, immunohistochemical analysis; AD, adenocarcinoma; SQ, squamous cell carcinoma; UL, undifferentiated large-cell carcinoma; P, positive; N, negative; Ab, aberrant expression; No, normal expression; ND, not done; NA, not available; NM, normal methylation; HM, hypermethylation; +, possible mutation; −, no mutation.
*Numbers indicate months of survival after surgery; + indicates patient is alive.
Immunohistochemistry
Antibodies
For immunohistochemical analysis the following antibodies (Abs) were used: C-20 (IgG rabbit polyclonal; residues 137 to 156 of the carboxy terminus of p16) (Santa Cruz Biotechnology, Santa Cruz, CA), F-12 (IgG2a mouse monoclonal; residues 1 to 167 representing the full length p16) (Santa Cruz Biotechnology), Rb1 (IgG1 mouse monoclonal; residues 375 to 658 of pRb) (Dako, Glostrup, Denmark), DO7 (IgG2b mouse monoclonal; residues 1 to 45 of p53) (Dako), and 1B10 (IgM mouse monoclonal; carboxy terminus of MDM2) (Novocastra Laboratories, New Castle, UK).
Method
Five-micron paraffin sections of the lesions were mounted on poly-l-lysine-coated slides, dewaxed, rehydrated, and incubated for 30 minutes with 0.3% hydrogen peroxide to quench the endogenous peroxidase activity. Unmasking of p16, pRb, p53, and MDM2 proteins was carried out with the heat-mediated antigen retrieval (HMAR) method, as previously described, 35,36 when the monoclonal antibodies (MAbs) F-12, Rb1, DO7, and 1B10 were applied. The immunohistochemical assay for polyclonal (P)Ab C-20 did not include the antigen retrieval step. The sections were incubated with the antibodies at a 1:100 dilution at 4°C overnight. Biotin-conjugated secondary antibody was added at a 1:200 dilution for 1 hour at room temperature (RT). For color development we used 3,3′-diaminobenzidine tetrahydrochloride (DAB) and hematoxylin as counterstain.
Controls
The HeLa and Lo Vo cell lines with well defined p16 and pRb status, respectively, and lung carcinomas specimens from previous studies overexpressing p53 and MDM2 proteins were used as positive controls. 35,36 Mouse IgG1 MAb of unrelated specificity and the IgG fraction of normal rabbit serum were used as negative controls. Furthermore, the specificity of p16 staining with the PAb C-20 was tested by incubating the latter with the control peptide, C-20P, against which it was raised and applying it on the sections. Elimination of staining verified p16 positivity.
Evaluation: p16-pRb
For scoring the p16 and pRb staining patterns we used previously published criteria. 22,40 Cytoplasmic reactivity was disregarded, and only nuclear staining above any cytoplasmic background was considered as evidence of expression of the p16 and pRb proteins. The samples were divided into two categories: 1) p16 or pRb normal (No), when more than 90% of the tumor nuclei were stained and 2) p16 or pRb abnormal (Ab), when there was absence of nuclear staining in a portion of (heterogeneous) or in the entire (homogeneous) tumor section, whereas admixed non-neoplastic cells showed nuclear reactivity. A mosaic pattern of staining with absence of p16 or pRb reactivity in a proportion of tumor cells was not interpreted as abnormal.
Evaluation: p53-MDM2
Tumors were considered p53 or MDM2 positive (P) when more than 20% of tumor cells showed nuclear staining. 36 The other tumors were scored as p53 negative (N) or MDM2 negative (N).
Microdissection and DNA Extraction
DNA was extracted from adjacent 5-μm sections of frozen tumor specimens. Contiguous 5-μm sections were processed, and the first section was stained with hematoxylin and eosin to visualize the extent of the tumor cells within each sample. The boundaries of the cancerous tissue were delineated microscopically, and excess normal tissues were removed with sterile surgical blades, as previously described. 41 The remaining neoplastic material was digested in 500 μl of lysis solution: 50 mmol/L Tris/HCl (pH 8.0), 150 mmol/L NaCl, 5 mmol/L EDTA, containing 1% SDS and proteinase K (Boehringer Mannheim, Mannheim, Germany) at a final concentration of 100 μg/ml. Lysis was carried out at 55°C for 48 hours. Additional proteinase K (50 μg/ml) was added on each day of lysis. DNA was extracted using the phenol/chloroform/isoamyl alcohol method. 42
Microsatellite Alteration (MA) Analysis of the Chromosomal Region 9p21–22
Method
DNA from normal and cancerous tissue was compared by PCR analysis of a tight cluster of highly polymorphic microsatellite markers spanning 11cM across 9p21–22 with the following order from telomeric to centromeric 9p (Figure 2a) ▶ : D9S162 (172 to 196 bp), IFNA (138 to 150 bp), D9S171 (159 to 177 bp), and D9S126 (226 to 250 bp). The 50-μl reaction mixture contained 100 ng of DNA, 10 mmol/L Tris/HCl, pH 8.8, 50 μmol/L KCl, 2.5 mmol/L MgCl2, 0.1% Triton X-100, 200 μmol/L each dNTP (dATP, dCTP, dGTP, and dTTP), 1 μmol/L each primer pair (Research Genetics, Huntsville, AL) and 1.5 U of Taq DNA polymerase (Promega, Madison WI). The thermal cycle profile was denaturation at 95°C for 10 minutes before the addition of Taq polymerase followed by 30 cycles of 40 seconds at 95°C, 35 seconds at 56°C, and 30 seconds at 72°C. The products were analyzed on 10% polyacrylamide gels containing 10% glycerol and visualized by silver staining. 42
Control
To determine whether the PCR product was a microsatellite region or not, electrophoresis on denaturing polyacrylamide gels was used. The bands that correspond to the microsatellite regions, due to the higher existence of dinucleotide repeats, are separated according to their molecular weight. Furthermore, all samples showing changes in microsatellite sequences were verified by subjecting the corresponding DNA sample to a second, independent PCR analysis. This as well as control PCR reactions lacking DNA were done to eliminate the chance of false positives due to PCR artifacts or sample contamination. Finally, for microsatellite instability (MI) determination we used previously published criteria. 43
Methylation Analysis of the CDKN2/p16ink4a Gene
Primers
A 239-bp fragment of exon 1a was amplified using the following primers, employing the Oligo Software v.4.01.: p16AU, 5′-GGAGAGGGGGAGAGCAGGCA-3′, and p16AD, 5′-CTCCAGAGTCGCCCGCCATC-3′.
Method
The inability of some restriction enzymes to cut methylated sequences was used to examine the methylation status of exon 1a. There are two HpaII sites and one KspI site (both restriction enzymes are methylation sensitive) within exon 1a. One microgram of genomic DNA from matched normal and tumor samples was digested overnight with HpaII and KspI as previously described. 44 Five microliters of the digested DNA solutions was used as template in a 50-μl reaction mixture containing 10 μmol/L Tris/HCl, pH 8.8, 50 μmol/L KCl, 1.5 mmol/L MgCl2, 0.1% Triton X-100, 5% dimethylsulfoxide, 5% glycerol, 200 μmol/L each dNTP (dATP, dCTP, dGTP, and dTTP), 1 μmol/L each primer, and 1.5 U of Taq DNA polymerase (Promega). The thermal cycle profile was denaturation at 95°C for 10 minutes before the addition of Taq polymerase followed by 30 cycles of 1 minute at 95°C, 45 seconds at 57°C, and 45 seconds at 72°C. PCR products were electrophoresed in 2% agarose gels stained with ethidium bromide.
Evaluation–Controls
When exon 1a of the CDKN2 gene is methylated, then the methylation-sensitive restriction enzyme fails to cut and a PCR product is obtained. For each specimen analyzed, undigested and non-methyl-sensitive MspI-digested samples served as positive and negative controls, respectively, as previously described. 44 Finally, we evaluated as methylation-positive samples only the cases with reproducible results.
Single-Strand Conformation Polymorphism Analysis of CDKN2/p16ink4a Gene
Primers
The primers used for methylation analysis of exon 1a and the following primers, designed by using the Oligo Software v.4.01., for exon 2 of CDKN2/P16ink4a (419-bp PCR-amplified fragment) were used for mutation analysis: p16BU, 5′-GGCTCTGACCATTCTGTTCTCTC-3′, and p16BD, 5′-GGCTGAACTTTCTGTGCTGG-3′.
Method
Single-strand conformation polymorphism (SSCP) was performed on normal and tumor-tissue-derived DNA as previously described. 45
Nested-PCR/SSCP Analysis of the p53 Gene
The method was performed on matched normal and tumor DNA as previously described. 45 Briefly, a 2.9-kb p53 gene fragment, which contains exons 4 to 9, was amplified, 36,37 as previous studies have shown that the majority of p53 mutations, in lung carcinomas, are found in this region. Individual exons 4–9 were further amplified by PCR, and nested products were analyzed with SSCP. Exons that showed mobility shifts were further analyzed with automated sequencing.
Automated Sequencing of the p53 Gene
PCR Amplification
Exons that harbored putative mutations were amplified with primers previously described 37 that were tailed with M13 forward and reverse primers. Reactions were performed on a PEC 480 thermal cycler block in 100-μl reactions (10 mmol/L Tris/HCl, pH 9.0, 50 mmol/L KCl, 2.5 mmol/L MgCl2, 0.1% Triton X-100, 200 μmol/L dNTPs, 1 μmol/L each primer, 2.5 U of Taq (Promega), and 100 ng of genomic DNA) for 30 cycles of 1 minute at 94°C, 40 seconds at 56°C (for exons 4 and 7; 50°C for exons 5, 6, and 8; and 44°C for exon 9), and 40 seconds at 72°C.
Purification of PCR Product
The PCR products were purified with QIAquick Gel Extraction kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions and then quantitated by agarose gel electrophoresis after comparison with known amounts of standard DNA fragments.
Automated Sequencing
Cycle sequencing was performed on GeneAmp PCR System 9600 (Perkin-Elmer, Norwalk, CT) using fluorescently labeled M13 forward and reverse primers with AmpliTaq DNA polymerase, FS (Taq-FS, Perkin-Elmer/Applied Biosystems Division, Foster City, CA) according to the manufacturer’s directions. Briefly, purified DNA (0.1 to 1 mg) was mixed with 2 μl of 10X FS buffer (125 mmol/L Tris/HCl, pH 9.5, 50 mmol/L (NH4)2SO4, 150 mmol/L MgCl2), 2 pmol of Cy-5-labeled primer, and 1 μl of FS enzyme in 20-μl reactions. Five-microliter aliquots of the above were mixed with 2 μl of each termination solution (a mixture of deoxy- and dideoxy-ribonucleotides provided by Perkin-Elmer). Sequencing reactions were run under the following conditions: 2 minutes of initial denaturation at 95°C followed by 25 cycles at 95°C for 30 seconds and 50°C for 30 seconds. At the end, 4 μl of formamide stop solution was added, and samples were heated at 95°C for 2 minutes and loaded on ALF Express sequencing gels (Pharmacia, Uppsala, Sweden). Gels were run on an automated DNA sequencer (ALF Express, Pharmacia).
Statistical Analysis
Statistical analysis was based on χ 2 with Yates’ correction. An additional two-tailed Fisher’s exact test was used only when the number of samples in any cell of a given statistical table was five or fewer. Multiple regression analysis was performed to examine the possible relationship between the biologically relevant and combined immunoprofiles and clinicopathological parameters. The computations were carried out using the Statistica software package. The survival curves were calculated by the Kaplan-Meier method using the SPSS software package. The statistical difference was considered significant if the P value was <0.05.
Results
Immunohistochemistry
Immunohistochemical Expression of p16, pRb, p53, and MDM2 Proteins and Relationship with Various Clinicopathological Parameters
Immunohistochemical (IHC) analysis revealed aberrant expression of p16 and pRb in 33 (49%) and 27 (40%) of the 67 carcinomas, respectively (Figures 3 and 4) ▶ ▶ . There was concordance of p16 staining with both antibodies used. Reactivity for p53 and MDM2 was observed in 39 (58%) and 47 (70%) of the tumors, respectively (Figure 5) ▶ . P16, pRb, p53, and MDM2 results and their relationship with smoking habits, histology, lymph node status, and stage are presented in Table 2 ▶ . As shown in Table 2 ▶ , the percentages of p16, pRb, and MDM2 expression are comparable between SCLCs and adenocarcinomas, whereas p53 staining in adenocarcinomas is lower (47%) as compared with SCLCs (70%). Statistical analysis did not reveal any significant correlations with the exception of a probable association between p53 overexpression and smoking (35/54 (65%) versus 4/13 (30%); P = 0.054).
Figure 3.

Squamous cell lung carcinoma with heterogeneous aberrant p16 expression (see Materials and Methods). Streptavidin-biotin peroxidase technique with F-12 anti-p16 antibody (see Materials and Methods) and hematoxylin counterstain; magnification, ×500.
Figure 4.
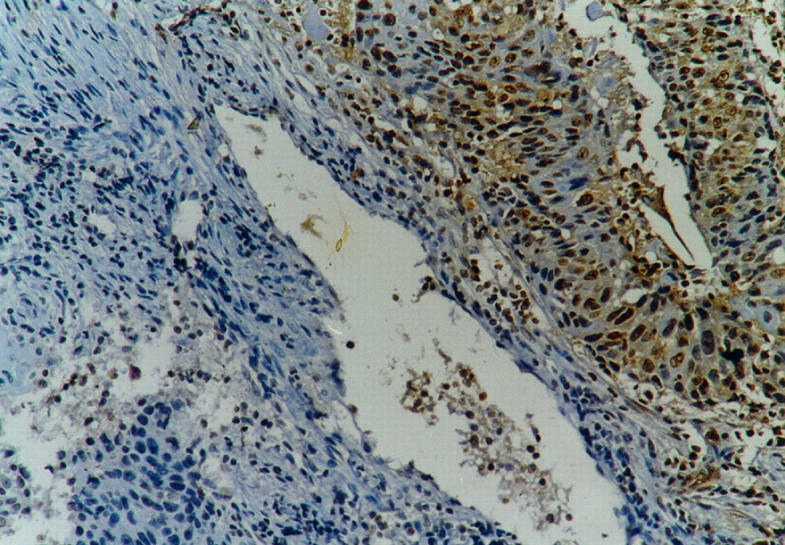
Squamous cell lung carcinoma with heterogeneous aberrant pRb expression (see Materials and Methods). Streptavidin-biotin peroxidase technique with Rb1 anti-pRb antibody (see Materials and Methods) and hematoxylin counterstain; magnification, ×125.
Figure 5.

Squamous cell lung carcinoma with MDM2 protein overexpression. Streptavidin-biotin peroxidase technique with 1B10 anti-MDM2 antibody (see Materials and Methods) and hematoxylin counterstain; magnification, ×500.
Table 2.
Relationship of the Immunohistochemical Results with Various Clincopathological Parameters
| IHC | Clinicopathological data | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Smoking | Histology | LN | Stage | |||||||||||
| Yes | No | P | SQ | AD | UL | P | Yes | No | P | I | II | III | P | |
| p16 | ||||||||||||||
| No | 26 | 8 | 0.57 | 14 | 17 | 3 | 0.8011 | 16 | 18 | 0.537 | 16 | 9 | 9 | 0.9 |
| Ab | 28 | 5 | 16 | 15 | 2 | 19 | 14 | 14 | 9 | 10 | ||||
| Total | 54 | 13 | 30 | 32 | 5 | 35 | 32 | 30 | 18 | 19 | ||||
| pRb | ||||||||||||||
| No | 32 | 8 | 0.86 | 18 | 19 | 3 | 0.9986 | 21 | 19 | 0.843 | 17 | 12 | 11 | 0.77 |
| Ab | 22 | 5 | 12 | 13 | 2 | 14 | 13 | 13 | 6 | 8 | ||||
| Total | 54 | 13 | 30 | 32 | 5 | 35 | 32 | 30 | 18 | 19 | ||||
| p53 | ||||||||||||||
| N | 19 | 9 | 0.054 | 9 | 17 | 2 | 0.1817 | 13 | 15 | 0.57 | 16 | 7 | 5 | 0.2 |
| P | 35 | 4 | 21 | 15 | 3 | 22 | 17 | 14 | 11 | 14 | ||||
| Total | 54 | 13 | 30 | 32 | 5 | 35 | 32 | 30 | 18 | 19 | ||||
| MDM2 | ||||||||||||||
| N | 15 | 5 | 0.67 | 9 | 11 | 0 | 0.2951 | 8 | 12 | 0.297 | 11 | 4 | 5 | 0.5 |
| P | 39 | 8 | 21 | 21 | 5 | 27 | 20 | 19 | 14 | 14 | ||||
| Total | 54 | 13 | 30 | 32 | 5 | 35 | 32 | 30 | 18 | 19 |
Ab, abnormal expression; No, normal expression; SQ, squamous cell carcinoma; AD, adenocarcinoma; UL, undifferentiated large-cell carcinoma; P, positive; N, negative.
Relationship of the Expression of the Biologically Relevant Molecules (p16/pRb, p53/MDM2, MDM2/pRb, and p53/pRb)
The relationship of the biologically relevant molecules examined in the present work is illustrated in Figure ▶ 1. Statistical analysis of the immunostaining patterns showed that 1) MDM2 overexpression was accompanied by p53 overexpression (MDM2(P)/p53(P) 32/39 (82%) versus MDM2 (P)/p53(N)15/28 (54%); P = 0.02) and 2) abnormal detection of pRb was significantly associated with increased production of MDM2 and p53 proteins, respectively (pRb(Ab)/MDM2(P) 24/47 (51%) versus pRb(Ab)/MDM2(N) 3/20 (15%); P = 0.013; and pRb(Ab)/p53(P) 21/39 (54%) versus pRb(Ab)/p53(N) 6/28 (21%); P = 0.01). Although p16-pRb expression patterns did not reveal any significant correlations, an inverse relationship between p16 and pRb was observed in 26 (39%) of the 67 carcinomas (p16(Ab)/pRb(No), 10/67 (15%), or p16(No)/pRb(Ab), 16/67 (24%)). Furthermore, coincident aberrant expression of p16 and pRb was observed in 17 cases (25%).
Relationship of the Biologically Relevant Immunoprofiles with Various Clinicopathological Parameters
The relationship of the biologically relevant immunophenotypes, p16/pRb, p53/MDM2, MDM2/pRb, and p53/pRb with smoking habits, histology, lymph node status, and stage are summarized in Table 3 ▶ . Statistical analysis showed a probably significant association between the p53(P)/MDM2(P) pattern and smoking (30/54 (56%) versus 2/13 (15%); P = 0.04, Table 3 ▶ ). Next we examined the association between p16/pRb/p53/MDM2 profile and clinicopathological characteristics of the carcinomas. The tumors were divided into four groups according to the number of alterations detected (Table 4) ▶ .
Table 3.
Relationship of the Biologically Relevant Immunoprofiles with Various Clinicopathological Parameters
| IHC | Clinicopathological data | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Smoking | Histology | LN | Stage | |||||||||||
| Yes | No | P | SQ | AD | UL | P | Yes | No | P | I | II | III | P | |
| p16/pRb | ||||||||||||||
| No/No | 19 | 5 | 0.7 | 9 | 12 | 3 | 0.5321 | 11 | 13 | 0.77 | 12 | 7 | 5 | 0.8 |
| No/Ab | 7 | 3 | 6 | 4 | 0 | 5 | 5 | 4 | 2 | 4 | ||||
| Ab/No | 13 | 3 | 9 | 7 | 0 | 10 | 6 | 5 | 5 | 6 | ||||
| Ab/Ab | 15 | 2 | 6 | 9 | 2 | 9 | 8 | 9 | 4 | 4 | ||||
| Total | 54 | 13 | 30 | 32 | 5 | 35 | 32 | 30 | 18 | 19 | ||||
| p53/MDM2 | ||||||||||||||
| N/N | 10 | 3 | 0.04 | 4 | 8 | 0 | 0.4863 | 5 | 8 | 0.59 | 8 | 4 | 1 | 0.1 |
| P/N | 5 | 2 | 5 | 2 | 1 | 3 | 4 | 3 | 0 | 4 | ||||
| N/P | 9 | 6 | 5 | 9 | 1 | 8 | 7 | 8 | 3 | 4 | ||||
| P/P | 30 | 2 | 16 | 13 | 3 | 19 | 13 | 11 | 11 | 10 | ||||
| Total | 54 | 13 | 30 | 32 | 5 | 35 | 32 | 30 | 18 | 19 | ||||
| pRb/MDM2 | ||||||||||||||
| No/N | 12 | 4 | 0.9 | 7 | 9 | 0 | 0.8247 | 5 | 11 | 0.15 | 10 | 3 | 3 | 0.4 |
| No/P | 19 | 4 | 10 | 10 | 3 | 15 | 8 | 7 | 9 | 7 | ||||
| Ab/N | 3 | 1 | 2 | 2 | 0 | 3 | 1 | 1 | 1 | 2 | ||||
| Ab/P | 20 | 4 | 11 | 11 | 2 | 12 | 12 | 12 | 5 | 7 | ||||
| Total | 54 | 13 | 30 | 32 | 5 | 35 | 32 | 30 | 18 | 19 | ||||
| p53/pRb | ||||||||||||||
| N/No | 16 | 6 | 0.871 | 8 | 12 | 2 | 0.4952 | 11 | 11 | 0.757 | 11 | 6 | 5 | 0.395 |
| P/No | 16 | 2 | 10 | 7 | 1 | 10 | 8 | 6 | 6 | 6 | ||||
| N/Ab | 3 | 3 | 1 | 5 | 0 | 2 | 4 | 5 | 1 | 0 | ||||
| P/Ab | 19 | 2 | 11 | 8 | 2 | 12 | 9 | 8 | 5 | 8 | ||||
| Total | 54 | 13 | 30 | 32 | 5 | 35 | 32 | 30 | 18 | 19 |
Ab, abnormal expression; No, normal expression; SQ, squamous cell carcinoma; AD, adenocarcinoma; UL, undifferentiated large-cell carcinoma; P, positive; N, negative.
Table 4.
Relationship of the Combined Immunoprofile with Various Clinicopathological Parameters
| p16/pRb/p53/MDM2 pattern | Clinicopathological data | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Smoking | Histology | LN | Stage | |||||||||||
| Yes | No | P | SQ | AD | UL | P | Yes | No | P | I | II | III | P | |
| Alterations (0) | 6 | 0 | 3 | 3 | 0 | 2 | 4 | 4 | 2 | 0 | ||||
| Alterations (1+) | 10 | 8 | 5 | 11 | 2 | 8 | 10 | 9 | 3 | 6 | ||||
| Alterations (2+) | 12 | 2 | 0.03 | 8 | 5 | 1 | 0.57 | 8 | 6 | 0.6 | 6 | 5 | 3 | 0.5 |
| Alterations (3+) | 13 | 2 | 8 | 7 | 0 | 10 | 5 | 4 | 5 | 6 | ||||
| Alterations (4+) | 13 | 1 | 6 | 6 | 2 | 7 | 7 | 7 | 3 | 5 | ||||
| Total | 54 | 13 | 30 | 32 | 5 | 35 | 32 | 30 | 18 | 19 |
SQ, squamous cell carcinoma; AD, adenocarcinoma; UL, undifferentiated large-cell carcinoma.
As shown, alterations in three and four components of the network (Figure 1) ▶ were observed in 15 (22%) and 14 (21%) of the cases, respectively. A probably significant correlation was found between the p16(Ab)/pRb(Ab)/p53(P)/MDM2(P) immunoprofile and smoking (13/54 (24%) versus 1/13 (8%); P = 0.03, Table 4 ▶ ).
Microsatellite Alteration Analysis of the Chromosomal Region 9p21–22
For microsatellite alteration (MA) analysis we used a tight cluster of highly polymorphic microsatellite markers spanning 11cM across 9p21–22 with the following order from telomeric 9p to centromeric 9p (Figure 2a) ▶ : D9S162, IFNA, D9S171, and D9S126.
Of 68 cases, 50 (74%) were informative for D9S162, 42 (62%) for IFNA, 43 (63%) for D9S171, and 29 (43%) for D9S126. Thirty-five (51%) NSCLCs displayed loss of heterozygosity (LOH) at one or more markers (Figures 6 and 7) ▶ ▶ . The deletion frequencies for each marker are presented in Figure 2b ▶ . The results were further categorized into three groups: group LOH(c), the cases that demonstrated a central LOH, at the IFNA and/or D9S171 loci indicating a possible deletion of the CDKN2 gene; group LOH(f), the patients with chromosomal abnormalities at the flanks upstream of IFNA and/or downstream of D9S171; and group LOH(−), the cases with no indication of LOH and/or noninformative (NI) samples. Of the 68 patients, 27 (40%) were placed in group LOH(c), 9 (13%) in group LOH(f), and 32 (47%) in group LOH(−) (Figure 8) ▶ .
Figure 6.

Representative results of deletion mapping and microsatellite instability analysis (MI) with chromosome markers IFNA and D9S171. a: Lane m , marker (PUC19/Sau3AI); lanes 1, 2, 7, and 8, matched normal-tumor samples (cases 28 and 31) with no polymorphism at the IFNA locus; lanes 3 and 4, matched normal-tumor samples (case 49) with polymorphism at the IFNA locus (a1 and a2 alleles; Hd, heteroduplex but no indication of loss of heterozygosity (LOH); lanes 5 and 6, matched normal-tumor samples (case 61) with polymorphism at the IFNA locus and LOH. ax: Magnification of lanes 5 and 6; LOH is shown with arrowhead. b: Lane m, marker (PUC19/Sau3AI); lanes 3 to 6, matched normal-tumor samples (cases 49 and 54) with polymorphism at the D9S171 locus but no indication of LOH; lanes 1 and 2, matched normal-tumor samples (case 15) with polymorphism at the D9S171 locus and LOH. bx: Magnification of lanes 1 and 2; LOH is shown with arrowhead. c: Lane m, marker (PUC19/Sau3AI); lanes 1 and 2, matched normal-tumor samples (case 3). The tumor sample showed at the IFNA locus MI, which is demonstrated as an aberrant band (a1′). The band at the position of the a1 allele is possibly due to DNA contamination from the non-neoplastic surrounding cells.
Figure 7.

Representative results of deletion mapping with chromosome markers D9S162 and D9S126. A: Lane M, marker (φχ174/HaeIII); lanes 1 and 2, matched normal-tumor samples (case 11) with polymorphism at the D9S162 locus and LOH (arrowhead). a1 and a2, alleles; Hd, heteroduplex). B: Lanes 1 and 2, with polymorphism at the D9S126 locus but no indication of LOH; lanes 3 and 4, matched normal-tumor samples (case 12) with polymorphism at the D9S126 locus and LOH (arrowhead). a1 and a2, alleles; Hd, heteroduplex.
Figure 8.

Relationship between p16 immunohistochemistry, microsatellite alterations at the 9p21–22 chromosome region and methylation of the CDKN2/p16ink4a gene.
Apart from chromosomal deletions, 11 (16%) patients demonstrated MAs, which appeared as an expansion or a compression of a single band or a ladder of bands (Figure 6) ▶ . These alterations were characterized as microsatellite instability (MI), as previously described. 43,47 MI was observed in all of the microsatellite loci examined, and it was more frequent in SCLCs (8/30, 27%) than in adenocarcinomas (3/32, 9%). In two of these cases, MI co-existed with LOH. Thus, in total, MAs (LOH plus MI) at the 9p21–22 chromosomal region were detected in 44 (65%) cases.
In relation to clinicopathological parameters we observed a probably significant association between smoking and MAs at the examined region (MAs/smoking(+), 36/55 (65%), versus MAs/smoking(−), 4/13 (31%); P = 0.04).
Interestingly, LOH and MAs were more frequent in the cases with abnormal pRb expression (LOH/pRb(Ab), 19/27 (70%), versus LOH/pRb(No), 16/40 (40%); P = 0.02, and MAs/pRb(Ab), 20/27 (74%), versus MAs/pRb(No), 19/40 (48%); P = 0.05). Finally, we examined the relationship between LOH(c) and MDM2 protein status because there is current evidence that demonstrates that p14ARF is involved in the stabilization of MDM2. 48,49 However, no significant association was observed (LOH(c)/MDM2(P), 21/48 (44%), versus LOH(c)/MDM2(N), 5/19 (26%); P = 0.29).
CDKN2/p16ink4a Gene Methylation Analysis
To address further the molecular basis of aberrant p16 expression we examined the methylation status of CDKN2/p16ink4a exon 1a, which has been shown to correlate strongly with transcriptional silencing of the gene. 50 Briefly, after DNA digestion with methylation-sensitive and -insensitive restriction enzymes, PCR amplification was performed. When exon 1a is methylated, a PCR product is obtained. Using this procedure, we found that 18 (26%) of 68 carcinomas analyzed exhibited de novo methylation of exon 1a (Figure 9) ▶ . Methylation patterns did not vary between HpaII and KspI. Furthermore, all normal tissues were unmethylated within the examined regions.
Figure 9.

Representative results of the PCR-based methylation assay. a: De novo methylation of CDKN2/p16INK4a exon 1a in a undifferentiated NSCLC (case 4). Lane m, marker (PUC19/Sau3AI); lane 1, undigested PCR product from HeLa cell line included as control; lanes 2 and 3, undigested PCR products from matched normal-tumor sample; lanes 4 and 5, digestion with methylation-sensitive enzyme HpaII revealed that CDKN2/p16ink4a in nontumor tissue is unmethylated as no amplification by PCR is seen, whereas in the tumor, CDKN2/p16ink4a is methylated as digestion does not affect amplification; lanes 6 and 7, digestion with non-methylation-sensitive enzyme MspI affects amplification in normal and tumor tissue. b: De novo methylation of CDKN2/p16ink4a exon 1a in a squamous cell lung carcinoma (case 11). Lane m, marker (PUC19/Sau3AI); lanes 1 and 2, undigested PCR products from matched normal-tumor sample; lanes 3 and 4, digestion with methylation-sensitive enzyme KspI revealed that CDKN2/p16ink4a in nontumor tissue is unmethylated as no amplification by PCR is seen, whereas in the tumor, CDKN2/p16ink4a is methylated as digestion does not affect amplification; lanes 5 and 6, digestion with non-methylation-sensitive enzyme MspI affects amplification in normal and tumor tissue. c: An adenocarcinoma with no indication of CDKN2/p16ink4a methylation (case 25). Lane m, marker (PUC19/Sau3AI); lane 1, undigested PCR product from HeLa cell line; lanes 2 and 3, undigested PCR products from the sample; lanes 4 and 5, digestion with methylation-sensitive enzyme KspI; lanes 6 and 7, digestion with non-methylation-sensitive enzyme MspI.
SSCP Analysis of the p53 and CDKN2/p16ink4a Genes
To assess point mutations and short nucleotide sequence insertions or deletions of the p53 gene, we analyzed with SSCP exons 4 to 9, obtained by nested PCR, from tumor-derived and normal-tissue-derived DNA. The same procedure was carried out with exons 1a and 2 of the CDKN2/p16ink4a gene.
Tumor-specific mobility shifts were observed in 24 (61.5%) and 7 (25%) of the p53 immunohistochemically positive and negative cases, respectively. A highly significant association was observed between p53 gene mutations and p53-positive staining (p53 mt(+)/p53 IHC(+), 24/39 (61.5%), versus p53 mt(+)/p53 IHC(−), 7/28 (25%); P = 0.006). The data from the SSCP and sequencing analysis (Figure 10) ▶ are presented in detail in Table 1 ▶ .
Figure 10.

Representative automated sequencing in a squamous cell lung carcinoma (case 18) with SSCP mobility shift in exon 7. The position of the p53 mutation is located at codon 230 and changes ACC (T) to AAC (N).
No tumor-specific migrational shifts were observed in any of the NSCLCs by PCR-SSCP analysis of exons 1a and 2 of CDKN2/p16ink4a.
Relationship between p16 Immunohistochemical Findings, Microsatellite Alterations at the 9p21–22 Locus, and Methylation Status of the CDKN2 Gene (Figure 8) ▶
The cases with abnormal p16 (Ab) expression were placed in group A and the cases with normal p16 staining (No) in group B. Group A was further divided into four subgroups according to the LOH(c) and methylation analysis results and group B into three subgroups according to the LOH status.
Statistical analysis revealed a strong correlation between aberrant expression of p16 protein, LOH, and MAs at the 9p21–22 locus, respectively (LOH/p16(Ab), 24/33 (73%), versus LOH/p16(No), 11/34 (32%); P = 0.002, and MAs/p16(Ab), 25/33 (76%), versus MAs/p16(No), 14/34 (41%); P = 0.008. This association was even higher in the patients of group LOH(c) (LOH(c)/p16(Ab), 21/33 (64%), versus LOH(c)/p16(No), 5/34 (15%); P = 0.0001). All of the cases with de novo CDKN2/p16ink4a methylation were accompanied with abnormal p16 staining (subgroups A2 and A3). In six methylated samples of subgroup A3 (cases 4, 27, 37, 42, 46, and 58) we did not notice microsatellite alterations in the 9p21–22 chromosomal region whereas in subgroup A2 de novo methylation of CDKN2/p16ink4a was accompanied by LOH(c). Interestingly, the samples that comprised subgroup A4 did not show either LOH(c) or SSCP tumor-specific shifts or abnormal methylation status of CDKN2/p16ink4a.
Survival Analysis
The survival analysis (follow-up duration up to 25 months) using the Kaplan-Meier method did not demonstrate any statistically significant association between patient outcome, individual abnormalities of the network proteins (including p53 mutations), various immunoprofiles, and alterations at the 9p21–22 chromosome region, respectively.
Discussion
To the best of our knowledge, the information of our study, which deals with alterations of the p16/pRb/p53/MDM2 protein network (Figure 1) ▶ in NSCLCs and examines its relationship with the patients’ outcome, has not been reported so far. Furthermore, we investigated possible abnormalities of the chromosomal region 9p21–22, which harbors at least three candidate tumor suppressor genes, CDKN2, p15ink4b, and methylthioadenosine phosphorylase (MTAP), and correlated the findings with p16 protein expression. 2,51,52
The p16-pRb pathway was investigated by IHC, which allows evaluation of the protein expression at a single-cell level. 12,22,40,53 In our approach (see Materials and Methods) a mosaic pattern of staining was not interpreted as abnormal as even in p16- or pRb-positive cell lines a subset of nuclei remained unstained due to cell cycle fluctuations. 54,55 In our study, solely cytoplasmic reactivity of p16 was disregarded. Its significance is unclear to date and has been observed by others. 53,56,57 If cytoplasmic reactivity proves to be specific, one possible explanation is that alteration in subcellular localization can represent a mechanism of p16 inactivation. Wild-type p53 protein has an analogous mechanism of loss of function. 58
Aberrant p16 was observed in 49% of the carcinomas. This is consistent with certain studies reporting abnormal p16 protein staining in 47% to 51% of NSCLCs, 20,22,25 although other studies reported percentages ranging from 27% to 67%. 13,21,24 This discrepancy may be due to technical parameters of the assays and/or criteria of positivity used by the authors.
To investigate the mechanisms that underlie CDKN2/p16ink4a inactivation we examined the chromosomal region 9p21–22 by microsatellite analysis (Figure 2a) ▶ and the methylation and structural status of the CDKN2/p16ink4a gene by a PCR-based methylation assay and PCR-SSCP, respectively, and then correlated the findings with the results of IHC. A highly significant association was observed between LOH(c) and abnormal p16 protein staining (P = 0.0001), suggesting that deletions may represent an important mode of CDKN2/p16ink4a gene inactivation. Several groups have investigated CDKN2/p16ink4a gene deletions with controversial results. 13,15-17,21,23,25 These differences may be due to contamination of the homogenates by non-neoplastic cells and aneuploidy of tumor cells. By using a microdissection technique we ameliorated the approach, but we did not completely solve the problem. Furthermore, we observed that 52% of the NSCLCs with p16 loss by IHC were methylated at the 5′CpG island of the first exon of the CDKN2/p16ink4a gene (subgroups A2 and A3, Figure 8 ▶ ). This is in agreement with previous results showing a correlation between p16 staining and exon 1 methylation status in primary NSCLCs and NSCLC cell lines. 25,26 PCR-SSCP analysis of exons 1 and 2 in our study did not reveal any minor structural alterations, providing support to previous evidence that intragenic CDKN2/p16ink4a alterations are infrequent events in NSCLCs. 15-20,23,25 Thus, overall, 30 of 33 cases (91%) with abnormal p16 protein staining had LOH(c) and/or abnormal methylation status of the CDKN2/p16ink4a gene (Figure 8) ▶ . Therefore, taking together our data and those so far reported it appears that deletions and transcriptional silencing by methylation might be the predominant mechanisms that inactivate CDKN2/p16ink4a gene in NSCLCs. The three cases of subgroup A4 with loss of p16 protein expression do not show obvious genetic and epigenetic alterations (Figure 8) ▶ . One possible explanation is that these microsatellite markers were not sufficient to detect small deletions in the CDKN2/p16ink4a region (case 17). Alternatively, the presence of normal contaminating DNA may contribute to a retention pattern in cases with homozygous deletion (cases 17, 29, and 45). A third possibility that could account for loss of protein expression is the presence of mutations outside the examined region; such is the case of the mutation being in the second intron splice donor site, which results in a smaller p16 protein with a reduced half-life, possibly undetectable by IHC. 59 The retention of p16 protein expression in the cases of subgroup B3 suggests either alterations of other TSGs located in the 9p21–22 region and/or a compensatory mechanism by the remaining CDKN2/p16ink4a allele (cases 30 and 59).
Deletion mapping of the 9p21–22 region in NSCLCs has been performed by several groups. The frequency of genetic alterations detected ranged from 48% to 68%. These percentages are higher than those reported for CDKN2/p16ink4a gene deletions. 13,15-17,21,23,25 Similarly, in the present study LOH was observed in 51% of the samples, which is consistent with the findings presented by other groups 60-63 and, moreover, higher than the frequency of LOH(c) (38%). These results suggest that another TSG(s) might reside in 9p21–22 besides CDKN2, p15ink4b, and methylthioadenosine phosphorylase (MTAP), which are placed between D9S171 and IFNA loci. This hypothesis is further supported by our deletion-mapping analysis, which revealed LOH at the loci D9S126 and D9S162 in 41% and 32% of the informative cases, respectively. Both loci lie 1 cM and 6 cM far from the aforementioned TSGs, respectively (Figure 2a) ▶ and recently have been proposed to lie near the location of putative TSGs. 38,64,65 The talin gene, also, has recently been mapped in this region. 66 Talin is a critical molecule for the formation of focal adhesions, and its inactivation disassembles many of these structures. 67 The talin gene, therefore, is a candidate for a role in NSCLC carcinogenesis. An interesting finding of our study was that 16% of the carcinomas showed MI. This was found more frequently in SCLCs (27%) than in adenocarcinomas (9%) and co-existed in two cases with LOH (Figure 8) ▶ . MI provides a marker for replication error phenotype (RER+), a recently defined manifestation of genetic instability observed in a wide range of tumors. 68 Cellular populations with RER+ phenotype exhibit elevated mutational rates, which may lead to oncogene and/or oncosuppressor gene deregulation and therefore contribute to tumor development. The data concerning MI in NSCLCs are conflicting with respect to the frequency and the pattern. 43,69,70 Considering the frequency, our percentage is closer to that of Fong et al, 43 who found MI in 6.5% of the cases examined. In that study MI was associated with extensive, concurrent molecular changes in K-ras and p53. In our study 55% of the cases with MI had p53 mutations. In two other reports MI occurred in 2% 69 and 34% 70 of NSCLCs. As far as the pattern is concerned, we observed that in most cases affected, MI was restricted to one marker each time. This is in agreement with some studies 43,69 but differs from another study where MI affected multiple markers concurrently. 70 Therefore, more studies are needed to clarify whether MI in NSCLCs merely reflects extensive genetic damage or plays a more important role similar to that observed in hereditary nonpolyposis colorectal cancer (HNPCC). 71
The incidence of aberrant pRb expression in our analysis was 40% and is the highest reported so far. 12,20-22,25,27-30 Serrano et al has proposed a negative feedback model, showing that inactivation of pRb during G1 leads to increased p16 expression to limit CDK4 activity. 4 Moreover, several studies in various tumors, including NSCLCs, have demonstrated a reciprocal relationship between pRb and p16. 16,20,21,25,72,73 However, in this cohort we noticed an inverse distribution of p16 and pRb proteins in 26 (39%) cases whereas 24 (36%) of the carcinomas showed coincident aberrant expression of both proteins. Simultaneous abnormal p16 and pRb expression has also been reported by Hangaishi et al in primary lymphoid malignancies. 74 As several lines of evidence suggest that functional pRb is essential for cell-cycle inhibition by p16, 75 inactivation of p16 in a cell without functional pRb is not likely to confer additional growth advantage to the cancerous cell. In carcinomas with double hits one possible explanation is that p16 inactivation is an early event and precedes Rb mutation. In the latter case neutralization of pRb could contribute to advantageous tumor growth by canceling all of the inhibitory effects of the other cyclin-dependent kinase inhibitors (CDKIs) via pRb. 76 The report of Kishimoto et al, who detected specific allelic loss in IFNA and D9S171 loci in preneoplastic lesions accompanying NSCLCs, enforces the assumption for an early CDKN2/p16ink4a deletion in NSCLC carcinogenesis. 77 In this respect, our findings that p16 inactivation was noticed in almost one-half of the carcinomas and its frequency was higher than loss of pRb, besides the lack of association with tumor stage, suggest an early involvement of CDKN2/p16ink4a alterations in NSCLC development. This can be supported by several studies reporting no correlation between loss of p16 and clinical stage in primary NSCLCs. 21 Early implication of p16 has been also suggested in head and neck squamous carcinomas (HNSC), which share many etiological and morphological features with NSCLCs. 78 In contrast to this hypothesis are the results of Okamoto et al, who detected CDKN2/p16ink4a mutations only in metastatic and not primary NSCLCs. 14 Alternatively, p16 may participate in other yet unknown pRb-independent pathways. This can be supported by the findings that p16 inactivation causes activation of cyclins D2 and D3, which may act on other substrates besides pRb. 76
In this cohort of NSCLCs we found co-expression of p53 and MDM2 proteins in ∼48% of the cases, thereby confirming our previous findings. 35,36 A highly significant association was also observed between p53 mutations and p53 IHC (P = 0.006), verifying previous reports that show that p53-positive staining in the carcinomas of the upper and lower respiratory tract is strongly indicative of p53 mutations. 46 Simultaneous expression of p53 and MDM2 has been increasingly reported in a wide variety of tumors. 33,34 Co-detection of MDM2 and mutant p53 (mt p53) protein might represent a mutant p53 gain of function phenotype. Recently, we provided evidence that certain p53 mutants maintain the ability to transactivate the p53 responsive element (p53RE) of the MDM2 gene. 37 Similar evidence has been reported also by others. 79 Furthermore, we have observed that in certain cases this effect is limited to the p53RE of growth-promoting genes. 80 Alternatively, in cases with inactive or absent p53, the MDM2 protein might be induced via a p53-independent manner. MDM2 gene amplification is unlikely to represent this p53-independent mechanism as it is a very rare event in lung carcinomas. 35,36,81 The activation of the MDM2 p53-independent promoter P1 may account for MDM2 overexpression in these cases. 33,34,37 On the other hand, we observed elevated levels of MDM2 and wild-type (wt) p53 protein in 19% of the carcinomas (Table 1) ▶ . As MDM2 and wt p53 form a negative feedback loop, this immunophenotype might reflect a complex formation between the two molecules that down-regulates the effects of wt p53. 33 However, we should keep in mind that immunohistochemical co-expression of p53 and MDM2 does not necessary imply this complex formation because the proportion of p53 bound to MDM2 is controlled by complicated post-translational events. 82,83 Moreover, recent reports suggest an MDM2-promoted degradation mechanism of wt and mt p53 and a p14ARF-dependent regulation mechanism of MDM2, making the picture of the p53-MDM2 relationship much more complex. 48,49,84 Finally, one cannot exclude the possibility that MDM2 overexpression may simply reflect the activity of wt p53, accumulated in the hypoxic environment of the proliferating cancerous cells. 33,34,46
After having evaluated independently the p16-pRb and p53-MDM2 pathways we addressed the issue of possible interrelations among impairments of these four molecules. Interestingly, we found two statistically significant associations. Abnormal expression of pRb was correlated with elevated levels of MDM2 (P = 0.013) and p53 (P = 0.01), respectively. In the first case, MDM2 overexpression could represent an additional mechanism of pRb inactivation, as MDM2 inhibits pRb regulatory function by interacting physically with it. 32 It is unlikely that aberrant pRb staining represents a masking effect due to MDM2-pRb complex formation because the anti-pRb antibody we used and MDM2 recognize different epitopes of pRb. 32 The significance of the association between p53 overexpression and abnormal pRb staining is unclear. Shiio et al showed that wt p53 suppresses RB1 transcription (Figure 1) ▶ and proposed that reduction of pRb would relieve the load of CDKs, thus creating another pathway to down-regulate the activity of pRb and promoting cells to enter S phase. 31 In a similar manner, inactivation of p53 in a cancerous cell might possibly facilitate loss of the RB1 gene or, vice versa, abrogation of pRb, which has a known anti-apoptotic effect, might lead to alteration of the p53 gene to bypass induction of apoptosis by wt p53. 85 This can be supported by findings in various malignant tumors. 86-88 Furthermore, Kinoshita and co-workers showed that deregulation of the p16/pRb pathway might synergistically increase proliferation activity with altered p53 protein. 21 However, more studies are required to address this point. One of the most important findings of the present study is that multiple disruption (three and four molecules affected) of the p16/pRb/p53/MDM2 network occurred in a large (43%) proportion of NSCLCs. Also, the absence of correlation with clinical stage of the carcinomas suggests that multiple hits of this network may be a relatively early event in the development of a subset of NSCLCs.
The relationship of the alterations detected at the 9p21–22 locus and in each one of the four network proteins, with the patients’ clinicopathological parameters and outcomes revealed two probably significant correlations: between smoking, p53 staining (P = 0.05), and MAs (P = 0.04), respectively. There has been no previous report, to the best of our knowledge, of a correlation between smoking and alterations at the 9p21–22 chromosomal region, although Kishimoto et al observed a tendency for an increased frequency of LOH on 9p in smokers with NSCLCs. 77 A number of studies on NSCLCs have examined the association of p16, pRb, and p53, independently, with smoking habits, histology, lymph node status, stage, and survival, reporting controversial findings. Indeed p16 and/or pRb abnormalities were not associated with adverse prognostic factors in several studies, 12,20,21,25,28,29 although correlation was found in others. 18,22,24,25,30 As far as p53 is concerned, many studies have investigated the implication of p53 in NSCLC carcinogenesis, and it appears that the prognostic significance of p53 alterations is rather weak (reviewed in Ref. 89 ). The only certain fact is that p53 is frequently affected in NSCLCs and it represents a major target for tobacco derivatives, a finding confirmed in our study as well. 46 Finally, with regard to MDM2, besides our previous studies, there are no available data by other groups. 35,36 We have found no statistical correlation between the various biologically relevant immunophenotypes, with the clinicopathological features and survival of the patients except for two probably significant relationships, between smoking and the p53(P)/MDM2(P) (P = 0.04) and p16(Ab)/pRb(Ab)/p53(P)/MDM2(P) (P = 0.03) patterns, respectively. The significance of these statistical relationships is not clear and needs more studies to be determined. However, in view of the wide spectrum of genetic targets identified in NSCLCs, 90 the overlapping and compensatory pathways linking most of them and the role of smoking as an indisputable major causal factor in lung carcinogenesis, it is tempting to suggest that, in a subset of NSCLCs, simultaneous deregulation of the members of this network may represent one way of initiating the oncogenic procedure whereas in other NSCLC subsets alternative molecules may play this role.
Acknowledgments
We thank Dr. P. Kanavaros for helpful criticism on the manuscript. Also, we thank Professor M. Oren, Dr. E. Haupt, and Dr, D. Mitropoulos for critical advice.
Footnotes
Address reprint requests to Dr. Gorgoulis Vassilis, Antaiou 53, Lamprini, Ano Patisia, Athens Greece, GR 11146.
References
- 1.Linnoila RI, Aisner SC: Pathology of lung cancer: an exercise in classification. Johnson BE Johnson DH eds. Lung Cancer. 1995, :pp 73-95 Wiley-Liss, New York [Google Scholar]
- 2.Cordon-Cardo C: Mutations of cell cycle regulators. Am J Pathol 1995, 147:545-560 [PMC free article] [PubMed] [Google Scholar]
- 3.Haber DA: Splicing into senescence: the curious case of p16 and p19ARF. Cell 1997, 91:551-558 [DOI] [PubMed] [Google Scholar]
- 4.Serrano M, Hannon G, Beach D: A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366:704-707 [DOI] [PubMed] [Google Scholar]
- 5.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV: A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994, 264:436-440 [DOI] [PubMed] [Google Scholar]
- 6.Knudson AG: Antioncogenes and human cancer. Proc Natl Acad Sci USA 1993, 90:10914-10921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA: Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994, 368:753-756 [DOI] [PubMed] [Google Scholar]
- 8.Cairns P, Mao L, Merlo A: Rates of the p16 (MTS1) mutations in primary tumours with 9p loss. Science 1994, 265:415-416 [DOI] [PubMed] [Google Scholar]
- 9.Okamoto A, Demetrick DJ, Spillare EA, Hagiwara K, Hussain SP, Bennet WP, Forrester K, Gerwin B, Serrano M, Beach DH, Herris CC: Mutations and altered expression of p16INK4 in human cancer. Proc Natl Acad Sci USA 1994, 91:11045-11049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spruck CH, III, Gonzalez-Zulueta M, Shibata A, Simoneau AR, Lin MF, Gonzales F, Tsai YC, Jones PA: p16 gene in uncultured tumours. Nature 1994, 370:183-184 [DOI] [PubMed] [Google Scholar]
- 11.Zhang S-Y, Klien-Szanto AJP, Sauter ER, Shafarenko M, Mitsunaga S, Nobori T, Carson DA, Ridge JA, Goodrow TL: Higher frequency of the alterations in the p16/CDKN2 gene in squamous cell carcinoma cell lines than in primary tumours of the head and neck. Cancer Res 1994, 54:5050-50503 [PubMed] [Google Scholar]
- 12.Reissmann PT, Koga H, Takahashi R, Figlin RA, Holmes C, Piantadosi S, Cardon-Cardo C, Slamon DJ: The Lung Cancer Study Group: Inactivation of the retinoblastoma susceptibility gene in non-small-cell lung cancer. Oncogene 1993, 8:1913-1919 [PubMed] [Google Scholar]
- 13.Sharipo GI, Edwards CD, Kobzik L, Godleski J, Richards W, Sugarbaker DJ, Rollins BJ: Reciprocal Rb inactivation and p16INK4 expression in primary lung cancers and cell lines. Cancer Res 1995, 55:505-509 [PubMed] [Google Scholar]
- 14.Okamoto A, Hussain SP, Hagiwara K, Spillare EA, Rusin MR, Demetrick DJ, Serrano M, Hannon GJ, Shiseki M, Zariwala M, Xiong Y, Beach DH, Yokota J, Harris CC: Mutations in the p16INK4 and p15INKB in primary and metastatic lung cancer. Cancer Res 1995, 55:1448-1451 [PubMed] [Google Scholar]
- 15.Shimizu T, Sekiya T: Loss of heterozygosity at 9p21 loci and mutations of the MTS1 and MTS2 genes in human lung cancers. Int J Cancer 1995, 63:616-620 [DOI] [PubMed] [Google Scholar]
- 16.Xiao S, Joseph DL, Vijg J, Fletcher JA: Codeletion of p15 and p16 genes in primary non-small cell lung carcinoma. Cancer Res 1995, 55:2968-2971 [PubMed] [Google Scholar]
- 17.Washimi O, Nagatake M, Osada H, Ueda R, Koshikawa T, Seki T, Takahashi T: In vivo occurrence of p16 (MTS1) and p15 (MTS2) alterations preferentially in non-small cell lung cancers. Cancer Res 1995, 55:514-517 [PubMed] [Google Scholar]
- 18.Nakagawa K, Conrad NK, Williams JP, Johnson BE, Kelley MJ: Mechanism of inactivation of CDKN2/P16ink4a and MTS2 in non-small cell lung cancer and association with advanced stage. Oncogene 1995, 11:1843-1851 [PubMed] [Google Scholar]
- 19.Rusin MR, Okamoto A, Chorazy M, Czyzewski K, Harasim J, Spillare EA, Hagiwara K, Hussain P, Xiong Y, Demetrick DJ, Harris CC: Intragenic mutations of the p16INK4, p15INKB and p18 genes in primary non-small-cell lung cancers. Int J Cancer 1996, 65:734-739 [DOI] [PubMed] [Google Scholar]
- 20.Sakaguchi M, Fujii Y, Hirabayashi H, Yoon H-E, Komoto Y, Oue T, Kusafuka T, Okada A, Matsuda H: Inversely correlated expression of p16 and Rb protein in non-small cell lung cancers: an immunohistochemical study. Int J Cancer 1996, 65:442-445 [DOI] [PubMed] [Google Scholar]
- 21.Kinoshita I, Dosaka-Akita H, Mishina T, Akie K, Nishi M, Hiroumi H, Hommura F, Kawakami Y: Altered p16INK4 and retinoblastoma protein status in non-small cell lung cancer: potential synergistic effect with altered p53 protein on proliferative activity. Cancer Res 1996, 56:5557-5562 [PubMed] [Google Scholar]
- 22.Kratzke RA, Greatens TM, Rubins JB, Maddaus MA, Niewoehner DE, Niehans GA, Geradts J: Rb and p16INK4a expression in resected non-small cell lung tumors. Cancer Res 1996, 56:3415-3420 [PubMed] [Google Scholar]
- 23.Marchetti A, Buttitta F, Pellegrini S, Bertacca G, Chella A, Carnicelli V, Tognoni V, Filardo A, Angeletti CA, Bevilacqua G: Alterations of p16(MTS1) in node-positive non-small cell lung carcinomas. J Pathol 1997, 181:178-182 [DOI] [PubMed] [Google Scholar]
- 24.Taga S, Osaki T, Ohgami A, Imoto H, Yoshimatsu T, Yoshino I, Yano K, Nakanishi R, Ichiyoshi Y, Yasumoto K: Prognostic value of the immunohistochemical detection of p16INK4 expression in non-small cell lung carcinoma. Cancer 1997, 80:389-395 [DOI] [PubMed] [Google Scholar]
- 25.Betticher DC, White GRM, Vonlanthen S, Liu X, Kappeler A, Altermatt HJ, Thatcher N, Heighway J: G1 control gene status is frequently altered in resectable non-small cell lung cancer. Int J Cancer 1997, 74:556-562 [DOI] [PubMed] [Google Scholar]
- 26.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sydranski D: 5′ CpG island methylation is associated with transcriptional silencing of the tumor suppressor p16/CDKN2/MTS1 in human cancers. Nature Med 1995, 1:686-692 [DOI] [PubMed] [Google Scholar]
- 27.Geradts J, Hu S-X, Lincoln CE, Benedict WF, Xu H-J: Aberrant Rb gene expression in routinely processed, archival tumor tissues determined by three different anti-Rb antibodies. Int J Cancer 1994, 58:161-167 [DOI] [PubMed] [Google Scholar]
- 28.Higashiyama M, Doi O, Kodama K, Yochouchi H, Tateishi R: Retinoblastoma protein expression in lung cancer: an immunohistochemical analysis. Oncology 1994, 51:544-551 [DOI] [PubMed] [Google Scholar]
- 29.Shimizu E, Coxon A, Otterson GA, Steinberg SM, Kratzke RA, Kim YW, Fedorko J, Oie H, Johnson BE, Mulshine JL, Minna JD, Gazdar AF, Kaye FJ: Rb protein status and clinical correlation from 171 cell lines representing lung cancer, extrapulmonary small carcinoma, and mesothelioma. Oncogene 1994, 9:2441-2448 [PubMed] [Google Scholar]
- 30.Xu H-L, Hu S-X, Cagle PT, Moore GE, Benedict WF: Absence of retinoblastoma protein expression in primary non-small cell lung carcinomas. Cancer Res 1991, 51:2735-2739 [PubMed] [Google Scholar]
- 31.Shiio Y, Yamamoto T, Yamaguchi N: Negative regulation of Rb expression by the p53 gene product. Proc Natl Acad Sci USA 1992, 89:5206-5210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao ZX, Chen J, Levine AJ, Modjtahedi N, Xing J, Sellers WR, Livingston DM: Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature 1995, 375:694-698 [DOI] [PubMed] [Google Scholar]
- 33.Piette J, Neel H, Marechal V: Mdm2: keeping p53 under control. Oncogene 1997, 15:1001-1010 [DOI] [PubMed] [Google Scholar]
- 34.Haines DS: The mdm2 proto-oncogene. Leuk Lymphoma 1996, 26:227-238 [DOI] [PubMed] [Google Scholar]
- 35.Gorgoulis V, Rassidakis G, Karameris A, Papastamatiou H, Trigidou R, Veslemes M, Rassidakis A, Kittas C: Immunohistochemical and molecular evaluation of the MDM2 gene product in bronchogenic carcinoma. Mod Pathol 1996, 9:544-554 [PubMed] [Google Scholar]
- 36.Gorgoulis V, Zoumpourlis V, Rassidakis G, Karameris A, Rassidakis A, Spandidos DA, Kittas C: A molecular and immunohistochemical study of the MDM2 protein isoforms and p53 gene product in bronchogenic carcinoma. J Pathol 1996, 180:129-137 [DOI] [PubMed] [Google Scholar]
- 37.Gorgoulis VG, Zacharatos PV, Manolis E, Ikonomopoulos JA, Damalas A, Lamprinopoulos C, Rassidakis GZ, Zoumpourlis V, Kotsinas A, Rassidakis AN, Halazonetis TD, Kittas C: Effects of p53 mutants derived from lung carcinomas on the p53 responsive element (p53RE) of the MDM2 gene. Br J Cancer 1998, 77:378-384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiest JS, Franklin WA, Otstot JT, Forbey K, Varella-Garcia M, Rao K, Drabkin H, Gemmill R, Ahrent S, Sidransky D, Saccomanno G, Fountain JW, Anderson MW: Identification of a novel region of homozygous deletion on chromosome 9p in squamous cell carcinoma of the lung: the location of a putative tumor suppressor gene. Cancer Res 1997, 57:1-7 [PubMed] [Google Scholar]
- 39.: World Health Organization: The World Health Organization histologic typing of lung tumours. Am J Clin Pathol 1982, 77:123-136 [DOI] [PubMed] [Google Scholar]
- 40.Geradts J, Wilson PA: High frequency of aberrant p16ink4A expression in human breast cancer. Am J Pathol 1996, 149:15-20 [PMC free article] [PubMed] [Google Scholar]
- 41.Whetsell L, Maw G, Nadon N, Ringer D, Schaefer F: Polymerase chain reaction microanalysis of tumors from stained histological slides. Oncogene 1995, 7:581-585 [PubMed] [Google Scholar]
- 42.Davis LG, Dibner MD, Battey JF: Basic Methods in Molecular Biology. 1986. Elsevier Science Publishing, Amsterdam
- 43.Fong KM, Zimmermann PV, Smith PJ: Microsatellite instability and other molecular abnormalities in non-small cell lung cancer. Cancer Res 1995, 55:28-30 [PubMed] [Google Scholar]
- 44.Chaubert P, Guillou L, Kurt A-M, Bertholet M-M, Metthez G, Leisinger H-J, Bosman F, Shaw F: Frequent p16INK4 (MTS1) gene inactivation in testicular germ cell tumors. Am J Pathol 1997, 151:859-865 [PMC free article] [PubMed] [Google Scholar]
- 45.Gorgoulis V, Zoumpourlis V, Rassidakis G, Karameris A, Barbatis C, Spandidos DA, Kittas C: Molecular analysis of p53 gene in laryngeal pre-malignant and malignant lesions: p53 protein immunohistochemical expression is positively related to proliferating cell nuclear antigen labelling index. Virchows Arch 1995, 426:339-344 [DOI] [PubMed] [Google Scholar]
- 46.Greenblatt MS, Bennett WP, Hollstein M, Harris CC: Mutations in the p53 tumour suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1995, 54:4855-4878 [PubMed] [Google Scholar]
- 47.Merlo A, Mabry M, Gabrielson E, Vollmer R, Baylin SB, Sidransky D: Frequent microsatellite instability in primary small cell lung cancer. Cancer Res 1993, 54:2098-2101 [PubMed] [Google Scholar]
- 48.Pomerantz J, Shreiber-Agus N, Liegeois N, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee H-W, Cordon-Cardo C, DePindo RA: The ink4a tumor supppressor gene product, p19ARF, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92:713-723 [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Xiong Y, Yarbrourgh WG: ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92:725-734 [DOI] [PubMed] [Google Scholar]
- 50.Otterson GA, Kleif SN, Chen W, Coxon AB, Kaye FJ: CDKN2 gene silencing in lung cancer by DNA hypermethylation, and kinetics of p16INK4 protein induction by 5-aza 2′ deoxycytidine. Oncogene 1995, 11:1211-1216 [PubMed] [Google Scholar]
- 51.Haber DA: Splicing into senescence: the curious case of p16 and p19ARF. Cell 1997, 91:555-558 [DOI] [PubMed] [Google Scholar]
- 52.Olopade OI, Pomykala HM, Hagos F, Sveen LW, Espinosa R, III, Dreyling MH, Gursky S, Stadler WM, Le Beau MM, Bolander SK: Construction of a 2.8-megabase yeast artificial chromosome contig and cloning of the human methylthioadenosine phosphorylase gene from the tumor suppressor region on 9p21. Proc Natl Acad Sci USA 1995, 92:6489-6493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geradts J, Kratzke RA, Niehans GA, Lincoln CE: Immunohistochemical detection of the cyclin dependent kinase inhibitor 2/multiple suppressor gene 1 (CDKN2/MTS1) product p16ink4A in archival human solid tumors: correlation with retinoblastoma protein expression. Cancer Res 1995, 55:6006-6011 [PubMed] [Google Scholar]
- 54.Tam SW, Stay JW, Pagano M: Differential expression and cell cycle regulation of the cyclin-dependent kinase 4 inhibitor p16INK4. Cancer Res 1994, 54:5816-5820 [PubMed] [Google Scholar]
- 55.Xu H-J, Hu S-X, Benedict WF: Lack of nuclear RB protein staining in G0/middle G1 cells: correlation to changes in total RB protein level. Oncogene 1991, 6:1139-1146 [PubMed] [Google Scholar]
- 56.Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, Peters G, Bartek J: Retinoblastoma protein dependent cell-cycle inhibition by the tumour suppressor p16. Nature 1995, 375:503-506 [DOI] [PubMed] [Google Scholar]
- 57.Okamoto A, Detnetrick DJ, Spillare EA, Hagiwara K, Hussain SP, Bennet WP, Forrester K, Gerwin B, Serrano M, Beach DH, Harris CC: Mutations and altered expression of p16INK4 in human cancer. Proc Natl Acad Sci Usa 1994, 91:11045-11049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zambetti GP, Levine AJ: A comparison of the biological activities of wild type and mutant p53. FASEB J 1993, 7:855-865 [DOI] [PubMed] [Google Scholar]
- 59.Shapiro GI, Park JE, Edwards CD, Mao L, Merlo A, Sidransky D, Ewen ME, Rollins BJ: Multiple mechanisms of p16INK4A inactivation in non-small cell lung lines. Cancer Res 1995, 55:6200-6209 [PubMed] [Google Scholar]
- 60.Merlo A, Gabrielson E, Mabry M, Vollmer R, Baylin SB, Sydranski D: Homozygous deletion on chromosome 9p and loss of heterozygosity on 9p, 6p, and 6q in primary human small cell lung cancer. Cancer Res 1994, 54:2322-2326 [PubMed] [Google Scholar]
- 61.Packenham JP, Taylor JA, White CM, Anna CH, Barrett JC, Devereux TR: Homozygous deletions at chromosome 9p21 and mutation analysis of p16 and p15 in microdissected primary non-small cell lung cancers. Clin Cancer Res 1995, 1:687-690 [PubMed] [Google Scholar]
- 62.Shimizu T, Sekiya T: Loss of heterozygosity at 9p21 loci and mutations of the MTS1 and MTS2 genes in human lung cancers. Int J Cancer 1995, 63:616-620 [DOI] [PubMed] [Google Scholar]
- 63.Mead LJ, Gillespie MT, Hung JY, Rane US, Rayeroux KC, Irving LB, Campbell LJ: Frequent loss of heterozygosity in early non-small cell lung cancers at chromosome 9p21 proximal to the CDKN2a gene. Int J Cancer 1997, 71:213-217 [DOI] [PubMed] [Google Scholar]
- 64.Vos S, Miller C, Takeuchi S, Combart A, Cho S, Koeffler H: Alteration of CDKN2 (p16) in non-small cell lung cancer. Genes Chromosomes & Cancer 1995, 14:164-170 [DOI] [PubMed] [Google Scholar]
- 65.Neville EM, Stewart M, Myskow M, Donnelly RJ, Field JK: Loss of heterozygosity at 9p23 defines a novel locus in non-small cell cancer. Oncogene 1995, 11:581-585 [PubMed] [Google Scholar]
- 66.Gilmore AP, Ohanian V, Spurr NK, Critchley DR: Localisation of the human gene encoding the cytoskeletal protein talin to chromosome 9p. Hum Genet 1995, 96:221-224 [DOI] [PubMed] [Google Scholar]
- 67.Albiges-Rizo C, Frachet P, Block MR: Down regulation of talin alters cell adhesion and the processing of the α5β1 integrin. J Cell Sci 1995, 108:3317-3329 [DOI] [PubMed] [Google Scholar]
- 68.Jiricny J: Colon cancer and DNA repair: have mismatches met their match? Trends Genet 1994, 10:164-168 [DOI] [PubMed] [Google Scholar]
- 69.Peltomaki P, Lothe RA, Aaltonen LA, Pylkkanen L, Nystrom-Lathi M, Secura R, David L, Holm R, Ryberg D, Haugen A, Brogger A, Borresen A-L, de la Chapelle A: Microsatellite instability is associated with tumors that characterize the hereditary non-polyposis colorectal carcinoma syndrome. Cancer Res 1993, 53:5853-5855 [PubMed] [Google Scholar]
- 70.Shridhar V, Siegfried J, Hunt J, del Mar Alonso M, Smith DI: Genetic instability of microsatellite sequences in many non-small cell lung carcinomas. Cancer Res 1994, 54:2084-2087 [PubMed] [Google Scholar]
- 71.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 72.Yeager T, Stadler W, Belair C, Puthenveettil J, Olopade O, Reznikoff C: Increased p16 levels correlate with pRb alterations in human urothelial cells. Cancer Res 1995, 55:493-497 [PubMed] [Google Scholar]
- 73.Otterson GA, Kratzke RA, Coxon A, Kim YW, Kaye FJ: Absence of p16INK4 protein is restricted to the subset of lung cancer lines that retains wild type RB. Oncogene 1994, 9:3375-3378 [PubMed] [Google Scholar]
- 74.Hangaishi A, Ogawa S, Imamura N, Miyawaki S, Miura Y, Uike N, Shimazaki C, Emi N, Takeyama K, Hirosawa S, Kamada N, Kobayashi Y, Takemoto Y, Kitani T, Toyama K, Ohtake S, Yazaki Y, Ueda R, Hirai H: Inactivation of multiple tumor-suppressor genes involved in negative regulation of the cell cycle, MTS1/p16INK4A, MTS2/p15INK4B, p53, and Rb genes in primary lymphoid malignancies. Blood 1996, 87:4949-4958 [PubMed] [Google Scholar]
- 75.Medema RH, Herrera RE, Lam F, Weinberg RA: Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc Natl Acad Sci USA 1995, 92:6289-6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bernards R: E2F: a nodal point in cell cycle regulation. Biochem Biophys Acta 1997, 1333:M33-M40 [DOI] [PubMed] [Google Scholar]
- 77.Kishimoto Y, Sugio K, Hung JY, Virmani AK, McIntire DD, Minna JD, Gazdar AF: Allele-specific loss in chromosome 9p loci in preneoplastic lesions accompanying non-small-cell lung cancers. J Natl Cancer Inst 1995, 87:1224-1229 [DOI] [PubMed] [Google Scholar]
- 78.El-Naggar AK, Lai S, Clayman G, Lee J-KJ, Luna MA, Goepfert H, Batsakis JG: Methylation, a major mechanism of p16/CDKN2 gene inactivation in head and neck squamous carcinoma. Am J Pathol 1997, 151:1767-1774 [PMC free article] [PubMed] [Google Scholar]
- 79.Lianes P, Orlow I, Zhang Z-F, Oliva MR, Sarkis AS, Reuter VE, Cordon-Cardo C: Altered patterns of MDM-2 expression in human bladder cancer. J Natl Cancer Inst 1994, 86:1325-1330 [DOI] [PubMed] [Google Scholar]
- 80.Zacharatos P, Gorgoulis V, Kotsinas A, Kanavaros P, Zoumpourlis V, Veslemes M, Halazonetis T, Kittas C: Modulation of wild-type p53 activity by mutant p53 R273H depends on the p53 responsive element (p53RE): a comparative study between the p53REs of the MDM2, WAF1/Cip1 and Bax genes in lung cancer environment. Abstract presented at the 9th p53 Workshop, Crete, May 9 to 14, 1998
- 81.Marchetti A, Buttitta F, Pellegrini S, Merlo G, Chella A, Angeletti CA, Bevilacqua G: mdm2 gene amplification and overexpression in non-small cell lung carcinoma with accumulation of the p53 protein in the absence of p53 gene mutation. Diagn Mol Pathol 1995, 4:93-97 [DOI] [PubMed] [Google Scholar]
- 82.Momand J, Zambetti GP: Analysis of the proportion of p53 bound to mdm-2 in cells with defined growth characteristics. Oncogene 1996, 12:2279-2289 [PubMed] [Google Scholar]
- 83.Shieh S-Y, Ikeda M, Taya Y, Prives C: DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91:325-334 [DOI] [PubMed] [Google Scholar]
- 84.Haupt Y, Maya R, Kazaz A, Oren M: Mdm2 promotes the rapid degradation of p53. Nature 1997, 387:296-299 [DOI] [PubMed] [Google Scholar]
- 85.White E: p53, guardian of Rb. Nature 1994, 371:21-22 [DOI] [PubMed] [Google Scholar]
- 86.Murakami Y, Hirayashi K, Hirohashi S, Sekiya T: Aberrations of the tumor suppressor p53 and the retinoblastoma genes in human hepatocellular carcinomas. Cancer Res 1991, 51:5520-5525 [PubMed] [Google Scholar]
- 87.Cheng J, Hass M: Frequent mutations in the p53 tumor suppressor gene in human leukemia T-cell lines. Mol Cell Biol 1990, 10:5502-5509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T: Cooperative tumorigenic effects of germline mutations in Rb and p53. Nature Genet 1994, 7:480-484 [DOI] [PubMed] [Google Scholar]
- 89.Kalemkerian GP: Biology of lung cancer. Curr Opin Oncol 1994, 6:147-155 [DOI] [PubMed] [Google Scholar]
- 90.Groeger AM, Esposito V, Mueller MR, Caputi M, Kaiser HE, Giordano A: Advances in the understanding of lung cancer. Anticancer Res 1997, 17:2519-2522 [PubMed] [Google Scholar]