

Abstract
We examined functional contributions of interdomain contacts within the nicotinic receptor ligand binding site using single channel kinetic analyses, site-directed mutagenesis, and a homology model of the major extracellular region. At the principal face of the binding site, the invariant αD89 forms a highly conserved interdomain contact near αT148, αW149, and αT150. Patch-clamp recordings show that the mutation αD89N markedly slows acetylcholine (ACh) binding to receptors in the resting closed state, but does not affect rates of channel opening and closing. Neither αT148L, αT150A, nor mutations at both positions substantially affects the kinetics of receptor activation, showing that hydroxyl side chains at these positions are not hydrogen bond donors for the strong acceptor αD89. However substituting a negative charge at αT148, but not at αT150, counteracts the effect of αD89N, demonstrating that a negative charge in the region of interdomain contact confers rapid association of ACh. Interpreted within the structural framework of ACh binding protein and a homology model of the receptor ligand binding site, these results implicate main chain amide groups in the domain harboring αW149 as principal hydrogen bond donors for αD89. The specific effect of αD89N on ACh association suggests that interdomain hydrogen bonding positions αW149 for optimal interaction with ACh.
Keywords: acetylcholine receptor, ligand binding site, single channel kinetics, hydrogen bond, structural model
INTRODUCTION
Chemical recognition of neurotransmitter is the first step in activation of post-synaptic receptors, and is accomplished by specialized structures within the major extracellular region of the receptor protein. In the Cys-loop superfamily of receptors, these specialized structures are formed at interfaces between subunits where multiple domains, spaced far apart along the protein chains of the subunits, converge to form the recognition site. In the muscle nicotinic receptor, recognition domains A–C originate from the α subunit and domains D–G from the δ, γ, or ɛ subunits (for review see Sine, 2002). Atomic-scale insight into the structure of the agonist recognition site emerged with x-ray structures of acetylcholine binding protein (AChBP; Brejc et al., 2001; Celie et al., 2004), a structural homologue to Cys-loop receptors, and with homology models derived from it (Sine et al., 2002a; Le Novere et al., 2002; Molles et al., 2002; Schapira et al., 2002).
In the muscle nicotinic receptor, the ACh binding site is a specialized pocket of aromatic and hydrophobic residues, as shown by AChBP, homology models of the major extracellular region, affinity labeling, and mutagenesis combined with functional measurements (for reviews see Corringer et al., 2000; Karlin, 2002; Sine, 2002). Among the multiple aromatic residues in the binding pocket, αW149 emerged as a strong candidate for stabilizing the quaternary ammonium moiety of ACh (Zhong et al., 1998), which was confirmed by the x-ray structure of AChBP with bound carbamylcholine (Celie et al., 2004). Contained within recognition domain B of the binding site, αW149 is part of a larger, highly conserved structural motif that includes hydroxyl side chains at positions αT148 and αT150 and the closely juxtaposed domain A, which harbors the invariant αD89 (Fig. 1 D). In AChBP, the Asp equivalent to αD89 hydrogen bonds with hydroxyl side chains of Ser and Thr residues flanking the Trp equivalent to αW149, as well as with the main chain amide of the residue at the position equivalent to αT150 (Celie et al., 2004; Fig. 1 C). These hydrogen bonds are likely critical for stabilizing the protein main chain of recognition domain B and consequently for determining the orientation and mobility of the key binding site Trp.
Figure 1.

Sequence alignment and relationship between recognition domains A and B in AChBP and our structural model of receptor ligand binding domain. (A) Alignment of local regions of AChBP and α subunits from different species. (B) Mutagenesis-based homology model of the human muscle receptor ligand binding domain (Sine et al., 2002a). The α subunit is highlighted in magenta and ɛ subunit in yellow, with the remaining subunits in gray. The box encloses recognition domains A (orange) and B (green) of the α subunit. Close up views of the box in B for AChBP (C) and receptor (D) with side chains of key residues rendered in ball and stick representation. Structures in C and D are views from inside the central vestibule. Dotted lines indicate hydrogen bonds with the following distances. AChBP: D85 oxygen1 to T144 hydroxyl, 2.7 Å; D85 oxygen1 to T144 main chain amide, 2.9 Å; D85 oxygen2 to S142 hydroxyl, 2.6 Å. Receptor: αD89 oxygen1 to αW149 main chain amide, 2.7 Å; αD89 oxygen1 to αT150 main chain amide, 2.5 Å.
To determine whether αD89 contributes to the function of the muscle nicotinic receptor by interacting with recognition domain B, we generated receptors containing mutations of αD89, αT148, and αT150, recorded single channel currents, and measured rate constants underlying elementary steps in receptor activation. We find that αD89 plays a key role in receptor activation by maintaining a high rate of ACh association and optimizing the rate of ACh dissociation; this is achieved by serving as a strong hydrogen bond acceptor for donors in domain B that in turn may optimize αW149 for interaction with the agonist.
MATERIALS AND METHODS
Construction of Wild-type and Mutant AChRs
Human α, β, δ, and ɛ subunit cDNAs subcloned in the CMV-based mammalian expression vector pRBG4 (Lee et al., 1991) were described elsewhere (Ohno et al., 1996). Site-directed mutations were made using the QuickChange site-directed mutagenesis kit (Stratagene). The presence of each mutation and absence of unwanted mutations was confirmed by sequencing the entire cDNA insert.
Mammalian Cell Expression
All experiments were performed using the BOSC 23 cell line (Pear et al., 1993; Wang et al., 2000), which is a variant of the 293 HEK cell line. Cells were maintained in the Dulbecco's Modified Eagle Medium (DMEM) containing FBS (10% vol/vol) at 37°C until they reached ∼50-70% confluence. Wild-type or mutant AChR cDNAs were then transfected by calcium-phosphate precipitation using final cDNA concentrations of 0.68 μg/ml, except the α-subunit cDNA, which was 1.36 μg/ml. Patch-clamp and [125I]α-bungarotoxin binding measurements were performed 2 and 3 d after transfection, respectively.
Patch-clamp Recordings
To record single channel currents, cells transfected with wild-type or mutant AChR cDNAs were rinsed with and maintained in the following bath solution (in mM): KCl 142, NaCl 5.4, CaCl2 1.8, MgCl2 1.7, and HEPES 10 (pH was adjusted to 7.4). The same solution was used to fill patch pipettes. ACh (Sigma-Aldrich) was kept as a 100 mM stock, dissolved in bath solution and stored at −80°C until use. Glass micropipettes (type 7052; Garner Glass Co.) were coated with Sylgard 184 (Dow Corning Co.) and heat polished to yield resistances of 5–8 MΩ. Single channel currents were recorded in the cell-attached configuration at 21°C using the Axopatch 200A (Axon Instruments, Inc.) at a membrane potential of −70 mV. Data were collected from two to four different patches for each ACh concentration, choosing only recordings in which channel activity was low enough to allow clear identification of activation episodes resulting from a single channel. The current signal was low pass filtered at 50 kHz and recorded to hard disk at 200 kHz using the program Acquire (Bruxton Co.).
Single Channel Kinetic Analysis
The digitized current signal was filtered using a 10-kHz digital Gaussian filter (Colquhoun and Sigworth, 1983), and channel events were detected by the half-amplitude threshold criterion using the program TAC using a dead time of 10 μs (Bruxton Co.). Precise determination of dwell time at threshold was achieved by cubic spline interpolation of the digital signal and correcting the measured dwell time at threshold for the effects of the Gaussian filter as previously described (Colquhoun and Sigworth, 1983). Open and closed time histograms were fitted by the sum of exponentials by maximal likelihood using the program TACFit (Bruxton Co.). Openings corresponding to a single receptor channel were identified by assigning a critical closed time defined as the intersection of the closed time component that depended on ACh concentration with the succeeding concentration-independent component due to rapid desensitization (Ohno et al., 1996). Because this method removes most but not all closed dwell times due to rapid desensitization, analyses were also performed in which the critical closed time was defined as the intersection of the longest closed time component, due to slow onset desensitization and the number of channels in the patch, with the adjacent component due to intermediate onset desensitization. To obtain kinetically homogeneous data, defined single channel episodes containing at least five openings were analyzed for open probability, mean open time, and mean closed time, and episodes within two standard deviations of the mean were accepted for further analysis (Wang et al., 1997). It should be mentioned that this processing removes a small population of brief openings flanked by long closed periods (Sine and Steinbach, 1987). For the receptors examined in this study, this selection typically retained ∼85–95% of the detected channel events, yielding 4,000–8,000 events per single patch recording. Kinetic analysis was performed on data obtained across a range of ACh concentrations simultaneously using MIL software (QuB suite, State University of New York), which employs a maximum likelihood method, corrects for missed events, and gives error estimates of the fitted parameters (Qin et al., 1996). An instrument dead time of 22 μs was uniformly applied to all recordings. For a given wild-type or mutant receptor, our global analysis included data from two to four patches for each ACh concentration, ACh concentrations spaced at half log-unit intervals and a 10- to 333-fold range of the ACh concentration. For each receptor mutant, the range of the ACh concentration spanned from the minimum to the maximum channel open probability (see Fig. 7).
Figure 7.

Channel open probability (Popen) is plotted against ACh concentration for receptors containing the indicated α subunits. Symbols represent experimentally measured Popen from the mean fraction of time the channel is open within activation episodes from single receptor channels. Each symbol and error bar corresponds to mean ± SD from two to four different patches. Curves represent predicted Popen calculated from fitted rate constants in Tables I and II. The concentration dependence of Popen for αD89N receptors shifts to high ACh concentrations relative to wild type, but is partially reversed by combination with the αT148D mutation.
[125I]α-Bungarotoxin Binding Measurements
Cell surface expression of wild-type and mutant receptors was determined by measuring [125I]α-bungarotoxin (125I-α-Btx; PerkinElmer) binding to intact cells. 3 d after transfection, cells were harvested using PBS containing EDTA (5 mM), and resuspended in high potassium Ringer solution buffered with 25 mM HEPES and supplemented with 0.3% BSA. The cell suspension was incubated with 5 nM 125I-α-Btx for 1 h and harvested (Brandel Inc.) to Whatman GF-B glass fiber filters (1 μm cutoff; Brandel Inc.). Nonspecific binding was determined in the presence of 100 μM d-tubocurarine. Total specific binding for each mutant was normalized to that of the wild-type receptor.
RESULTS
Conserved Structural Motif Formed by Recognition Domains A and B in the α Subunit
At the ligand binding site of AChBP, recognition domain A corresponds to the linker spanning β-strands 3 and 4, while recognition domain B corresponds to the linker spanning β-strands 7 and 8 (Brejc et al., 2001). These recognition domains are stabilized by hydrogen bonds between the carboxylate moiety of D85 in the β3-4 linker and hydroxyl side chains of S142 and T144 and the main chain amide of T144 in the β7-8 linker (Celie et al., 2004; Fig. 1 C). Hydrogen bonding between these domains likely affects the orientation and mobility of both the main chain carbonyl oxygen and indole side chain of the key binding site residue W143. All nicotinic receptor α subunits contain Asp at the position equivalent to D85, while in most subtypes and species of α subunits, residues with hydroxyl side chains are found at positions equivalent to those flanking the key W143 (Fig. 1 A). A structural motif containing four residues equivalent to these is also present in our homology model of the major extracellular region from the human muscle receptor (Sine et al., 2002a; Fig. 1 D), but whether hydrogen bonds between domains A and B form and are functionally significant is not known. By analogy to AChBP, the αD89 side chain in the receptor is a candidate for linking domains A and B through hydrogen bonding, which could affect conformation of αW149 and the local main chain and thus the kinetics of receptor activation by ACh.
Single Channel Kinetic Analysis of the Adult Human Receptor
To provide a frame of reference for comparison with mutant receptors examined in this study, we first describe kinetic analysis of ACh-evoked single channel currents through the wild-type human receptor. To record these currents, we cotransfected α, β, ɛ, and δ subunit cDNAs in BOSC 23 cells and employed the cell-attached mode of the patch-clamp (see materials and methods). Application of high concentrations of ACh in the patch pipette causes clustering of current pulses into epochs during which only one channel is active (Sakmann et al., 1980; Sine and Steinbach, 1987; Colquhoun and Ogden, 1988). At progressively increasing concentrations of ACh, closed periods between openings of a single receptor become briefer, and a corresponding closed time component shifts from long to brief times (Fig. 2). Distributions of open times change from bi- to mono-exponential as the ACh concentration is increased, but the mean duration of the major component of openings is relatively insensitive to ACh concentration. These qualitative features of the concentration dependence of dwell times are similar to those described previously (Sine et al., 1990; Zhang et al., 1995; Ohno et al., 1996; Hatton et al., 2003).
Figure 2.
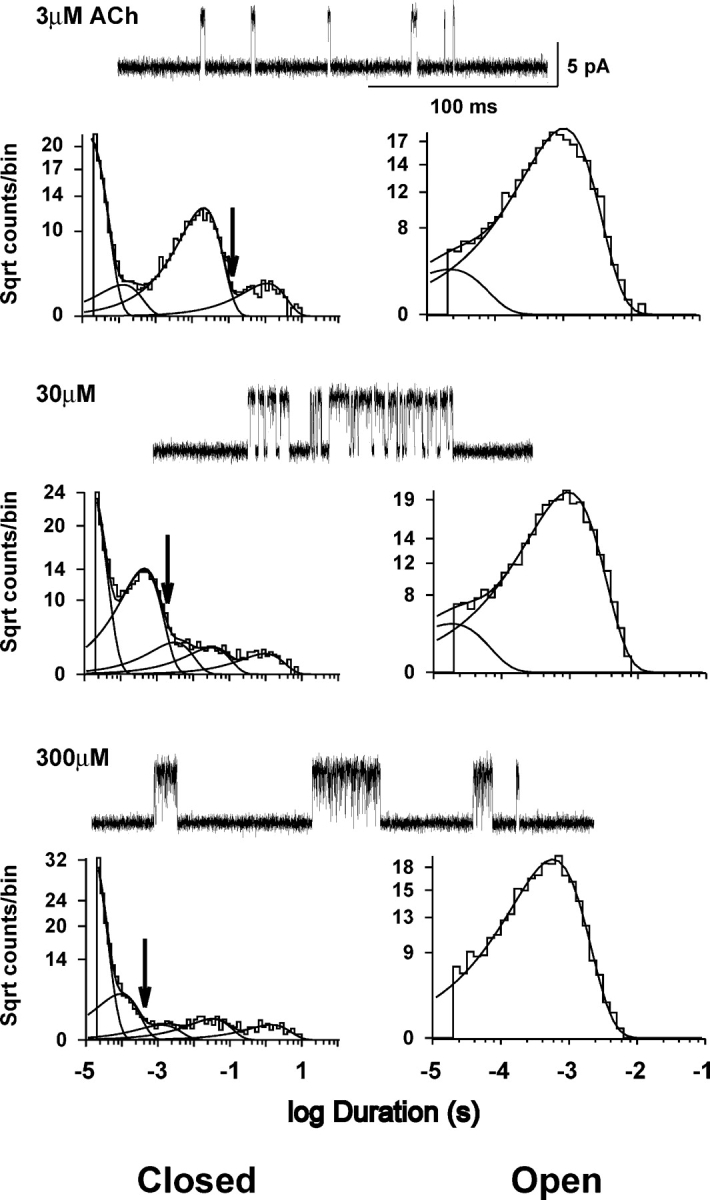
ACh-evoked single channel currents and dwell time histograms from human wild-type receptors expressed in BOSC 23 cells. Traces of single channel current clusters at the indicated concentrations of ACh are shown at a bandwidth of 10 kHz with openings upward deflections. Closed (left) and open (right) time histograms are shown with logarithmic axes and fitted to the sum of exponential components (smooth curves). Arrows indicate critical closed time used to distinguish closed times due to activation from those due to fast, intermediate, and slow desensitization processes.
The first step in kinetic analysis is to identify single channel events due to receptor activation and remove closed intervals due to receptor desensitization. At concentrations of ACh approaching saturation, closed intervals due to desensitization appear as three distinct components with small amplitudes (Fig. 2), corresponding to fast, intermediate, and slow desensitization processes (Elenes and Auerbach, 2002). Our previous analyses of the adult human receptor identified channel events due to receptor activation as a series of openings flanked by closed intervals greater than a defined critical closed time (Ohno et al., 1996; Wang et al., 1999; Sine et al., 2002b); this was taken as the point of intersection of the component that depended on ACh concentration with the succeeding component due to fast desensitization (Fig. 2, arrow). Using this method to identify channel events due to receptor activation, we applied maximum likelihood analysis of the closed and open intervals according to kinetic schemes (Qin et al., 1996; see materials and methods). The first scheme depicts receptor activation as sequential binding of ACh to receptors in the resting, closed state, followed by conformational change to the open state, and then block of the doubly occupied open receptor by ACh (Fig. 3, Scheme I). The second scheme also depicts sequential binding of ACh, but allows the initial binding event to occur at either binding site (Fig. 3, Scheme II). In both schemes, mono-liganded receptors can open, but in Scheme II only one of two possible mono-liganded openings is allowed; this restriction is necessary to obtain well-defined rate constants because only two components are present in the open time histograms (Fig. 2).
Figure 3.

Kinetic schemes for receptor activation. Receptors in the resting closed state are symbolized by R, the open state by R*, and blocked state by Rb. Agonist (A) associates with rate constants k+1, k+2, and k+b and dissociates with rate constants k−1, k−2, and k−b. Channels open with rate constants β1 and β2, and close with rate constants α1 and α2.
Both Schemes I and II can be fitted to receptor activation episodes obtained over a wide range of ACh concentrations, referred to as global fitting, yielding probability density functions that describe the entire set of open and closed time histograms for the human wild-type receptor (Fig. 4). The likelihood of Scheme I is greater than that of Scheme II, perhaps because the diagonal binding steps in Scheme II are constrained to be equal to reduce the number of free parameters (Table I). The fitted rate constants in Table I are generally similar to those obtained in previous analyses of the adult human receptor (Ohno et al., 1996; Wang et al., 1999; Sine et al., 2002b; Hatton et al., 2003), and demonstrate rapid and efficient opening of the doubly occupied receptor, distinct association and dissociation rate constants for each binding site, and transient, low affinity block of the open receptor channel by ACh.
Figure 4.
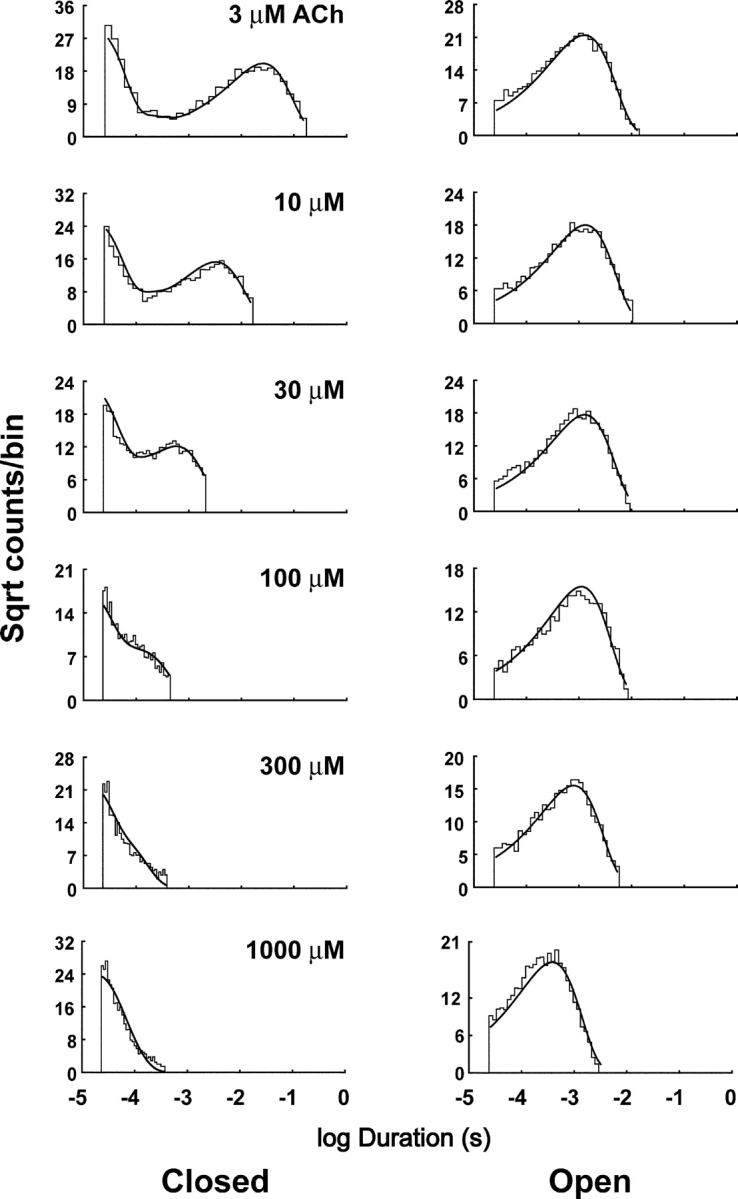
Kinetic analysis of the human wild-type receptor. Results of global fitting are shown by superimposing probability density functions (pdf; smooth curves) computed from the fitted rate constants (Scheme I, Table I) on dwell time histograms generated from currents elicited by the indicated concentrations of ACh. Fitted pdfs from Scheme II were indistinguishable from those from Scheme I.
TABLE I.
Kinetic Analysis of the Wild-type Human Receptor
| Schemes | k+1 | k−1 | K1 | k+2 | k−2 | K2 | β1 | α1 | Θ1 | β2 | α2 | Θ2 | k+b | k−b | KB | Log likelihood |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μM | μM | mM | ||||||||||||||
| I | 271 ± 16 |
7190 ± 545 |
26 | 192 ± 6 |
19300 ± 309 |
101 | 67 ± 8 |
2190 ± 160 |
0.03 | 43700 ± 999 |
1650 ± 32 |
26 | 26 ± 1 |
113000 ± 1690 |
4.3 | 345549 |
| II | 126 ± 7 |
4500 ± 399 |
35 | 182 ± 11 |
14000 ± 525 |
77 | 262 ± 29 |
2310 ± 200 |
0.11 | 44600 ± 1010 |
1670 ± 33 |
27 | 24 ± 1 |
110000 ± 1940 |
4.6 | 342585 |
| Ia | 229 ± 17 |
5180 ± 447 |
23 | 149 ± 5 |
17900 ± 289 |
120 | 62 ± 8 |
2210 ± 214 |
0.03 | 46500 ± 1060 |
1600 ± 33 |
29 | 25 ± 1 |
114000 ± 1670 |
4.6 | 458281 |
| IIa | 137 ± 8 |
5110 ± 587 |
37 | 174 ± 12 |
12600 ± 840 |
72 | 120 ± 22 |
2440 ± 246 |
0.05 | 43400 ± 1031 |
1460 ± 30 |
30 | 25 ± 1 |
111000 ± 1580 |
4.4 | 445053 |
Kinetic parameters and error estimates are derived from global fitting of kinetic schemes to data obtained over a wide range of ACh concentrations (materials and methods). Units are μM−1s−1 for the association rate constants, s−1 for all others. Gating equilibrium constants (Θ) are ratios of channel opening (β) to closing rate (α) constants.
Results of fitting Scheme I or Scheme II modified to contain two desensitized states linked to the open state (see text). Fitted desensitization rate constants, k+d/k−d, in units of s−1, are D1 (41/529) and D2 (65/30).
The analysis just described does not account for the small number of closed intervals arising from rapid and intermediate desensitization that fall below the critical closed time used to define channel events due to receptor activation. To see whether these closed intervals due to desensitization affect the estimated rate constants, we applied maximum likelihood analyses to modifications of Schemes I and II in which two desensitized states are each connected to the doubly occupied open state (A2R*). In this analysis, activation episodes are defined as a series of openings separated by closed intervals greater than the intersection of the longest closed time component with the adjacent closed time component (Fig. 2). Fitting modified versions of either Scheme I or II to the global data again yields excellent descriptions of closed and open interval histograms and reveals only minor changes in the rate constants underlying receptor activation (Table I). Thus the small number of misclassified closed intervals due to rapid and intermediate desensitization has little effect on the estimated rate constants for receptor activation.
Functional Contribution of αD89
To determine whether αD89 contributes to the kinetics of receptor activation, we constructed the mutation αD89N, cotransfected it with complementary β, ɛ, and δ subunit cDNAs in BOSC 23 cells and recorded ACh-evoked single channel currents. The Asn substitution introduces a relatively conservative structural change, neutralizing the negative charge but changing only the hydroxyl group of the original side chain. Compared with wild-type receptors, the mutant receptors show markedly prolonged closed intervals between successive openings of a single channel, indicating impaired kinetics of receptor activation (compare Figs. 2 and 5). However when the concentration of ACh is increased sufficiently, closed intervals from single mutant receptors approach the brief durations observed for single wild-type receptors. These qualitative observations are born out in the closed time histograms where the shift of the distributions from long to short closed times requires greater concentrations of ACh for αD89N compared with wild-type receptors (compare Figs. 4 and 5). Thus, αD89 plays a pivotal role in receptor activation, predominantly affecting the dependence of receptor activation kinetics on ACh concentration.
Figure 5.
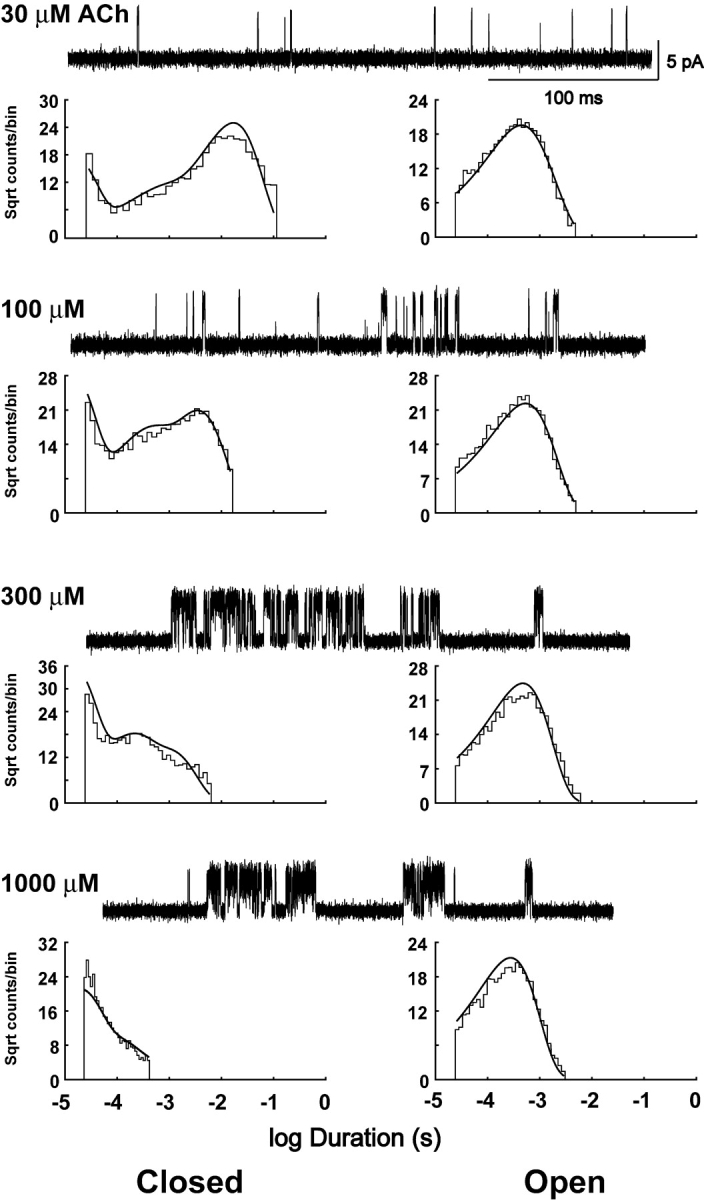
Clusters of single channel currents and kinetic analysis of receptors harboring the αD89N mutation. Current traces obtained at the indicated ACh concentrations are shown at a bandwidth of 10 kHz. Dwell time histograms are shown with pdfs generated from the Scheme II rate constants superimposed (see Table II). Fitting Scheme II yielded a higher log likelihood, 555,963, than Scheme I, 555,594.
To identify rate constants altered by αD89N, we analyzed open and closed dwell times according to Schemes I and II, as described for the wild-type receptor. Unlike the wild-type receptor, Scheme II yields a substantially higher likelihood than Scheme I (see legend to Fig. 5). The fitted rate constants show that αD89N dramatically slows the rate of ACh association with receptors in the resting closed state, but has a smaller effect on the rates of ACh dissociation; the net result is a 10- to 20-fold reduction in ACh affinity at the two binding sites (Table II).
TABLE II.
Kinetic Analysis of Mutant Receptors
| Mutant | k+1 | k−1 | K1 | k+2 | k−2 | K2 | β1 | α1 | Θ1 | β2 | α2 | Θ2 | k+b | k−b | KB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μM | μM | mM | |||||||||||||
| Wild type | 126 ± 7 |
4500 ± 399 |
35 | 182 ± 11 |
14000 ± 525 |
77 | 262 ± 29 |
2310 ± 200 |
0.11 | 44600 ± 1010 |
1670 ± 33 |
27 | 24 ± 1 |
110000 ± 1940 |
4.6 |
| αD89N | 3 ± 0.1 |
1380 ± 52 |
460 | 37 ± 1 |
51800 ± 1420 |
1400 | 2070 ± 64 |
3200 ± 72 |
0.64 | 40000a | 2500 ± 30 |
16 | 20 ± 1 |
97400 ± 1480 |
4.9 |
| αD89E | 14 ± 0.6 |
1160 ± 65 |
83 | 99 ± 3 |
33700 ± 744 |
340 | 962 | 3250 | 0.30 | 47500 ± 1620 |
2570 ± 63 |
18 | 21 ± 1 |
111000 ± 2500 |
5.3 |
| αD89Tb | 0.8 ± 0.03 |
910 ± 35 |
1140 | 44 ± 4 |
57000 ± 4680 |
1300 | 222 ± 23 |
3070 ± 323 |
0.07 | 49000 ± 6080 |
1380 ± 102 |
36 | 22 ± 1 |
107000 ± 2220 |
4.9 |
| αT148L | 114 ± 5 |
3600 ± 325 |
32 | 236 ± 11 |
16500 ± 626 |
70 | 249 ± 29 |
2450 ± 165 |
0.10 | 39400 ± 779 |
1680 ± 28 |
23 | 22 ± 1 |
109000 ± 1590 |
5.0 |
| αT150A | 334 ± 19 |
8480 ± 507 |
29 | 118 ± 3 |
26100 ± 778 |
221 | 287 ± 21 |
2220 ± 77 |
0.13 | 58000 ± 1860 |
2010 ± 52 |
29 | 20 ± 1 |
103000 ± 1860 |
5.2 |
| αT148L + T150A | 186 ± 18 |
2460 ± 252 |
13 | 105 ± 5 |
24400 ± 1080 |
232 | ND | ND | – | 39200 ± 1490 |
1270 ± 34 |
31 | 25 ± 1 |
101000 ± 1640 |
4.0 |
| αT148D | 28 ± 1 |
6060 ± 618 |
216 | 130 ± 10 |
27500 ± 1890 |
212 | 918 ± 116 |
2830 ± 162 |
0.32 | 31500 ± 1330 |
1500 ± 44 |
21 | 23 ± 1 |
99500 ± 1880 |
4.3 |
| αD89N + T148D | 89 ± 4 |
6530 ± 553 |
73 | 118 ± 5 |
29900 ± 1120 |
253 | ND | ND | – | 30100 ± 953 |
2320 ± 37 |
13 | 20 ± 1 |
109000 ± 2010 |
5.5 |
| αT150D | 50 ± 6 |
19500 ± 2550 |
390 | 89 ± 14 |
31500 ± 4850 |
354 | 232 ± 42 |
6870 ± 718 |
0.12 | 13900 ± 604 |
1730 ± 31 |
8 | 21 ± 2 |
122000 ± 4390 |
5.8 |
| αD89N + T150D | 59 ± 10 |
15300 ± 4650 |
259 | 82 ± 16 |
32100 ± 5120 |
391 | 249 ± 72 |
4980 ± 363 |
0.05 | 9580 ± 682 |
2060 ± 27 |
5 | 33 ± 3 |
138000 ± 4060 |
4.7 |
Kinetic parameters and error estimates are derived from global fitting of Scheme II to data obtained over a wide range of ACh concentrations (materials and methods). Units are μM−1s−1 for the association rate constants, s−1 for all others. Gating equilibrium constants (Θ) are ratios of channel opening (β) to closing rate (α) constants. ND, a second open time component was not detectable.
Opening rate constant is a lower bound (see text and Fig. 6).
Fitting was achieved using Scheme II modified to include two desensitized states linked to the open state. Fitted desensitization rate constants for αD89T, k+d/k−d, in units of s−1 are D1 (41/529) and D2 (65/30).
In contrast to the marked effect of αD89N on ACh binding steps, channel gating steps are not discernibly affected by αD89N (Table II). The rate constant for channel opening is given as a lower bound because a plot of log likelihood against channel opening rate plateaus at ∼30,000–40,000 s−1 (Fig. 6). The difficulty in placing an upper bound on the opening rate constant is likely due to bandwidth limitations; rapid dissociation of ACh, combined with rapid channel opening, predicts similar mean closed times for doubly liganded and ACh-blocked receptors, both of which approach the instrument dead time. The rate constant for channel closing, on the other hand, is well defined, although its magnitude depends on the choice of the opening rate constant beyond the lower bound. Thus αD89N, although markedly affecting the kinetics of ACh binding, has no discernable effect on the kinetics of channel gating.
Figure 6.

Plot of log likelihood (LL) against the channel opening rate constant β2 obtained from a global fit of Scheme II to the data for receptors containing αD89N. LL plateaus at a β2 of around 40,000 s−1 (open symbol and arrow).
The overall consequences of αD89N are further illustrated by plotting open probability within clusters of single receptor openings against ACh concentration. The mutation shifts the Popen curve 13-fold toward higher ACh concentrations (Fig. 7). The curves through the data are computed from the rate constants determined from kinetic fitting, and their close alignment with the data further supports the rate constant estimates. The overall findings show that αD89 is essential for optimizing ACh binding to receptors in the resting closed state through a prominent effect on rate constants for ACh association and a smaller effect on rate constants for ACh dissociation (Table II).
Side Chain Requirements at αD89
We substituted a series of residues with polar and nonpolar side chains at αD89 and measured cell surface expression with radiolabeled α-bungarotoxin (Fig. 8 A). The Ala substitution abolishes receptor expression on the cell surface, while Glu, Asn, Ser, Thr, His, and Lys all give low but measurable expression. Of these, the αD89T and αD89E mutations were examined for their effects on single channel kinetics. The Thr substitution, which neutralizes the negative charge but maintains a small side chain, impairs the kinetics of receptor activation even more than αD89N; it causes a more pronounced slowing of the rate of ACh association but does not affect the kinetics of channel gating (Fig. 9; Table II). For the αD89T mutation, adequate fitting could only be achieved by explicitly including two desensitized states, using the method described for the wild-type receptor, presumably because of the more pronounced slowing of ACh association and consequent overlap of closed time components due to activation with those due to desensitization.
Figure 8.
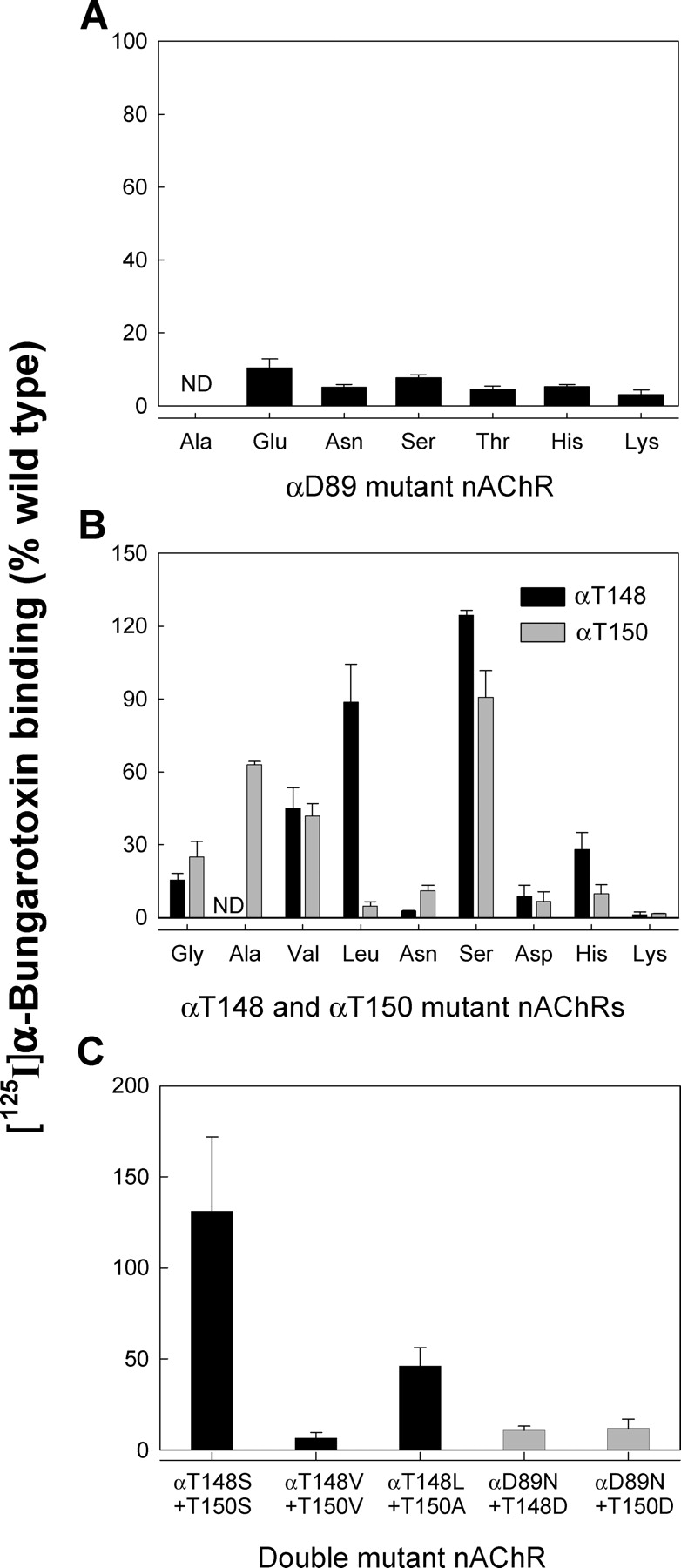
[125I]α-Bungarotoxin binding to wild-type and mutant AChRs expressed in BOSC 23 cells. (A) Cell surface expression of receptors containing mutations at αD89. (B) Cell surface expression of receptors containing mutations at αT148 and αT150. (C) Cell surface expression of receptors containing double mutations. Combination of the optimal single mutations yielded the hydrophobic pair with optimal cell surface expression, αT148L plus αT150A.
Figure 9.

Clusters of single channel currents and kinetic analysis of receptors harboring the αD89T mutation. Current traces obtained at the indicated ACh concentrations are shown at a bandwidth of 10 kHz. Dwell time histograms are shown with pdfs generated from rate constants for Scheme II modified to include fast and intermediate desensitized states superimposed (see Table II).
The Glu substitution has a smaller effect on ACh binding steps compared with αD89N and αD89T. The kinetics of channel gating are not substantially affected by αD89E, as observed for αD89N and αD89T (Table II). Thus kinetic analyses of αD89N, αD89T, and αD89E receptors show that the negative charge is the most important structural feature at position 89 of the α subunit, but side chain length is also essential for proper kinetics of receptor activation.
Main Chain Amide Groups in Recognition Domain B Are Hydrogen Bond Donors for αD89
The full negative charge of the αD89 side chain makes it a strong hydrogen bond acceptor. Hydrogen bond donors accessible to αD89 include the hydroxyl side chains of αT148 and αT150 and the main chain amide groups of αW149 and αT150 (Fig. 1 D). To determine whether the hydroxyl side chains of αT148 and αT150 hydrogen bond to αD89, as do the side chains of residues at equivalent positions in AChBP (Celie et al., 2004; Fig. 1 C), we substituted residues with hydrophobic side chains at these positions and measured rate constants for receptor activation. To determine optimal substitutions, we constructed a series of hydrophobic mutations at each position (Fig. 8 B). Substitution of Leu is strongly preferred at αT148, whereas Ala is preferred at αT150, suggesting that the environment of αT148 is more hydrophobic than that of αT150, in accord with the structures of AChBP and our model of the ligand binding site (Fig. 1, C and D). The mutation αT148L shows kinetics like that of the wild-type receptor, while αT150A has relatively modest effects on binding and gating steps (Fig. 10; Table II), showing that neither of these hydroxyl side chains is the sole hydrogen bond donor for αD89. We then replaced hydroxyl side chains of both αT148 and αT150 with nonpolar ones by combining the optimal hydrophobic substitutions at both positions to yield the double mutation αT148L plus αT150A, which expresses ∼40% of cell surface receptors compared with wild type (Fig. 8 C). Single channel kinetic analysis reveals only modest changes in binding and gating rate constants for the αT148L plus αT150A double mutation (Fig. 10; Table II). Thus hydroxyl side chains of αT148 and αT150 do not form functionally important hydrogen bonds to αD89. The remaining hydrogen bond donors accessible to αD89 are the main chain amide groups of αW149 and αT150 (Fig. 1 D).
Figure 10.

Clusters of single channel currents and kinetic analysis of receptors harboring the hydrophobic substitutions αT148L and αT150A. Current traces obtained at the indicated ACh concentrations are shown at a bandwidth of 10 kHz. Dwell time histograms are shown with pdfs generated from the Scheme II rate constants superimposed (see Table II). The hydrophobic substitutions cause little change from wild-type receptor activation kinetics (see Figs. 2 and 4).
Charge Reversal of the Kinetic Consequences of αD89N
To gain further insight into the structural contribution of αD89, we retained the αD89N mutation but substituted Asp at positions proximal to the main chain of binding domain B. Restoring the local negative charge at αT148 substantially reverses the slowing of the ACh association rate constants produced by αD89N alone (Fig. 11; Table II). It also counteracts the 13-fold shift in the plot of Popen against ACh concentration, yielding an overall threefold shift for the αD89N plus αT148D double mutant compared with the wild-type receptor (Fig. 7). Rate constants for ACh association are increased markedly by the double mutation compared with αD89N alone, with k+1 showing the greatest increase. By contrast, combining the negative charge at αT150 with αD89N shows no reversal of the plot of Popen against ACh concentration (Fig. 7), and smaller increases in association rate constants (Table II).
Figure 11.
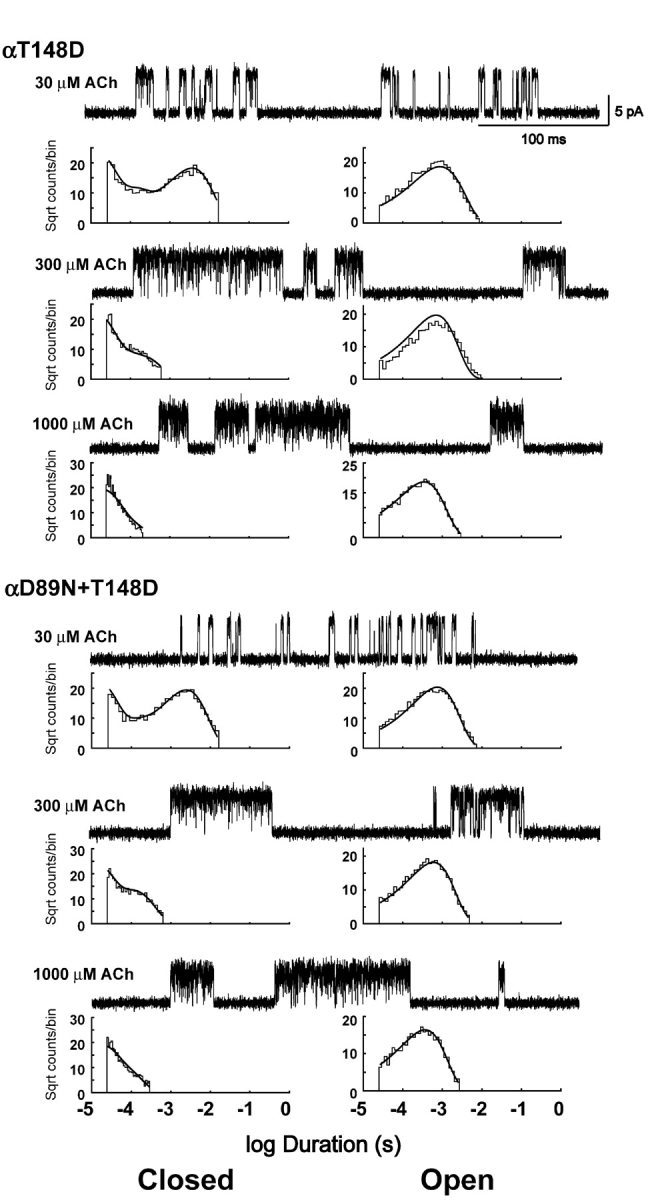
Clusters of single channel currents and kinetic analysis of receptors showing charge reversal of the effects of αD89N. Current traces at the indicated ACh concentrations are shown at a bandwidth of 10 kHz. Dwell time histograms are shown with pdfs generated from the Scheme II rate constants superimposed (see Table II). Combination of αT148D counteracts the change in receptor activation kinetics compared with αD89N alone (see Figs. 5 and 7).
The mutation αT148D by itself slows ACh association to the first binding site, although much less than αD89N, so if the two mutations were independent, the effect of combining them would be a further slowing of k+1. The observation that k+1 is nearly reversed in the double mutant indicates that the two mutations are interdependent, which, together with their close proximity, suggests they contribute to ACh association through a common mechanism. Normal or near normal ACh association is achieved only when a single negatively charged residue is present at either α89 or α148; association is greatly impaired when there is no negative charge (αD89N), and moderately impaired when two charges are present (αT148D or αT150D). Thus the common structural feature associated with fast association is a single negative charge at either α89 or α148. This explanation is readily accommodated by the AChBP structure and our structural model of the ligand binding site (Fig. 1, C and D). αT148 and the equivalent S142 in AChBP are situated in hydrophobic environments proximal to αD89 or D85 and behind the main chain of recognition domain B (Fig. 1, C and D), enabling the negatively charged side chain to accept hydrogen bonds from the main chain amide donors αW149 and αT150. This hydrogen bonding whether via Asp at α89 or α148 restricts rotation of the main chain of recognition domain B. The negative charge may also polarize the main chain carbonyl moiety of αW149, which may further stabilize the positive charge of the agonist (Celie et al., 2004).
The mutation αT150D by itself also slows k+1, and should produce a further slowing when combined with αD89N if the two residues are independent. The double mutant shows a greater association rate than expected from independence, indicating that these too are interdependent (Table II). However, the hydroxyl side chain of αT150 is far from αD89, and is in a hydrophilic rather than a hydrophobic environment (Fig. 8 B), both of which make it an unlikely hydrogen bond acceptor for the main chain amide group of αW149 or Asn substituted at αD89. Also the extent of reversal of k+1 is not as great as observed with αT148D, and αT150D alone or in combination with αD89N has multiple effects not shown by αT148D; αT150D speeds ACh dissociation from the first binding site and slows channel opening (Table II). The interdependence between αT150D and αD89N may be due to the different ways in which they stabilize the quaternary ammonium moiety of the agonist. A negative charge introduced at αT150 may enhance ACh association electrostatically, whereas a negative charge, at either α89 or α148, stabilizes the main chain of recognition domain B relative to domain A, polarizes the main chain carbonyl group of αW149, or both. Stabilization of domain B is likely essential for positioning the indole side chain and main chain carboxyl groups of αW149 for efficient encounter with ACh.
DISCUSSION
We show that the invariant aspartic acid residue αD89 contributes to the kinetics of receptor activation by maintaining high rates of association between ACh and the two binding sites of the muscle nicotinic receptor, as well as by optimizing rates of ACh dissociation. Channel gating rate constants, on the other hand, are not affected by mutations of αD89. Comparison of our structural model of the major extracellular domain of the receptor with the structure of AChBP suggests that recognition domains A and B are linked by hydrogen bonds between the carboxylate side chain of αD89 and hydroxyl side chains of αT148 and αT150. This tertiary structural fold encompassing domains A and B contains highly conserved residues, and may represent a primordial structural motif essential for binding ACh. However contrary to expectations from the AChBP structure, we find that substituting residues with hydrophobic side chains at αT148 and αT150 does not substantially affect the kinetics of receptor activation, pointing to main chain amide groups in recognition domain B as the most likely hydrogen bond donors. In accord with our structural model (Sine et al., 2002a), main chain amide groups of αW149 and αT150 are in close proximity to the αD89 side chain, suggesting a mechanism by which αD89 contributes to a high rate of ACh association. Hydrogen bonds between the carboxylate moiety of αD89 and main chain amide groups of αW149 and αT150 both positions αW149 and restricts its mobility to promote efficient encounter between the binding site and ACh. Stabilization of the main chain by αD89 may both position and polarize the main chain carbonyl of αW149, which may further stabilize the quaternary ammonium moiety of ACh. Further support for stabilization of domain B by a negative charge is our charge reversal of the effect of αD89N by substitution of Asp at αT148; in our structural model, αT148 is appropriately positioned for interaction with the main chain of domain B.
Patch-clamp recordings show that αD89N prolongs the latency between successive openings of single receptor channels at low and intermediate concentrations of ACh, but this latency is comparable to that of wild-type receptors at high concentrations of agonist. Analyzed within the framework of kinetic schemes, these observations lead to the conclusion that αD89 contributes to agonist binding steps but not to channel gating steps. The association rate constants for ACh are affected most, slowing 10- to 20-fold, while the dissociation rate constants are modestly affected, slowing twofold at one binding site and increasing threefold at the other site. Differences in functional consequences for ACh binding at the two sites likely owe to nonequivalence of the alpha-epsilon and alpha-delta interfaces. The selective effect of a mutation on agonist binding is often used as a criterion for direct contact between the agonist and the mutated site, but this is clearly not true for the muscle receptor studied here. αD89 is located at the periphery of the binding site and has not been identified as a site of labeling by covalent affinity reagents, contrary to the labeling of aromatic residues within the binding pocket, including αY93, αW149, αY190, αY198, and γW55 (Dennis et al., 1988; Abramson et al., 1989; Galzi et al., 1990; Chiara et al., 1998). Further, the mutational consequences of αD89N are like those of a small subset of functionally significant mutations of the receptor that selectively affect agonist binding such as αG153S, αN217K, and ɛL221F (Sine et al., 1995; Wang et al., 1997; Hatton et al., 2003). This group of binding-specific mutations is distinct from mutations of aromatic residues in the binding pocket, which alter binding as well as channel gating (Chen et al., 1995; Akk et al., 1999; Akk, 2001).
The AChBP structure first revealed differences in secondary and tertiary structures between principal and complementary faces of the ACh binding site (Brejc et al., 2001). The principal face is composed of loops that separate β strands, whereas the complementary face is composed primarily of β strands. Thus, side chain disposition of residues on the principal face is not constrained by periodic main chain hydrogen bonds, whereas side chain disposition on the complementary face is strictly regulated by main chain hydrogen bonds that stabilize the β sheet secondary structure (Sine et al., 2002a). The absence of a regular secondary structure on the principal face suggests that alternative linkages might be expected to form between conserved residues in proximal loops, such as those described here between the strong hydrogen bond acceptor αD89 in recognition domain A and donors in domain B. The recent higher resolution x-ray structure of AChBP confirms the presence of hydrogen bonds between the homologous D85 and hydroxyl side chain donors in domain B (Celie et al., 2004; Fig. 1 C). The present data show profound functional consequences of mutating αD89, suggesting that it interacts with chemical groups in domain B. However, unlike AChBP, the hydroxyl side chains of αT148 and αT150 in the receptor do not donate functionally significant hydrogen bonds to the negatively charged carboxylate group of αD89, but rather the donors are main chain amide groups of αW149, αT150, or both.
The strategy employed here takes advantage of new opportunities for establishing structure–function relationships based upon the AChBP structure (Brejc et al., 2001). Now one can test predicted atomic-scale interactions for their functional significance, rather than relying on the linear sequence, residue conservation, ligand interactions, and solvent accessibility used before the AChBP structure was known. The present results for the muscle nicotinic receptor show that predicted hydrogen bonds between the invariant αD89 and hydroxyl side chains of residues flanking αW149 do not contribute to receptor function, but rather hydrogen bonding to main chain amide groups of αW149 and αT150 is most likely. Possible reasons the predicted hydrogen bonds are not evident include (a) the local structure of the receptor differs slightly from that of AChBP, (b) the hydrogen bonds are actually present in the receptor but not functionally significant despite the high degree of conservation, (c) the predicted hydrogen bonds form in the crystal but not in solution, and (d) hydrogen bonds form to both the main chain amide groups and the hydroxyl side chains in domain B, but these switch back and forth in a dynamic manner. These possibilities emerging from the present data cannot be distinguished by the current body of structural or functional knowledge. However, the overall results underscore the deeper understanding that can be achieved by testing the functional significance of predicted atomic-scale interactions in other parts of the muscle nicotinic receptor as well as in other members of the Cys-loop receptor superfamily.
The invariant αW149 has long stood as the most likely candidate for the primary docking site for the quaternary ammonium moiety of ACh because the Phe substitution at this position profoundly impairs ligand binding affinity and the kinetics of receptor activation (Dennis et al., 1988; Zhong et al., 1998; Akk, 2001). The recent x-ray structure of AChBP in complex with carbamylcholine conclusively shows docking of the quaternary ammonium moiety of the agonist with the indole ring of the homologous W143, but also suggests that the negative charge of D85 polarizes the carbonyl group of W143 for additional stabilization of agonist (Celie et al., 2004). Within our structural model (Fig. 1 D; Sine et al., 2002a), the carboxylate moiety of αD89 is 2.5–3 Å away from the main chain amides of αW149 and αT150, suggesting a structural explanation for the selective effect of αD89N on elementary steps underlying ACh binding. Neutralizing the negative charge eliminates hydrogen bonding to the main chain of domain B, increasing mobility of the indole group of αW149 and reducing polarization of its carbonyl group, which together impede the association of ACh. Our findings suggest future studies aimed at identifying additional functionally relevant contacts between proximal domains of the ACh binding site.
Acknowledgments
We thank Nina Bren for outstanding technical contributions.
This work was supported by National Institutes of Health grant NS31744 to S.M. Sine.
Olaf S. Andersen served as editor.
Abbreviation used in this paper: AChBP, acetylcholine binding protein.
References
- Abramson, S.N., Y. Li, P. Culver, and P. Taylor. 1989. An analog of lophotoxin reacts covalently with tyrosine 190 in the α subunit of the nicotinic receptor. J. Biol. Chem. 264:12666–12672. [PubMed] [Google Scholar]
- Akk, G. 2001. Aromatics at the murine nicotinic receptor agonist binding site: mutational analysis of the αY93 and αW149 residues. J. Physiol. 535:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk, G., M. Zhou, and A. Auerbach. 1999. A mutational analysis of the acetylcholine receptor channel transmitter binding site. Biophys. J. 76:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc, K., W. van Dijk, R. Klassen, M. Schuurmans, J. van der Oost, A. Smit, and T. Sixma. 2001. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 411:269–276. [DOI] [PubMed] [Google Scholar]
- Celie, P.H.N., S.E. van Rossum-Fikkert, W.J. van Dijk, K. Brejc, A.B. Smit, and T.K. Sixma. 2004. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 41:907–914. [DOI] [PubMed] [Google Scholar]
- Chen, J., Y. Zhang, G. Akk, S. Sine, and A. Auerbach. 1995. Activation kinetics of recombinant mouse nicotinic acetylcholine receptors with mutations at α subunit residue tyrosine 190. Biophys. J. 69:849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara, D.C., R.E. Middleton, and J.B. Cohen. 1998. Identification of tryptophan 55 as the primary site of [3H]nicotine photoincorporation in the γ-subunit of the Torpedo nicotinic acetylcholine receptor. FEBS Lett. 423:223–226. [DOI] [PubMed] [Google Scholar]
- Colquhoun, D., and D.C. Ogden. 1988. Activation of ion channels in the frog end-plate by high concentrations of acetylcholine. J. Physiol. 395:131–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun, D., and F. Sigworth. 1983. Fitting and statistical analysis of single channel records. Single Channel Recording. B. Sakmann and E. Neher, editors. Plenum Publishing Corp., New York. 191–264.
- Corringer, J.P., N. Le Novere, and J.P. Changeux. 2000. Nicotinic receptors at the amino acid level. Annu. Rev. Pharmacol. Toxicol. 40:431–458. [DOI] [PubMed] [Google Scholar]
- Dennis, M., J. Giraudat, F. Kotzyba-Hibert, M. Goeldner, C. Hirth, J.Y. Chang, C. Lazure, M. Chretien, and J.P. Changeux. 1988. Amino acids of the Torpedo marmorata acetylcholine receptor α subunit labeled by a photoaffinity ligand for the acetylcholine binding site. Biochemistry. 27:2346–2357. [DOI] [PubMed] [Google Scholar]
- Elenes, S., and A. Auerbach. 2002. Desensitization of diliganded mouse muscle nicotinic acetylcholine receptor channels. J. Physiol. 541:367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galzi, J.L., F. Revah, D. Black, M. Goeldner, C. Hirth, and J.P. Changeux. 1990. Identification of a novel amino acid α-tyrosine 93 within the cholinergic ligands-binding sites of the acetylcholine receptor by photoaffinity labeling. Additional evidence for a three-loop model of the cholinergic ligands-binding sites. J. Biol. Chem. 265:10430–10437. [PubMed] [Google Scholar]
- Hatton, C.J., C. Shelley, M. Brydson, D. Beeson, and D. Colquhoun. 2003. Properties of the human muscle nicotinic receptor, and of the slow-channel myasthenic syndrome mutant ɛL221F, inferred from maximum likelihood fits. J. Physiol. 547:729–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin, A. 2002. The emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci. 3:102–114. [DOI] [PubMed] [Google Scholar]
- Le Novere, N., T. Grutter, and J.P. Changeux. 2002. Models of the extracellular domain of the nicotinic receptors and of agonist- and Ca2+-binding sites. Proc. Natl. Acad. Sci. USA. 99:3210–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, B.S., R.B. Gunn, and R.R. Kopito. 1991. Functional differences among nonerythroid anion exchangers expressed in a transfected human cell line. J. Biol. Chem. 266:11448–11454. [PubMed] [Google Scholar]
- Molles, B.E., I. Tsigelny, P. Nguyen, S. Gao, S.M. Sine, and P. Taylor. 2002. Residues in the ɛ subunit of the nicotinic acetylcholine receptor interact to confer selectivity of Waglerin-1 for the α–ɛ subunit interface site. Biochemistry. 41:7895–7906. [DOI] [PubMed] [Google Scholar]
- Ohno, K., H.-L. Wang, M. Milone, N. Bren, J.M. Brengman, S. Nakano, P. Quiram, J.N. Pruitt, S.M. Sine, and E.G. Engel. 1996. Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor ɛ subunit. Neuron. 17:157–170. [DOI] [PubMed] [Google Scholar]
- Pear, W.S., G.P. Nolan, M.L. Scott, and D. Baltimore. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 90:8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira, M., R. Abagyan, and M. Totrov. 2002. Structural model of nicotinic acetylcholine receptor isotypes bound to acetylcholine and nicotine. BMC Struct. Biol. 2:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, F., A. Auerbach, and F. Sachs. 1996. Estimating single channel kinetic parameters from idealized patch clamp data containing missed events. Biophys. J. 70:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmann, B., J. Patlak, and E. Neher. 1980. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 286:71–73. [DOI] [PubMed] [Google Scholar]
- Sine, S.M. 2002. The nicotinic receptor ligand binding domain. J. Neurobiol. 3:431–446. [DOI] [PubMed] [Google Scholar]
- Sine, S.M., and J.H. Steinbach. 1987. Activation of acetylcholine receptors in clonal BC3H-1 cells by high concentrations of agonist. J. Physiol. 385:325–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine, S.M., T. Claudio, and F. Sigworth. 1990. Activation of Torpedo acetylcholine receptors expressed in mouse fibroblasts: single channel current kinetics reveal distinct agonist binding affinities. J. Gen. Physiol. 96:395–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine, S.M., K. Ohno, C. Bouzat, A. Auerbach, M. Milone, J.N. Pruitt, and A.G. Engel. 1995. Mutation of the acetylcholine receptor α subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 15:229–239. [DOI] [PubMed] [Google Scholar]
- Sine, S.M., H.L. Wang, and N. Bren. 2002. a. Lysine scanning mutagenesis delineates structural model of the nicotinic receptor ligand binding domain. J. Biol. Chem. 277:29210–29223. [DOI] [PubMed] [Google Scholar]
- Sine, S.M., X.M. Shen, H.L. Wang, K. Ohno, W.Y. Lee, A. Tsujino, J. Brengmann, N. Bren, J. Vajsar, and A.G. Engel. 2002. b. Naturally occurring mutations at the acetylcholine receptor binding site independently alter ACh binding and channel gating. J. Gen. Physiol. 120:483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H.-L., A. Auerbach, N. Bren, K. Ohno, A. Engel, and S. Sine. 1997. Mutation in the M1 domain of the acetylcholine receptor α subunit decreases the rate of agonist dissociation. J. Gen. Physiol. 109:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H.L., M. Milone, K. Ohno, X.M. Shen, A. Tsujino, A.P. Batocchi, P. Tonali, J. Brengman, A.G. Engel, and S.M. Sine. 1999. Acetylcholine receptor M3 domain: stereochemical and volume contributions to channel gating. Nat. Neurosci. 2:226–233. [DOI] [PubMed] [Google Scholar]
- Wang, H.L., K. Ohno, M. Milone, J. Brengman, A. Evoli, A.P. Batocchi, L. Middleton, K. Christodoulou, A.G. Engel, and S.M. Sine. 2000. Fundamental gating mechanism of nicotinic receptor channel gating revealed by mutation causing a congenital myasthenic syndrome. J. Gen. Physiol. 116:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., J. Chen, and A. Auerbach. 1995. Activation of recombinant mouse acetylcholine receptors by acetylcholine, carbamylcholine and tetramethylammonium. J. Physiol. 486:189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, W., J.P. Gallivan, Y. Zhang, L. Li, H.A. Lester, and D.A. Dougherty. 1998. From ab initio quantum mechanics to molecular neurobiology: a cation-pi binding site in the nicotinic receptor. Proc. Natl. Acad. Sci. USA. 95:12088–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]