

Abstract
The five KCNE genes encode a family of type I transmembrane peptides that assemble with KCNQ1 and other voltage-gated K+ channels, resulting in potassium conducting complexes with varied channel-gating properties. It has been recently proposed that a triplet of amino acids within the transmembrane domain of KCNE1 and KCNE3 confers modulation specificity to the peptide, since swapping of these three residues essentially converts the recipient KCNE into the donor (Melman, Y.F., A. Domenech, S. de la Luna, and T.V. McDonald. 2001. J. Biol. Chem. 276:6439–6444). However, these results are in stark contrast with earlier KCNE1 deletion studies, which demonstrated that a COOH-terminal region, highly conserved between KCNE1 and KCNE3, was responsible for KCNE1 modulation of KCNQ1 (Tapper, A.R., and A.L. George. 2000 J. Gen. Physiol. 116:379–389.). To ascertain whether KCNE3 peptides behave similarly to KCNE1, we examined a panel of NH2- and COOH-terminal KCNE3 truncation mutants to directly determine the regions required for assembly with and modulation of KCNQ1 channels. Truncations lacking the majority of their NH2 terminus, COOH terminus, or mutants harboring both truncations gave rise to KCNQ1 channel complexes with basal activation, a hallmark of KCNE3 modulation. These results demonstrate that the KCNE3 transmembrane domain is sufficient for assembly with and modulation of KCNQ1 channels and suggests a bipartite model for KCNQ1 modulation by KCNE1 and KCNE3 subunits. In this model, the KCNE3 transmembrane domain is active in modulation and overrides the COOH terminus' contribution, whereas the KCNE1 transmembrane domain is passive and reveals COOH-terminal modulation of KCNQ1 channels. We furthermore test the validity of this model by using the active KCNE3 transmembrane domain to functionally rescue a nonconducting, yet assembly and trafficking competent, long QT mutation located in the conserved COOH-terminal region of KCNE1.
Keywords: MinK-related protein, voltage-gated, basal activation, ER retention, chromanol 293B
INTRODUCTION
The KCNE type I transmembrane peptides (∼125 residues) are a family of tissue-specific β-subunits that associate with and fine tune the electrical properties of several voltage-gated K+ channels (Abbott et al., 1999, 2001; Yu et al., 2001; Zhang et al., 2001; Grunnet et al., 2003; McCrossan et al., 2003; Lewis et al., 2004). In expression systems, KCNQ1 (Q1) K+ channels have affinity for all five KCNE peptides forming K+ conducting complexes with different voltage activation, gating kinetics, unitary conductance, and pharmacology (Barhanin et al., 1996; Sanguinetti et al., 1996; Sesti and Goldstein, 1998; Schroeder et al., 2000; Tinel et al., 2000; Angelo et al., 2002; Grunnet et al., 2002). Of these complexes, the physiological roles of the Q1-KCNE1 (E1) and Q1-KCNE3 (E3) complexes have been well established. In the heart, Q1–E1 complexes constitute the slowly activating and deactivating cardiac IKs current (Barhanin et al., 1996; Sanguinetti et al., 1996). Mutations in either Q1 or E1 that disrupt channel function and prolong the cardiac action potential have been implicated in several inherited forms of long QT syndrome (Splawski et al., 2000). Q1–E3 complexes have been detected in the basolateral membranes of several epithelial tissues where these apparently constitutively open channels are critical for cAMP-stimulated chloride secretion in colon and lung (Schroeder et al., 2000; Grahammer et al., 2001). Given their connectivity to chloride secretion, Q1–E3 complexes may be potential targets for cystic fibrosis therapies (Cowley and Linsdell, 2002; Cuthbert and MacVinish, 2003).
Numerous studies have examined the molecular interactions between E1 and Q1. Two independent cysteine mutagenesis studies have demonstrated that the E1 peptides are intimate with the ion conducting pore unit, yet these studies disagree on the peptides' exact location (Wang et al., 1996; Tai and Goldstein, 1998; Tapper and George, 2001). Deletion studies of E1 have shown that the transmembrane segment is sufficient for assembly with Q1, whereas a COOH-terminal region of ∼25 amino acids proximal to the cytoplasmic membrane is necessary for Q1 modulation (Takumi et al., 1991; Tapper and George, 2000). These results are in sync with the long QT mutations found in E1 that reside within this same region (Splawski et al., 1997; Bianchi et al., 1999). However, recent studies with KCNE1/3 chimeras suggest that a triplet of amino acids within the transmembrane segment of either KCNE is sufficient for modulation of Q1, and it was proposed that the COOH terminus, which is so well conserved between E1 and E3, is merely required for proper anchoring or positioning of the transmembrane domain (Melman et al., 2001). In a subsequent report, this modulation specificity was further rendered down to a single amino acid within the triplet: T58/V72 in E1/E3 (Melman et al., 2002a). To date, however, the interaction between E3 and Q1 has not been directly examined.
We sought to determine the regions of E3 responsible for assembly with and modulation of Q1 channels by constructing a panel of E3 truncation mutants and coexpressing them with Q1 channels. We find a double truncation mutant composed essentially of the E3 transmembrane domain assembles with Q1 channels, producing K+ channel complexes that are open at hyperpolarizing potentials. This mutant directly demonstrates that the E3 transmembrane domain is sufficient for assembly with Q1; by analogy, we predict this will be the case for all KCNE peptides. These results suggest a bipartite model for modulation of Q1 channels by E3 and E1 where the transmembrane domain is either active in modulation (E3) and masks the contribution of the COOH terminus, or is passive (E1) and reveals COOH-terminal modulation of Q1. We further corroborate the bipartite model by using an “active” E3 transmembrane domain to override a nonfunctional long QT mutation located in the COOH terminus of E1.
MATERIALS AND METHODS
Mutagenesis and cRNA Preparation
KCNQ1 and KCNE3 were subcloned into vectors containing the 5′ and 3′ UTRs from the Xenopus β-globin gene for optimal protein expression. KCNE3 mutations were introduced using Quikchange (Stratagene) and confirmed by DNA sequencing. The HA tag, PYDVPDYA, was incorporated into the NH2 terminus of E3 between residues 11 and 12 (Abbott et al., 2001). Plasmids were linearized with MluI, and RNA synthesized by run-off transcription using SP6 (Promega).
Oocyte Preparation and cRNA Injection
Mature oocytes were surgically extracted from anesthetized Xenopus laevis. Isolated oocytes were mechanically separated and bathed in OR2 (in mM): 82.5 NaCl, 2.5 KCl, 1 MgCl2, 5 HEPES, pH 7.4 + 2 mg/ml collagenase (Worthington Biochemical Corp.) for 85–95 min to remove ovarian material and follicles. After rinsing, oocytes were incubated overnight at 18°C in ND96-bathing (ND96B) solution + gentamicin (in mM): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, 50 μg/ml gentamicin (Sigma-Aldrich), pH 7.4. Oocytes were microinjected with RNA 12–50 h after extraction with 27.6 nl total volume of cRNA solution. Oocytes were injected with (ng/oocyte) Q1, 15; Q1/E3, 7.5/3.5; Q1/E3 truncation, 5/3.5; Q1/KCNE D→N mutant, 7.5/18.5. For experiments in which E3 cRNA injection ratios were compared, Q1 was kept constant at 7.5 ng/oocyte, while the quantity of E3 was varied, 18.5, 3.0, 0.7, and 0.4 ng/oocyte. Likewise, in experiments using HA-tagged E3 constructs, Q1 was kept constant at 7.5 ng/oocyte, and was coinjected with E3 (3.0 or 0.4 ng/oocyte). In controls where only Q1 or E3 was injected, an equivalent volume of DEPC water was also injected. Injected oocytes were incubated at 18°C in ND96B + gentamicin for 2–7 d before conducting experiments.
Electrophysiology
Currents were measured using two-electrode voltage clamp (OC-725-C; Warner Instrument Corp.) and data acquired with Digidata 1322A (Axon Instruments) running Clampex 8.2 (Axon Instruments) at RT. Electrodes were filled with 3 M KCl, 5 mM EGTA, 10 mM HEPES, pH 7.6, and had resistance between 0.2 and 1.5 MΩ. A home-made gravity-fed perfusion chamber was used, in which complete solution exchange occurred within ∼10 s.
Q1–E3 channels are open at negative membrane potentials, and thus several steps were taken to ensure that the currents measured are from K+ selective currents and not from nonspecific leak. Oocytes injected with wild-type (WT) and mutant E3 cRNA were only recorded from if the resting membrane potential was more negative than −75 mV in ND96, since oocytes with more positive resting potentials routinely had nonspecific leak and/or were dead. ND96 (in mM): 96 NaCl, 2 KCl, 0.3 CaCl2, 1 MgCl2, 5 HEPES, pH 7.0. For experiments using solutions with K+ concentrations greater than ND96, the current at −80 mV was measured in ND96 before and after each experiment. Data was only included if current at −80 mV returned to the original level in ND96 after treatment with high K+ solution. The majority of the E3 mutants had positive current at −80 mV due to outward “leak” of potassium, presumably through the Q1–E3 channels. For Q1, the leak current at −80 mV was between −0.01 and −0.04 μA in ND96. All traces shown were not leak subtracted.
Tail currents for activation curves were measured in KD98 (in mM): 98 KCl, 0.3 CaCl2, 1 MgCl2, 5 HEPES, pH 7.0. Upon switching the external solution to KD98, oocytes were held at −80 mV until the inward current plateaued. The tail current protocol was a series of command pulses to potentials between −100 and +60 mV in 10-mV increments for 2 s, after which oocytes were returned to −80 mV. The interpulse interval was 13 s.
Reversal potentials were measured at several external potassium concentrations, each isotonically balanced with sodium. The reversal potential protocol was a holding potential of −80 mV, depolarizing to +40 mV for 2 s, followed by repolarization to potentials around the Nernst-predicted value of EK in 2-mV steps to determine the reversal potential.
Chromanol 293B inhibition was measured in ND96C (in mM): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, pH 7.4. Chromanol 293B (Tocris) was added to ND96C from a 10 mM stock in DMSO. The final concentration of DMSO in ND96C was 0.1%. Oocytes were held at −80 mV and subjected to +40 mV depolarizations for 2 s every 30 s until the current level stabilized (≤5 min). After the current stabilized, 10 μM chromanol 293B was perfused until inhibition was complete (∼5 min), and the remaining current was measured.
Oocytes expressing complexes with HA-tagged E3 proteins became appreciably inactivated during incubation at 18°C in ND96B. Cumulative inactivation was removed by holding at −80 mV in ND96 with periodic test depolarization to +40 mV to monitor relief of inactivation. After relieving inactivation, oocytes were subjected to a family of depolarizations from −100 to +40 mV in 20-mV steps with a 13-s interpulse interval. Cumulative inactivation was monitored using a 2-s test depolarization from −80 to +40 mV at 30-s intervals. At first, isochronal current measurements were taken 100 ms before the respective capacitive transients at −80 and +40 mV in ND96 solution. Then, the bath solution was changed to KD98, and the current was then measured until there was no change in current between test depolarizations. A family of currents was then measured in KD98 using the same depolarization protocol used with ND96. Recovery from cumulative inactivation was monitored by returning the bath solution to ND96 and measuring the current at −80 mV and +40 mV as described above.
To detect the voltage-dependent inactivation of homotetrameric Q1 channels with the HA-tagged E3 constructs, the oocytes were held at −40 mV and bathed in RD10 solution, which minimized cumulative inactivation, yet enabled the visualization of inward currents (in mM): 88 NaCl, 10 RbCl, 0.3 CaCl2, 1 MgCl2, 5 HEPES, pH 7.4. The pulse protocol used a holding potential of −40 mV, a 2-s test pulse between −100 and +60 in 20-mV increments, followed by a tail pulse to −80 mV for 1.5 s. The interpulse interval was 11.5 s.
Data Analysis
The amplitude of the tail currents were measured 3 ms after return to −80 mV and plotted versus the depolarized potential. The resultant activation curves were initially fit to a Boltzmann function, y=A2+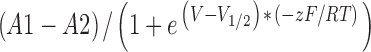 , where V1/2 is the midpoint of activation and z is the maximum slope. The upper and lower asymptotes, A1 and A2, respectively, were left to vary, allowing data to be fit in cases where channels did not fully close or were not fully activated in the voltage ranges that can be used with oocytes (−100 to +60 mV). After the initial fit, the tail current amplitudes were normalized such that the upper asymptote (A1) was equal to 1. These data were refit to the Boltzmann function, and the lower asymptote (A2) of the second fit was used to compare basal activation of the wild type and mutant Q1–E3 complexes. Deactivation time constants were measured by fitting the current at −80 mV following a (40–60 mV) depolarization to a single or double exponential. Time constants of deactivation were not determined in samples where recovery from inactivation obscured the fast component of deactivation.
, where V1/2 is the midpoint of activation and z is the maximum slope. The upper and lower asymptotes, A1 and A2, respectively, were left to vary, allowing data to be fit in cases where channels did not fully close or were not fully activated in the voltage ranges that can be used with oocytes (−100 to +60 mV). After the initial fit, the tail current amplitudes were normalized such that the upper asymptote (A1) was equal to 1. These data were refit to the Boltzmann function, and the lower asymptote (A2) of the second fit was used to compare basal activation of the wild type and mutant Q1–E3 complexes. Deactivation time constants were measured by fitting the current at −80 mV following a (40–60 mV) depolarization to a single or double exponential. Time constants of deactivation were not determined in samples where recovery from inactivation obscured the fast component of deactivation.
Oocyte Membrane Preparation
3–5 d after injection with 7.5 ng Q1 and E3 (3.0 or 0.4 ng cRNA/oocyte), several oocytes were checked for functional K+ conducting complexes, and each batch of oocytes was homogenized at 4°C in 0.9 ml homogenization buffer, 100 mM HEPES, pH 7.6, 1 mM EDTA, 50 mM DTT, and 100 μg/ml PMSF. Subsequent steps were performed at 4°C. The cell debris from the homogenate was pelleted (3,000 g, 10 min), and the supernatant was collected. The pellet was vigorously resuspended in homogenization buffer (0.9 ml) and cell debris was pelleted again (3,000 g, 10 min). The supernatants were combined and overlaid on a 15% sucrose cushion, prepared with 100 mM HEPES, pH 7.6, and 10 mM N-ethyl maleimide. Samples were subjected to ultracentrifugation (175,000 g, 75 min), and pelleted membranes were detergent solubilized at RT in 1% SDS, 150 mM NaCl, 100 mM DTT, 10 mM TRIS-HCl, pH 7.5. Since a proportionally smaller volume of solubilization buffer per oocyte was used for oocytes injected with 0.4 ng of mutant E3 cRNA, these membranes were solubilized with 5% SDS to aid in solubilization.
SDS-PAGE and Western Blot
SDS-solubilized membranes were diluted with 1/5 volume of 6× SDS-PAGE loading buffer, and the proteins were separated on either a 15% SDS-PAGE (TRIS/glycine) or 16.5% (TRIS/tricine) (Schagger and von Jagow, 1987). Proteins were transferred to nitrocellulose (0.2 μm pore size) at constant amperage (0.2–0.3 A) for 1 h. Blots were blocked for 45 min at RT in blocking buffer (5% nonfat milk in Western wash: 150 mM NaCl, 10 mM TRIS-HCl, pH 7.4, and 0.2% Tween-20). The blots were incubated overnight at 4°C with 100 ng/ml rat monoclonal α-HA (Roche) in blocking buffer. Blots were washed in Western wash (3 × 5 min) and incubated for 45 min at RT with 200 ng/ml goat α-rat HRP (Santa Cruz Biotechnology, Inc.) in blocking buffer. After washing (3 × 5 min) in Western wash, the blot was developed in SuperSignal West Dura Extended Duration Substrate (Pierce) for 5 min, and the immunoreactive bands were captured using a Fujifilm LAS-3000 CCD Camera.
PNGase F and Endo H Analysis
SDS-solubilized membranes as described above were diluted to 0.5% SDS with an equal volume of water and then raised to 1% NP-40 and 50 mM sodium citrate, pH 5.5, or 50 mM sodium phosphate, pH 7.5, and digested with Endo Hf or PNGase F (New England Biolabs), respectively, at 37°C for 1 h. After digestion, samples were raised to a total of 75 mM DTT and 3.5% SDS and then mixed with 6× gel loading buffer, and the proteins were resolved on SDS-PAGE and analyzed by Western blot as described above.
Cell Surface Labeling and Luminometry
The level of cell surface HA-tagged E3 proteins was measured by luminometry as previously reported (Zerangue et al., 1999). Oocytes were injected with 7.5 ng Q1 and WT or mutant E3–HA (3.0 or 0.4 ng). Control oocytes were injected with only 7.5 ng Q1 cRNA. On day 5 post-injection, a few oocytes were checked for functioning channel complexes, and then the oocytes were cooled to 4°C in ND96B to prevent further trafficking. Oocytes were blocked for 30 min with 1% BSA in ND96B. Oocytes were then incubated for 1 h with 1 μg/ml rat monoclonal α-HA (Roche) in 1% BSA in ND96B. Oocytes were washed (8 × 5 min) with 1% BSA in ND96B and incubated for 40 min with secondary α-rat F(ab)2 antibody (Jackson ImmunoResearch Laboratories) added at 2 μg/ml in 1% BSA in ND96B. Secondary antibody was removed and washed once with 1% BSA ND96B for 1 h followed by a second wash in ND96B for 1 h. Oocytes were individually placed in wells with 50 μl of ND96B solution, mixed with 50 μl of SuperSignal ELISA Femto Maximum Sensitivity Substrate (Pierce Chemical Co.), and the signal at 405 nm was integrated for 15 s using a Mediators PhL Luminometer (Aureon Biosystems). Data is given in relative light units, RLU, and comes from the same batch of oocytes.
RESULTS
Expression of homomeric Q1 channels (Fig. 1 A, left) gives rise to outwardly rectifying K+ currents that undergo inactivation, which can be readily detected upon repolarization as a hook in the tail currents since the rate of recovery from inactivation is faster than deactivation (Pusch et al., 1998). In contrast, coexpression of Q1 with E3 produces a K+ conducting complex that displays no inactivation and has large standing currents at hyperpolarizing potentials (Fig. 1 A, right). The standing currents are caused by the large basal activation of the complex, which is compared with homomeric Q1 channels using the voltage–activation relationship in Fig. 1 B. Using this comparative analysis, the basal activation of the Q1–E3 complex is ∼30%, whereas Q1 channels are relatively closed (3%). To directly determine the regions of E3 required for assembly with and modulation of Q1 channels, we constructed a panel of E3 truncation mutants (Fig. 2), coexpressed them with Q1 channels in Xenopus oocytes, and analyzed them using two-electrode voltage clamp (TEVC).
Figure 1.

KCNQ1/KCNE3 channels have a large standing current at negative membrane potentials. (A) Two-electrode voltage clamp recordings from Xenopus oocytes injected with Q1 (left) or Q1/E3 (right). Traces were recorded in KD98 with a 13-s interpulse interval. Dashed line indicates zero current. Inset, “Activation Curve Protocol” of 2-s depolarizations used to elicit currents shown. (B) Voltage–activation curves from Q1 and Q1/E3 channels. Tail currents were measured 3 ms after repolarization, fit to a Boltzmann, and the data normalized such that the upper asymptote was equal to 1. Squares, Q1; circles, Q1/E3. Data were averaged from six oocytes each ± SEM. Scale bars represent 1 μA and 0.5 s.
Figure 2.

KCNE3 truncation mutants. Solid lines depict which amino acid residues remain in E3 truncation mutants. Numbers across the top denote amino acid residue number. TM, transmembrane domain; N, putative N-linked glycosylation site.
We first examined whether removal of the NH2 terminus altered E3′s ability to assemble with and modulate Q1 channels. A series of overlapping NH2-terminal partial truncation mutants were constructed such that every NH2-terminal amino acid, except for the first four residues, was absent in at least one construct. All NH2-terminal mutants shown in Fig. 3 A rapidly activate and deactivate, lack inactivation, and have large standing currents at negative potentials in response to families of command voltages. Glycosylation of E3 appears to be required for complex assembly and/or function since severe NH2-terminal mutants that removed every putative N-linked glycosylation site did not express current when coinjected with Q1 (unpublished data). Like WT E3, expression of NH2-terminal truncation mutants alone did not yield any appreciable current over uninjected controls (unpublished data). In fact, all E3 mutants studied did not afford currents above background when injected without Q1 RNA. Functional Q1–E3 NH2-terminal truncation complexes were compared using voltage activation curves (Fig. 3 B). This comparison is valid, provided that the conducting complexes can be nearly maximally activated to accurately fit the upper asymptote of the Boltzmann equation, as is the case for Δ5-40 and Δ10-51. In the case of the Δ41-55 mutant, the curve is markedly right shifted and not amenable to this comparative analysis. Nonetheless, it is clear from the families of current traces that the NH2-terminal sequence (barring glycosylation) is not required for Q1–E3 complex assembly and function.
Figure 3.

KCNE3 NH2-terminal truncation mutants produce standing currents at negative potentials when coexpressed with Q1. (A) TEVC recordings from Xenopus oocytes injected with Q1 and either Δ5-40 (left), Δ10-51 (center), or Δ41-55 (right). Traces were recorded in KD98 with a 13-s interpulse interval. Dashed line indicates zero current. Inset, “Activation Curve Protocol” of 2-s depolarizations used to elicit currents shown. (B) Voltage–activation curves from E3 NH2-terminal truncation mutants. Activation curve data were fit to a Boltzmann, and the data renormalized as described in materials and methods. Squares, Δ5-40; circles, Δ10-51; triangles, Δ41-55. Dotted lines indicate the voltage-independent activation of Q1 and Q1–E3 from the lower asymptotes of Q1 the corresponding Boltzmann fits. Data were averaged from four to six oocytes ± SEM. Scale bars represent 1 μA and 0.5 s.
Given the high degree of sequence homology between the COOH termini of E1 and E3, we next determined whether the COOH terminus of E3 is active in modulation of and assembly with Q1 channels. Fig. 4 shows representative data from a COOH-terminal truncation mutant (Δ84) and a double truncation (Δ10-51/Δ84) mutant coexpressed with Q1. From these data, it is clear that E3 peptides lacking their COOH termini can assemble with Q1 channels and form conducting complexes that have standing currents at hyperpolarizing potentials with unaltered K+ selectivity (Fig. 4 C). However, removal of the COOH terminus does come with a cost; these mutants display some inactivation, slower activation/deactivation kinetics, and a reduction in basal activation. Moreover, roughly equal nanogram amounts of Q1 and COOH-terminal mutant RNA were required to afford E3-like currents shown in Fig. 4 A; typical injection ratios (1/0.1 Q1/KCNE) afforded currents with more Q1 character (vide infra). The electrophysiological properties of COOH-terminal truncation mutants in Table I are compared with WT E3 and the NH2-terminal truncations, which were also injected at this near equal RNA ratio for comparison.
Figure 4.

KCNE3 COOH-terminal and combined NH2- and COOH-terminal truncation mutants also exhibit standing currents at negative potentials when coexpressed with Q1. (A) TEVC recordings from Xenopus oocytes injected with Q1 and either Δ84 (left) or Δ10-51/Δ84 (right). Traces were recorded in KD98 with a 13-s interpulse interval. Dashed line indicates zero current. Scale bars represent 1 μA and 0.5 s. Inset, “Activation Curve Protocol” of 2-s depolarizations used to elicit currents shown. (B) Voltage–activation curves from KCNE3 COOH-terminal and double truncation mutants. Data were fit to a Boltzmann, and normalized as described in materials and methods. Squares, Δ84; circles, Δ10-51/Δ84. Dotted lines indicate the voltage-independent activation of Q1 or Q1–E3 (E3). Data were averaged from four oocytes each ± SEM. (C) External K+ concentration was varied and the reversal potential was measured. log[K+]ext is plotted against observed reversal potential. WT and E3 truncation mutants coexpressed with Q1 produced linear fits within error of 53 mV per decade. Data was averaged from four to six oocytes ± SEM.
TABLE I.
Electrophysiological Properties of KCNE3 Truncation Mutants
| τdeactivation (ms) | ||||||
|---|---|---|---|---|---|---|
| Construct | V1/2 (mV) | z | Basal Activation, A2 | fast | slow | |
| Q1 | −23.8 ± 0.6 | 1.39 ± 0.03 | 0.03 ± 0.01 | 221 ± 10 | ||
| E3 | 23.5 ± 2.5 | 0.75 ± 0.03 | 0.31 ± 0.02 | 14.9 ± 0.3 | 124 ± 5 | |
| Δ5-40 | 25.3 ± 5.1 | 0.81 ± 0.07 | 0.35 ± 0.03 | 19.1 ± 1.2 | 130 ± 5 | |
| Δ10-51 | 24.6 ± 5.5 | 1.00 ± 0.05 | 0.30 ± 0.02 | 14.4 ± 0.6 | 102 ± 4 | |
| Δ41-55 | 86.4 ± 7.4 | 0.51 ± 0.01 | 0.12 ± 0.01 | 10.9 ± 0.2 | 115 ± 3 | |
| Δ84 | 10.1 ± 1.3 | 0.67 ± 0.02 | 0.07 ± 0.01 | ND | ND | |
| Δ10-51/Δ84 | −10.0 ± 0.7 | 1.31 ± 0.05 | 0.14 ± 0.02 | ND | ND | |
| D90N | 27.8 ± 4.1 | 0.59 ± 0.02 | 0.28 ± 0.02 | 13.7 ± 0.4 | 156 ± 28 | |
| E1/E3 TM D76N | 4.5 ± 4.3 | 0.82 ± 0.03 | 0.08 ± 0.01 | ND | ND | |
Data from individual activation curves and deactivation time constants in KD98, obtained from four to six oocytes. Activation curves were fit to a Boltzmann function as described in materials and methods. Values are mean ± SEM. Basal activation (A2) for all activation curves was determined by normalizing the data after the Boltzmann fit such that the maximal current fitting parameter (A1) was equal to 1. V1/2 is the voltage of half-maximal activation and z is the slope factor. Time constants of deactivation were fit to a single or double exponential as described in materials and methods. ND, not determined.
Since the E3 transmembrane domain is able to modulate Q1 channels without the presence of a COOH-terminal domain, we next asked whether a dysfunctional mutation within the conserved COOH terminus would affect Q1–E3 function. E1 peptides containing the long QT–causing mutation D76N have been previously shown to assemble with Q1 channels and traffic to the plasma membrane, but are essentially nonconducting complexes (Fig. 5 A, left) (Wang and Goldstein, 1995). Incorporation of the corresponding mutation into E3 (D90N) and coexpression with Q1 results in currents nearly indistinguishable from wild type (Fig. 5 A, right). This functional rescue can be solely attributed to the E3 transmembrane domain since an E1/E3 chimera possessing the E3 transmembrane sequence can also functionally rescue the D76N mutation, albeit with slower activation and deactivation kinetics (Fig. 5 B). Although these mutants produce functional Q1–E3 complexes, they required injecting more mutant E3 RNA than Q1 to produce currents with primarily E3 character; injection of lower amounts of E3 resulted in currents with more Q1 character, as was previously observed with the E3 D90N mutation (Fig. 6 A) (Abbott and Goldstein, 2002). These results demonstrate that the E3 transmembrane domain is capable of functionally overriding the modulatory effects of the KCNE COOH terminus.
Figure 5.

A long QT mutation in the COOH terminus of KCNE1 is masked by the KCNE3 transmembrane domain. Inset, “Activation Curve Protocol” of 2-s depolarizations used to elicit currents shown. Scale bar represents 1 μA and 0.5 s for all recordings. (A) TEVC recording from Xenopus oocytes injected with Q1 and either E1 D76N (left) or E3 D90N (right). (B) Representative TEVC recordings from oocytes coinjected with Q1 and a chimeric partner protein, E1 D76N with the E3 transmembrane (TM) sequence. All oocytes were injected with equal amounts and ratios of Q1 and E1 or E3 RNA, and were recorded in KD98 using a 13-s interpulse interval. Dashed lines indicate zero current.
Figure 6.

KCNE3 COOH-terminal mutants demonstrate a functional dependence on cRNA injection ratios. (A) Representative two-electrode voltage clamp recordings taken from oocytes injected with varying amounts of E3, between 2.5x and 0.05x of Q1. Currents were recorded in KD98, using a 13-s interpulse interval. The Q1:E3 ratio is labeled across the top. Top row, E3 (square); second row, Δ10-51 (circle); third row, Δ84 (diamond); bottom row, D90N (triangle). Dashed lines indicate zero current. Scale bars represent 1 μA and 0.5 s. Inset, “Activation Curve Protocol” of 2-s depolarizations used to elicit currents shown. (B) Percent current remaining after inhibition with 10 μM chromanol 293B. Oocytes were held at −80 mV and depolarized for 2 s to + 40 mV with a 28-s interpulse interval. Current levels were allowed to stabilize for ≤5 min before 10 μM chromanol 293B was perfused in. Chromanol inhibition typically reached equilibrium within 5 min of wash in. Dashed line indicates the percentage of current remaining in oocytes expressing Q1 alone. Data was averaged from 3–11 oocytes ± SEM.
To afford the characteristic Q1–E3 currents with the D90N and COOH-terminal truncation mutants, an unusually high injection ratio of Q1:E3 RNA was required, suggesting that the COOH terminus might be important for cellular assembly and trafficking of the Q1–E3 complex. Supporting this notion was the presence of inactivation in the tail currents with the Δ84 and D90N mutants at the lower injection ratios (Fig. 6 A). This inactivation was not present in either WT Q1–E3 complexes or in complexes where the majority of the E3 NH2 terminus is removed (Δ10-51). Therefore, its presence with the Δ84 and D90N mutants indicated that a mixed population of Q1 channels (unpartnered and E3 partnered) was functioning at the cell surface. To confirm that unpartnered Q1 channels were present and functioning at the plasma membrane, we used chromanol 293B, which inhibits Q1–E3 complexes at a 10-fold lower concentration than homomeric Q1 channels (Schroeder et al., 2000). In the presence of 10 μM chromanol 293B, WT and Δ10-51 mutant Q1–E3 complexes were consistently inhibited at ∼85%, independent of the ratio of RNA injected (Fig. 6 B). Conversely, as less D90N or Δ84 RNA was injected, chromanol 293B inhibition decreased, approaching the amount of inhibition observed with unpartnered Q1 channels at 10 μM chromanol 293B (Fig. 6 B, dotted line). Vehicle (0.1% DMSO) alone had no effect on WT or mutant Q1–E3 complexes (unpublished data). At the highest RNA injection ratio, there was consistently less total current remaining for the Δ84 and D90N mutant than with either WT or the NH2-terminal truncation mutant (Δ10-51). Attempts to inject even more E3 mutant DNA to determine whether unpartnered Q1 channels remained at the cell surface with the 1/2.5 Q1/E3 ratio were thwarted by extensive oocyte death. Nonetheless, lowering the Q1:E3 RNA injection ratio with the COOH-terminal and D90N mutants decreased chromanol 293B inhibition and mirrored the increase in inactivation, suggesting that unpartnered Q1 channels were functioning at the plasma membrane. These results implicate the COOH terminus in Q1–E3 complex assembly.
We next determined whether the COOH-terminal and D90N mutant protein was failing to form properly assembled complexes with Q1 and traffic to the plasma membrane, or whether the mutant proteins were merely being degraded. To distinguish injected E3 protein from endogenous KCNE peptides recently identified in Xenopus oocytes (Anantharam et al., 2003), we used an HA-tagged version of E3 that has been previously used for biochemical experiments (Abbott et al., 2001; McCrossan et al., 2003; Lewis et al., 2004). Incorporation of the NH2-terminal HA tag caused a unique form of K+-enhanced, cumulative inactivation in all Q1–E3 complexes, including wild type (Fig. 7, A and B). Initial recordings in low external K+ (ND96) required holding at −80 mV for several minutes using test depolarizations to monitor recovery from inactivation. Perfusion of 98 mM K+ slowly inactivated the HA-tagged Q1–E3 complex, which could not be reversed with hyperpolarizing pulses (Fig. 7 B). Since the E3 HA-tagged Q1 complex does not recover from inactivation with voltage in high external K+, no recovery “hooks” in the tails were observed upon repolarization. Subsequent perfusion with ND96 could partially relieve cumulative inactivation, but the currents never returned to their original values. To ameliorate K+-enhanced cumulative inactivation of the HA-tagged Q1–E3 complexes, we replaced the external K+ with 10 mM Rb+ (Fig. 7 C). At this concentration of Rb+, we could readily detect the “hook tails” associated with homotetrameric Q1 inactivation since inward currents are about threefold larger with external Rb+ in comparison to K+ (Seebohm et al., 2003). Using these conditions, Q1 complex assembly with the COOH-terminal truncation mutant (Δ84) remained dependent on the ratio of RNA injected and homotetrameric Q1 channels could be detected at both injection ratios. Remarkably, addition of the epitope tag to the D90N mutant essentially rescued the assembly defect of this mutant (Fig. 7 C).
Figure 7.
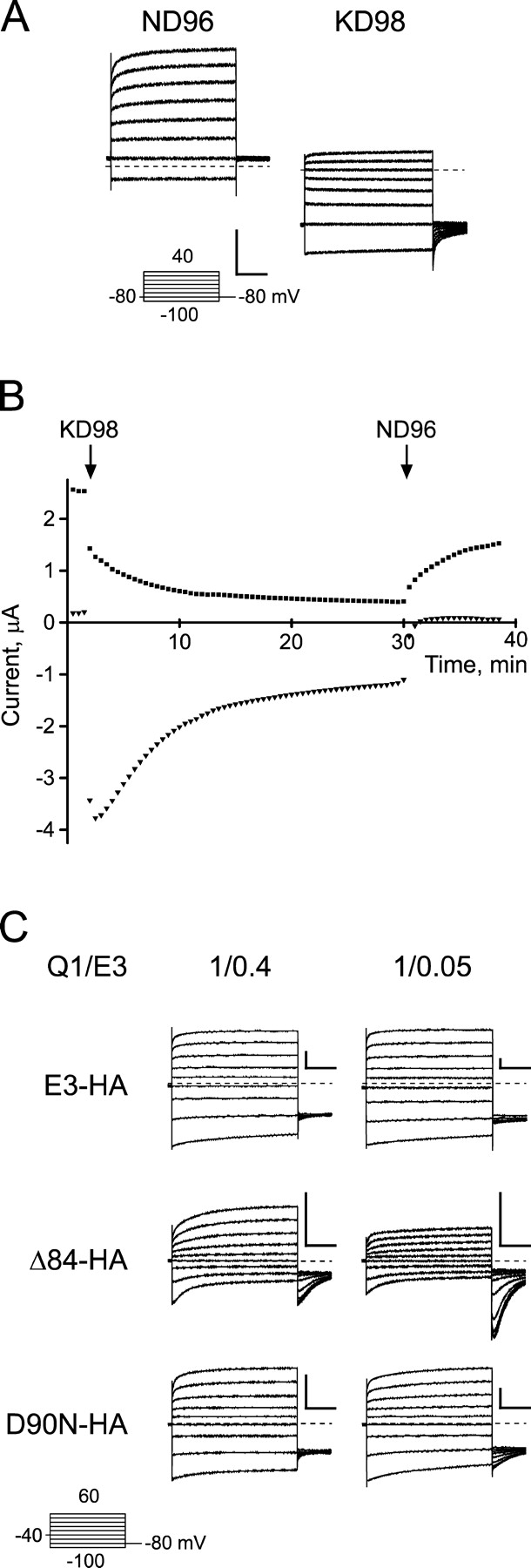
Functional characterization of KCNQ1/KCNE3–HA complexes. (A) Q1 complexes formed with HA-tagged E3 exhibit potassium-dependent inactivation. TEVC recordings of oocytes expressing Q1 and E3-HA were bathed in ND96 (left) and KD98 (right). Both recordings are shown at the same scale, and use a 13-s interpulse interval. Insets, “Activation Curve Protocol” of 2-s depolarizations used to elicit currents shown. (B) Current versus time plotted for potassium concentration changes for the Q1–E3–HA complex shown in A. Oocytes were held at −80 mV, and depolarized for 2 s to + 40 mV, with a 28-s interpulse interval. Squares denote the current measured at +40 mV, triangles at −80 mV 100 ms before the respective capacitive transients. (C) HA-tagged E3 shows altered dose dependence for the COOH-terminal truncation mutant. TEVC measurements of oocytes injected with 0.4x (left) or 0.05x (right) E3–HA relative to Q1. Currents were recorded with 10 mM Rb+ (RD10), and oocytes were held at −40 mV, with an 11.5-s interpulse interval. Top, E3–HA; middle, Δ84–HA; bottom, D90N–HA. Scale bars represent 1 μA and 0.5 s.
Since Q1 complex assembly with the HA-tagged Δ84 mutant remained dependent on the amount of RNA injected in functional experiments, we next determined whether the E3 COOH-terminal mutants were proteolytically stable. HA-tagged E3 proteins were isolated from crude oocyte membranes after 3–5 d of coexpression with Q1. Three oocytes worth of membranes from each sample at the 1/0.4 Q1/E3 ratio were analyzed by SDS-PAGE and transferred to nitrocellulose for E3 detection. Immunoblots in Fig. 8 A showed a similar complex pattern of bands for both WT E3 and D90N, which is consistent with several N-linked glycoforms. The Δ84 COOH-terminal truncation mutant protein also afforded an abundant signal on the immunoblot; however, only a single, intense band was observed at 20 kD. The Δ84 protein was also detected at the lower 1/0.05 Q1/E3 ratio (Fig. 8 B).
Figure 8.

WT and COOH-terminal mutant HA-tagged KCNE3 peptides are glycosylated and not proteolytically degraded in oocytes. Crude membranes for the immunoblots were prepared 3–5 d after coinjection with Q1 and the indicated E3 construct. (A) Immunoblot of HA-tagged E3 peptides from SDS-solubilized membranes isolated from oocytes injected at a Q1/E3 ratio of 1/0.4. Membranes from three oocytes were loaded in each lane and resolved with a 15% Tris-glycine SDS gel. Molecular weight standards are labeled on the left for both blots. (B) Immunoblots of enzymatically deglycosylated E3 and Δ84 HA-tagged proteins were separated on 16.5% Tris-tricine SDS gels. E3 (four oocytes/lane) and Δ84 (six oocytes/lane) were digested with endoglycosylase Hf (Endo H) or PNGase F. Lane marked (−) represents untreated samples. Loaded in the lane marked 1/0.05 are solubilized membranes from 17 oocytes expressing Q1/Δ84 injected at the lowest Q1/E3 ratio examined. Membranes from uninjected oocytes are denoted and were loaded at the left (4 oocytes) and right (17 oocytes). Mature and immature glycosylation is denoted, as determined by enzymatic deglycosylation. (C) Cell surface expression of HA-tagged E3 proteins was quantitated by single oocyte chemiluminescence. Oocytes were injected with a 1/0.4 Q1/E3 ratio, allowed to incubate for 5 d, and the cell surface–tagged proteins were labeled with an anti-HA antibody followed by a secondary HRP-conjugated antibody. Single oocyte luminescence was quantitated in a luminometer and is reported in relative light units (RLU). Q1 sample is from control oocytes injected with only Q1 RNA. Error bars are standard error measurement from 15–19 oocytes.
The markedly different pattern of protein bands for WT and Δ84 suggested that the COOH-terminal truncation mutant was largely residing in the ER, which could be directly tested using E3′s N-linked glycosylation. N-glycosylated membrane proteins in the ER possess immature glycans (∼3 kD/glycan), whereas the N-linked glycans on membrane proteins that have trafficked through the Golgi mature and often increase in size due to the various trimming and additions of glycosides. To identify the maturity of the N-linked glycans on WT and Δ84, two glycosidases were used: endoglycosidase H (Endo H), which cleaves only the immature glycoform, and PNGase F, which removes all forms of N-linked glycosylation. SDS-solubilized membranes containing WT and Δ84 protein were enzymatically degylcosylated and separated on Tris-Tricine gels to resolve the ∼10–20-kD unglycosylated proteins (Fig. 8 B). Endo H treatment of WT E3 removes only a single band at 23 kD. Although the deglycosylated product cannot be observed at this exposure, extended exposures revealed a faint band at 17 kD, suggesting that the majority of the protein has mature N-linked glycosylation (unpublished data). PNGase F treatment of WT E3 demonstrated that the remaining, slower mobility bands were due to mature N-linked glycosylation, as all bands collapsed to a single species at ∼17 kD. In contrast, both glycosidases equally acted on the single band in the Δ84 sample, identifying the N-linked glycan as the immature form found in the ER. Given the small size of the Δ84 mutant, the slight difference in protein mobility for the differently deglcosylated samples is presumably due to the remaining monosaccharides left behind after Endo H treatment. PNGase F, in contrast, removes all of the carbohydrate from each N-glycosylation site, resulting in a protein with a smaller molecular weight. These results demonstrate that the COOH terminus is important for assembling Q1–E3 complexes that efficiently exit the ER.
As shown in Fig. 8 B, the PNGase F–treated Δ84 samples were routinely stronger in intensity than when treated with Endo H. This difference suggested that some mature glycoforms of Δ84 may have been present, but were not detectable in the immunoblot due to the heterogeneity of mature glycosylation that often causes an underrepresentation of the protein signal. To ensure that a small portion of the HA-tagged Δ84 was reaching the plasma membrane, we used whole oocyte cell surface labeling in combination with chemiluminescence to detect the extracellularly HA-tagged E3 proteins on the cell surface (Zerangue et al., 1999). Whole oocytes coexpressing Q1 with HA-tagged E3 proteins were labeled with anti-HA antibody followed by labeling with a secondary HRP-conjugated antibody. The amount of secondary antibody on the cell surface of each oocyte was quantitated using a luminometer. Strong cell surface signals were observed for WT and D90N (Fig. 8 C). Cell surface expression of Δ84 was easily detected over Q1-injected background controls, but was noticeably less than WT and D90N. These cell surface expression data are in agreement with the biochemistry and electrophysiological experiments, which in total demonstrate that the E3 COOH terminus aides in cellular assembly and trafficking of the Q1–E3 complex.
DISCUSSION
In this study, we have used the dramatic changes in Q1 channel function to assess Q1–E3 complex assembly, and by definition, the region of E3 responsible for basal activation of the complex. Through a series of truncation mutants, exemplified by a 41-residue double truncation mutant that is active, yet lacks the majority of its NH2 and COOH termini, we have shown that the transmembrane domain of E3 is sufficient for assembly with and modulation of Q1 channels. A comparative analysis of gating revealed that truncation of the majority of the NH2 terminus does not appreciably alter Q1–E3 basal activation as long as one putative N-linked glycosylation site is retained. The importance of N-linked glycosylation in Q1–KCNE complex formation and function has been previously observed with E1 (Takumi et al., 1991; Freeman et al., 2000). A requirement of using tail current analysis to compare the basal activation of these mutants is that the channel complexes must be sufficiently activated past their midpoint of activation to fit the data accurately to a Boltzmann. Any uncertainty in the upper asymptote of the fit will greatly affect the value determined for the basal activation. The Δ41-55 highlights this caveat since its basal activation is apparently reduced; however, this mutant clearly forms complexes with Q1 channels that are open at hyperpolarizing potentials, which cause a substantial lowering of the oocyte resting potential similarly to WT E3.
Mutants that allow unpartnered Q1 channels to escape the ER and function at the plasma membrane would also affect the measurement of basal activation. The lower basal activation of the COOH-terminal truncation mutants may be due in part to unpartnered Q1 channels affecting the activation curve analysis, which was supported by the following three observations in electrophysiological experiments where the Q1/E3 RNA injection ratio was systematically varied. (1) The COOH-terminal truncation and D90N point mutants required injecting more E3 RNA than Q1 to afford the characteristic Q1–E3 currents. Moreover, as relatively less E3 RNA was injected, the resulting currents resembled unpartnered Q1 channels. In contrast, Q1 complexes with WT and the NH2-terminal E3 truncation mutant (Δ10-51) always afforded Q1–E3-like currents over a similar 50-fold range of E3 RNA. (2) The presence of homotetrameric Q1 inactivation, as observed by the hook in the tail currents upon repolarization, was detected only with the COOH-terminal mutants and increased as less E3 RNA was injected. (3) The chromanol 293B sensitivity, an indirect measurement of Q1–E3 complex assembly, was reduced for the COOH-terminal mutants and was also dependent on the Q1/E3 RNA injection ratio. These observations strongly imply that a heterogeneous population of Q1 channels is at the cell surface when coexpressed with E3 COOH-terminal mutants; however, these observations do not rule out the possibility that there are more than two populations of Q1 channels functioning at the plasma membrane. The diminished assembly capacity of the COOH-terminal mutants, which allows unpartnered Q1 channels to traffic to the plasma membrane, also explains an apparent inconsistency with a previous investigation of the D90N mutant. Injection at more typical Q1/KCNE RNA ratios (1/0.1) results in currents with primarily Q1 character, as was previously observed with this mutant (Abbott and Goldstein, 2002). It is not until a large excess of D90N RNA is injected that Q1–E3 currents are consistently observed.
Biochemical examination of the E3 COOH-terminal mutant proteins necessitated the use of an epitope-tagged E3 construct since oocytes contain endogenous KCNE peptides (Anantharam et al., 2003). Although this particular HA-tagged E3 construct has been previously used in biochemical experiments (Abbott et al., 2001; McCrossan et al., 2003; Lewis et al., 2004), in our hands, we observed a unique form of inactivation when it assembled with Q1 that was enhanced by high external K+ and not reversible with hyperpolarizing pulses under these high K+ conditions. Surprisingly, incorporation of the HA tag into the D90N mutant enabled this assembly-defective mutant to readily assemble with Q1 channels. We observed efficient Q1–D90N–HA complex assembly in all three assays tested: RNA titration, maturation of glycosylation, and cell surface expression. The HA tag had little effect on the Δ84 mutant, as it remained assembly impaired and dependent on RNA injection ratios. While the addition of the HA tag does add two extra negative charges per KCNE subunit, and it is has been shown that these charges are in the vicinity of the outer vestibule of the Q1 channel (Chen et al., 2003), any proposed mechanisms for either of these two observed phenomena would be conjecture.
The finding that the majority of the Δ84 mutant protein possesses the immature form of N-linked glycosylation indicates that the COOH terminus assists in the proper assembly of Q1–E3 complexes that can efficiently exit the ER. In addition, the fact that only the immature core oligosaccharide is observed for the Δ84 mutant, and not the unglycosylated peptide, demonstrates that COOH-terminal truncation mutant is recognized by oligosaccharyltransferase and is treated as a bona fide substrate. The presence of the immature glycan on Δ84 is also consistent with Q1–KCNE complex formation occurring in the ER, which is in agreement with several recent studies that suggest Q1–KCNE complex formation occurs in the ER (Bianchi et al., 2003; Krumerman et al., 2004). Although no mature form of Δ84 was directly observed in the Western blots, cell surface labeling of individual oocytes confirmed that at least a small portion of the Δ84 mutant protein was indeed at the cell surface. The presence of immature Δ84 protein in both the low and high RNA injection ratios indicates that the protein resides in the ER, but is unable to prevent unpartnered Q1 channels from escaping the ER. These results, however, do not directly address whether the majority of the Δ84 protein is retained in the ER, and not assembled with Q1, or whether the assembled Q1–E3 complexes lacking an E3 COOH terminus are not passing the protein quality control (QC) mechanisms in the ER. The most likely QC proteins involved in ER retention are calnexin/calreticulin since the Δ84 mutant lacks a COOH terminus, eliminating the possibility of a cytoplasmic ER retention signal (Ellgaard and Helenius, 2003). The lack of an ER retention signal also argues that Δ84 mutant is retained in the ER and not refluxing back from the cis-Golgi since COPI proteins require these motifs for retrograde trafficking (Ellgaard and Helenius, 2003).
Our findings directly bear on the controversy regarding the locations of the assembly and modulation domains of E1 and E3. The current debate revolves around assigning assembly and modulatory properties to two distinct locations: the transmembrane domain and a highly conserved region within the COOH terminus. Deletion analysis of E1 initially demonstrated that the transmembrane domain was sufficient for assembly with Q1 channels, but the COOH terminus was required for modulation (Tapper and George, 2000). More recent E1/E3 chimeras suggest exactly the opposite: the transmembrane sequence houses the modulatory domain while the COOH terminus anchors and properly positions the KCNE peptide within the Q1 channel complex (Melman et al., 2001). In the context of these previous reports, the results from our E3 truncation and point mutants suggest a bipartite model for assembly and modulation. In this model (Fig. 9), the transmembrane domain and the conserved COOH-terminal region both play a role in assembly. The transmembrane domain is the primary player in Q1–KCNE complex assembly, but the COOH terminus also aids in assembling competent complexes, as evidenced by the “assembly-challenged” E3 COOH-terminal truncation mutants, E3 D90N mutant and D76N E1/E3 chimera. For modulation, the KCNE transmembrane sequence is either active or passive in basal activation. In the case of E3, the transmembrane domain is active and overrides the COOH terminus, affording a Q1 channel complex with large basal activation. Conversely, the E1 transmembrane domain is passive and reveals COOH-terminal modulation of Q1, producing slowly activating and deactivating IKs currents. The bipartite model also predicts that a dysfunctional COOH terminus would have no effect on E3 modulation of Q1, but would be detrimental to Q1–E1 complex function. Incorporation of such mutation into E3 (D90N) had no effect on Q1–E3 complex function, whereas this same long QT mutation in E1 renders the complex nonfunctional. And with some ghoulish tinkering (placing the E3 transmembrane sequence into the E1 D76N mutant), we demonstrated that the E3 transmembrane domain alone was sufficient to mitigate the disruptive effects of this COOH-terminal mutation.
Figure 9.

A bipartite model for modulation of KCNQ1 by KCNE1 and KCNE3. Net diagrams of the transmembrane domains of E1 and E3 are aligned; amino acid residues, denoted as circles, are shaded based on conservation. Black circles, identical amino acids; gray circles, similar amino acids; horizontally striped circles, high-impact amino acids identified by chimeric studies; vertically striped circles, D76 and D90; no fill, nonsimilar amino acids; TM, transmembrane domain.
Is bipartite assembly and modulation exclusive to E1 and E3? Based on sequence homology, previous deletion studies with E1 (Tapper and George, 2000) and our E3 truncation mutants show that the KCNE transmembrane segment is a general, minimal Q1 assembly domain. Although the KCNE COOH terminus is not as conserved as the transmembrane domain throughout the entire family, there is some evidence that the E1 COOH terminus may also be important for Q1–E1 complex assembly. Previous E1 deletion studies initially suggested that the COOH terminus was solely involved in modulation, since removing it did not prevent complex formation with Q1 channels (Tapper and George, 2000). However, complex formation was not readily apparent from the current traces (the raw data was nearly indistinguishable from unpartnered Q1 currents) and a cysteine residue had to be incorporated into the E1 transmembrane domain so that Q1–E1 complex formation could be detected by cadmium inhibition. Although cadmium inhibition was clearly detected in these mutants, it was noticeably less than WT Q1–E1 complexes (Tapper and George, 2000), suggesting that a population of unpartnered Q1 channels may have also been present and functioning at the cell surface. Further examination of the COOH terminus of E1, as well as the remaining KCNE peptides, will reveal whether this region facilitates Q1–KCNE complex formation in the entire KCNE family.
Transmembrane modulation, and thus the bipartite model, may be limited to KCNE modulation that involves altering the basal activation of Q1 channels. Given the reported proximity of the KCNE transmembrane segment to the S6 helix (Wang et al., 1996; Tai and Goldstein, 1998; Tapper and George, 2001), we view E3 transmembrane modulation of Q1 similarly to the constitutively activated S6 mutants in the Shaker K+ channel described by Swartz and coworkers (Hackos et al., 2002). Whether the basal activation of the Q1–E3 complex is due to a subconducting leaky closed state, a perturbed closed to open equilibrium, or a supplantation of an obliterated cytoplasmic gate by an alternative voltage-activated gating mechanism is yet to be determined. This perspective of channel gating provides a rationalization of the E1/E3 chimera data where a single point mutation (Melman et al., 2002a,b) can essentially convert E1 into E3 by inducing basal activation by one of the aforementioned gating mechanisms.
Our discovery of COOH-terminal E3 mutants that are assembly compromised brings up an important issue that has not been directly addressed in the KCNE literature. Several elegant studies have shown significant electrophysiological changes when KCNE peptides are coexpressed with many types of voltage-gated K+ channels; however, the percent of functioning K+ channels assembled with KCNE peptides at the cell surface was never directly determined. While we could show that the COOH-terminal E3 mutants were assembly challenged using RNA ratio titration experiments in combination with chromanol 293B pharmacology, we could not accurately determine the fraction of unpartnered Q1 channels at the cell surface. The development of new methods that directly determine the ratio of partnered and unpartnered K+ channels functioning at the plasma membrane would be of great value in structure/function studies of K+ channel–KCNE complexes and may help corroborate the physiological relevance of many of these newly identified complexes.
Acknowledgments
We are grateful to Drs. Nick Rhind, Steve Haase, and Christopher Miller for suggestions on the manuscript. We thank Geoffrey Abbott (Weill Medical College of Cornell University, New York, NY) for sending us the HA-tagged E3 clone.
W.R. Kobertz thanks the Burroughs-Wellcome Foundation for a Career Award in the Biomedical Sciences that supported this work.
David Gadsby served as editor.
Abbreviations used in this paper: E1, KCNE1; E3, KCNE3; Q1, KCNQ1; TEVC, two-electrode voltage clamp; WT, wild type.
References
- Abbott, G.W., and S.A. Goldstein. 2002. Disease-associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J. 16:390–400. [DOI] [PubMed] [Google Scholar]
- Abbott, G.W., F. Sesti, I. Splawski, M.E. Buck, M.H. Lehmann, K.W. Timothy, M.T. Keating, and S.A. Goldstein. 1999. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 97:175–187. [DOI] [PubMed] [Google Scholar]
- Abbott, G.W., M.H. Butler, S. Bendahhou, M.C. Dalakas, L.J. Ptacek, and S.A. Goldstein. 2001. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell. 104:217–231. [DOI] [PubMed] [Google Scholar]
- Anantharam, A., A. Lewis, G. Panaghie, E. Gordon, Z.A. McCrossan, D.J. Lerner, and G.W. Abbott. 2003. RNA interference reveals that endogenous Xenopus MinK-related peptides govern mammalian K+ channel function in oocyte expression studies. J. Biol. Chem. 278:11739–11745. [DOI] [PubMed] [Google Scholar]
- Angelo, K., T. Jespersen, M. Grunnet, M.S. Nielsen, D.A. Klaerke, and S.P. Olesen. 2002. KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys. J. 83:1997–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin, J., F. Lesage, E. Guillemare, M. Fink, M. Lazdunski, and G. Romey. 1996. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 384:78–80. [DOI] [PubMed] [Google Scholar]
- Bianchi, L., Z. Shen, A.T. Dennis, S.G. Priori, C. Napolitano, E. Ronchetti, R. Bryskin, P.J. Schwartz, and A.M. Brown. 1999. Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum. Mol. Genet. 8:1499–1507. [DOI] [PubMed] [Google Scholar]
- Bianchi, L., S.M. Kwok, M. Driscoll, and F. Sesti. 2003. A potassium channel-MiRP complex controls neurosensory function in Caenorhabditis elegans. J. Biol. Chem. 278:12415–12424. [DOI] [PubMed] [Google Scholar]
- Chen, H., L.A. Kim, S. Rajan, S. Xu, and S.A. Goldstein. 2003. Charybdotoxin binding in the I(Ks) pore demonstrates two MinK subunits in each channel complex. Neuron. 40:15–23. [DOI] [PubMed] [Google Scholar]
- Cowley, E.A., and P. Linsdell. 2002. Characterization of basolateral K+ channels underlying anion secretion in the human airway cell line Calu-3. J. Physiol. 538:747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert, A.W., and L.J. MacVinish. 2003. Mechanisms of anion secretion in Calu-3 human airway epithelial cells by 7,8-benzoquinoline. Br. J. Pharmacol. 140:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard, L., and A. Helenius. 2003. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4:181–191. [DOI] [PubMed] [Google Scholar]
- Freeman, L.C., J.J. Lippold, and K.E. Mitchell. 2000. Glycosylation influences gating and pH sensitivity of I(sK). J. Membr. Biol. 177:65–79. [DOI] [PubMed] [Google Scholar]
- Grahammer, F., R. Warth, J. Barhanin, M. Bleich, and M.J. Hug. 2001. The small conductance K+ channel, KCNQ1: expression, function, and subunit composition in murine trachea. J. Biol. Chem. 276:42268–42275. [DOI] [PubMed] [Google Scholar]
- Grunnet, M., T. Jespersen, H.B. Rasmussen, T. Ljungstrom, N.K. Jorgensen, S.P. Olesen, and D.A. Klaerke. 2002. KCNE4 is an inhibitory subunit to the KCNQ1 channel. J. Physiol. 542:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunnet, M., H.B. Rasmussen, A. Hay-Schmidt, M. Rosenstierne, D.A. Klaerke, S.P. Olesen, and T. Jespersen. 2003. KCNE4 is an inhibitory subunit to Kv1.1 and Kv1.3 potassium channels. Biophys. J. 85:1525–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackos, D.H., T.H. Chang, and K.J. Swartz. 2002. Scanning the intracellular S6 activation gate in the shaker K+ channel. J. Gen. Physiol. 119:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumerman, A., X. Gao, J.S. Bian, Y.F. Melman, A. Kagan, and T.V. McDonald. 2004. An LQT mutant minK alters KvLQT1 trafficking. Am. J. Physiol. Cell Physiol. 286:C1453–C1463. [DOI] [PubMed] [Google Scholar]
- Lewis, A., Z.A. McCrossan, and G.W. Abbott. 2004. MinK, MiRP1, and MiRP2 diversify Kv3.1 and Kv3.2 potassium channel gating. J. Biol. Chem. 279:7884–7892. [DOI] [PubMed] [Google Scholar]
- McCrossan, Z.A., A. Lewis, G. Panaghie, P.N. Jordan, D.J. Christini, D.J. Lerner, and G.W. Abbott. 2003. MinK-related peptide 2 modulates Kv2.1 and Kv3.1 potassium channels in mammalian brain. J. Neurosci. 23:8077–8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melman, Y.F., A. Domenech, S. de la Luna, and T.V. McDonald. 2001. Structural determinants of KvLQT1 control by the KCNE family of proteins. J. Biol. Chem. 276:6439–6444. [DOI] [PubMed] [Google Scholar]
- Melman, Y.F., A. Krumerman, and T.V. McDonald. 2002. a. A single transmembrane site in the KCNE-encoded proteins controls the specificity of KvLQT1 channel gating. J. Biol. Chem. 277:25187–25194. [DOI] [PubMed] [Google Scholar]
- Melman, Y.F., A. Krummerman, and T.V. McDonald. 2002. b. KCNE regulation of KvLQT1 channels: structure-function correlates. Trends Cardiovasc. Med. 12:182–187. [DOI] [PubMed] [Google Scholar]
- Pusch, M., R. Magrassi, B. Wollnik, and F. Conti. 1998. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti, M.C., M.E. Curran, A. Zou, J. Shen, P.S. Spector, D.L. Atkinson, and M.T. Keating. 1996. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 384:80–83. [DOI] [PubMed] [Google Scholar]
- Schagger, H., and G. von Jagow. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368–379. [DOI] [PubMed] [Google Scholar]
- Schroeder, B.C., S. Waldegger, S. Fehr, M. Bleich, R. Warth, R. Greger, and T.J. Jentsch. 2000. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 403:196–199. [DOI] [PubMed] [Google Scholar]
- Seebohm, G., M.C. Sanguinetti, and M. Pusch. 2003. Tight coupling of rubidium conductance and inactivation in human KCNQ1 potassium channels. J. Physiol. 552:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti, F., and S.A. Goldstein. 1998. Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol. 112:651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski, I., M. Tristani-Firouzi, M.H. Lehmann, M.C. Sanguinetti, and M.T. Keating. 1997. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 17:338–340. [DOI] [PubMed] [Google Scholar]
- Splawski, I., J. Shen, K.W. Timothy, M.H. Lehmann, S. Priori, J.L. Robinson, A.J. Moss, P.J. Schwartz, J.A. Towbin, G.M. Vincent, and M.T. Keating. 2000. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 102:1178–1185. [DOI] [PubMed] [Google Scholar]
- Tai, K.K., and S.A. Goldstein. 1998. The conduction pore of a cardiac potassium channel. Nature. 391:605–608. [DOI] [PubMed] [Google Scholar]
- Takumi, T., K. Moriyoshi, I. Aramori, T. Ishii, S. Oiki, Y. Okada, H. Ohkubo, and S. Nakanishi. 1991. Alteration of channel activities and gating by mutations of slow ISK potassium channel. J. Biol. Chem. 266:22192–22198. [PubMed] [Google Scholar]
- Tapper, A.R., and A.L. George Jr. 2000. MinK subdomains that mediate modulation of and association with KvLQT1. J. Gen. Physiol. 116:379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper, A.R., and A.L. George Jr. 2001. Location and orientation of minK within the I(Ks) potassium channel complex. J. Biol. Chem. 276:38249–38254. [DOI] [PubMed] [Google Scholar]
- Tinel, N., S. Diochot, M. Borsotto, M. Lazdunski, and J. Barhanin. 2000. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 19:6326–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K.W., and S.A. Goldstein. 1995. Subunit composition of minK potassium channels. Neuron. 14:1303–1309. [DOI] [PubMed] [Google Scholar]
- Wang, K.W., K.K. Tai, and S.A. Goldstein. 1996. MinK residues line a potassium channel pore. Neuron. 16:571–577. [DOI] [PubMed] [Google Scholar]
- Yu, H., J. Wu, I. Potapova, R.T. Wymore, B. Holmes, J. Zuckerman, Z. Pan, H. Wang, W. Shi, R.B. Robinson, et al. 2001. MinK-related peptide 1: a β subunit for the HCN ion channel subunit family enhances expression and speeds activation. Circ. Res. 88:E84–E87. [DOI] [PubMed] [Google Scholar]
- Zerangue, N., B. Schwappach, Y.N. Jan, and L.Y. Jan. 1999. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 22:537–548. [DOI] [PubMed] [Google Scholar]
- Zhang, M., M. Jiang, and G.N. Tseng. 2001. minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as β subunit of cardiac transient outward channel? Circ. Res. 88:1012–1019. [DOI] [PubMed] [Google Scholar]