

Abstract
The phosphorylation profile of ciliary proteins under basal conditions and after stimulation by extracellular ATP was investigated in intact tissue and in isolated cilia from porcine airway epithelium using anti-phosphoserine and anti-phosphothreonine specific antibodies. In intact tissue, several polypeptides were serine phosphorylated in the absence of any treatment (control conditions). After stimulation by extracellular ATP, changes in the phosphorylation pattern were detected on seven ciliary polypeptides. Serine phosphorylation was enhanced for three polypeptides (27, 37, and 44 kD), while serine phosphorylation was reduced for four polypeptides (35, 69, 100, and 130 kD). Raising intracellular Ca2+ with ionomycin induced identical changes in the protein phosphorylation profile. Inhibition of the NO pathway by inhibiting either NO syntase (NOS), guanylyl cyclase (GC), or cGMP-dependent protein kinase (PKG) abolished the changes in phosphorylation induced by ATP. The presence of PKG within the axoneme was demonstrated using a specific antibody. In addition, in isolated permeabilized cilia, submicromolar concentrations of cGMP induced protein phosphorylation. Taken together, these results suggest that the axoneme is an integral part of the intracellular NO pathway. The surprising observation that ciliary activation is accompanied by sustained dephosphorylation of ciliary proteins via NO pathway was not detected in isolated cilia, suggesting that the protein phosphatases were either lost or deactivated during the isolation procedure. This work reveals that any pharmacological manipulation that abolished phosphorylation and dephosphorylation also abolished the enhancement of ciliary beating. Thus, part or all of the phosphorylated polypeptides are likely directly involved in axonemal regulation of ciliary beating.
Keywords: axoneme, airway epithelium, extracellular ATP, cGMP, phosphatases
INTRODUCTION
The mucociliary system is responsible for transporting mucus and entrapped materials over the epithelia. This transport is extremely efficient due to the cooperative beating of the cilia and their ability to robustly enhance activity in response to various stimuli. Several groups have established that ciliary beat frequency (CBF) enhancement, which is the hallmark of the mucociliary systems, is mediated by various second messengers, including Ca2+, cAMP, and cGMP (Verdugo, 1980; Verdugo et al., 1980; Tamaoki et al., 1989; Villalon et al., 1989; Di Benedetto et al., 1991; Lansley et al., 1992; Korngreen and Priel, 1994; Geary et al., 1995; Salathe and Bookman, 1995; Tarasiuk et al., 1995; Yang et al., 1996; Levin et al., 1997; Braiman et al., 1998; Uzlaner and Priel, 1999; Zhang and Sanderson, 2003). Recently it was shown that elevated cytosolic concentrations of Ca2+, cAMP, and cGMP are essential in order to achieve a physiologically significant enhancement of CBF (Uzlaner and Priel, 1999; Ma et al., 2002; Zagoory et al., 2002). Moreover, the results of Ma et al. (2002) strongly suggest that robust CBF enhancement is due to direct interaction of these three second messengers with the axoneme, the machinery that drives ciliary beating and controls the frequency and pattern of beating. It is well known that hormones and neurotransmitters, acting through second messengers, mediate physiological actions by altering the phosphorylation of specific target proteins, providing important regulatory mechanisms in various cellular processes, including motility. Thus, the results of Ma et al. (2002) raise the possibility that one or more of the second messengers lead to the phosphorylation of proteins within the axoneme to achieve ciliary activation. To test this idea, and to further elucidate the agonist-induced molecular events that lead to CBF enhancement, studies must focus on the mechanisms of protein modulation within the axoneme in response to CBF enhancers and on the physiological consequences of such modulation.
The function of most of the >200 different polypeptides that make up the axoneme has not yet been elucidated. Despite the complexity of the axoneme, considerable progress has been made in the identification of various proteins, enzymes, and phosphorylated substrates assembled on the axoneme of unicells and flagella (Gibbons, 1995; Hamasaki et al., 1995; Porter and Sale, 2000). The general structure of the axoneme appears to be conserved throughout evolution. Yet, the pattern of ciliary beating varies considerably among flagella, single cell organisms, and mucociliary epithelium, reflecting the unique physiological function of each ciliated cell type (Tamm and Terasaki, 1994). The differences in beat pattern between these cell types indicate that the mechanisms controlling the axoneme may also be different. Consequently, a straightforward extrapolation of data obtained in single cell organisms and flagella to mucociliary systems may be misleading. To the best of our knowledge, only two studies investigated the phosphorylation of axonemal proteins in a mucociliary system and showed that PKC and PKA phosphorylate axonemal proteins in sheep airway epithelium, suggesting possible involvement of these protein kinases in mucociliary regulation (Salathe et al., 1993a,b).
To link agonist-induced cytosolic events that lead to CBF enhancement to molecular events consequently evoked on the axoneme, several basic questions have to be resolved. For example, (a) do the same signal transduction pathways that lead to CBF enhancement also induce phosphorylation of axonemal proteins, (b) can intracellular Ca2+, a powerful CBF enhancer, induce phosphorylation of axonemal proteins, and (c) are axonemal PKG and PKA the main target enzymes for the cytosolic cyclic nucleotides? These and other fundamental questions remain open partly due to technical limitations. Protein phosphorylation assays in isolated cilia can provide valuable information regarding the modification of axonemal proteins by various second messengers, yet correlating the biochemical findings obtained in isolated cilia with physiological processes involved in ciliary beating is not often straightforward. In an attempt to overcome these limitations, we adapted an assay of protein phosphorylation in intact tissue that uses monoclonal antibodies that specifically recognize phosphorylated forms of serine or threonine residues (Heffetz et al., 1991; Naz, 1999). The modified assay enables us to study inducible changes in phosphorylation of ciliary and axonemal proteins in intact porcine airway epithelium. Extracellular ATP or ionomycin (which raises intracellular Ca2+) were found to induce sustained phosphorylation of three ciliary proteins and sustained dephosphorylation of four other ciliary proteins. These changes in protein phosphorylation levels are mediated by the NO pathway, a pathway recently shown to mediate robust mucociliary activation (Uzlaner and Priel, 1999; Braiman et al., 2000). Consistent with this observation, axonemal PKG was found to play a pivotal role in the sustained phosphorylation and sustained dephosphorylation. Thus, the same agonist-activated pathway that leads to CBF enhancement (Uzlaner and Priel, 1999; Braiman et al., 2000) also induces axonemal phosphorylation/dephosphorylation. These findings will ultimately make it possible to single out axonemal proteins involved in the mechanism of CBF enhancement.
MATERIALS AND METHODS
Materials
Sucrose, sodium acetate, magnesium sulfate, tris(hydroxymethyl) aminomethane (Tris), dithiothreitol (DTT), HEPES, EGTA, di(Tris)-p-nitrophenylphosphate (pNPP), β-glycerophosphate, BSA, Triton X-100, ATP, and guanosine 3′,5′-cyclic monophosphate (cGMP) were from Sigma-Aldrich. Aprotinin, N-(trans-epoxysuccinyl)-l-leucine-4-guanidinobutylamine (E-64), leupeptin, EDTA, and 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF) were from Applichem. Protein assay kit was from Bio-Rad Laboratories. Protease inhibitor cocktail and okadaic acid were obtained from Calbiochem-Novabiochem. NaCl, KCl, CaCl2, MgCl2, and glucose were from Merck. N ω-nitro-l-arginine methyl ester (L-NAME), KT-5823, and 6-anilinoquinoline-5,8-quinone (LY-83583) were from BIOMOL Research Laboratories, Inc. Dulbecco's PBS, FCS, RPMI-1640 culture medium, and antibiotics were obtained from Biological Industries. Fura-2/AM was from either Molecular Probes or from Teflabs. The dye was stored in solid form at −20°C, and fresh solutions were made before each experiment in dry DMSO.
E-64 and KT-5823 were dissolved in a minimal volume of DMSO. Stock solution of LY-83583 was prepared in ethanol. The final concentrations of DMSO or ethanol in assay buffers never exceeded 0.1%.
Antibodies
Monoclonal anti-phosphoserine and anti-phosphothreonine antibodies used in the present study were obtained from Sigma-Aldrich. These antibodies are widely used in immunoblotting methods (Heffetz et al., 1991; Abu-Lawi and Sultzer, 1995; Bousquet et al., 1997; Brustle et al., 2001; Liu et al., 2001) and have been reported to specifically bind to phosphoserine/phosphothreonine (Heffetz et al., 1991; Abu-Lawi and Sultzer, 1995; Naz, 1999).
Monoclonal anti-phosphoserine antibody (mouse IgG1 isotype, clone PSR-45) specifically reacts against phosphorylated serine both as free or conjugated amino acid. It does not react with nonphosphorylated serine, phosphorylated tyrosine or threonine, AMP, or ATP. Monoclonal anti-phosphothreonine antibody (mouse IgG2b isotype, clone PTR-8) specifically reacts against phosphorylated threonine both as a free or a conjugated amino acid. It does not react with nonphosphorylated threonine, phosphorylated tyrosine or serine, AMP, or ATP. Monoclonal anti–α-tubulin (mouse IgG1 isotype, clone B-5-1-2), also obtained from Sigma-Aldrich, specifically recognizes an epitope located in the COOH-terminal end of the α-tubulin isoform in a variety of organisms (e.g., humans, sea urchins, Chlamydomonas, and others). cGKIα (N-16) was obtained from Santa Cruz Biotechnology, Inc. cGKIα (cGMP-dependent protein kinase type I, isoform α) is an affinity-purified polyclonal antibody raised against a peptide mapping near the NH2 terminus of cGKIα of human origin. cGKIα is specific for detection of mouse, rat, and human origins.
Anti-mouse IgG (H+L) HRP conjugate used as secondary antibody for immunoblotting analysis with monoclonal antibodies was obtained from Promega. Anti-goat IgG+HRP used as secondary antibody for immunoblotting analysis with polyclonal antibody was obtained from Delta Biolabs.
Preparation of Mucociliary Epithelial Tissue
Pig trachea were obtained from a local slaughterhouse, washed quickly with ice-cold PBS, and transferred to the laboratory on ice in the same buffer. Epithelial tissue was carefully removed on ice with a sharp surgical blade and washed again with ice-cold PBS. The tissue was either used immediately for cilia isolation or was incubated with test compounds under different experimental conditions before cilia isolation. Moreover, a primary epithelial cell culture was established from the tissue pieces.
Protein Phosphorylation Assay for Isolated Cilia
Cilia were first isolated from intact untreated epithelial tissue and then used in protein phosphorylation reactions as described below. Cilia isolation was performed according to the method of Anderson (1974) with modifications. About 10 g of the PBS-washed pieces of epithelial tissue (see above) were incubated in 100 ml of isolation buffer containing 0.5 M sodium acetate (pH 5.5), 0.25 M sucrose, 20 mM CaCl2, 0.05% Triton X-100, supplemented with phosphatase inhibitors (0.5 mM EDTA, 0.5 mM AEBSF, 1 μM E-64, 1 μM leupeptin, and 0.15 μM aprotinin) for 1.5 h at 4°C under continuous stirring. The extract containing isolated cilia was separated from tissue by low-speed centrifugation (1,000 g, 10 min, 4°C). Cilia were then pelleted by high-speed centrifugation (20,000 g, 30 min, 4°C) and resuspended in a storage buffer containing 0.25 M sucrose, 40 mM HEPES (pH 7.4), and 1 mM EGTA. The suspension was snap frozen in liquid nitrogen and stored at −70°C. Protein content was measured by the method of Bradford (1976) using BSA as a standard. Thin sections of the ciliary pellet were examined by electron microscopy (see method below). The electron micrograph presented in Fig. 1 demonstrates a highly enriched ciliary preparation showing the characteristic features of intact axoneme.
Figure 1.

Electron micrograph showing a highly enriched preparation of cilia isolated from porcine airway epithelium. Thin section of the ciliary pellet demonstrates the features of intact axonemes (nine outer doublet microtubules and an inner pair). Bar, 1 μm.
For the detection of phosphorylated proteins in isolated cilia we used the procedure described by Salathe et al. (1993a) with modifications. Cilia (40–50 μg protein) were preincubated in the reaction mixture containing 10 mM Tris-HCl (pH 7.4), 1 mM DTT, 5 mM MgCl2, 1 mM EGTA, 1 mM total Ca2+ (750 nM free Ca2+), 0.012% Nonidet P-40, and various test compounds for 5 min at 32°C. The rational for choosing the above-mentioned free Ca2+ concentration was as follows. Using confocal microscopy, it was recently shown that strong and prolonged CBF stimulation is accompanied by a sustained rise in [Ca2+]i in close proximity to the cilia, ranging between 0.6 and 1 μM (Braiman and Priel, 2001). Therefore, 750 nM [Ca2+]i was chosen. The reaction was then initiated by adding 100 μM ATP and the samples were further incubated for various periods of time. The reaction was stopped by adding sample buffer (Laemmli, 1970) followed by heating at 100°C for 3 min. Phosphorylated proteins were detected by Western blotting (see below).
Protein Phosphorylation Assay for Intact Tissue
Protein phosphorylation assay for intact epithelial tissue was developed in order to observe changes in the phosphorylation profile of ciliary proteins induced in intact system. It enabled us to correlate the pattern of the phosphorylated ciliary proteins and physiological results measured in our current and previous studies (Korngreen and Priel, 1996; Uzlaner and Priel, 1999; Braiman et al., 2000).
The phosphorylation was induced in pieces of intact epithelial tissue, which was immediately used after its isolation from porcine tracheas. The phosphorylation processes were stimulated by incubation of tissue pieces with different reagents. Cilia were then immediately isolated, and the phosphorylated proteins were visualized by Western blotting.
The following procedure was performed at 32°C, and found to be the optimal temperature for protein phosphorylation responses in intact tissue. About 2 g of PBS-washed pieces of epithelium (1 × 2 cm) were preincubated for 5 min in 25 ml Erlenmeyer flasks in 20 ml of incubation buffer containing 10 mM HEPES (pH 7.4), 118 mM NaCl, 4.4 mM KCl, 2.5 mM CaCl2, 1.1 mM MgSO4, and 11 mM glucose. The incubation buffer content used in the present study was identical to that used in the CBF measurements (Korngreen and Priel, 1996; Uzlaner and Priel, 1999; Braiman et al., 2000) to provide a correlation of phosphorylation experiments with our previous physiological studies. All experiments were performed at 32°C, as it was found to be the optimal temperature for phosphorylation experiments on intact tissue. After preincubation, the test compounds were added for an additional 0.5–20 min. After the drug treatment, the buffer was removed and the tissue was rapidly washed three times with ice-cold PBS. Cilia were then prepared using sodium acetate extraction, as described above, except that all incubations were performed at 4°C in the presence of phosphatase inhibitors (2 mM sodium orthovanadate, 40 mM β-glycerophosphate, and 20 mM p-nitrophenylphosphate) to prevent protein dephosphorylation during cilia preparation. The final cilia pellet was resuspended in 10 mM Tris-HCl (pH 7.4) followed by protein determination.
To determine whether the detected phosphorylated proteins are part of the axoneme, isolated cilia were further extracted with Triton X-100 to separate axoneme from ciliary membrane as described previously (Salathe et al., 1993a) with modifications. Cilia (50–70 μg protein) in 50 μl 10 mM Tris-HCl (pH 7.4) were incubated with an equal volume of the same buffer containing 1% Triton X-100 for 30 min on ice with intermittent vortexing. Detergent-insoluble axonemal fraction was then pelleted by centrifugation at 100,000 rpm, for 30 min in a Beckman Coulter airfuge. The supernatant was used as a membrane fraction. Cilia, axoneme, and membrane fractions were suspended in sample buffer (Laemmli, 1970), and phosphorylated proteins were detected by Western blotting.
Western Blot Assay
Equal amounts of samples (70 μg protein) were subjected to SDS-PAGE in 10% gels and transferred to a Protrain BA 83 cellulosenitrate membrane (Schleicher & Schuell). As a protein loading control, the membrane was stained with Ponceau S to determine the efficiency of the protein loading and transfer. Immunoblotting was not performed if different protein levels were seen in the different lanes by the stain. The membrane was washed in Tris-buffered saline using 0.1% Tween 20 (TBST), blocked with 3% BSA in TBST for 1 h, and blotted for 1 h with specific monoclonal anti-phosphoserine (ratio 1:1,000), monoclonal anti-phosphothreonine (ratio 1:1,000), polyclonal cGKIα antibody (ratio 1:200), or monoclonal anti–α-tubulin antibody (ratio 1:1,000). After 1 h incubation with anti-mouse IgG (H+L) HRP conjugate secondary antibody (for all monoclonal antibodies) or anti-goat IgG+HRP secondary antibody (for polyclonal antibody), the protein bands were detected using a chemiluminescence assay system (NEN Life Science Products) and visualized on a Kodak X-Omat LS film. The OD of each band was quantified using image-analyzing software (Image Gauge V3.122, Fuji Photo Film).
Cilia Preparation for Electron Microscopy
An aliquot of freshly obtained cilia was encapsulated in 3% agarose and immediately fixed in 0.1 M phosphate buffer (pH 7.0) containing 3% gluteraldehyde, 2% tannic acid, and 10% sucrose for 2 h at room temperature. The cilia were washed with 0.1 M phosphate buffer (pH 6.0) containing 10% sucrose (washing buffer) and post-fixed with 1% osmium tetra-oxide for 2 h on ice followed by one washing step. The dehydration procedure was done gradually with increasing concentrations of ethyl alcohol (from 30 to 100%). The samples were embedded in araldite (CY 212) and polymerized at 60°C overnight. Ultrathin sections (silver/silver gray sections) for electron microscopy were mounted on copper grids and counterstained at room temperature with 1% uranyl acetate for 2 h and with 0.3% lead citrate for 2 min (Fig. 1).
Tissue Culture
Monolayer tissue cultures were established from porcine tracheal epithelium using the procedure described previously (Korngreen and Priel, 1996). In brief, the freshly isolated ciliary epithelium was cut into small pieces. Two or three tissue pieces were placed on an ethanol-sterilized glass coverslip and were incubated in plastic Petri dishes (Nunk) in RPMI-1640 medium supplemented with 7.5% FCS, 20 U/ml penicillin, 2.5 U/ml nystatin, and 20 μg/ml streptomycin at 37°C in an atmosphere of 95% air and 5% CO2. The medium was replaced every 2 d. Measurements were performed on 3–5-d-old tissue cultures when the epithelial monolayers were large enough to be used in the experiments.
Simultaneous Measurement of [Ca2+]i and Ciliary Beating
System Description.
We used the method of simultaneous measurement of [Ca2+]i and ciliary beating that has been previously extensively described (Korngreen and Priel, 1994). In brief, [Ca2+]i was measured with the fluorescent indicator Fura-2. The dye-loaded cells were epi-illuminated with light from a 150-W xenon lamp (Oriel Corp.) filtered by 340- and 380-nm interference filters (Oriel) mounted on a four-position rotating filter wheel. The fluorescence, emitted at 510 nm, was detected by a photon-counting photomultiplier (H3460-53; Hamamatsu). The 340/380 fluorescence ratio, averaged over a period of 1 s, was stored in a computer. CBF was measured by trans-illuminating the same cells with light at 600 nm (so as not to interfere with the Fura-2 fluorescence at 510 nm). The light scattering from the beating cilia created amplitude modulations of the 600-nm light that were detected by a photomultiplier (R2014; Hamamatsu).
The [Ca2+]i was calculated using a calibration curve that correlates Fura-2 fluorescence ratio to calcium concentration. The calibration curve was constructed from measurements of the fluorescence ratio obtained from calibration solutions composed of 115 mM KCl, 20 mM NaCl, 5 mM MgCl2, 5 mM d-glucose, 5 mM HEPES, 10 mM EGTA, 1 μM K2Fura-2, and CaCl2 at a range of concentrations (Grynkiewicz et al., 1985). The [Ca2+]i concentration was calculated directly from the calibration curve using a standard algorithm. The frequency response was calibrated by (1) oscillating lamp in the range of 2 to 40 Hz, and (2) vibrating needle between 2 and 40 Hz. The response was linear in these dynamic ranges.
Experimental Procedure and Data Presentation.
Cells were preloaded with Fura-2 by incubating the tissue in growth medium containing 8.5 μM Fura-2/AM for 60 min at 37°C in a rotating water bath. The cells were then rinsed with Ringer solution composed of (in mM) 150 NaCl, 2.5 KCl, 1.5 CaCl2, 1.5 MgCl2, 5 d-glucose, 5 HEPES (pH 7.4), and maintained at room temperature for 30 min to allow full de-esterification and equilibration of the dye in the cytoplasm. Only evenly fluorescent cells were used, i.e., those which did not display bright spots and had a steady CBF. In each experiment, basal CBF (Fo) and basal [Ca2+]i ([Ca2+]io) were measured for 2–5 min in the appropriate solution. These were taken as reference values. The test chemical was then introduced by exchanging the bath solution, and the frequency (F) and [Ca2+]i were monitored in the same ciliary area for 10–15 min. Each experimental condition was tested on native cultures to avoid possible bias arising from previous manipulations. Beat frequencies are presented as the fold increase in CBF (F/Fo). Changes in [Ca2+]i are presented as the difference between the observed [Ca2+]i and the basal [Ca2+]i ([Ca2+]i − [Ca2+]io). The experiments were performed on 3–15 tissue cultures from at least 2–4 animals.
RESULTS
This work explored whether axonemal proteins are phosphorylated in response to mucociliary cell stimulation by extracellular ATP or ionomycin, and whether the NO pathway is involved in this process. To analyze the phosphorylation status of ciliary proteins, an assay was developed (see materials and methods) that enabled the detection of proteins phosphorylated after stimulation of intact cells, under conditions similar to those used for CBF measurement (Korngreen and Priel, 1996; Uzlaner and Priel, 1999; Braiman et al., 2000). ATP was chosen as a stimulant since it is well known that extracellular ATP induces significant CBF enhancement, accompanied by a rise in [Ca2+]i, in almost all examined vertebrate mucociliary systems: frog palate (Ovadyahu et al., 1988), rabbit oviduct (Villalon et al., 1989), frog esophagus (Weiss et al., 1992), rabbit trachea (Korngreen and Priel, 1996), dog trachea (Wong and Yeates, 1992), and human polyps (Mason et al., 1991). Furthermore, the signal transduction pathway activated by ATP in tissue cultures of mammalian airway epithelium is well characterized (Korngreen and Priel, 1996; Uzlaner and Priel, 1999; Braiman et al., 2000).
Extracellular ATP Induced CBF Enhancement and a Rise in [Ca2+]i
Porcine airway epithelium was selected for these experiments since a large number of airways could be obtained from the local abattoir within minutes of death and thus sufficient epithelium could be collected for the phosphorylation assays. It was first necessary, however, to determine whether porcine airway cilia cells respond similarly to extracellular ATP as other airway epithelium. Fig. 2 shows results from the simultaneous measurements of [Ca2+]i and CBF from a cilia cell in an explant of porcine airway epithelium. Extracellular ATP (100 μM) induced a rise in [Ca2+]i and in CBF, indicating that porcine airway cilia cells express purinergic receptors, and that upon activation these receptors give rise to a physiological response characteristic of other mucociliary systems.
Figure 2.

Effect of extracellular ATP on [Ca2+]i and CBF enhancement. Representative experiment of time course responses of [Ca2+]i (A) and CBF enhancement (B) to 100 μM ATP in porcine tracheal tissue cultures. [Ca2+]i and CBF were sampled from the same cell every second, but only each third data point is displayed for purposes of clarity. A representative of 10 similar experiments is shown.
Extracellular ATP Induced Sustained Ciliary Protein Phosphorylation and Dephosphorylation
A key to the successful determination of protein phosphorylation in intact cells was the requirement to conduct the experiments on the same day of tissue collection. The mucociliary epithelium was immediately cleaned and used within 1 h after its isolation from the body. Ciliary protein phosphorylation was determined by Western blotting using monoclonal anti-phosphoserine (clone PSR-45) or anti-phosphothreonine antibodies (clone PTR-8), which specifically react against phosphoserine or phosphothreonine, respectively (Heffetz et al., 1991; Abu-Lawi and Sultzer, 1995; Naz, 1999). Although a set of ciliary polypeptides was detected using anti-phosphothreonine antibody under basal conditions (control), there were no significant changes induced by extracellular activation (unpublished data). Therefore, it appears that serine residues represent the major stimulus-responsive phosphorylation sites in ciliary proteins. Thus, in all the presented experiments, the anti-phosphoserine antibody was used. The specificity of the anti-phosphoserine antibody was confirmed by immunoadsorption. No significant phosphorylated bands were detected after overnight incubation of antibody at 4°C with phosphoserine, 1:10 antibody:peptide ratio, confirming the specificity of the used antibody (unpublished data).
Several phosphorylated polypeptides were detected in untreated cells, the majority of which did not significantly change their phosphorylation level upon stimulation by ATP (Fig. 3 A). However, purinergic activation of mucociliary cells with 10 μM or 100 μM ATP resulted in a substantial enhancement in phosphorylation of three ciliary polypeptides: 27, 37, and 44 kD, as compared with untreated (control) cells (Fig. 3 A). Occasionally, enhanced phosphorylation of a 17-kD polypeptide was also observed. The 27- and 44-kD polypeptides exhibited a relatively high level of basal phosphorylation (Fig. 3 A, lane 1), while the 37-kD polypeptide was not observed in control tissue. For this and all subsequent experiments, an error of protein molecular weight calculation did not exceed 2%.
Figure 3.

Extracellular ATP induces phosphorylation and dephosphorylation of ciliary polypeptides in intact epithelial tissue. (A) Typical pattern of phosphorylated polypeptides in cilia isolated from epithelial tissue exposure to extracellular ATP (100 μM) for 3 min (lane 2) or 5 min (lane 3), as compared with untreated control (lane 1). Western blot analysis using anti-phosphoserine antibody indicates an increase in phosphorylation of 27, 37, and 44 kD polypeptides, and a decrease in phosphorylation of 35, 69, 100, and 130 kD polypeptides. (B) Reprobing of the nitrocellulose membrane presented in A with anti–α-tubulin antibody demonstrates equal protein loading. Representative blot of at least 10 similar experiments is shown. (C) Time course of extracellular ATP-dependent ciliary polypeptide phosphorylation. (D) Time course of extracellular ATP-dependent ciliary polypeptide dephosphorylation. Data represents a quantitative analysis of the magnitude of protein phosphorylation (C) and dephosphorylation (D) induced by extracellular ATP (100 μM). The optical density of protein bands on Western blots was measured, as described under materials and methods. The data indicate similar time dependences for the phosphorylation (C) of three major polypeptides (44, 37, and 27 kD) and dephosphorylation (D) of three additional polypeptides (130, 100, and 69 kD), which can be seen in A. The maximal phosphorylation and dephosphorylation effects were obtained at 1–5 min. Each point is the mean ± SEM of 4–10 experiments.
Surprisingly, ATP treatment also considerably reduced the level of phosphorylation of four ciliary polypeptides (35, 69, 100, and 130 kD) that exhibited relatively high levels of phosphorylation in unstimulated cells. The observed changes in the level of phosphorylation were not due to differences in the amount of protein loaded per lane as determined by either Ponceau S protein staining (not depicted) or by immunodetection of the housekeeping ciliary protein, α-tubulin (Fig. 3 B). The extent of dephosphorylation of the 35-kD polypeptide could not accurately be determined due to the proximity of the 37-kD polypeptide (after ATP stimulation) and the 33-kD polypeptide (in control). Therefore, in all further quantitative displays, the phosphorylation intensity of the 35-kD polypeptide is not presented, though it should be kept in mind that dephosphorylation of the 35-kD polypeptide was consistently observed.
Despite the apparent effect of extracellular ATP on the phosphorylation status of ciliary polypeptides, the initial results were confusing. It became apparent over time that there is a large variability in the response among pools of tracheas, ranging from a strong response to no response. The implications of this finding were twofold. First, for each experiment, it was necessary to determine to what extent ciliary proteins are phosphorylated by ATP. Second, comparison among various manipulations of the tissues (treatment with activators or inhibitors) was limited to each experiment, a requirement that greatly slowed the progress. Thus, extracellular ATP (100 μM) was selected as a stimulating solution to test the viability of each preparation. Only preparations in which a clear (visualized by a naked eye) change in phosphorylation of ciliary polypeptides was induced by extracellular ATP were used to investigate the effects of activators and/or inhibitors. Overall, in about one third of the experiments (46/144), extracellular ATP failed to induce a detectable change in the phosphorylation profile. In the remaining responsive preparations, ATP induced the same profile of phosphorylation. It is noteworthy that this phenomenon appears to be a general pattern and is not limited to the present biochemical study (see discussion).
Extracellular ATP-dependent protein phosphorylation and dephosphorylation was a time-dependent process. As demonstrated in Fig. 3 (C and D), the maximal magnitude of phosphorylation or dephosphorylation was achieved within 3–5 min after ATP application. In accordance with the obtained time course of the ATP effect, in all subsequent experiments the intact tissue was exposed to extracellular ATP for times ranging from 3 to 5 min.
Ionomycin Induces a Change in the Phosphorylation Pattern Similar to that of ATP
Recently it was shown that extracellular ATP induces a strong and sustained rise in intracellular Ca2+ concentration ([Ca2+]i) near the apical membrane, in proximity to the cilia (Paradiso et al., 1995; Braiman and Priel, 2001). It is well known that elevated [Ca2+]i may activate various protein kinases and/or phosphatases via numerous different signaling pathways. Hence, to assess whether the effect of extracellular ATP on protein phosphorylation is mediated by Ca2+, the purinergic receptor was bypassed by directly elevating [Ca2+]i with ionomycin. Application of ionomycin (1 μM) to intact epithelial tissue for 0.5 or 1 min induced phosphorylation of 27-, 37-, and 44-kD polypeptides and dephosphorylation of 35-, 69-, 100-, and 130-kD polypeptides. Thus, the pattern of changes in the phosphorylation profile was identical to that produced by extracellular ATP (compare Fig. 4 with Fig. 3 A).
Figure 4.

Ionomycin treatment induces phosphorylation and dephosphorylation of the same polypeptides that are modified by extracellular ATP. Typical pattern of phosphorylated polypeptides in cilia isolated from epithelial tissue after exposure to ionomycin (1 μM) for 0.5 min (lane 2) or 1 min (lane 3), as compared with untreated control (lane 1). Western blot analysis using anti-phosphoserine antibody indicates an increase in phosphorylation of 27, 37, and 44 kD polypeptides, and a decrease in phosphorylation of 35, 69, 100, and 130 kD polypeptides. A representative blot of eight similar experiments is shown.
Extracellular ATP Induces Changes in Ciliary Protein Phosphorylation Via the NO Pathway
It is well known that the binding of Ca2+ to calmodulin activates the NO pathway. Moreover, it has been shown that the NO pathway plays a pivotal role in robust mucociliary stimulation (Uzlaner and Priel, 1999; Braiman et al., 2000). To assess whether the NO pathway mediates the observed changes in ciliary protein phosphorylation, we examined the effect of extracellular ATP in the presence of L-NAME, an NO syntase (NOS) inhibitor. Pretreating the tissue with L-NAME (50 μM) for 10 min abolished the changes in phosphorylation and dephosphorylation induced by ATP (Fig. 5, A and B). Similarly, L-NAME prevented the changes in phosphorylation profile induced by ionomycin (unpublished data). Conversely, incubating the cells for 5 min with the NOS substrate l-arginine (1 mM) moderately augmented the changes in phosphorylation and dephosphorylation induced by extracellular ATP, while having no detectable effect on the basal levels of phosphorylation (unpublished data). These results indicate that NOS is involved in the changes in protein phosphorylation induced by extracellular ATP.
Figure 5.

Inhibition of NOS abolishes extracellular ATP-dependent phosphorylation (A) and dephosphorylation (B) of ciliary polypeptides in intact tissue. Epithelial tissue was preincubated for 10 min with or without 50 μM L-NAME followed by stimulation with extracellular ATP (100 μM) for 5 min. Cilia were then isolated and subjected to Western blot analysis, as described under materials and methods. Quantitative analysis of the magnitude of phosphorylation shows the complete elimination of phosphorylation and dephosphorylation induced by ATP (100 μM) in the presence of L-NAME (50 μM). Each column represents the mean ± SEM averaged over six experiments.
The step that follows NOS stimulation in the NO pathway is the activation of guanylyl cyclase (GC) by NO, resulting in the elevation of intracellular cGMP, which in turn activates PKG. It has previously been shown that the activation of PKG in the presence of elevated [Ca2+]i results in robust mucociliary activity (Uzlaner and Priel, 1999). To examine whether inhibition of GC or PKG would affect the changes in protein phosphorylation induced by extracellular ATP, epithelial tissue was preincubated for 10 min with either 50 μM LY-83583 or 1 μM KT-5823, the inhibitors of GC and PKG, respectively. As shown in Fig. 6, both inhibitors blocked the ATP-induced enhancement in ciliary protein phosphorylation. It should be mentioned that all applied inhibitors, except KT-5823, did not affect the basal level of phosphorylation of ciliary proteins (Figs. 5 and 6). Preincubation of mucociliary tissue with KT-5823 resulted in a moderate increase in phosphorylation of the 44-kD polypeptide to ∼130–160% of pretreated value (=100%) (Fig. 6, lane 5). The reason for this effect is unclear. However, this phosphorylation was not altered by ATP addition (Fig. 6, compare lanes 5 and 6). It is reasonable to assume from these results that the phosphorylation induced by extracellular ATP is directly mediated by PKG; however, the involvement of PKA or other protein kinases cannot be excluded. Interestingly, the inhibition of NOS, GC, or PKG also abolished the dephosphorylation induced by extracellular ATP, suggesting that PKG activates one or more phosphatases (Figs. 5 and 6). Taken together, these results strongly indicate that activation of the NO pathway is essential for axonemal phosphorylation and dephosphorylation induced by extracellular ATP.
Figure 6.

Inhibition of GC or PKG prevents ATP-dependent polypeptide phosphorylation and dephosphorylation in intact tissue. Epithelial tissue was preincubated for 10 min in the absence (lanes 1 and 2) or in the presence of either 50 μM LY-83583 (GC inhibitor, lanes 3 and 4) or 1 μM KT-5823 (PKG inhibitor, lanes 5 and 6) followed by stimulation with extracellular ATP (100 μM) for 5 min (lanes 2, 4, and 6). Cilia were then isolated and subjected to Western blot analysis, as described under materials and methods. Application of LY-83583 did not alter the basal level of phosphorylation (lane 3), although preincubation with KT-5823 resulted in a moderate phosphorylation of 44-kD polypeptide (lane 5), which was not influenced by the ATP addition (compare lanes 5 and 6). A representative of seven similar experiments is shown.
Dephosphorylation of Ciliary Polypeptides Is Not Observed in Experiments on Isolated Cilia
The phosphorylation of axonemal proteins in isolated cilia has been reported in various ciliary preparations, including flagella, unicells, and mammalian airway epithelium (Miglietta and Nelson, 1988; Bonini and Nelson, 1990; Hamasaki et al., 1991; Salathe et al., 1993a; Gibbons, 1995; Bracho et al., 1998; Naz, 1999; Porter and Sale, 2000; Christensen et al., 2001). However, to the best of our knowledge, there has been no report of sustained protein dephosphorylation in ciliated cells induced by protein kinase stimulation. One possible explanation for the observed difference between the present results and previous work might be that dephosphorylation requires intact cells. To test this possibility, experiments were performed on isolated cilia. Cilia detached from untreated epithelium were incubated with either 0.1 μM or 1 μM cGMP, as described under materials and methods. Two of the polypeptides phosphorylated in the intact tissue (27 and 44 kD) were also phosphorylated by cGMP in the experiments on isolated cilia (Fig. 7, A and B). However, while the 35-, 69-, 100-, and 130-kD polypeptides were dephosphorylated as a result of the PKG stimulation in intact tissue, the phosphorylation of these polypeptides was significantly enhanced by cGMP in the detached cilia (Fig. 7, A and B). In addition, the phosphorylation of the 37-kD polypeptide was not observed in detached cilia, while it was markedly phosphorylated in intact tissue (compare Fig. 3 A, Figs. 4–6, and Fig. 7 A). Furthermore, additional polypeptides that did not appear to be regulated by the NO pathway in intact cells were phosphorylated by cGMP in the detached cilia. These results might indicate that the protein phosphorylation assay in the intact tissue more closely tracks the physiological changes in protein modification pattern accompanying ciliary stimulation.
Figure 7.

cGMP induces phosphorylation of ciliary polypeptides in cilia isolated from untreated tissue. (A) Isolated cilia were incubated with or without 1 μM cGMP for 3 min, as described under materials and methods, followed by Western blot analysis with anti-phosphoserine antibody. The data represent cGMP-stimulated phosphorylation (lane 2) as compared with control (lane 1). A representative of five similar experiments is shown. (B) Quantitative analysis of the magnitude of polypeptide phosphorylation induced by 0.1 μM cGMP (empty bars) and 1 μM cGMP (hatched bars). The data indicate that cGMP modifies the major polypeptides, which are phosphorylated by extracellular ATP or ionomycin in intact tissue experiments (Figs. 3 and 4). Each column represents the mean ± SEM averaged over five to seven experiments.
PKG and Its Potential Protein Substrates Are Colocalized Mainly in the Axoneme
The main effect of extracellular ATP or ionomycin is to significantly enhance phosphorylation and dephosphorylation of ciliary polypeptides, which are already phosphorylated to some degree in untreated cells (Figs. 3 and 4). We took advantage of this phenomenon (phosphorylation status of untreated cilia) to assess whether the detected proteins are axonemal or are associated with the ciliary membrane. For this purpose, cilia were incubated with a high concentration of Triton X-100, and the detergent-insoluble axoneme was separated from the solubilized membrane proteins (see materials and methods). The profiles of polypeptide phosphorylation in intact cilia, ciliary membrane fraction, and axonemal fraction were compared. As can be seen in Fig. 8 A, the polypeptides that were found to be further phosphorylated or dephosphorylated in response to extracellular ATP or ionomycin (Figs. 3 and 4) are predominantly located in the axoneme (Fig. 8 A, lane 3) with the exception of the 69-kD polypeptide that is mainly associated with the ciliary membrane fraction (Fig. 8 A, lane 2). The 37-kD polypeptide was not observed either in whole cilia or in the separated fractions because it appeared de novo after stimulation of intact cells. Therefore, the location of the 37-kD polypeptide is yet undetermined. Based on these experiments, we conclude that multiple axonemal polypeptides are regulated (phosphorylated and dephosphorylated) during ciliary activation by extracellular ATP or ionomycin.
Figure 8.
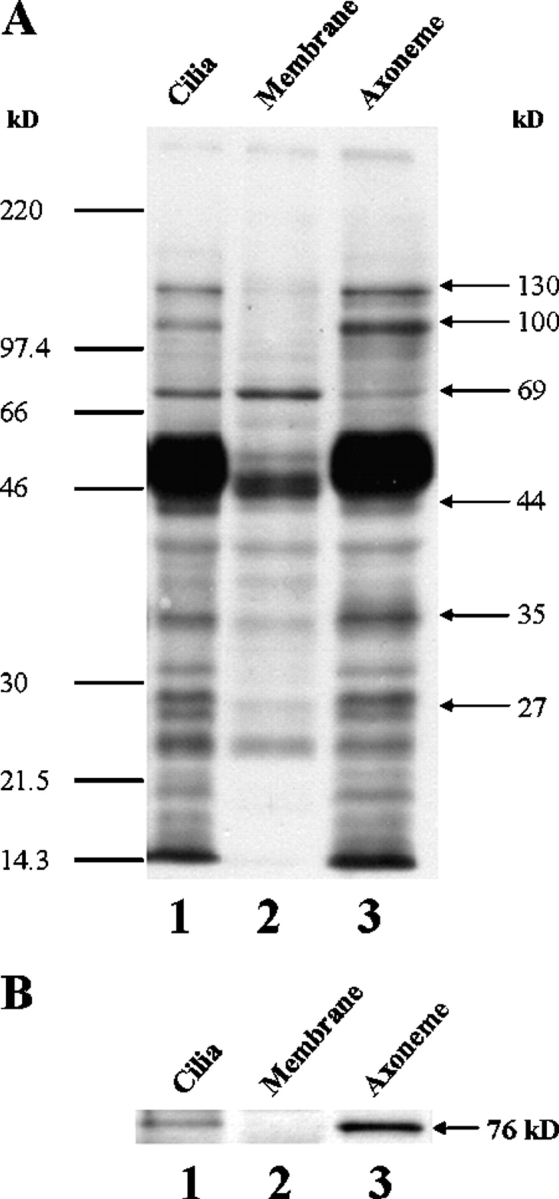
The ciliary polypeptides found to be modified by extracellular ATP (A) and PKG (B) are predominantly located in the axoneme. (A) Phosphorylated polypeptide profiles of untreated cilia and its fractions separated after detergent treatment. Cilia were isolated from untreated epithelial tissue (lane 1), separated to detergent-soluble membrane (lane 2) and detergent-insoluble axonemal (lane 3) fractions, and subjected to Western blot analysis, as described under materials and methods. The data indicate that the major polypeptides found to be phosphorylated (27 and 44 kD) and dephosphorylated (35, 100, and 130 kD) after exposure to extracellular ATP (Fig. 3) are predominantly located in the axoneme, while the 69-kD polypeptide appears mainly in the membrane fraction. Note that the 37-kD polypeptide is not presented either in whole cilia or in the separated fractions. This is consistent with the above data (Fig. 3 A, lane 1; Fig. 4, lane 1; Fig. 6, lane 1), showing the absence of the 37-kD band under control conditions (in unstimulated cilia). Moreover, enrichment of the axonemal fraction in all polypeptides, as compared with whole cilia, can be seen. A representative of seven similar experiments is shown. (B) PKG is an integral axonemal protein. Reprobing of the nitrocellulose membrane presented in A with cGKIα antibody demonstrates the polypeptide of 76 kD typical for PKG isoform in both whole cilia and the axonemal fraction. An enrichment of the axonemal fraction in PKG, as compared with whole cilia, can be seen. A representative of three similar blots is shown.
In the experiments on detached cilia, protein phosphorylation was achieved by submicromolar concentrations of cGMP (Fig. 7 B), a concentration range in which cGMP selectively activates PKG (Lincoln and Cornwell, 1993). Thus it is reasonable to assume that PKG is an integral axonemal protein. To confirm that PKG is integrated within the axoneme, the polyclonal anti-cGKIα antibody raised against a peptide mapping near the NH2 terminus of PKG (regulatory region) was used. The presence of PKG was checked in intact cilia and after separation to the axonemal and ciliary membrane fractions. Fig. 8 B shows that a polypeptide with a molecular weight of 76 kD, typical for PKG isoform, is detected by the antibody in whole cilia and in the axonemal fraction. Since equal amounts of protein were loaded in each lane, the quantitative amplification of the indicated polypeptides in axonemal fraction as compared with whole cilia reveals that the axonemal fraction is enriched for the phosphorylated polypeptides and PKG (Fig. 8, A and B, compare lanes 1 and 3). Taken together, the result demonstrates that PKG is an integral protein of the axoneme, and that upon stimulation, PKG mediates the phosphorylation and dephosphorylation of axonemal proteins.
DISCUSSION
The Intact Tissue Assay Reveals Sustained Protein Dephosphorylation During Ciliary Activation
Most of the biochemical research on ciliates, including the two works on mucociliary cells (Salathe et al., 1993a,b), were performed on detached cilia. These studies helped establish the importance of protein phosphorylation in the regulation of ciliary function and have identified proteins that may be important in this process. As is true for any experimental procedure, the protein phosphorylation assay on detached cilia has drawbacks. In the test tube, it is currently not possible to mimic the spatio-temporal changes in cellular components within the cilia that accompany ciliary stimulation. Yet, the sequence and/or the local concentrations of these compounds might determine the function of the cilia. In addition, the isolation procedure might lead to the loss of essential regulatory components and/or cause changes in the conformation of proteins within the axoneme, masking physiologically important regulatory sites. Therefore, an immunochemical method was adapted that enables one to identify agonist-induced ciliary protein phosphorylation in intact cells, and to reveal the intracellular molecular pathway(s) that lead to these ciliary/axonemal phosphorylations and dephosphorylations. The described assay allows us to correlate between phosphorylated proteins and physiological results obtained in our present and previous studies (Korngreen and Priel, 1996; Uzlaner and Priel, 1999; Braiman et al., 2000). The sensitivity of immunochemical detection of phosphoproteins is probably lower than that of the radioactive assay using metabolic labeling with 32P. Unfortunately, it was impossible to apply a radioactive protein phosphorylation assay in intact airway epithelium, since this assay requires ∼16 h incubation of the tissue with 32P (Yin et al., 2000). Such a long incubation period was found in our hands to drastically reduce release of NO induced by extracellular ATP and/or phosphorylation of axonemal polypeptides. Since the specificity of anti-phosphoamino acid antibodies is widely debatable, we employed anti-phosphoserine and anti-phosphothreonine antibodies, which were previously successfully used in biochemical analysis and established as specifically recognizing only the phosphorylated forms of serine and threonine amino acids, respectively (Heffetz et al., 1991; Abu-Lawi and Sultzer, 1995; Naz, 1999). Furthermore, we confirmed the specificity of the used antibody in an immunoadsorption experiment with free phosphoserine, which completely abolished the immunoreactivity of the antibody.
Despite the limitations of the immunochemical technique to monitor the phosphorylation status of the ciliary proteins, marked differences in phosphorylation and dephosphorylation were observed between intact tissue and detached cilia preparations (compare Fig. 3 with Fig. 7). In particular, sustained dephosphorylation of ciliary proteins was absent in the detached cilia experiments, suggesting that one or more phosphatases were either lost or deactivated during the isolation of the cilia. Indeed, protein phosphatases were recently shown to be lost in Paramecium during axonemal isolation (Noguchi et al., 2003), which may explain why sustained dephosphorylation of ciliary proteins was not previously shown to accompany protein phosphorylation. Another likely reason why dephosphorylation of ciliary polypeptides was overlooked is that much of the previous biochemical work aimed at identified proteins that are phosphorylated by second messengers was performed on detached cilia in the presence of various phosphatase inhibitors. Recently, the presence of a number of phosphatases as an integral part of human cilia axonemes was confirmed in the first systematic analysis of the protein composition of the mammalian axoneme (Ostrowski et al., 2002).
Elevated [Ca2+]i Induces Ciliary Protein Phosphorylation and Dephosphorylation Accompanied by Robust CBF Enhancement
It is well established that a rise in [Ca2+]i induces strong and sustained CBF enhancement. On the other hand, whether elevated [Ca2+]i by itself can enhance CBF (Lansley and Sanderson, 1999; Salathe and Bookman, 1999; Zhang and Sanderson, 2003) or whether a rise in [Ca2+]i activates and synchronizes additional intracellular pathway(s) that are essential for CBF enhancement is yet debatable. In favor of the latter, we recently showed that elevated [Ca2+]i does not directly activate the axoneme and that additional cytosolic second messengers are required, mainly cAMP and cGMP (Ma et al., 2002). Furthermore, results obtained using either ciliary tissue cultures, intact ciliary cells, or intact ciliary tissues, using a range of methods, all support this conclusion, suggesting that these findings are general (Uzlaner and Priel, 1999; Braiman et al., 2000; Zagoory et al., 2001, 2002; Ma et al., 2002). The present work supports our previous findings and extends our knowledge of the functions of elevated [Ca2+]i in the ciliary cells. It demonstrates that direct elevation of [Ca2+]i by ionomycin induces profound changes in the phosphorylation profile of ciliary proteins (Fig. 4), identical to those induced by extracellular ATP (compare Fig. 4 with Fig. 3 A). This suggests that the effect of extracellular ATP is mainly mediated by elevation of [Ca2+]i. Moreover, inhibition of NOS abolished the changes in the phosphorylation of ciliary proteins induced by ionomycin or extracellular ATP (Fig. 5), as well as CBF enhancement in the presence of elevated [Ca2+]i (Uzlaner and Priel, 1999; Braiman et al., 2000), indicating that phosphorylation and/or dephosphorylation of ciliary proteins is essential in the mechanism of CBF enhancement.
Agonist-induced Phosphorylation of Ciliary Proteins and CBF Enhancement Requires Viable NOS
The present study demonstrates that the NO pathway, which induces CBF enhancement, also leads to sustained phosphorylation/dephosphorylation of ciliary polypeptides. This correlation suggests that changes in the phosphorylation status of ciliary proteins are a prerequisite to CBF enhancement. Over the last decade, there is increasing evidence that the NO pathway plays an important role in CBF stimulation in mammalian cells. Expression of NOS was demonstrated in human airway epithelium (Guo et al., 1997; Robbins et al., 1997), and release of NO was reported in rabbit trachea mucosa (Tamaoki et al., 1995). Inhibition of NOS was shown to attenuate CBF stimulation induced by isoprenaline in bovine airway epithelium (Jain et al., 1993), by β-adrenergic stimulation in rabbit trachea (Tamaoki et al., 1995) and by methacholine in cultured human adenoid explants (Yang et al., 1996). Moreover, a correlation between the nasal NO concentration and mucociliary function in patients suffering from respiratory tract diseases such as chronic sinusitis or recurrent pneumonia was demonstrated (Lindberg et al., 1997). Similarly, in healthy subjects, a correlation was established between nasal NO concentration and nasal airway resistance or saccharin transport rate (Imada et al., 2002). Furthermore, we have shown that activated NO is a necessary condition to induce CBF stimulation by rise of [Ca2+]i (Uzlaner and Priel, 1999).
Based on the strong correlation between NO concentrations and CBF enhancement revealed in various mammalian ciliary systems, we hypothesize that the relatively high percentage (∼30%) of irresponsive porcine trachea preparations revealed in the present study is due to impaired NOS activity. In agreement with this idea, a similar percentage of nonresponsive porcine tracheas were observed in experiments designed to detect NO release induced by extracellular ATP (Lozinsky et al., 2004). While the reason(s) for the strong variability of NO· production in mammalians is yet unknown, it may depend on age, disease state, environment agents and/or isolation procedure. It must be emphasized that NO· is a short living radical that can easily be deactivated by various chemicals, thus representing a weak link in the chain of molecular events that leads to the changes in phosphorylation status of ciliary polypeptides and CBF enhancement.
The Pivotal Role of Axonemal PKG in Ciliary Protein Phosphorylation and Dephosphorylation Induced by Extracellular ATP
The sequences of molecular events that activate the NO pathway and at least part of its upstream elements are located within the ciliary cell. Therefore, the question of whether the activated NO pathway directly interacts with the axoneme or whether additional molecular intermediates are involved remains unresolved. Nevertheless, there are strong indications that PKG, the final element in the NO pathway, is located within the cilium and is involved in CBF stimulation (Ma et al., 2002). The presence of PKG within the axoneme revealed in this work (Fig. 8 B) is consistent with this conclusion. Moreover, the same six polypeptides (with one exception, 37 kD) that were phosphorylated and dephosphorylated in intact tissue were also phosphorylated in the detached and permeabilized cilia in response to submicromolar concentration of cGMP (Fig. 7), which strongly suggests the involvement of axonemal PKG in these processes.
Robust ciliary activation requires elevated [Ca2+]i and activated PKA and PKG (Braiman et al., 2000; Ma et al., 2002; Zagoory et al., 2002). The rise in [Ca2+]i inevitably leads to the activation of axonemal PKG via the NO pathway and may also activate PKA by stimulating Ca2+/calmodulin-sensitive adenylyl cyclase. Furthermore, cross-talk between the cAMP- and cGMP-dependent pathways was recently revealed in ciliary cells (Zagoory et al., 2002). Hence, it is not surprising that elevating [Ca2+]i by ionomycin resulted in changes in the phosphorylation profile of ciliary polypeptides (Fig. 4). At present, it is not clear which of the multiple Ca2+-activated cellular processes underlies the observed protein phosphorylations. Nevertheless, it is apparent that axonemal PKG plays a pivotal role in these phosphorylations. In the presence of elevated [Ca2+]i, the condition that should activate all Ca2+-sensitive protein kinases, inhibiting axonemal PKG prevented protein phosphorylation (Fig. 6). An identical dependence on PKG was reported for mucociliary activity (Uzlaner and Priel, 1999; Braiman et al., 2000; Ma et al., 2002; Zagoory et al., 2002). It can be concluded from these results that if the observed protein phosphorylations are induced by protein kinases other than PKG, these kinases must be activated by axonemal PKG. Surprisingly, the sustained protein dephosphorylations observed in this study are also under the control of axonemal PKG, since inhibiting PKG or any step in the pathway leading to PKG stimulation prevented these dephosphorylations. These results indicate that axonemal PKG directly or indirectly regulates one or more phosphatases activated during ciliary stimulation. Further work is needed to dissect apart the distinct functions of PKA and PKG in the phosphorylation and dephosphorylation processes, to identify the involved phosphatases, and to resolve the unique role of axonemal PKG in regulating both the phosphorylation and dephosphorylation of ciliary proteins.
Basal Protein Phosphorylation and Basal Activity
Using the patch-clamp technique to directly control the composition of the intracellular environment in native dissociated airway mucociliary cells, we recently showed that cytosolic second messengers are not needed to support spontaneous (basal) ciliary beating (Ma et al., 2002). In the present study, it was found that several proteins are serine phosphorylated in unstimulated (control) cilia. An intriguing possibility is that some or all of these basally phosphorylated polypeptides control spontaneous cilia beating. The same underlying pathway (NO pathway) that leads to robust CBF enhancement also leads to phosphorylation and dephosphorylation of ciliary polypeptides, supporting this suggestion. Indeed, it was shown in axonemes of Paramecium that one of the phosphorylated proteins (29 kD) copurifies with the outer dynein arm and seems to be a dynein light chain. Moreover, after cAMP-dependent phosphorylation of the 29-kD protein, purified dynein increased translocation of microtubules in vitro (Hamasaki et al., 1991). In Chlamydomonas flagella, an intermediate chain of inner dynein arm (138 kD) was identified as a critical polypeptide for regulation of motility. Phosphorylation and dephosphorylation of 138 kD inhibits and restores, respectively, wild-type microtubule sliding in experiments in vitro (Habermacher and Sale, 1997). The question of whether the 27-kD polypeptide and the 130-kD polypeptide described in the present study are outer dynein light chain and intermediate chain of inner arm dynein, respectively, in the mucociliary system remains to be determined. It is noteworthy that in flagella and in cytoplasmic dynein, heavy chains are mainly serine phosphorylated in vivo (Dillman and Pfister, 1994; King and Witman, 1994), consistent with the present study, which reveals that agonist-induced phosphorylation and dephosphorylation of ciliary polypeptides predominantly occur on serine residues.
Molecular Events Induced by Extracellular ATP that Lead to Ciliary Phosphorylation/Dephosphorylation and Enhanced CBF
Based on the present findings and on previous work (Uzlaner and Priel, 1999; Braiman et al., 2000; Ma et al., 2002), we propose that the following cascade of molecular events underlies robust CBF enhancement (Fig. 9). Stimulation by extracellular ATP leads to activation of phospholipase C and mobilization of Ca2+ from intracellular stores. In parallel, extracellular ATP activates Ca2+ influx by a yet unknown mechanism. The combined effect is a large and sustained rise in [Ca2+]i (Paradiso et al., 1995; Korngreen and Priel, 1996; Braiman and Priel, 2001). The Ca2+ binds to calmodulin to form Ca2+/calmodulin complex, which in turn activates NOS. Activated NOS produces NO·, which excites GC to produce cGMP. The increased level of cGMP activates axonemal PKG, while the stimulated PKG in the presence of elevated [Ca2+]i enhances the phosphorylation of three ciliary polypeptides (27, 37, and 44 kD) either directly or by activating one or more additional kinases. In parallel, PKG induces activation of one or more phosphatases that dephosphorylate three axonemal polypeptides (35, 100, and 130 kD) and one ciliary membrane polypeptide (69 kD). We assumed that some or all of the polypeptides that undergo sustained phosphorylation or dephosphorylation are directly involved in the mechanism of robust ciliary enhancement. Accordingly, the concerted action of phosphorylated and/or dephosphorylated proteins in the presence of elevated [Ca2+]i results in strong CBF enhancement. It remains to be determined whether axonemal PKG directly phosphorylates the axonemal polypeptides and activates the phosphatases responsible for the sustained dephosphorylations, or whether additional intermediates are involved in both these processes. To conclude, a rise in [Ca2+]i leads to both changes in ciliary phosphorylation and to CBF enhancement by stimulating identical pathways, suggesting that the phosphorylation and/or dephosphorylation of ciliary polypeptides is a necessary condition for CBF enhancement.
Figure 9.

Molecular events underlying ATP-induced CBF enhancement via phosphorylation and dephosphorylation of ciliary proteins. See details in the text.
Acknowledgments
This work was supported by the Israeli Science Foundation (grant 262/00) founded by the Israeli Academy of Sciences and Humanities.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: CBF, ciliary beat frequency; E-64, N-(trans-epoxysuccinyl)-l-leucine-4-guanidinobutylamine; GC, guanylyl cyclase; L-NAME, N ω-nitro-l-arginine methyl ester; LY-83583, 6-anilinoquinoline-5,8-quinone; NOS, NO syntase.
References
- Abu-Lawi, K.I., and B.M. Sultzer. 1995. Induction of serine and threonine protein phosphorylation by endotoxin-associated protein in murine resident peritoneal macrophages. Infect. Immun. 63:498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, R.G. 1974. Isolation of ciliated or unciliated basal bodies from the rabbit oviduct. J. Cell Biol. 60:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini, N.M., and D.L. Nelson. 1990. Phosphoproteins associated with cyclic nucleotide stimulation of ciliary motility in Paramecium. J. Cell Sci. 95:219–230. [DOI] [PubMed] [Google Scholar]
- Bousquet, C., D.W. Ray, and S. Melmed. 1997. A common pro-opiomelanocortin-binding element mediates leukemia inhibitory factor and corticotropin-releasing hormone transcriptional synergy. J. Biol. Chem. 272:10551–10557. [DOI] [PubMed] [Google Scholar]
- Bracho, G.E., J.J. Fritch, and J.S. Tash. 1998. Identification of flagellar proteins that initiate the activation of sperm motility in vivo. Biochem. Biophys. Res. Commun. 242:231–237. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. [DOI] [PubMed] [Google Scholar]
- Braiman, A., and Z. Priel. 2001. Intracellular stores maintain stable cytosolic Ca2+ gradients in epithelial cells by active Ca2+ redistribution. Cell Calcium. 30:361–371. [DOI] [PubMed] [Google Scholar]
- Braiman, A., O. Zagoory, and Z. Priel. 1998. PKA induces Ca2+ release and enhances ciliary beat frequency in a Ca2+-dependent and -independent manner. Am. J. Physiol. 275:C790–C797. [DOI] [PubMed] [Google Scholar]
- Braiman, A., N. Uzlaner, and Z. Priel. 2000. The role of cyclic nucleotide pathways and calmodulin in ciliary stimulation. Computational Modeling in Biological Fluid Dynamics. Vol. 124. L.J. Fauci and S. Gueron, editors. Springer-Verlag, New York. 53–64.
- Brustle, B., S. Kreissl, D.L. Mykles, and W. Rathmayer. 2001. The neuropeptide proctolin induces phosphorylation of a 30 kDa protein associated with the thin filament in crustacean muscle. J. Exp. Biol. 204:2627–2635. [DOI] [PubMed] [Google Scholar]
- Christensen, S.T., C. Guerra, Y. Wada, T. Valentin, R.H. Angeletti, P. Satir, and T. Hamasaki. 2001. A regulatory light chain of ciliary outer arm dynein in Tetrahymena thermophila. J. Biol. Chem. 276:20048–20054. [DOI] [PubMed] [Google Scholar]
- Di Benedetto, G., F.S. Manara-Shediac, and A. Mehta. 1991. Effect of cyclic AMP on ciliary activity of human respiratory epithelium. Eur. Respir. J. 4:789–795. [PubMed] [Google Scholar]
- Dillman, J.F., III, and K.K. Pfister. 1994. Differential phosphorylation in vivo of cytoplasmic dynein associated with anterogradely moving organelles. J. Cell Biol. 127:1671–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary, C.A., C.W. Davis, A.M. Paradiso, and R.C. Boucher. 1995. Role of CNP in human airways: cGMP-mediated stimulation of ciliary beat frequency. Am. J. Physiol. 268:L1021–L1028. [DOI] [PubMed] [Google Scholar]
- Gibbons, I.R. 1995. Dynein family of motor proteins: present status and future questions. Cell Motil. Cytoskeleton. 32:136–144. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz, G., M. Poenie, and R.Y. Tsien. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- Guo, F.H., K. Uetani, S.J. Haque, B.R. Williams, R.A. Dweik, F.B. Thunnissen, W. Calhoun, and S.C. Erzurum. 1997. Interferon γ and interleukin 4 stimulate prolonged expression of inducible nitric oxide synthase in human airway epithelium through synthesis of soluble mediators. J. Clin. Invest. 100:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermacher, G., and W.S. Sale. 1997. Regulation of flagellar dynein by phosphorylation of a 138-kD inner arm dynein intermediate chain. J. Cell Biol. 136:167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki, T., K. Barkalow, J. Richmond, and P. Satir. 1991. cAMP-stimulated phosphorylation of an axonemal polypeptide that copurifies with the 22S dynein arm regulates microtubule translocation velocity and swimming speed in Paramecium. Proc. Natl. Acad. Sci. USA. 88:7918–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki, T., K. Barkalow, and P. Satir. 1995. Regulation of ciliary beat frequency by a dynein light chain. Cell Motil. Cytoskeleton. 32:121–124. [DOI] [PubMed] [Google Scholar]
- Heffetz, D., M. Fridkin, and Y. Zick. 1991. Generation and use of antibodies to phosphothreonine. Methods Enzymol. 201:44–53. [DOI] [PubMed] [Google Scholar]
- Imada, M., S. Nonaka, Y. Kobayashi, and J. Iwamoto. 2002. Functional roles of nasal nitric oxide in nasal patency and mucociliary function. Acta Otolaryngol. 122:513–519. [DOI] [PubMed] [Google Scholar]
- Jain, B., I. Rubinstein, R.A. Robbins, K.L. Leise, and J.H. Sisson. 1993. Modulation of airway epithelial cell ciliary beat frequency by nitric oxide. Biochem. Biophys. Res. Commun. 191:83–88. [DOI] [PubMed] [Google Scholar]
- King, S.M., and G.B. Witman. 1994. Multiple sites of phosphorylation within the α heavy chain of Chlamydomonas outer arm dynein. J. Biol. Chem. 269:5452–5457. [PubMed] [Google Scholar]
- Korngreen, A., and Z. Priel. 1994. Simultaneous measurement of ciliary beating and intracellular calcium. Biophys. J. 67:377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korngreen, A., and Z. Priel. 1996. Purinergic stimulation of rabbit ciliated airway epithelia: control by multiple calcium sources. J. Physiol. 497:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- Lansley, A.B., and M.J. Sanderson. 1999. Regulation of airway ciliary activity by Ca2+: simultaneous measurement of beat frequency and intracellular Ca2+. Biophys. J. 77:629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansley, A.B., M.J. Sanderson, and E.R. Dirksen. 1992. Control of the beat cycle of respiratory tract cilia by Ca2+ and cAMP. Am. J. Physiol. 263:L232–L242. [DOI] [PubMed] [Google Scholar]
- Levin, R., A. Braiman, and Z. Priel. 1997. Protein kinase C induced calcium influx and sustained enhancement of ciliary beating by extracellular ATP. Cell Calcium. 21:103–113. [DOI] [PubMed] [Google Scholar]
- Lincoln, T.M., and T.L. Cornwell. 1993. Intracellular cyclic GMP receptor proteins. FASEB J. 7:328–338. [DOI] [PubMed] [Google Scholar]
- Lindberg, S., A. Cervin, and T. Runer. 1997. Low levels of nasal nitric oxide (NO) correlate to impaired mucociliary function in the upper airways. Acta Otolaryngol. 117:728–734. [DOI] [PubMed] [Google Scholar]
- Liu, Y.F., K. Paz, A. Herschkovitz, A. Alt, T. Tennenbaum, S.R. Sampson, M. Ohba, T. Kuroki, D. LeRoith, and Y. Zick. 2001. Insulin stimulates PKCζ -mediated phosphorylation of insulin receptor substrate-1 (IRS-1). A self-attenuated mechanism to negatively regulate the function of IRS proteins. J. Biol. Chem. 276:14459–14465. [DOI] [PubMed] [Google Scholar]
- Lozinsky, E.M., L.V. Martina, A.I. Shames, N. Uzlaner, A. Masarwa, G.I. Likhtenshtein, D. Meyerstein, V.V. Martin, and Z. Priel. 2004. Detection of nitric oxide from pig trachea by a fluorescence method. Anal. Biochem. 326:139–145. [DOI] [PubMed] [Google Scholar]
- Ma, W., S.D. Silberberg, and Z. Priel. 2002. Distinct axonemal processes underlie spontaneous and stimulated airway ciliary activity. J. Gen. Physiol. 120:875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason, S.J., A.M. Paradiso, and R.C. Boucher. 1991. Regulation of transepithelial ion transport and intracellular calcium by extracellular ATP in human normal and cystic fibrosis airway epithelium. Br. J. Pharmacol. 103:1649–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miglietta, L.A., and D.L. Nelson. 1988. A novel cGMP-dependent protein kinase from Paramecium. J. Biol. Chem. 263:16096–16105. [PubMed] [Google Scholar]
- Naz, R.K. 1999. Involvement of protein serine and threonine phosphorylation in human sperm capacitation. Biol. Reprod. 60:1402–1409. [DOI] [PubMed] [Google Scholar]
- Noguchi, M., J.Y. Sasaki, H. Kamachi, and H. Inoue. 2003. Protein phosphatase 2C is involved in the cAMP-dependent ciliary control in Paramecium caudatum. Cell Motil. Cytoskeleton. 54:95–104. [DOI] [PubMed] [Google Scholar]
- Ostrowski, L.E., K. Blackburn, K.M. Radde, M.B. Moyer, D.M. Schlatzer, A. Moseley, and R.C. Boucher. 2002. A proteomic analysis of human cilia: identification of novel components. Mol. Cell. Proteomics. 1:451–465. [DOI] [PubMed] [Google Scholar]
- Ovadyahu, D., D. Eshel, and Z. Priel. 1988. Intensification of ciliary motility by extracellular ATP. Biorheology. 25:489–501. [DOI] [PubMed] [Google Scholar]
- Paradiso, A.M., S.J. Mason, E.R. Lazarowski, and R.C. Boucher. 1995. Membrane-restricted regulation of Ca2+ release and influx in polarized epithelia. Nature. 377:643–646. [DOI] [PubMed] [Google Scholar]
- Porter, M.E., and W.S. Sale. 2000. The 9 + 2 axoneme anchors multiple inner arm dyneins and a network of kinases and phosphatases that control motility. J. Cell Biol. 151:F37–F42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, R.A., J.H. Sisson, D.R. Springall, K.J. Nelson, J.A. Taylor, N.A. Mason, J.M. Polak, and R.G. Townley. 1997. Human lung mononuclear cells induce nitric oxide synthase in murine airway epithelial cells in vitro: role of TNFα and IL-1β. Am. J. Respir. Crit. Care Med. 155:268–273. [DOI] [PubMed] [Google Scholar]
- Salathe, M., and R.J. Bookman. 1995. Coupling of [Ca2+]i and ciliary beating in cultured tracheal epithelial cells. J. Cell Sci. 108:431–440. [DOI] [PubMed] [Google Scholar]
- Salathe, M., and R.J. Bookman. 1999. Calcium and the regulation of mammalian ciliary beating. Protoplasma. 206:234–240. [Google Scholar]
- Salathe, M., M.M. Pratt, and A. Wanner. 1993. a. Cyclic AMP-dependent phosphorylation of a 26 kD axonemal protein in ovine cilia isolated from small tissue pieces. Am. J. Respir. Cell Mol. Biol. 9:306–314. [DOI] [PubMed] [Google Scholar]
- Salathe, M., M.M. Pratt, and A. Wanner. 1993. b. Protein kinase C-dependent phosphorylation of a ciliary membrane protein and inhibition of ciliary beating. J. Cell Sci. 106:1211–1220. [DOI] [PubMed] [Google Scholar]
- Tamaoki, J., M. Kondo, and T. Takizawa. 1989. Effect of cAMP on ciliary function in rabbit tracheal epithelial cells. J. Appl. Physiol. 66:1035–1039. [DOI] [PubMed] [Google Scholar]
- Tamaoki, J., A. Chiyotani, M. Kondo, and K. Konno. 1995. Role of NO generation in β-adrenoceptor-mediated stimulation of rabbit airway ciliary motility. Am. J. Physiol. 268:C1342–C1347. [DOI] [PubMed] [Google Scholar]
- Tamm, S.L., and M. Terasaki. 1994. Visualization of calcium transients controlling orientation of ciliary beat. J. Cell Biol. 125:1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasiuk, A., M. Bar-Shimon, L. Gheber, A. Korngreen, Y. Grossman, and Z. Priel. 1995. Extracellular ATP induces hyperpolarization and motility stimulation of ciliary cells. Biophys. J. 68:1163–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzlaner, N., and Z. Priel. 1999. Interplay between the NO pathway and elevated [Ca2+]i enhances ciliary activity in rabbit trachea. J. Physiol. 516:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdugo, P. 1980. Ca2+-dependent hormonal stimulation of ciliary activity. Nature. 283:764–765. [DOI] [PubMed] [Google Scholar]
- Verdugo, P., N.T. Johnson, and P.Y. Tam. 1980. β-Adrenergic stimulation of respiratory ciliary activity. J. Appl. Physiol. 48:868–871. [DOI] [PubMed] [Google Scholar]
- Villalon, M., T.R. Hinds, and P. Verdugo. 1989. Stimulus-response coupling in mammalian ciliated cells. Demonstration of two mechanisms of control for cytosolic [Ca2+]. Biophys. J. 56:1255–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, T., L. Gheber, V. Shoshan-Barmatz, and Z. Priel. 1992. Possible mechanism of ciliary stimulation by extracellular ATP: involvement of calcium-dependent potassium channels and exogenous Ca2+. J. Membr. Biol. 127:185–193. [DOI] [PubMed] [Google Scholar]
- Wong, L.B., and D.B. Yeates. 1992. Luminal purinergic regulatory mechanisms of tracheal ciliary beat frequency. Am. J. Respir. Cell Mol. Biol. 7:447–454. [DOI] [PubMed] [Google Scholar]
- Yang, B., R.J. Schlosser, and T.V. McCaffrey. 1996. Dual signal transduction mechanisms modulate ciliary beat frequency in upper airway epithelium. Am. J. Physiol. 270:L745–L751. [DOI] [PubMed] [Google Scholar]
- Yin, X., P.T. Jedrzejewski, and J.X. Jiang. 2000. Casein kinase II phosphorylates lens connexin 45.6 and is involved in its degradation. J. Biol. Chem. 275:6850–6856. [DOI] [PubMed] [Google Scholar]
- Zagoory, O., A. Braiman, L. Gheber, and Z. Priel. 2001. Role of calcium and calmodulin in ciliary stimulation induced by acetylcholine. Am. J. Physiol. 280:C100–C109. [DOI] [PubMed] [Google Scholar]
- Zagoory, O., A. Braiman, and Z. Priel. 2002. The mechanism of ciliary stimulation by acetylcholine: roles of calcium, PKA, and PKG. J. Gen. Physiol. 119:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L., and M.J. Sanderson. 2003. The role of cGMP in the regulation of rabbit airway ciliary beat frequency. J. Physiol. 551:765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]