

Abstract
The putative first intracellular and third extracellular linkers are known to play important roles in defining the transport properties of the type IIa Na+-coupled phosphate cotransporter (Kohler, K., I.C. Forster, G. Stange, J. Biber, and H. Murer. 2002b. J. Gen. Physiol. 120:693–705). To investigate whether other stretches that link predicted transmembrane domains are also involved, the substituted cysteine accessibility method (SCAM) was applied to sites in the predicted first and fourth extracellular linkers (ECL-1 and ECL-4). Mutants based on the wild-type (WT) backbone, with substituted novel cysteines, were expressed in Xenopus oocytes, and their function was assayed by isotope uptake and electrophysiology. Functionally important sites were identified in both linkers by exposing cells to membrane permeant and impermeant methanethiosulfonate (MTS) reagents. The cysteine modification reaction rates for sites in ECL-1 were faster than those in ECL-4, which suggested that the latter were less accessible from the extracellular medium. Generally, a finite cotransport activity remained at the end of the modification reaction. The change in activity was due to altered voltage-dependent kinetics of the Pi-dependent current. For example, cys substitution at Gly-134 in ECL-1 resulted in rate-limiting, voltage-independent cotransport activity for V ≤ −80 mV, whereas the WT exhibited a linear voltage dependency. After cys modification, this mutant displayed a supralinear voltage dependency in the same voltage range. The opposite behavior was documented for cys substitution at Met-533 in ECL-4. Modification of cysteines at two other sites in ECL-1 (Ile-136 and Phe-137) also resulted in supralinear voltage dependencies for hyperpolarizing potentials. Taken together, these findings suggest that ECL-1 and ECL-4 may not directly form part of the transport pathway, but specific sites in these linkers can interact directly or indirectly with parts of NaPi-IIa that undergo voltage-dependent conformational changes and thereby influence the voltage dependency of cotransport.
Keywords: mutagenesis site directed, electrophysiology, phosphate transport proteins, electrogenic, Xenopus laevis
INTRODUCTION
The task of reabsorbing inorganic phosphate (Pi) at the brush border membrane of the renal proximal tubule is largely fulfilled by the type IIa Na+/Pi cotransporter (NaPi-IIa) (SLC34A1) and consequently this protein plays a central role in mammalian phosphate homeostasis (for review see Murer et al., 2000, 2003). NaPi-IIa isoforms from several species have been cloned and their transport kinetics characterized by expression in Xenopus laevis oocytes using isotope tracer and electrophysiological assays (for review see Forster et al., 2002). The key functional features are as follows: Na+-dependent Pi cotransport (3:1 Na+:Pi stoichiometry), ordered substrate binding (Na+–Pi 2−–2Na+), substrate specificity (divalent Pi is the preferred species), and voltage-dependent transport (one net charge transfer per transport cycle); uniport or uncoupled Na+-dependent leak in the absence of Pi; and intrinsic pH dependency (suppressed Pi transport with external acidification) (Forster et al., 2002). Elucidation of the structure–function relationships between molecular entities and kinetic properties of NaPi-IIa is an ongoing challenge that we are currently addressing using the substituted cysteine accessibility method (SCAM) (e.g., Karlin and Akabas, 1998).
The predicted topology of NaPi-IIa, derived from studies on the rat isoform, suggests a protein with eight transmembrane domains, a large extracellular loop with two N-glycosylation sites and intracellular NH2 and COOH termini (Magagnin et al., 1993; Murer et al., 2000) (Fig. 1 A). Antibody accessibility studies (Lambert et al., 1999b) and cysteine scanning approaches (Lambert et al., 1999a, 2000; Kohler et al., 2002a,b) also support this model. Using SCAM we have identified sites in the putative third extracellular linker (ECL-3) (Lambert et al., 1999a, 2001) and the putative first intracellular linker (ICL-1) (Kohler et al., 2002a,b) that when modified by methanethiosulfonate (MTS) reagents result in block of cotransport mode activity. Given that NaPi-IIa is a functional monomer (Kohler et al., 2000), and taking into account our previous SCAM findings, we currently postulate that these two topologically opposed linker regions associate with one another to constitute the transport pathway (Kohler et al., 2002a,b). To examine if other external linker regions may also define NaPi-IIa transport properties, we performed SCAM on sites in the first and fourth predicted linker regions, designated ECL-1 and ECL-4, respectively (Fig. 1 A). As shown in Fig. 1 B, these regions are very well conserved among several isoforms of the SLC34A family and therefore we might expect them to play common functional roles.
Figure 1.

(A) Topological representation of the rat type IIa Na+/Pi cotransporter comprises a backbone of eight putative membrane-spanning domains (TMD-1 to -8) and corresponding linker regions. A functionally essential cysteine bridge is formed between Cys-306 and Cys-334 in the large second extracellular linker (ECL-2) and an additional bridge between Cys-225 and either Cys-520 or Cys-597 has also been recently proposed (Kohler et al., 2003). The cluster of functionally important (MTS-accessible) sites previously identified by SCAM in the third extracellular linker (ECL-3) (Lambert et al., 2001) and two sites in the first intracellular linker (ICL-1) (Kohler et al., 2002a) are indicated (filled squares). Sites 130–140 in the putative first extracellular linker (ECL-1) and 532–538 in the predicted fourth extracellular linker (ECL-4) were mutated to cysteines in this study (gray-filled squares) (B) Comparison of the amino acid sequences for the predicted transmembrane domains flanking ECL-1 (TMD-1 and -2) and ECL-4 (TMD-7 and -8) show a high degree of homology between different isoforms of the type II Na+/Pi cotransporter family (SLC34). Bold lettered amino acids in ECL-1 and ECL-4 for the rat isoform were mutated to cysteines for the present study. Amino acids are named using the single letter code.
Cysteine substitution at nine sites in ECL-1 and four sites in ECL-4 was well tolerated, and these mutants displayed no significant deviations from wild-type (WT) behavior, based on a two-point screening assay for substrate and cosubstrate activation. In both linker regions, altered kinetic phenotypes were documented for membrane-impermeant MTS reagents, which established that these sites were accessible from the extracellular milieu, in agreement with the current NaPi-IIa secondary topology. Estimated rates of modification suggested that ECL-1 was more accessible than ECL-4. Some mutations in each linker showed significantly modified voltage dependency before and after exposure to MTS reagents. Of particular significance were the complementary changes that resulted from in situ molecular manipulations of certain residues in ECL-1 and ECL-4. Our results suggest that these linkers are most likely not directly part of the transport pathway, but they contain specific sites that may interact directly or indirectly with voltage-sensitive elements of the NaPi-IIa protein.
MATERIALS AND METHODS
Mutagenesis
Mutations were introduced following the Stratagene Quickchange Site-Directed Mutagenesis Kit manual. 10 ng of the plasmid (pSPORT; GIBCO BRL) containing the WT rat NaPi-IIa cDNA was amplified with 2.5 U of Pfu Turbo DNA polymerase (Stratagene), in the presence of primers (200 nM). PCR amplification was performed with 20 cycles, with each cycle consisting of one 95°C (30 s), one 55°C (1 min), and one 68°C (15 min) step. Afterwards, 10 U of Dpn I was added directly to the amplification reaction, and the sample was incubated for 1 h at 37°C to digest the parental DNA. XL1-blue supercompetent cells were transformed with 1 μl of the reaction mixture and plated onto LB-ampicillin plates. The sequence was verified by sequencing (Microsynth) and the plasmid linearized by digestion with NotI. In vitro synthesis of capped cRNA was done following the Ambion MEGAscript T7 kit manual (Ambion). In brief, 1 μg linearized DNA encoding the rat NaPi-IIa constructs was incubated in the presence of 40 U of T7 RNA polymerase and Cap Analogue (New England Biolabs, Inc.) at 37°C for 4 h. Oocytes were injected with either 50 nl water or 50 nl of water containing 10 ng cRNA.
Reagents and Solutions
All standard reagents were obtained from either Sigma-Aldrich or Fluka. Methanethiosulfonate (MTS) reagents were obtained from Toronto Research Chemicals (Downsview). The solution compositions (in mM) were as follows. (a) Oocyte incubation (modified Barth's solution): NaCl (88), KCl (1), CaCl2 (0.41), MgSO4 (0.82), NaHCO3 (2.5), Ca(NO3)2, HEPES (7.5), adjusted to pH 7.6 with TRIS and supplemented with antibiotics (10 mg/l gentamycin, streptomycin). (b) Control superfusate (ND100): NaCl (100); KCl (2); CaCl2 (1.8); MgCl2 (1); HEPES-TRIS (10) at pH 7.4. Solutions with intermediate Na+ concentrations were prepared by mixing ND0 and ND100 in the appropriate proportions. (c) Substrate test solutions: inorganic phosphate (Pi) was added to the control superfusate from 1 M K2HPO4 and KH2PO4 stocks that were mixed to give the required pH (7.4 or 6.2). Phosphonoformic acid (PFA) was added to ND100 from frozen 100 mM stock (in H2O) to yield a final concentration of 1 mM. (d) MTS reagents, 2-aminoethyl MTS hydrobromide (MTSEA), 2-(trimethylammonium)ethyl MTS bromide (MTSET), and sodium (2-sulfonatoethyl) MTS (MTSES) were prepared from dry stock in DMSO to give 1 M stock solutions. These were stored at −20°C until required and added to ice-cold ND100 solution. The final concentration of DMSO did not exceed 0.1%, and DMSO alone at this concentration was confirmed not to alter the kinetic characteristics of the expressed constructs.
Immunoblotting of Oocyte Homogenates: Western Blotting
Yolk-free homogenates were prepared 3 d after injection of cRNA. Pools of eight oocytes were lysed together with 160 μl homogenization buffer (1% eluent [Calbiochem] in 100 mM NaCl, 20 mM Tris/HCl, pH 7.6) by pipetting the oocytes up and down (Turk et al., 1996). To pellet the yolk proteins, samples were centrifuged at 16,000 g for 3 min at 22°C. 10 μl supernatant was mixed with 10 μl 2× loading buffer (4% SDS, 2 mM EDTA, 20% glycerol, 0.19 M Tris/HCl, pH 6.8, 2 mg/ml bromphenol blue, 200 mM dithiothreitol [DTT]), denatured for 2 min at 95°C and separated on a 10% SDS-PAGE gel. Separated proteins were transferred onto a nitrocellulose membrane (Schleicher & Schuell). The membrane was then processed according to standard procedures (Sambrook et al., 1989) using a rabbit polyclonal antibody raised against a synthetic peptide from the NH2 termini of the rat NaPi-IIa cotransporter (dilution 1:2,000). The specificity of the antibody has been demonstrated previously (Custer et al., 1994). Immunoreactive proteins were detected with a swine anti-rabbit Ig horseradish peroxidase–linked F(ab′)2 secondary antibody (dilution 1:5,000) (Amersham Biosciences) and visualized with a chemiluminescence system (Pierce Chemical Co.).
Streptavidin Precipitation of MTSEA-biotinylated Protein
Groups of eight oocytes per NaPi-IIa mutant were incubated for 10 min in 1 mM MTSEA-Biotin, prepared as above. Oocytes expressing the S460C or the WT protein were taken as positive and negative control, respectively. Biotin-streptavidin precipitation was performed as previously described (Lambert et al., 1999a). In brief, after homogenization in 160 μl of homogenization buffer and centrifugation (see immunoblot of oocyte homogenates), a sample for Western blotting was taken. The rest of the supernatant (∼120 μl) was incubated for 2 h with 60 μl ImmunoPure immobilized streptavidin beads (Pierce Chemical Co.) on a rotator. After five washing steps with homogenization buffer, precipitated proteins were eluted with 2× loading buffer (including DTT) at 95°C for 5 min. Samples were loaded on a 10% SDS gel and immunoblotted after protein separation.
Functional Assays and Data Analysis
Radiolabeled Pi Uptake.
This procedure has been described in detail elsewhere (Werner et al., 1990). 32Pi uptake was measured 3 d after injection in water-injected (control) and cRNA-injected oocytes (n ≥ 10).
Electrophysiology.
The standard two-electrode voltage clamp technique was used as previously described (Forster et al., 1998). The voltage clamp was a laboratory-built system with membrane current measured using a virtual ground bath electrode and with active series resistance compensation to reduce clamp errors. Oocytes were mounted in a small recording chamber (100 μl volume) and continuously superfused (5 ml/min) with test solutions precooled to ∼20°C. Data were acquired online using Digidata 1200 hardware and compatible pClamp8 software (Axon Instruments, Inc.). Recorded currents were prefiltered at a bandwidth less than twice the sampling rate (typically 20 Hz for fixed voltage recording and 200 Hz for I–V determination).
Steady-state Current-Voltage (I–V) Relations.
These were determined by using a voltage staircase from −120 mV to +40 mV with 100-ms-long steps, as previously described (Forster et al., 1998), and subtracting the response in the absence of substrate (Pi or PFA) from that in the presence of substrate. Only single sweeps were used.
Incubation with MTS Reagents and Determination of Rate of Modification.
Freshly prepared MTSEA/ET/ES (see above) was delivered to the oocyte chamber using a stainless steel cannula (inner diameter 0.3 mm) positioned near the cell and fed by gravity. Reagents were always applied in the presence of ND100 at a holding potential V h = −50 mV. Incubation was followed by a 1-min washout period before applying Pi (1 mM) as the test assay. A further 1-min washout period was allowed before reincubation at the next concentration of MTS reagent to ensure complete removal of substrate and return of response to a steady-state holding current.
Cys Modification Reaction Rate.
The effective second order reaction rates were determined by fitting a single decaying exponential to the peak Pi-dependent current after a cumulative exposure time t, (I Pi t) determined following each successive MTS exposure:
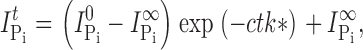 |
(1) |
where I Pi 0 is the Pi response at t = 0; I Pi ∞ is the response at t = ∞; c is the concentration of MTS reagent (assumed to be in excess), and k* is the effective second order rate constant (Zhang and Karlin, 1997; Karlin and Akabas, 1998).
All curve fitting was performed using GraphPad Prism version 3.02/4.02 for Windows (GraphPad Software).
RESULTS
Expression of Mutants and Identification of MTS-accessible Sites
Fig. 1 A shows the currently proposed secondary structure of the rat NaPi-IIa protein to indicate the location of the residues in the predicted first and fourth extracellular loops (ECL-1/-4) that were mutated individually to cysteines (indicated in bold in Fig. 1 B). We injected Xenopus laevis oocytes with cRNA coding for each mutant and screened them for functional transport by using 32Pi uptake (Fig. 2 A) and a basic electrophysiological assay (ND100 ± 1 mM Pi, holding potential [V h] = −50 mV) (Fig. 2 B). For all mutants that gave functional expression, we detected a main band in the Western blots, the position of which corresponded to the expected molecular weight of the WT (80–100 kD) (Fig. 3). We assumed that mutant Q537C in ECL-4 was not expressed based on the Western blot data, as well as the lack of both detectable 32Pi uptake (Fig. 2 A) and electrogenic response (not depicted). Taken together, these findings indicated that for the functional mutants, Cys mutagenesis did not affect the basic transport function and these constructs were indistinguishable from the WT in terms of the size of the expressed protein.
Figure 2.

Identification of functionally important sites in ECL-1 and ECL-4. (A) 32Pi uptake of WT and mutants before (filled bars) and after (unfilled bars) incubation for 10 min in 1 mM MTSEA (n ≥ 10 oocytes per group). (B) Remaining electrogenic activity (expressed as % of the initial response) after 3–5 min exposure to 1 mM MTSEA for n ≥ 3 oocytes/construct, measured at V h = −50 mV, ND100 ± 1 mM Pi. WT response, filled bar; mutants, open bars. ND (not determined) refers to mutants that gave an electrogenic response <−15 nA or no measurable response to 1 mM Pi in at least three batches of oocytes from different donor frogs. Insets show representative recordings from three oocytes voltage clamped to −50 mV that expressed the WT (1), G134C (2), and F137C (3), respectively, before (−) and after (+) incubation for 3 min in 1 mM MTSEA. The response of each oocyte to a 20-s application of 1 mM Pi (open bars) and 1 mM PFA (filled bars) was recorded. Dotted lines indicate peak of Pi-dependent response and holding current in ND100 to aid comparison. Asterisks indicate mutants that showed statistically significant deviations from the WT, as reported by an unpaired t test (P < 0.05), applied to the normalized data. (C) Comparison of activity after MTSEA exposure for mutants for which reliable electrophysiological data was available (ECL-1, empty squares; ECL-4, gray-filled squares; WT, filled square) normalized to control (−MTSEA) condition. Vertical error bars indicate SEM for normalized uptake data, horizontal error bars indicate SEM from data in B. Gray bar represents mean ± SEM to indicate change of electrogenic activity observed for WT-expressing oocytes after MTSEA exposure.
Figure 3.

Accessibility of cysteine mutants in ECL-1 (A) and ECL-4 (B) by MTSEA-Biotin. Western blots show streptavidin precipitates of MTSEA-biotinylated oocytes (top) and the corresponding whole cell lysates (bottom) from oocytes that expressed the indicated mutants probed with an antibody raised against the NH2 terminus of NaPi-IIa. Biotinylation data have been combined from several assays made from different batches of oocytes. In each assay, WT and water (H2O) injected oocytes were used as negative controls, and oocytes injected with cRNA coding for the mutant S460C (Lambert et al., 1999a) were used as a positive control. The band in the range 80–100 kD confirms that the labeled protein is NaPi-IIa. White lines indicate that intervening lanes have been spliced out.
We assayed the functional activity of oocytes that expressed each mutant before and after exposure to 1 mM of the Cys-modifying reagent MTSEA for 10 min by 32Pi uptake (Fig. 2 A) and electrophysiology (ND100 ± 1 mM Pi at V h = −50 mV) (Fig. 2 B), to determine if the Cys substitutions were made at sites that might directly or indirectly alter basic transport function. The electrogenic response to Pi superfusion varied among mutants and typically fell within the range −30 to −100 nA at −50 mV. Comparison of these datasets indicated that the trends revealed by the two assays were similar; i.e., those mutants for which MTSEA treatment led to a reduced 32Pi activity also showed reduced Pi-dependent currents (I Pi), as indicated in Fig. 2 C, in which both datasets have been normalized to the respective activities before exposure to MTSEA. A notable exception to this general behavior was mutant G134C, which showed the opposite behavior in the two assays; i.e., after MTS exposure, we observed a decreased 32Pi uptake, but an increased I Pi. Depending on the oocyte batch, the uptake assays often showed a large spread in response that we attributed to variability in expression level. Therefore we focused on the electrophysiological assays to study the mutants on an individual cell basis. We also confirmed that the change in electrogenic activity after MTS incubation could be reversed by incubation in the reducing agent DTT (10 mM, 5 min) for selected mutants, whereas no change in WT activity was documented (unpublished data). This finding confirmed that the change in activity most likely resulted from the introduction of a reducible covalent bond between the novel cysteine and MTS reagent. Moreover, the rapidity of the reversal in activity indicated that the number of active transporters in the membrane had not changed as a result of the MTS treatment.
As shown in Fig. 2 B, mutants displayed three types of behavior, depending on their response to 1 mM Pi before and after MTSEA exposure: (1) WT-like (see Fig. 2 B, inset 1) in which little change in activity occurred, found for five mutants in ECL-1 (G130C, V132C, D135C, K138C, and D139C) and one in ECL-4 (S532C); (2) one mutant in ECL-1 (G134C) that showed increased activity (Fig. 2 B, inset 2); and (3) those in which a loss of activity occurred, like F137C (Fig. 2 B, inset 3) and others in ECL-1 (I136C and N140C) and ECL-4 (M533C, A534C, and A538C). In addition to cotransport mode (I Pi), the leak mode was assayed electrophysiologically by measuring the response to the blocker PFA (1 mM) (Forster et al., 1998) under the same experimental conditions. At V h = −50 mV, all the functional mutants showed a reduced holding current during PFA application (I PFA), which indicated that the leak mode was still viable. Two mutants in ECL-4 (G535C and W536C) gave 32Pi uptake significantly greater than H2O-injected cells (Fig. 2 A); however, the magnitude of the Pi-dependent currents was typically ≤15 nA using oocytes from three donor frogs. This low electrogenic activity and the absence of any detectable effect of MTSEA on 32Pi uptake led to the exclusion of these mutants from further investigation.
The invariance of I Pi before and after MTSEA exposure (G130C, V132C, D135C, K138C, and D139C in ECL1; S532C in ECL-4), could indicate that (a) the mutated sites were functionally unimportant, (b) other characteristics were modified that were not identified under the assay conditions, or (c) the sites were not accessible from the external medium by MTSEA. To test the latter possibility, we incubated mutant-expressing oocytes in MTSEA-Biotin and performed a streptavidin precipitation with immunodetection using an antibody raised against the NH2 terminus of the NaPi-IIa protein. We used the mutant S460C in ECL-3 (Fig. 1 A), which we have previously shown to be accessible by MTSEA (Lambert et al., 1999a), and WT-expressing oocytes as positive and negative controls, respectively. As shown in Fig. 3, except for D135C (ECL-1) and W536C and Q537C (ECL-4), MTSEA-biotin was able to bind to all mutants, which confirmed the accessibility of these linkers from the external medium.
Rate of MTS Modification Reveals Differences in Apparent Accessibility
We estimated the rate of the Cys modification reaction for mutants that showed a significant change of cotransport function after MTS exposure under standard assay conditions, by measuring I Pi after successive exposures to a fixed concentration of MTS reagent. The MTS reagent concentration was chosen initially by trial and error so that a 2-min exposure resulted in an intermediate change of activity, typically in the range 30–60% of the initial response. The test concentration was found to vary over two orders of magnitude, depending on the mutation site (Table I). Fig. 4 A shows a representative record from an oocyte that expressed mutant F137C (ECL-1) during repeated exposure to 10 μM MTSET. After each exposure and washout, the response to Pi was tested and the holding current was allowed to recover to the initial baseline before the next exposure. After normalization, these data were fit with a single exponential function (Eq. 1). Under the assumption that the MTS reagent was always in excess, we could estimate the effective second order reaction rate (k*) (Table I). Impermeant (MTSET) and semi-permeant (MTSEA) reagents were used to confirm the sidedness of the reaction, i.e., to confirm that modification occurred from the external medium in the present study. Fig. 4 (B and C) illustrates the behavior of selected mutants from ECL-1 and ECL-4, respectively.
TABLE I.
Reactivity of Mutated Sites
| I Pi ∞ | k* (M−1s−1) | |||||
|---|---|---|---|---|---|---|
| Linker | Site | c (mM) | MTSEA | MTSET | MTSEA | MTSET |
| ECL-1 | G-134 | 0.01 | 1.81 ± 0.03 | 2.10 ± 0.08 | 455 ± 42 | 206 ± 27 |
| I-136 | 0.01 | 0.58 ± 0.03 | 0.52 ± 0.03 | 835 ± 134 | 1170 ± 250 | |
| F-137 | 0.01 | 0.24 ± 0.02 | 0.07 ± 0.05 | 651 ± 67 | 1470 ± 300 | |
| N-140 | 0.1 | 0.60 ± 0.02 | 0.60 ± 0.13 | 122 ± 23 | 109 ± 21 | |
| ECL-4 | M-533 | 0.5 | 0.24 ± 0.01 | 0.19 ± 0.01 | 13.7 ± 0.9 | 37.1 ± 1.8 |
| A-534 | 0.1 | 0.54 ± 0.03 | 0.63 ± 0.06 | 60.1 ± 10.8 | 75.2 ± 24.4 | |
| A-538 | 1.0 | 0 | 0 | 2.67 ± 0.11 | 1.97 ± 0.08 | |
Figure 4.

Determination of MTS-Cys reaction rates for mutants that showed a change of activity after MTS exposure. (A) Representative recording of current from an oocyte that expressed mutant F137C after successive applications of MTSET (10 μM) (light gray bars). The cumulative exposure time (min) is indicated above each test response. Test substrates Pi (1 mM, black bars) and PFA (1 mM, dark gray bars) were applied for ∼20 s. (B and C) Pi-dependent currents at V h = −50 mV, normalized to the initial value plotted as a function of cumulative MTS reagent exposure time for selected mutants in ECL-1 (B) and ECL-4 (C). Cells were exposed to either MTSEA (filled squares) or MTSET (open squares) for the cumulative time indicated and at the concentrations given in Table I. Continuous line is a fitted single exponential function with plateau (Eq. 1) from which the reaction rate and plateau were estimated. Each data point is pooled from ≥3 cells. Broken lines indicate plateau levels (I Pi ∞) estimated from the fit of Eq. 1 (Table I).
Three features were readily apparent. First, there was a finite response to Pi after the reaction was complete, indicated by the nonzero plateau (I Pi ∞). I Pi ∞ varied depending on the site of mutation and the type of MTS reagent used. For mutant A538C (ECL-4), the time window used for the assay (15 min) did not allow the reaction to reach completion using 1 mM of reagent. Incubation at longer times indicated that the plateau was <10% of the initial amplitude (unpublished data). Second, with the exceptions of G134C, N140C (ICL-1), and A538C (ECL-4), MTSET incubation resulted in significantly faster reaction rates compared with MTSEA applied at the same concentration (Table I). Third, the reaction rates for mutants with cys substitutions in ECL-1 were faster than those in ECL-4.
We previously reported that oocytes that expressed the flanking mutations in ECL-4 (S532C and A538C) did not show a significant loss of transport function when exposed to MTSEA at 100 μM (Lambert et al., 1999a). However, in the present study, at 10-fold greater MTSEA concentration, significant loss of function was observed for A538C, but still not for S532C (Fig. 2 B). Additional electrophysiological assays (unpublished data) on mutant S532C showed ∼20% loss of response to 1 mM Pi (V h = −50 mV) when exposed to 1 mM MTSEA for 10 min and ∼40% after 25 min (unpublished data; n = 3). Because of its low reactivity compared with the other mutants in ECL-4, this site was considered only marginally accessible. At 1 mM concentration, it is possible that MTSEA was also present intracellularly, due to its reported membrane permeability (e.g., Holmgren et al., 1996), and therefore the loss of cotransport function may have been due to cytosolic action on other, internally accessible cysteines. However, exposure to 1 mM of MTSET also led to a comparable loss of activity for A538C, albeit with slower reaction rates (Fig. 4 C; Table I). This confirmed that the novel cysteines were accessible from the extracellular medium, in agreement with their predicted topology and the surface biotinylation assay.
Substrate Activation Unaffected by Cys Substitution and Cys Modification
The finite activity that remained at the completion of the MTS modification reactions suggested that kinetic properties of the mutated cotransporter were sensitive to cys modification. The most likely explanation would be a reduction (or increase, in the case of G134C) of the apparent substrate affinities at −50 mV. We therefore screened all the functional mutants to compare their substrate activation properties before and after a 5-min application of 1 mM MTSEA. To detect large deviations from the WT behavior of the apparent affinities for Pi (K m Pi) and Na+ (K m Na), we applied a two-point assay, because in the case of poorly expressing mutants, a complete dose response could be difficult to obtain due to contamination from endogenous currents at low substrate concentrations. We have previously applied these assays to identify a mutant in the first intracellular linker region that showed significantly decreased apparent substrate affinities compared with the WT (Kohler et al., 2002a).
Pi activation was assayed by comparing the ratio of the responses to 0.1 mM Pi and 1 mM Pi for superfusion in ND100, termed the Pi activation index. Similarly, a Na+ activation index, given by the ratio of the responses to 1 mM Pi for superfusion in ND50 and ND100, was used to quantitate the Na+ activation. We confirmed the sensitivity of using this assay with simulations in which we assumed the Pi and Na+ activation characteristics could be described by the modified Hill equation. For Pi activation (Fig. 5 A, left), the typical range of the index for the WT, based on assays performed in this study as well others (Lambert et al., 2000; Kohler et al., 2002a), should allow detection of changes in K m Pi outside the range 0.05 < K m Pi < 0.12 mM. For the Na+ activation (Fig. 5 A, right), we would conservatively expect to detect changes in K m Na outside the range 40 < K m Na < 60 mM. For example, if in the case of mutant G137C, the 60% decrease in the electrogenic response to 1 mM Pi after MTSEA exposure were due only to an increase in the apparent K m Pi from the typical WT value of 0.06 mM to 1.65 mM, the Pi activation index would decrease from ∼0.66 to 0.15, based on a simple Michaelian model for Pi activation and the same maximum transport rate ± MTSEA.
Figure 5.

Screening for deviations from WT behavior of substrate activation kinetics at −50 mV using a two-point assay. (A) Two-point assay sensitivity for Pi activation (left) and Na+ activation (right). Continuous lines are the Pi activation index (ratio of response in 100 mM Na+ to 0.1 mM Pi and 1 mM Pi) as a function of apparent affinity for Pi (K m Pi) and Na+ activation index (ratio of response to 1 mM Pi in 50 mM Na+ and 100 mM Na+) as a function of apparent affinity for Na+ (K m Na), respectively. Indices were determined as a function of K m Pi or K m Na using the modified Hill equation to describe the electrogenic response to Pi, I Pi = I Pi max (S nH)/(S nH + K m SnH), where S is the concentration of the variable substrate, K m S is the apparent affinity constant, n H is the Hill coefficient (n H = 1.0 for Pi activation, n H = 2.5 for Na+-activation; Forster et al., 1998, 1999), and I Pi max is the Pi-dependent change in holding current at the saturating limit. In general, K m S and I Pi max can also depend on the concentration of the invariant substrate and holding potential (V h). Lower test concentration was chosen close to previously reported estimates for the WT apparent affinity, and upper test concentration was chosen close to the saturation concentration for Pi or maximum usable Na+ concentration, for the Pi and Na+ activation screens, respectively. For each case, the gray bar represents range of index values observed for WT-expressing oocytes (n = 9, three donor frogs). Vertical lines indicate typical K m Pi for WT (0.06 mM) and K m Na for WT (50 mM) at −50 mV, previously reported (e.g., Forster et al., 1998), respectively. (B) Pi activation index (top) and Na+ activation index (bottom), where each point is mean ± SEM for ≥4 oocytes. Gray bars indicate typical range observed for WT-expressing oocytes measured under the same experimental conditions. ND, not determined; −MTS, filled squares; +MTS, empty squares.
As shown in Fig. 5 B, for all functional mutants, both activation indices lay within or close to the WT range. Moreover, the indices before and after MTS exposure were similar for individual mutants. This suggested that at −50 mV, assuming an invariant Na+:Pi 3:1 stoichiometry and that the degree of cooperativity of Na+ binding does not deviate significantly from the WT, neither the cys substitution nor MTS modification had a significant effect on the apparent substrate affinities (K m Pi and K m Na), and was most likely not the underlying reason for the altered transport activity.
Cys Substitution and Cys Modification Alter Voltage Dependency
Further insight into the mechanism underlying the changes in transport activity after cys modification was obtained from the current–voltage (I–V) relationships for Pi-dependent current. These were determined for superfusion with ND100 in response to the application of 1 mM Pi over voltage window −120 ≤ V ≤ +20 mV, in which contamination by endogenous currents was negligible. I–V data for four mutants that showed significantly changed activity after MTSEA treatment (1 mM, 3–5 min), G134C, I136C, F137C (ECL-1), and M533C (ECL-4), are shown in Fig. 6 A. Similar I–V data was also obtained using the positively charged MTSET and the negatively charged MTSES (unpublished data). To account for differences in expression levels between oocytes that expressed the same mutant, I–V data were normalized to the magnitude of I Pi at −100 mV (1 mM Pi) before MTSEA exposure for WT, I136C, F137C and M533C. In the case of G134C, data were normalized to the magnitude of I Pi after MTSEA exposure to aid comparison with the WT data. For the WT, MTSEA, MTSET, MTSES treatment at concentrations up to 10 mM resulted in insignificant changes to I Pi over the voltage window shown (unpublished data).
Figure 6.

Current–voltage (I–V) curves for selected mutants. (A) Mutants that showed significantly decreased electrogenic activity at −50 mV after MTSEA exposure: I136C (1), F137C (2) in ECL-1, and M533C (3) in ECL-4, before MTSEA exposure (filled squares) and after 3–5 min exposure to 1 mM MTSEA (empty squares) (n ≥ 4). G134C (4) in ECL-1 showed increased electrogenic activity at −50 mV after MTSEA exposure. Each data point is the change in holding current induced by 1 mM Pi (pH 7.4, in ND100), normalized to the magnitude of I Pi at −100 mV, 1 mM Pi, before MTSEA exposure. For G134C, the data were normalized to the magnitude of I Pi at −100 mV after MTSEA exposure. WT steady-state I–V data for I Pi (filled circles) for WT-expressing oocytes, normalized to the magnitude of I Pi at −100 mV (n = 4), are superimposed. SEMs smaller than symbol size are not shown. (B) I–V data showing the respective leak current (top) and Pi-dependent current (1 mM Pi) (bottom) for a representative oocyte that expressed M533C before (filled squares) and after (empty squares) MTSEA exposure; data points joined by dotted lines. The leak was estimated from the response to superfusion in 1 mM PFA (−I PFA). The Pi-dependent currents were leak corrected by adding the respective leak values at each test potential before MTSEA exposure (filled diamonds) and after MTSEA exposure (empty diamonds); data points joined by continuous lines. (C) I–V data showing the respective leak current (top) and Pi-dependent current (1 mM Pi) (bottom) for a representative oocyte that expressed G134C before (filled squares) and after MTSEA exposure (empty squares); data points joined by dotted lines. The Pi-dependent currents were leak corrected by adding the respective leak values at each test potential before MTSEA exposure (filled diamonds) and after MTSEA exposure (empty diamonds); data points joined by continuous lines.
The I–V curves of three mutants that showed a significant decrease in both 32Pi uptake (Fig. 2 A) and electrogenic activity at −50 mV after MTS exposure (Fig. 2 B), I136C (Fig. 6 A, 1), F137C (Fig. 6 A, 2), and M533C (Fig. 6 A, 3), each showed only a small deviation from the normalized WT I–V relation before MTS exposure. This indicated that the cys substitution was reasonably well tolerated at these sites. At 0 mV after MTS incubation, there was a loss of transport activity, compared with WT, with the relative residual activity of F137C being significantly lower than the other mutants. Interestingly, the I–V data for M533C + MTS showed a negative slope over the voltage window used for these assays, whereas I136C + MTS and F137C + MTS showed curvilinear I–V relationships. Mutant G134C (Fig. 6 A, 4), which gave a reduced 32Pi uptake (Fig. 2 A), but increased electrogenic activity at −50 mV (Fig. 2 B) also exhibited an I–V relationship with a negative slope before MTS exposure like M533C + MTS. After MTS exposure, the normalized I–V data for G134C showed a curvilinear I–V relationship compared with the WT.
The negative slopes for G134C − MTS and M533C + MTS were inconsistent with the expected behavior of an electrogenic secondary-active transport system in which one net positive charge is transported per cycle (Forster et al., 1998). However, we could reconcile this apparent anomaly when we took account of an error introduced by the subtraction procedure used to obtain I Pi. In these assays, we eliminated oocyte endogenous currents by subtracting the holding current in ND100 from the current in ND100 + Pi. However, if the leak and cotransport modes are assumed to be mutually exclusive (Kohler et al., 2002b), this procedure would underestimate the true cotransport mode current by an amount equal to the leak, if we assume that it is fully suppressed in the presence of 1 mM Pi. The leak mode activity was estimated by recording the change in holding current when superfusing in 1 mM PFA (−I PFA). Fig. 6 B shows the Pi-dependent and leak currents for a representative oocyte that expressed M533C (B) and G134C (C) before and after incubation in 1 mM MTSEA for 3 min. The effect of MTS exposure on the leak current was minimal over the voltage range −120 < V < 0. For V > 0 mV, we found that endogenous Cl− currents could also be induced by PFA in noninjected oocytes, and these data were therefore considered unreliable. When I Pi was corrected for the leak error by adding −I PFA at each potential, the leak-corrected I Pi data now showed a positive slope with voltage-independent rate-limiting behavior at hyperpolarizing potentials for M533C + MTS and G134C − MTS.
The unique behavior of the G134C steady-state I–V data before and after MTS exposure can also account for the apparent anomaly between the 32Pi uptake assay and the electrophysiological assay at V h = −50 mV for this mutant. As shown in Fig. 6 C there is a crossover of the leak-corrected G134C I–V data before and after MTS exposure at approximately −40 mV for this oocyte. Under voltage clamp conditions for V h < −50 mV, we would therefore predict an increased cotransport activity after MTS exposure. However, for the 32Pi assay conditions used in this study, the membrane potential (V m) was undefined. Therefore, externally applied Pi would induce a net inward positive charge movement (3 Na+ + 1 HPO4 2− per cycle) that would depolarize the oocyte membrane. If the shift in V m were more positive than the crossover potential, a reduced uptake would be expected, since 32Pi uptake and net translocated charge are tightly coupled for electrogenic type II Na+/Pi cotransporters (Forster et al., 1999).
To test this hypothesis, we measured the change in membrane potential (V m) with the voltage clamp loop open before and after MTS exposure. For the representative G134C-expressing oocyte in Fig. 6 C, application of 1 mM Pi induced a change in V m from −28 mV to +5 mV, whereas after MTS exposure, V m changed from −35 to −10 mV. For a given cell impedance, the Pi-dependent change in V m is proportional to the net charge translocated, which is a function of the number of transporters in the membrane (assumed constant) and the transporter turnover rate. It follows from these data that we would expect a reduced 32Pi uptake after MTS exposure, as we observed. We found that this result was also consistent with the corresponding currents at the V m reached during Pi application (−MTS, −56 nA; +MTS, −42 nA), which we found by interpolation of the leak-corrected I–V data of Fig. 6 C. The reduced depolarizing shift in V m after MTS exposure was confirmed for a batch (n = 9) of G134C-expressing oocytes, under the same conditions as for the tracer flux assay. The change in V m due to Pi superfusion was +31 ± 1 mV for −MTS, and this decreased to +19 ± 1 mV for +MTS. These findings serve to underscore the importance of defining the membrane potential in transport assays involving electrogenic membrane proteins.
From the I–V data of Fig. 6 A, it was obvious that the effect of cys substitution at Gly-134 and modification of Cys-136, Cys-137C, and Cys-533 was to reduce the transport activity induced by 1 mM Pi over a wide voltage range. By normalizing the leak-corrected I–V data to V = 0, the relative effect of hyperpolarizing membrane potentials on the cotransport activity before and after MTS exposure was determined (Fig. 7 A). These data showed that cys substitution at Ile-136 and Met-533 resulted in constructs with a voltage dependency that was indistinguishable from the WT. Cys modification at these sites increased (I136C + MTS) and decreased (M533 + MTS), respectively, the response to changes in membrane potential in the hyperpolarizing direction relative to the WT. Cys substitution at Phe-137 gave an increased voltage dependency compared with the WT that was further augmented after MTS exposure, like I136C + MTS. Finally, cys substitution at Gly-134 gave a reduced voltage dependency compared with the WT that was comparable to M533C + MTS. After MTS exposure, the voltage dependency of G134C was similar to that of I136C + MTS.
Figure 7.

Cysteine engineering at sites in ECL-1 and ECL-4 induces deviations from WT voltage dependency. (A) Voltage dependency of G134C (squares), I136C (triangles), F137C (inverted triangles), and M533C (diamonds), before (filled symbols) and after (open symbols) incubation in MTSEA (1 mM, 3 min). Pi-dependent currents, corrected for leak, were normalized to the response at 0 mV. The ordinate scale represents the relative change in I Pi as a function of membrane potential. Data pooled from n > 4 cells. WT data is represented by continuous line. (B) Voltage dependency index for all functional mutants given by ratio of response to 1 mM Pi at −100 mV to that at 0 mV (100 mM Na+) before (filled squares) and after (open squares) MTS treatment (1 mM MTSEA, 3 min). Continuous reference line indicates WT index. Dotted reference line indicates that G134C-MTS, S532C ± MTS, and M533C + MTS have the same index. Arrows indicate direction of voltage dependency change for selected mutants after MTS incubation.
To quantify changes in voltage dependency after MTS exposure of other functional mutants that had shown little or no change in electrogenic activity at −50 mV (Fig. 2 B), we assigned a voltage dependency index to each mutant. This was given by the ratio of leak-corrected I Pi at −100 mV to I Pi at 0 mV, before and after MTS exposure (Fig. 7 B). This index represents the change in cotransport activity for a voltage change from 0 to −100 mV. Cys substitution produced changes in this index relative to the WT for nearly all mutants. Mutants that showed no change in activity after MTS exposure also showed no significant change in their respective voltage dependency indices (G130C, V132C, D135C, K138C, D139C in ECL-1 and A534C, S532C in ECL-4); however, the cys substitution itself resulted in altered voltage dependencies compared with the WT. The general trend was for the voltage dependency index to increase after cys modification in both ECL-1 and ECL-4, with the notable exception of M533C in ECL-4.
DISCUSSION
In this study of the structure–function relationships of sites in putative external linkers ECL-1 and ECL-4 of the rat NaPi-IIa isoform, SCAM yielded both structural and functional information. First, it established the location of substituted sites in hydrophilic regions accessible from the external medium, in agreement with current topological predictions. Second, based on an analysis of the steady-state I–V relationships before and after cys modification, we show that cysteine engineering at sites in each linker determines the sensitivity of cotransport activity to changes in membrane potential.
To interpret our data, which is based on the introduction of novel Cys residues in the WT backbone, we made a tacit assumption that the target Cys residue was the novel Cys itself and not one or more of the 12 native Cys residues. Ideally, SCAM should be performed on a cys-less backbone to avoid ambiguities that could arise if the introduction of a novel Cys exposes a previously inaccessible and functionally important native Cys (Kaback et al., 2001; Kamdar et al., 2001). Unfortunately, a cys-reduced construct of NaPi-IIa shows significantly lower transport activity compared with the WT (Kohler et al., 2003) and this would exacerbate detailed kinetic analyses. Nevertheless, three pieces of evidence support the validity of the above assumption: (1) when removed singly, no native Cys appears to be critical for viable cotransport function (Lambert et al., 2000); (2) up to eight Cys can be removed, while retaining transport function (Kohler et al., 2003), the remaining four being involved in two disulfide bridges (Lambert et al., 2000; Kohler et al., 2003); and (3) introduction of a Cys at site 460, previously found to be functionally important for the WT backbone (Lambert et al., 1999b), shows the same behavior in the cys-reduced NaPi-IIa (Kohler et al., 2003).
Accessibility and Membrane Topology
Although we used MTSEA as the standard probe reagent, exposure to impermeant MTSET gave qualitatively similar results. This confirmed that the sites were accessible from the external medium only. Small differences in reactivity between MTSEA and MTSET were observed in the rates of modification (i.e., the rate of change of the electrogenic activity) at different sites in ECL-1 and ECL- 4 (Table I). The faster reaction rates of MTSET for most mutants most likely reflect the higher reactivity of this reagent with small thiols compared with MTSEA (Karlin and Akabas, 1998). For G134C and A538C, MTSEA was more reactive than MTSET, perhaps because the greater bulk of MTSET restricts its access to these sites. As summarized in Fig. 8, the effective second order reaction rate constants lay within the range of values that we have previously reported for Cys mutants in ECL-3 (Lambert et al., 2001) and establish conclusively that these three regions are accessible from the external aqueous environment. Rates of modification for extracellularly accessible sites in other transport proteins such as the excitatory amino acid transporter (EAAT1) (e.g., Leighton et al., 2002), the mitochondrial citrate transporter (Ma et al., 2004), and the serotonin transporter (SERT) (Chen et al., 1997) have been reported to lie in the same range as we found for NaPi-IIa. In the present study, the modification rates for mutants in ECL-1 were consistently faster than those in ECL-4. This would suggest that ECL-4 was less accessible from the extracellular medium. At one site in ECL-1 (Asp-135) and one site in ECL-4 (Trp-536) we were unable to detect the novel cysteines using the biotinylation assay, which indicated that these cysteines were inaccessible, either because of the bulk of MTSEA-Biotin or the specific folding of the linkers at these sites.
Figure 8.

Graphical representation of accessibility of three putative extracellular linkers based on effective second order reaction rate k* plotted on a logarithmic scale (filled squares). A larger k* indicates that the site has a greater accessibility from the external aqueous medium. Sites that were labeled with MTSEA-Biotin, for which the respective mutants showed no detectable change in activity, are indicated (empty squares). Data for ECL-1 and ECL-4 were obtained from this study (Table I); data for ECL-3 were replotted from a previous study (Lambert et al., 2001).
The functional consequences of exposure to MTS reagents varied considerably among neighbors. For example, Cys-135 could not be labeled with MTSEA-Biotin, and, consistent with this result, mutant D135C showed no change in its kinetics after MTS exposure. In contrast, we could detect its immediate neighbors (Cys-134 and Cys-136) with MTSEA-Biotin; however, G134C and I136C showed significantly altered voltage dependencies after MTS exposure. In other cases, cys substitution did not result in MTS-sensitive mutants, although we could successfully detect the novel cysteines with the biotinylation assay (e.g., Cys-130, Cys-132, Cys-138, and Cys-139). This suggested that these sites were not functionally critical. Finally, the striking behavior of mutant G134C indicated that the native Gly at this site is crucial for normal electrogenic behavior. Gly-134 is conserved among a number of NaPi-II isoforms, and its small size might confer a unique flexibility to this linker and the associated transmembrane domains that is restricted when replaced with a bulkier Cys.
Modulation of Steady-state Voltage-dependent Kinetics
In a previous SCAM study, a cluster of mutants was found in ECL-3, most of which showed full suppression of cotransport mode activity after MTS exposure (Lambert et al., 2001). These constructs yielded characteristically similar Pi and PFA responses at the end of the modification reaction, from which we concluded that Pi could interact with NaPi-IIa and thereby inhibit the leak mode, but the cotransport mode was suppressed. This led us to postulate that these sites are associated with the transport pathway itself (Lambert et al., 1999a, 2001; Kohler et al., 2002a,b). In the present study, all the MTS-sensitive mutants, except A538C, showed a Pi-dependent electrogenic response at −50 mV that asymptoted to a nonzero level, at the end of the modification reaction. This behavior suggested that they may not be directly associated with the transport pathway or the substrate recognition site as, in the latter case, kinetic analysis of these mutants under voltage clamp conditions revealed that apparent substrate affinities appeared unchanged, either from cys substitution or cys modification. On the other hand, cysteine engineering at sites in ECL-1 and ECL-4 resulted in significantly altered voltage-dependent activity compared with the WT, depending on the site of cys substitution or cys modification.
Mutants could be broadly classified according to whether there was increased, decreased, or unchanged activity compared with the WT for hyperpolarizing membrane potentials (0 to −100 mV) (Fig. 7 B). The finding that most mutants deviated from WT behavior suggested that the amino acid residues in ECL-1 and ECL-4 were critical determinants of the NaPi-IIa voltage-dependent kinetics. Several “hot spots” emerged from these assays, for which the voltage-dependent activity of the corresponding mutants deviated significantly from the WT, either due to cys substitution alone, cys modification, or both. For example, cys substitution at Ile-136 increased the voltage-dependent activity, whereas at the neighboring Phe-137, cys substitution was well tolerated. However, cys modification at both sites led to a significantly increased voltage-dependent activity. Interestingly, cys substitution at Gly-130 and Ile-136 led to the same increase in activity. This was mimicked by cys modification of Cys-134, intermediate in the amino acid sequence in ECL-1, and by cys substitution at Ala-534 and Ala-538 in ECL-4. Such similar behavior indicated that engineering at sites in each linker had induced the same voltage-sensitive conformational changes in the protein. Complementary behavior was also documented, in which the opposite effects on voltage dependency were found for cys substitution at Gly-134 in ECL-1 and Met-533 in ECL-4. G134C-MTS and M533C + MTS both exhibited a weakly voltage-dependent activity, whereas that of G134C + MTS exceeded the WT, and the voltage dependency of M533C − MTS was indistinguishable from the WT. Interestingly, cys substitution at the Ser-532 resulted in the same weak voltage-dependent activity as shown by M533C + MTS with little change in activity after MTS exposure. This suggested that the conformational change induced by modification of Cys-533 in ECL-4 was the same as that made by introducing a cysteine at the preceding site (Ser-532), or at Gly-134 in ECL-1. The novel complementary behavior of G134C and M533C is the subject of a more detailed kinetic analysis in the accompanying study (Ehnes et al., 2004).
In addition to relative changes in voltage-dependent activity, the I–V behavior revealed that for G134C − MTS and M533C + MTS, the maximum transport rate at the hyperpolarizing limit was also affected. In each case, the leak-corrected I–V curves showed rate-limiting behavior in the same voltage range where the WT, G134C + MTS, and M533C − MTS voltage-dependent activity was still approximately linear. The overall behavior of these mutants suggested that the cys substitution and cys modification at these sites had (a) slowed one or more voltage-independent rate constants in the transport cycle and (b) induced a depolarizing shift in voltage-dependent transitions associated with either the empty carrier, first Na+ binding transition, or both, in accordance with the current alternating access model for NaPi-IIa (Forster et al., 2002).
Are sites in ECL-1 and ECL-4 part of the voltage-sensing mechanism of NaPi-IIa? Three pieces of evidence suggest this is not the case. First, one might expect mobile charged or polar residues to constitute the voltage sensor and their removal would have a dramatic effect on the electrogenic behavior of the protein. Although ECL-4 has no charged residues, three sites in ECL-1 (D-135, K-138, and D-139) could be potential candidates, but when replaced by cysteines, the resulting mutants were still electrogenic (Fig. 7 B), albeit with only small deviations in voltage dependency from the WT. Similarly, substitution of the polar Ser-532 (ECL-4) with a nonpolar Cys weakened the voltage dependency (Fig. 7 B). These findings suggest that the particular charged or polar residues may certainly interact with the putative voltage-dependent elements to modulate the voltage dependency of transport, but the intrinsic voltage sensitivity of the protein remains intact. In another electrogenic cotransporter, the Na+-coupled glucose transporter (SGLT-1), polar residues have been shown to modulate charge movements and alter empty carrier kinetics without affecting the overall apparent valency of the cotransporter (Panayotova-Heiermann et al., 1994). Second, one member of the SLC34 family, the renal type IIc isoform, is electroneutral (Segawa et al., 2002; unpublished data). NaPi-IIc has a high homology with the electrogenic type IIa Na+/Pi cotransporter, with both charged residues in ECL-1 being conserved (Fig. 1 B). Third, the same MTS-induced changes to the voltage dependency occurred using either positively or negatively charged MTS reagents. This suggests that the charge introduced by these reagents either lies outside the transmembrane field or does not interact with the intrinsic voltage-sensing elements.
In summary, the outermost external linkers of the NH2- and COOH-terminal halves of NaPi-IIa contain sites that when occupied by Cys residues, are localized in an externally accessible, hydrophilic environment, which is consistent with hydropathy predictions. The native residues in these linkers are not absolutely essential for Na+-coupled Pi cotransport function; however, cys substitution or covalent modification of the novel cysteines by externally applied MTS reagents affected the voltage-dependent kinetics of the mutant transporters. As changes in membrane potential are expected to induce major conformational changes in the native protein during the transport cycle (e.g., Loo et al., 1998), the findings reported here suggest that linkers ECL-1 and ECL-4 contain specific sites that may interact directly or indirectly with mobile, voltage-sensitive elements of the NaPi-IIa protein.
Acknowledgments
We thank Dr. Leila Virkki (University of Zurich) for helpful comments on the manuscript.
This work was supported by grants to H. Murer from the Swiss National Science Foundation (31-46523), the Hartmann Müller-Stiftung (Zurich), the Olga Mayenfisch-Stiftung (Zurich), and the Union Bank of Switzerland (Zurich) (Bu 704/7-1).
Lawrence G. Palmer served as editor.
C. Ehnes and I.C. Forster contributed equally to this paper.
K. Kohler's present address is Laboratory of Morphogenesis and Cell Signaling, UMR144, Institut Curie, Paris, France.
Abbreviations used in this paper: DTT, dithiothreitol; MTS, methanethiosulfonate; MTSEA, 2-aminoethyl MTS hydrobromide; MTSES, sodium (2-sulfonatoethyl) MTS; MTSET, 2-(trimethylammonium)ethyl MTS bromide; NaPi-IIa, type IIa Na+/Pi cotransporter; PFA, phosphonoformic acid; SCAM, substituted cysteine accessibility method; WT, wild type.
References
- Chen, J.G., S. Liu-Chen, and G. Rudnick. 1997. External cysteine residues in the serotonin transporter. Biochemistry. 36:1479–1486. [DOI] [PubMed] [Google Scholar]
- Custer, M., M. Lotscher, J. Biber, H. Murer, and B. Kaissling. 1994. Expression of Na-P(i) cotransport in rat kidney: localization by RT-PCR and immunohistochemistry. Am. J. Physiol. 266:F767–F774. [DOI] [PubMed] [Google Scholar]
- Ehnes, C., I.C. Forster, K. Kohler, A. Bacconi, J. Biber, and H. Murer. 2004. Structure–function relations of the first and fourth extracellular linkers of the type IIa Na+/Pi cotransporter. II. Substrate interaction and voltage dependency of two functionally important sites. J. Gen. Physiol. 124:489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster, I., N. Hernando, J. Biber, and H. Murer. 1998. The voltage dependence of a cloned mammalian renal type II Na+/Pi cotransporter (NaPi-2). J. Gen. Physiol. 112:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster, I., D.D. Loo, and S. Eskandari. 1999. Stoichiometry and Na+ binding cooperativity of rat and flounder renal type II Na+-Pi cotransporters. Am. J. Physiol. 276:F644–F649. [DOI] [PubMed] [Google Scholar]
- Forster, I.C., K. Kohler, J. Biber, and H. Murer. 2002. Forging the link between structure and function of electrogenic transporters: the renal type IIa Na+/Pi cotransporter as a case study. Prog. Biophys. Mol. Biol. 80:69–108. [DOI] [PubMed] [Google Scholar]
- Holmgren, M., Y. Liu, Y. Xu, and G. Yellen. 1996. On the use of thiol-modifying agents to determine channel topology. Neuropharmacology. 35:797–804. [DOI] [PubMed] [Google Scholar]
- Kaback, H.R., M. Sahin-Toth, and A.B. Weinglass. 2001. The kamikaze approach to membrane transport. Nat. Rev. Mol. Cell Biol. 2:610–620. [DOI] [PubMed] [Google Scholar]
- Kamdar, G., K.M. Penado, G. Rudnick, and M.M. Stephan. 2001. Functional role of critical stripe residues in transmembrane span 7 of the serotonin transporter. Effects of Na+, Li+, and methanethiosulfonate reagents. J. Biol. Chem. 276:4038–4045. [DOI] [PubMed] [Google Scholar]
- Karlin, A., and M.H. Akabas. 1998. Substituted-cysteine accessibility method. Methods Enzymol. 293:123–145. [DOI] [PubMed] [Google Scholar]
- Kohler, K., I.C. Forster, G. Lambert, J. Biber, and H. Murer. 2000. The functional unit of the renal type IIa Na+/Pi cotransporter is a monomer. J. Biol. Chem. 275:26113–26120. [DOI] [PubMed] [Google Scholar]
- Kohler, K., I.C. Forster, G. Stange, J. Biber, and H. Murer. 2002. a. Identification of functionally important sites in the first intracellular loop of the NaPi-IIa cotransporter. Am. J. Physiol. Renal Physiol. 282:F687–F696. [DOI] [PubMed] [Google Scholar]
- Kohler, K., I.C. Forster, G. Stange, J. Biber, and H. Murer. 2002. b. Transport function of the renal type IIa Na+/Pi cotransporter is codetermined by residues in two opposing linker regions. J. Gen. Physiol. 120:693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler, K., I.C. Forster, G. Stange, J. Biber, and H. Murer. 2003. Essential cysteine residues of the type IIa Na+/Pi cotransporter. Pflugers Arch. 446:203–210. [DOI] [PubMed] [Google Scholar]
- Lambert, G., I.C. Forster, G. Stange, J. Biber, and H. Murer. 1999. a. Properties of the mutant Ser-460-Cys implicate this site in a functionally important region of the type IIa Na+/Pi cotransporter protein. J. Gen. Physiol. 114:637–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, G., M. Traebert, N. Hernando, J. Biber, and H. Murer. 1999. b. Studies on the topology of the renal type II NaPi-cotransporter. Pflugers Arch. 437:972–978. [DOI] [PubMed] [Google Scholar]
- Lambert, G., I.C. Forster, J. Biber, and H. Murer. 2000. Cysteine residues and the structure of the rat renal proximal tubular type II sodium phosphate cotransporter (rat NaPi IIa). J. Membr. Biol. 176:133–141. [DOI] [PubMed] [Google Scholar]
- Lambert, G., I.C. Forster, G. Stange, K. Kohler, J. Biber, and H. Murer. 2001. Cysteine mutagenesis reveals novel structure-function features within the predicted third extracellular loop of the type IIa Na+/Pi cotransporter. J. Gen. Physiol. 117:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton, B.H., R.P. Seal, K. Shimamoto, and S.G. Amara. 2002. A hydrophobic domain in glutamate transporters forms an extracellular helix associated with the permeation pathway for substrates. J. Biol. Chem. 277:29847–29855. [DOI] [PubMed] [Google Scholar]
- Loo, D.D., B.A. Hirayama, E.M. Gallardo, J.T. Lam, E. Turk, and E.M. Wright. 1998. Conformational changes couple Na+ and glucose transport. Proc. Natl. Acad. Sci. USA. 95:7789–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, C., R. Kotaria, J.A. Mayor, L.R. Eriks, A.M. Dean, D.E. Walters, and R.S. Kaplan. 2004. The mitochondrial citrate transport protein: probing the secondary structure of transmembrane domain III, identification of residues that likely comprise a portion of the citrate transport pathway, and development of a model for the putative TMDIII-TMDIII interface. J. Biol. Chem. 279:1533–1540. [DOI] [PubMed] [Google Scholar]
- Magagnin, S., A. Werner, D. Markovich, V. Sorribas, G. Stange, J. Biber, and H. Murer. 1993. Expression cloning of human and rat renal cortex Na/Pi cotransport. Proc. Natl. Acad. Sci. USA. 90:5979–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murer, H., N. Hernando, I. Forster, and J. Biber. 2000. Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol. Rev. 80:1373–1409. [DOI] [PubMed] [Google Scholar]
- Murer, H., N. Hernando, I. Forster, and J. Biber. 2003. Regulation of Na/Pi transporter in the proximal tubule. Annu. Rev. Physiol. 65:531–542. [DOI] [PubMed] [Google Scholar]
- Panayotova-Heiermann, M., D.D. Loo, M.P. Lostao, and E.M. Wright. 1994. Sodium/D-glucose cotransporter charge movements involve polar residues. J. Biol. Chem. 269:21016–21020. [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and A.M. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 18.66–18.75.
- Segawa, H., I. Kaneko, A. Takahashi, M. Kuwahata, M. Ito, I. Ohkido, S. Tatsumi, and K. Miyamoto. 2002. Growth-related renal type II Na/Pi cotransporter. J. Biol. Chem. 277:19665–19672. [DOI] [PubMed] [Google Scholar]
- Turk, E., C.J. Kerner, M.P. Lostao, and E.M. Wright. 1996. Membrane topology of the human Na+/glucose cotransporter SGLT1. J. Biol. Chem. 271:1925–1934. [DOI] [PubMed] [Google Scholar]
- Werner, A., J. Biber, J. Forgo, M. Palacin, and H. Murer. 1990. Expression of renal transport systems for inorganic phosphate and sulfate in Xenopus laevis oocytes. J. Biol. Chem. 265:12331–12336. [PubMed] [Google Scholar]
- Zhang, H., and A. Karlin. 1997. Identification of acetylcholine receptor channel-lining residues in the M1 segment of the beta-subunit. Biochemistry. 36:15856–15864. [DOI] [PubMed] [Google Scholar]