

Abstract
The highly cooperative nature of Ca2+-dependent exocytosis is very important for the precise regulation of transmitter release. It is not known whether the number of binding sites on the Ca2+ sensor can be modulated or not. We have previously reported that protein kinase C (PKC) activation sensitizes the Ca2+ sensor for exocytosis in pituitary gonadotropes. To further unravel the underlying mechanism of how the Ca2+ sensor is modulated by protein phosphorylation, we have performed kinetic modeling of the exocytotic burst and investigated how the kinetic parameters of Ca2+-triggered fusion are affected by PKC activation. We propose that PKC sensitizes exocytosis by reducing the number of calcium binding sites on the Ca2+ sensor (from three to two) without significantly altering the Ca2+-binding kinetics. The reduction in the number of Ca2+-binding steps lowers the threshold for release and up-regulates release of fusion-competent vesicles distant from Ca2+ channels.
Keywords: exocytosis, kinetic modeling, Ca2+ dependency, fusion, protein phosphorylation
INTRODUCTION
Neuron and endocrine cells release neurotransmitters and hormones by highly regulated exocytosis of secretory vesicles. Calcium plays a pivotal role in triggering exocytosis. The relationship between the magnitude of Ca2+ entry and neurotransmitter release has been estimated to be approximately third order (Dodge and Rahamimoff, 1967; Augustine et al., 1985; Wu and Wu, 2001). Direct measurement of the relationship between the rate of exocytosis and the concentration of Ca2+ near release sites in a variety of neuron and endocrine cells has further supported the third- or fourth-order cooperative nature of Ca2+-dependent exocytosis (Heidelberger et al., 1994; Xu et al., 1998; Schneggenburger and Neher, 2000; Voets, 2000; Beutner et al., 2001). This has been taken as indirect evidence that final Ca2+-triggered fusion requires binding of at least three Ca2+ ions to the Ca2+ sensor(s).
It has been suggested that the fourth or higher order relationship between Ca2+ and exocytosis may be a unique feature of neuronal exocytosis, which is necessary for synchronizing neurotransmitter release with presynaptic Ca2+ influx. On the other hand, studies on hormone release from nonneuronal secretory cells have demonstrated a shallower dependence of exocytosis on Ca2+ of only second to third order, suggesting that variation in the number of Ca2+-binding steps allows precise tuning of exocytosis. It has been shown that mutations in the surface residues of the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex cause a decrease in the number of sequential Ca2+-binding steps preceding exocytosis (Sorensen et al., 2002). However, it is not known whether the number of binding sites on the Ca2+ sensor can be physiologically modulated or not.
Recently, we have shown that PKC activation sensitizes the Ca2+ sensor for exocytosis in pituitary gonadotropes (Zhu et al., 2002). To further uncover the underlying mechanism of how the Ca2+ sensor is modulated by phosphorylation, we have performed kinetic modeling of the experimental data and addressed the question of how the kinetic parameters for Ca2+ triggered exocytosis are influenced by PKC. We propose that PKC sensitizes exocytosis by reducing the number of calcium binding sites on the Ca2+ sensor from three to two, without significantly altering the Ca2+-binding kinetics.
MATERIALS AND METHODS
Cell Culture and Solutions
Isolated gonadotropes were prepared as previously described (Zhu et al., 2002). In brief, the anterior pituitary was removed from 4- to 6-wk-old male Sprague–Dawley rats that had been castrated at week 2. Cells were used 2–4 d after dispersion. Experiments were done at room temperature in a bath solution containing 140 mM NaCl, 2.5 mM KCl, 1.3 mM CaCl2, 1 mM MgCl2, 10 mM Hepes, and 10 mM glucose (pH 7.4). Cells were identified as gonadotropes as previously described (Kaftan et al., 2000). We confirmed our selection by challenging the cell with gonadotropin-releasing hormone (GnRH, 5 nM) and verifying a rapid, hormonally induced increase of in the intracellular free Ca2+ concentration ([Ca2+]i).
Membrane Capacitance (Cm) Measurement
Conventional whole-cell recordings used sylgard-coated 2–3 MΩ pipettes. Series resistances, which ranged from 4 to 12 MΩ, were included in the analysis. An EPC-9 patch-clamp amplifier was used together with PULSE+LOCK-IN software. A 977-Hz, 20-mV peak-to-peak sinusoidal voltage stimulus was superimposed on a holding potential of −80 mV. Currents were filtered at 2.9 kHz and sampled at 15.6 kHz.
Ca2+ Uncaging and [Ca2+]i Measurement
Homogenous [Ca2+]i elevation was generated by photolysis of caged Ca2+, either nitrophenyl-EGTA (NP-EGTA) or DM-nitrophen (DMN). Flashes of UV light and fluorescence-excitation light were generated as described (Xu et al., 1997). The NP-EGTA–containing internal solutions consisted of 110 mM CsCl, 5 mM NP-EGTA, 2 mM NaCl, 4 mM CaCl2, 2 mM MgATP, 0.3 mM GTP, 0.2 mM fura-6F, and 35 mM Hepes. To obtain a high level of postflash [Ca2+]i, we used DMN-containing internal solutions consisting of 110 mM CsCl, 10 mM DMN, 2 mM NaCl, 9 mM CaCl2, 2 mM MgATP, 0.3 mM GTP, 0.2 mM furaptra, and 35 mM Hepes. The basal [Ca2+]i was measured to be ∼200 nM by fura-2. Internal solutions were adjusted to pH 7.2 with either HCl or CsOH. The [Ca2+]i was calculated from the fluorescence ratio as previously described (Grynkiewicz et al., 1985).
To determine the [Ca2+]i sensitivity of exocytosis, ramp [Ca2+]i increases were generated by 5–10 s steady UV (380 nm) illumination (TILL Monochromator; TILL Photonics) of caged Ca2+. The same light was also used to monitor [Ca2+]i increases using the Ca2+ indicator, fura-6F. [Ca2+]i was calculated as follows: [Ca2+]i = Kd * (Fmax − F)/(F − Fmax/β), where β = Fmax/Fmin. Kd of fura-6F is 5.3 μM as published by Molecular Probes. Because the basal [Ca2+]i is buffered at ∼200 nM, far below the Kd of fura-6F, the initial fluorescence value can be used as Fmax. We neither measured the apparent Kd of fura-6F under our conditions nor compensated for photobleaching.
Data Analysis and Modeling
Measured data were analyzed with IgorPro (Wavemetrics). The differential equations for models were solved using Igor's IntegrateODE function. The integrating routines were written and performed with IgorPro, compiled and run on Pentium PCs. For the fitting, we first assigned γ to either 120 s−1 for control cells, or 90 s−1 for cells pretreated with 100 nM PMA for 2–3 min, as determined from the experiment (see results for more details). The parameters α and β were then determined by fitting the experimental Cm trace to one of the kinetic models as shown in Scheme 1 and Scheme 2. By trial and error fitting, we found that α and β fluctuated around 14 μM−1s−1 and 51 s−1, respectively. To avoid subjective fitting errors and to obtain the best fit, we adopted an objective criterion to assess the fitting; we varied α over the range 13–15 in 0.2 μM−1s−1 increments and β over the range 45–55 in 1 s−1 increments. We then calculated the square of the difference according to the following:
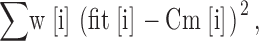 |
(1) |
where fit[i] is the calculated value according to the model for a given α and β pair, Cm[i] is the recorded raw Cm data, and w[i] is the corresponding weighting factor. Because the initial onset is sensitive to the Ca2+ stoichiometry, we set w[i] to 10 during the first 40 ms after the flash and 1 thereafter. The α and β pair that gave the minimal value according to Eq. 1 was considered the best fit. Values are given as the mean ± SEM. The Kolmogorov-Smirnov (KS) test was employed to assess the difference between cumulative histograms.
RESULTS
In response to a stepwise elevation in [Ca2+]i generated by flash photolysis of caged Ca2+, exocytosis proceeds with an initial, rapid exocytotic burst followed by a slower, sustained phase (Thomas et al., 1993; Xu et al., 1999). The kinetics of the burst component is steeply dependent on [Ca2+]i. At higher [Ca2+]i, the rate constant of the burst becomes faster. Our previous studies have demonstrated that pretreatment with PMA (100 nM) for 2–3 min accelerates the exocytotic burst and lowers the [Ca2+]i threshold required for exocytosis in gonadotropes. The effect of PMA was attributed to activation of PKC since it can be blocked by a specific PKC inhibitor and was not mimicked by an inactive phorbol ester analogue (Zhu et al., 2002). To better understand the mechanism of PKC action on the Ca2+-sensing process of exocytosis, we performed kinetic modeling of the exocytotic burst. We started with a kinetic model (see Fig. 2 C, Scheme 1) including three equivalent Ca2+-binding steps.
Figure 2.

Fitting of the exocytotic responses to appropriate kinetic models. (A) Representative Cm response from a control cell. The best fit was obtained using a three-step binding model (Scheme 1) giving the parameters α = 14 μM−1s−1, β = 51 s−1, and γ = 120 s−1 (solid line). The initial onset of the response after the flash is expanded in the inset to show the sigmoidal region in detail. The fit to a two-step binding model (Scheme 2) gives α = 8.8 μM−1s−1, β = 52 s−1, and γ = 120 s−1 (dashed line). (B) Cm response from a PMA-treated cell. The best fit to the model of Scheme 2 (dashed line) gives α = 14 μM−1s−1, β = 51 s−1, and γ = 90 s−1. Inset, comparison of the fits to Scheme 1 (solid line, α = 21.8 μM−1s−1, β = 50 s−1, γ = 90 s−1) and Scheme 2 for the initial onset. (C) Sequential Ca2+-binding models for exocytosis. (Top) Scheme 1 model with three equivalent Ca2+-binding sites. (Bottom) Scheme 2 model with only two equivalent Ca2+-binding sites.
We first estimated the irreversible fusion rate constant, γ, by inducing high level postflash [Ca2+]i. The rate constant for exocytosis is expected to approach γ at very high [Ca2+]i. We obtained the rate constants for exocytosis by fitting the burst component to a single exponential. The rate constants displayed a steep dependence on [Ca2+]i and ranged from 95 to 116 s−1 when the [Ca2+]i exceeded 100 μM (one example is shown in Fig. 1 A, see also Fig. 5 A). We therefore set the parameter γ to 120 s−1 accordingly, which is ∼5% more than the experimental range to be sure that γ is not underestimated. This number is ∼10-fold lower than that for chromaffin cells (Sorensen, 2004), but is consistent with previous findings in gonadotropes (Tse et al., 1997), probably reflecting a difference in the secretory machinery involved in the final fusion. Interestingly, we found the rate constants for exocytosis at higher [Ca2+]i after PMA treatment were consistently smaller than those for the control cells. The rate constants for PMA-treated cells ranged from 70 to 85 s−1 when the [Ca2+]i exceeded 100 μM (one example is shown in Fig. 1 B, see also Fig. 5 A). Hence, the parameter γ was set to 90 s−1 for PMA-treated cells.
Figure 1.

Determination of the irreversible fusion rate constant γ. We estimated γ by obtaining the rate constant for exocytosis from the single exponential fit of the initial burst component at very high postflash [Ca2+]i levels. Two example Cm responses from a control (A) and a PMA-treated cell (B) are displayed. Dashed lines represent the fit to a single exponential.
Figure 5.
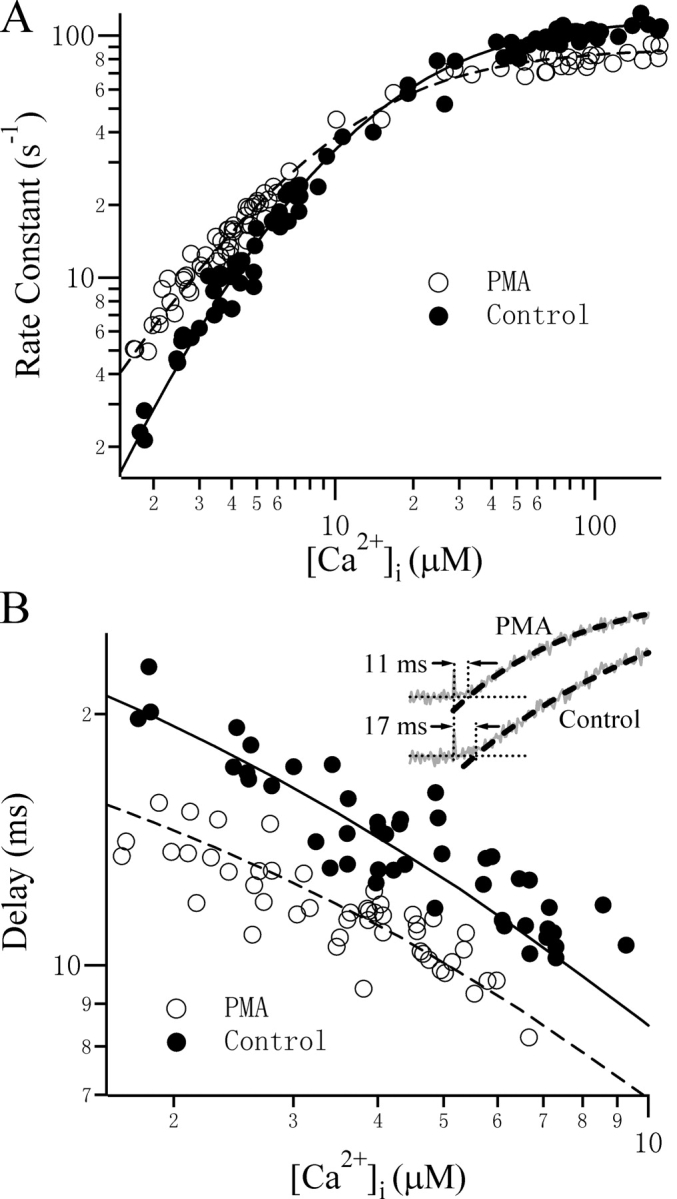
The Ca2+ dependence of the kinetics of exocytosis can be described by models with the appropriate number of Ca2+-binding sites. Rate constants (A) and the delays (B) for control (filled circles) and PMA-treated (open circles) cells were plotted as a function of the postflash [Ca2+]i level. Lines represent the simulated values using Scheme 1 (solid lines, α = 14 μM−1s−1, β = 51 s−1, γ = 120 s−1) and Scheme 2 (dashed lines, α = 14 μM−1s−1, β = 51 s−1, γ = 90 s−1). The delay was defined as the time interval between the flash and the time point at which the fitted curve (dashed line) crosses the preflash Cm level (Inset). From this value, 0.6 ms was subtracted to account for the delay required for Ca2+ to be released from the caged Ca2+. The postflash [Ca2+]i levels are 3.40 μM and 3.22 μM for the Control and PMA-treated cells, respectively.
After determining the parameter γ for control and PMA-treated cells, we next fitted the experimental responses to determine the rest of the parameters for the kinetic model(s). We found that the Cm responses to flash photolysis from control cells fit quite well to the model shown in Fig. 2 C, Scheme 1, giving values of around 14 μM−1·s−1 and 51 s−1 for α and β, respectively. Using a kinetic model with two equivalent Ca2+ binding steps (Fig. 2 C, Scheme 2) also yielded a reasonable fit to the traces for the overall time course of the burst component, albeit with lower α values. The exocytotic burst component always starts with a lag, resulting in a sigmoidal appearance of the Cm trace. It has been suggested that the binding of multiple Ca2+ ions contributes to this lag. Closer inspection of the onset of the response immediately after the flash shows that a model with three Ca2+-binding steps reproduces the sigmoidicity better than the model with two Ca2+-binding steps (Fig. 2 A, inset). To fit the Cm responses in the presence of PMA, we had to choose higher α values when using Scheme 1 (Fig. 2 B). However, when fitting the PMA-treated responses with two Ca2+-binding steps, similar α and β values were obtained as those for control responses when the three-step Ca2+-binding model was used. Also, the model with two Ca2+-binding steps seems to better describe the less sigmoidal feature of the onset of the PMA-treated responses, as shown in the inset of Fig. 2 B.
Clearly, it is not possible to exclude any particular Ca2+ stoichiometry from subtle difference in the fits to the onset of the flash responses. We therefore fitted all the experimental responses to both Scheme 1 and 2. The α and β values obtained are plotted in Fig. 3. The α values from 81 control cells display a narrow Gaussian distribution with a peak at 13.8 μM−1·s−1 when a three-step Ca2+ binding model (Scheme 1) was used, which is in contrast to the broader distribution of α when Scheme 2 was used (Fig. 3 A). This difference is significant (P < 0.01, KS test) when comparing the cumulative histogram plot. In contrast, in the presence of PMA, the estimated values for α using Scheme 1 displayed a broader distribution with a less clear peak (Fig. 3 B). When Scheme 2 was used to fit the data, a narrow Gaussian distribution of α was obtained with a clear peak at 14.2 μM−1·s−1. This difference is also clear from the cumulative histogram plot. Interestingly, the β value is less sensitive to the Ca2+ stoichiometry and follows a Gaussian distribution with a peak around 51 s−1 regardless of the number of Ca2+-binding steps chosen (Fig. 4). Similarly, no significant difference was observed in the cumulative histogram plots as assessed by KS test.
Figure 3.

Comparison of the fitting results for control and PMA-treated traces using different Ca2+-binding models. (A) The distribution and cumulative histogram of α values obtained from the fits for 81 control cells using Scheme 1 (gray bars) or Scheme 2 (open bars). Using Scheme 1, the distribution of α values displayed a sharp Gaussian distribution with a peak at 13.8 μM−1s−1 and a variation of 2.0 μM−1s−1, but was more broad when fitting to a model for only two Ca2+-binding sites. The difference in the two datasets is significant (KS test, P < 0.01). (B) The distribution and cumulative histogram of α values obtained from the fits for 95 PMA-treated traces using Scheme 1 (gray symbols) or Scheme 2 (open symbols). The distribution of α values showed a sharp Gaussian function for the model assuming two Ca2+-binding steps with a peak at 14.2 μM−1s−1 and a variation of 2.3 μM−1s−1.
Figure 4.

The value of β is not sensitive to the Ca2+ stoichiometry of the model chosen. The distribution (top) and the cumulative histogram (bottom) of β values obtained from the fits to two-step and three-step models was not significantly different (KS test, P > 0.05) for either control or PMA-treated cells. Gaussian fits to the four groups of β values yield similar peaks varying from 49.8 s−1 to 51.6 s−1.
We then examined whether our choice of Ca2+ stoichiometry and the kinetic parameters obtained could describe the observed Ca2+-dependent features of exocytosis. For this purpose, we plotted the rate constants of the burst components versus postflash [Ca2+]i. By assigning three Ca2+-binding steps (Scheme 1) for control cells and two Ca2+-binding steps (Scheme 2) for PMA-treated cells, we observed good accordance of the predicted and measured Ca2+ dependence of the rate constants for the burst component (Fig. 5 A). As explained above, the sigmoidal delay after the flash is another indication that multiple Ca2+-binding steps are required in order to trigger fusion. A higher degree of cooperativity results in a more strongly sigmoidal feature with a longer delay, whereas lower cooperativity usually results in an instantaneous response. We examined the dependence of the delay on [Ca2+]i and found that cells treated with PMA have a consistently shorter delay. The delay as a function of [Ca2+]i also fits well to the three and two Ca2+-binding stoichiometry for control and PMA-treated cells, respectively (Fig. 5 B).
To further confirm the modulatory mechanism of PMA on Ca2+ sensing, we employed the so-called “Ca2+ ramp” technique, where Ca2+ is liberated slowly from the caged Ca2+ over the course of 2–5 s by steady UV illumination. Since the K d of a single Ca2+ binding reaction is calculated to be around 4 μM, a Ca2+ ramp from several hundred nM to a few μM will be ideal for this purpose because the Ca2+ sensor is far from saturation over this range. The derivation of Ca2+ dependence of exocytosis from single ramp stimulation also minimizes cell-to-cell variation. The Cm responses to a Ca2+ ramp for control and PMA-treated cells are displayed in Fig. 6 (A and B). Based on the calculation of the local rate of the Cm change and the estimate of the size of the releasable pool, we calculated the rate of the release at different [Ca2+]i levels. Fig. 6 C compares the averaged rate of release from control and PMA-treated cells as a function of [Ca2+]i on a double logarithmic scale. It is evident that although PMA-treated cells exhibit larger rate constants, they have a shallower dependence on [Ca2+]i. The slope was 2.7 for control and 1.6 for PMA-treated cells. A satisfactory fit to the mean control data (Fig. 6 C, lower solid line) was achieved using Scheme 1 with the same kinetic parameters used in Fig. 5. To account for the shallower slope of the PMA-treated cells, we had to reduce the number of Ca2+-binding sites by one, as in Scheme 2, in order to yield a satisfactory fit (Fig. 6 C, upper dashed line). The time courses of the Cm response to a Ca2+ ramp also fitted well to Scheme 1 for control (Fig. 6 A, dashed line) and Scheme 2 for PMA-treated cells (Fig. 6 B, dashed line).
Figure 6.

PMA treatment reduces the steepness of the Ca2+ dependence of exocytosis revealed by ramp [Ca2+]i stimulation. Example Cm responses of a control (A) and PMA-treated (B) cells stimulated by ramp [Ca2+]i. The smooth dashed lines represent simulated responses using the models shown in Schemes 1 and 2 in the absence and presence of PMA, respectively. (C) Double logarithmic plot of the rate constants versus [Ca2+]i for 10 control (open circles) and 7 PMA-treated (filled circles) cells. The local rate constant was calculated as the exocytotic rate divided by the remaining pool size. The slope was 2.7 for the control and 1.6 for PMA-treated cells. Using the measured ramp [Ca2+]i as a forcing function, the control data were fitted to a three-step Ca2+-binding model (Scheme 1), whereas the PMA data were fitted to the two-step Ca2+-binding model (Scheme 2).
DISCUSSION
In the present study, we have taken advantage of the photolysis technique to elevate [Ca2+]i homogeneously and to study the relationship between the concentration of Ca2+ and the kinetics of exocytosis. The results obtained by fitting the experimental data to appropriate kinetic models for exocytosis suggest that sensitization of the secretory machinery by PKC phosphorylation involves reduction of the number of Ca2+-binding sites on the Ca2+ sensor for exocytosis without altering its binding kinetics.
PMA has been demonstrated to facilitate neurotransmitter release (Stevens and Sullivan, 1998; Yawo, 1999; Wu and Wu, 2001) as well as hormone secretion in endocrine cells (Ammala et al., 1994; Gillis et al., 1996; Billiard et al., 1997). The rate of release is proportional to the product of the size of the readily releasable pool (RRP) multiplied by the average probability of release. PMA has been suggested to augment the RRP size in both neurons (Stevens and Sullivan, 1998) and endocrine cells (Gillis et al., 1996). However, evidence has accumulated recently that PMA can also increase the probability of release by inducing the appearance of a highly Ca2+-sensitive pool (HCSP) of vesicles in a number of cell types (Yang et al., 2002; Zhu et al., 2002; Wan et al., 2004; Yang and Gillis, 2004). The steep dependence (third to fourth power) of exocytosis on [Ca2+]i means that even a small change in Ca2+ sensitivity could have large effects on the release rate. Indeed, our previous work has suggested that a twofold increase in the Ca2+ sensitivity of exocytosis caused by PMA predicts an approximately eightfold increase in the release rate at resting [Ca2+]i, similar to what has been previously reported (Billiard, et al., 1997). Although sensitization of the secretory machinery is regarded as a powerful way of augmenting the release, the mechanism underlying the sensitization (or the transition to HCSP) is unclear. We observed that the relationship between the rate of exocytosis of vesicles in the HCSP and the postflash [Ca2+]i after PMA treatment is not as steep as in control gonadotropes. Further analysis of the kinetics of exocytosis revealed that while a model incorporating three independent Ca2+-binding steps preceding the final fusion step is required to describe the control data, it could not adequately describe the HCSP after PMA treatment. Rather, the relatively shallow dependence of the rate constant for exocytosis on [Ca2+]i can be best modeled by two Ca2+-binding steps. Another finding of this study is that sensitization of the Ca2+ sensor by PMA is not associated with changes in the binding and dissociation kinetics of the Ca2+ to independent binding sites. By choosing kinetic models with appropriate Ca2+ stoichiometry, we found that the rate constants obtained from the fits, α and β, converged to the values 14 μM−1·s−1 and 51 s−1, respectively, regardless of whether PMA was present. Adequate measurement of these parameters has only been performed in chromaffin cells. Interestingly, we found that the value of β is not sensitive to the Ca2+ stoichiometry chosen and is almost identical to that estimated previously for chromaffin cells (β = 56 s−1; Voets, 2000). The value of α in gonadotropes is slightly higher than that for chromaffin cells and probably reflects the involvement of a higher affinity Ca2+ sensor in gonadotropes. In contrast, the value of γ in gonadotropes is much lower than that of the so-called rapidly releasable pool but comparable to that of the slowly releasable pool in chromaffin cells (Voets, 2000). Mutations in SNARE complexes also greatly reduce the value of γ without any apparent influence on the value of α and β (Sorensen et al., 2002). Interestingly, we found that PMA treatment slightly reduced the value of γ. The rate constants of exocytosis of control and PMA-treated cells actually crossed at a [Ca2+]i level of 14 μM as predicted from the model (Fig. 5 A). Thus, the effect of PMA on the kinetics of exocytosis can only be observed at low [Ca2+]i levels (i.e., a few μM). In contrast, at higher [Ca2+]i, it is not possible to distinguish any effect of PMA on the kinetics of exocytosis. Rather, the rate constants of exocytosis from PMA-treated cells should be slightly lower than that for control cells. This explains why a previous study using high postflash [Ca2+]i failed to detect any influence of PMA on the kinetics of the burst component (Gillis et al., 1996).
The fourth- to fifth-order dependence of exocytosis on [Ca2+]i is thought to be important for the characteristics of synchronous release. It also restricts the release sites to the Ca2+ entry sites. In contrast, the second- to third-order dependence of the rate of release on [Ca2+]i is consistent with the less demanding role of synchronization of hormone release from secretory cells. The reduction in the number of Ca2+-binding steps by PMA would further lower the threshold for release and tune the Ca2+ sensor to spatially averaged [Ca2+]i rather than nonoverlapping Ca2+ microdomains. Thus, more vesicles distant from Ca2+-channels will be released. Indeed, the majority of the RRP is found distant from the vicinity of Ca2+ channels in a number of endocrine cell types, including gonadotropes, chromaffin cells, and β cells (Moser and Neher, 1997; Voets, 2000; Barg et al., 2001; Yang et al., 2002; Zhu et al., 2002). Decreasing the order of [Ca2+]i dependence of release provides an efficient way of promoting release in these cells. In fact, a high affinity/low cooperativity exocytosis has been demonstrated in rod photoreceptors, permitting rods to respond linearly to the change of Ca2+ influx due to graded light responses (Thoreson et al., 2004). Sensitization of the Ca2+ sensor for exocytosis without apparent modulation of the RRP size has been suggested to occur in both endocrine cells (Yang et al., 2002; Zhu et al., 2002; Wan et al., 2004; Yang and Gillis, 2004) and neurons (Yawo 1999; Wu and Wu, 2001). The mechanism described in this study may thus apply to both endocrine cells and neurons. This is quite likely, as both these cell types utilize similar molecules in priming and fusion.
The targets of PMA action remain to be explored. Besides PKC, Munc13 has been suggested recently as an important receptor for phorbol esters. It is now believed that Munc13 is essential for the priming of vesicles by activating syntaxin in an open conformation (Brose et al., 2000; Silinsky and Searl, 2003; Junge et al., 2004). Activated syntaxin thus initiates the assembly of the primed core complex that makes vesicles fusion competent (Xu et al., 1999). It is suggested that the augmentation of neurotransmitter release by phorbol esters in hippocampal neurons might actually be due to the effects on Munc13 (Rhee et al., 2002). Binding of the C1 domain of Munc13 by phorbol esters is suggested to facilitate the priming and thus increase the RRP size. We have shown that the effect of PMA on the Ca2+ sensitivity of exocytosis is blocked by the PKC-specific inhibitor BIS, which binds to the ATP-binding domain (C3) of PKC. In contrast, Munc13 lacks a C3 domain. Moreover, the modulation we describe here applies to the final Ca2+-sensing steps of fusion, which is distinct from an effect on the RRP size caused by activation of Munc13. Thus, our results seem to point to a novel target of action, distinct from Munc13. Synaptotagmin has been proposed as the major Ca2+ sensor that mediates fusion, yet the precise mechanism of Ca2+ binding and the subsequent fusogenic action is unknown. The cytoplasmic region of synaptotagmin contains two C2 domains, C2A and C2B. It has been suggested that five highly conserved acidic residues in both the C2A and C2B domains coordinate the binding of three to four Ca2+ ions (Chapman et al., 2002; Sudhof, 2002). However, evidence also suggests that other auxiliary proteins may be involved in the Ca2+-sensing step. For instance, mutations in SNARE proteins change the calcium sensitivity and reduce the Ca2+ dependence of exocytosis in chromaffin cells, suggesting that the SNARE complex and synaptotagmin might constitute an integrated Ca2+ sensor for fusion. Indeed, double mutations in the two acidic and hydrophilic residues in the COOH-terminal end of SNAP-25, which has been postulated to coordinate divalent ion binding from the crystallized core SNARE complex, reduced the slope of Ca2+ dependence of exocytosis from 2.7 to 1.5, without significantly changing the Ca2+-binding kinetics (Sorensen et al., 2002). This result is in close agreement the PMA effect we observed in this study. It is not yet known whether an additional, unknown partner is required for the structural integrity of the Ca2+-binding sites, in which case phosphorylation of the auxiliary proteins of the Ca2+-sensing complex may lead to the loss of a Ca2+-binding site. Further experiments will be needed to elucidate the underlying mechanism of this modulation of the Ca2+-sensing steps that immediately precede the final fusion.
Acknowledgments
We thank Drs. Sarah Perrett and Bertil Hille for critical reading of the manuscript.
This work was supported by grants from the National Science Foundation of China (30025023, 30270363, 30130230, 30470448), the Major State Basic Research Program of P.R. China (G1999054000, 2004CB720000), the CAS Project (KSCX2-SW-224), the Li Foundation, and the Sino-German Scientific Center. The laboratory of T. Xu belongs to a Partner Group Scheme of the Max Planck Institute for Biophysical Chemistry, Göttingen.
Angus C. Nairn served as editor.
H. Yang and H. Liu contributed equally to this work.
Abbreviations used in this paper: DMN, DM-nitrophen; HCSP, highly Ca2+-sensitive pool; KS, Kolmogorov-Smirnov; NP-EGTA, nitrophenyl-EGTA.
References
- Ammala, C., L. Eliasson, K. Bokvist, P.O. Berggren, R.E. Honkanen, A. Sjoholm, and P. Rorsman. 1994. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic beta cells. Proc. Natl. Acad. Sci. USA. 91:4343–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine, G.J., M.P. Charlton, and S.J. Smith. 1985. Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. J. Physiol. 367:163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg, S., X. Ma, L. Eliasson, J. Galvanovskis, S.O. Gopel, S. Obermuller, J. Platzer, E. Renstrom, M. Trus, D. Atlas, et al. 2001. Fast exocytosis with few Ca2+ channels in insulin-secreting mouse pancreatic B cells. Biophys. J. 81:3308–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner, D., T. Voets, E. Neher, and T. Moser. 2001. Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron. 29:681–690. [DOI] [PubMed] [Google Scholar]
- Billiard, J., D.S. Koh, D.F. Babcock, and B. Hille. 1997. Protein kinase C as a signal for exocytosis. Proc. Natl. Acad. Sci. USA. 94:12192–12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose, N., C. Rosenmund, and J. Rettig. 2000. Regulation of transmitter release by Unc-13 and its homologues. Curr. Opin. Neurobiol. 10:303–311. [DOI] [PubMed] [Google Scholar]
- Chapman, A.L., M.B. Hampton, R. Senthilmohan, C.C. Winterbourn, and A.J. Kettle. 2002. Chlorination of bacterial and neutrophil proteins during phagocytosis and killing of Staphylococcus aureus. J. Biol. Chem. 277:9757–9762. [DOI] [PubMed] [Google Scholar]
- Dodge, F.A., Jr., and R. Rahamimoff. 1967. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J. Physiol. 193:419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis, K.D., R. Mossner, and E. Neher. 1996. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 16:1209–1220. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz, G., M. Poenie, and R.Y. Tsien. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- Heidelberger, R., C. Heinemann, E. Neher, and G. Matthews. 1994. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 371:513–515. [DOI] [PubMed] [Google Scholar]
- Junge, H.J., J.S. Rhee, O. Jahn, F. Varoqueaux, J. Spiess, M.N. Waxham, C. Rosenmund, and N. Brose. 2004. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell. 118:389–401. [DOI] [PubMed] [Google Scholar]
- Kaftan, E.J., T. Xu, R.F. Abercrombie, and B. Hille. 2000. Mitochondria shape hormonally induced cytoplasmic calcium oscillations and modulate exocytosis. J. Biol. Chem. 275:25465–25470. [DOI] [PubMed] [Google Scholar]
- Moser, T., and E. Neher. 1997. Rapid exocytosis in single chromaffin cells recorded from mouse adrenal slices. J. Neurosci. 17:2314–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, J.S., A. Betz, S. Pyott, K. Reim, F. Varoqueaux, I. Augustin, D. Hesse, T.C. Sudhof, M. Takahashi, C. Rosenmund, and N. Brose. 2002. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell. 108:121–133. [DOI] [PubMed] [Google Scholar]
- Schneggenburger, R., and E. Neher. 2000. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 406:889–893. [DOI] [PubMed] [Google Scholar]
- Silinsky, E.M., and T.J. Searl. 2003. Phorbol esters and neurotransmitter release: more than just protein kinase C? Br. J. Pharmacol. 138:1191–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen, J.B., U. Matti, S.H. Wei, R.B. Nehring, T. Voets, U. Ashery, T. Binz, E. Neher, and J. Rettig. 2002. The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc. Natl. Acad. Sci. USA. 99:1627–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen, J.B. 2004. Formation, stabilisation and fusion of the readily releasable pool of secretory vesicles. Pflugers Arch. 448:347–362. [DOI] [PubMed] [Google Scholar]
- Stevens, C.F., and J.M. Sullivan. 1998. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 21:885–893. [DOI] [PubMed] [Google Scholar]
- Sudhof, T.C. 2002. Synaptotagmins: why so many? J. Biol. Chem. 277:7629–7632. [DOI] [PubMed] [Google Scholar]
- Thomas, P., J.G. Wong, A.K. Lee, and W. Almers. 1993. A low affinity Ca2+ receptor controls the final steps in peptide secretion from pituitary melanotrophs. Neuron. 11:93–104. [DOI] [PubMed] [Google Scholar]
- Thoreson, W.B., K. Rabl, E. Townes-Anderson, and R. Heidelberger. 2004. A highly Ca2+-sensitive pool of vesicles contributes to linearity at the rod photoreceptor ribbon synapse. Neuron. 42:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse, F.W., A. Tse, B. Hille, H. Horstmann, and W. Almers. 1997. Local Ca2+ release from internal stores controls exocytosis in pituitary gonadotrophs. Neuron. 18:121–132. [DOI] [PubMed] [Google Scholar]
- Voets, T. 2000. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 28:537–545. [DOI] [PubMed] [Google Scholar]
- Wan, Q.F., Y.M. Dong, H. Yang, X.L. Lou, J.P. Ding, and T. Xu. 2004. Protein kinase activation increases insulin secretion by sensitizing the secretory machinery to Ca2+. J. Gen. Physiol. 124:653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X.S., and L.G. Wu. 2001. Protein kinase c increases the apparent affinity of the release machinery to Ca2+ by enhancing the release machinery downstream of the Ca2+ sensor. J. Neurosci. 21:7928–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, T., M. Naraghi, H. Kang, and E. Neher. 1997. Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells. Biophys. J. 73:532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, T., T. Binz, H. Niemann, and E. Neher. 1998. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat. Neurosci. 1:192–200. [DOI] [PubMed] [Google Scholar]
- Xu, T., B. Rammner, M. Margittai, A.R. Artalejo, E. Neher, and R. Jahn. 1999. Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell. 99:713–722. [DOI] [PubMed] [Google Scholar]
- Yang, Y., S. Udayasankar, J. Dunning, P. Chen, and K.D. Gillis. 2002. A highly Ca2+-sensitive pool of vesicles is regulated by protein kinase C in adrenal chromaffin cells. Proc. Natl. Acad. Sci. USA. 99:17060–17065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., and K.D. Gillis. 2004. A highly Ca2+-sensitive pool of granules is regulated by glucose and protein kinases in insulin–secreting INS-1 cells. J. Gen. Physiol. 124:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo, H. 1999. Protein kinase C potentiates transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J. Physiol. 515(Pt 1):169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H.L., B. Hille, and T. Xu. 2002. Sensitization of regulated exocytosis by protein kinase C. Proc. Natl. Acad. Sci. USA. 99:17055–17059. [DOI] [PMC free article] [PubMed] [Google Scholar]