

Abstract
Hyperpolarization-activated cyclic nucleotide-modulated (HCN) channels and cyclic nucleotide-gated (CNG) channels are activated by the direct binding of cyclic nucleotides. The intracellular COOH-terminal regions exhibit high sequence similarity in all HCN and CNG channels. This region contains the cyclic nucleotide-binding domain (CNBD) and the C-linker region, which connects the CNBD to the pore. Recently, the structure of the HCN2 COOH-terminal region was solved and shown to contain intersubunit interactions between C-linker regions. To explore the role of these intersubunit interactions in intact channels, we studied two salt bridges in the C-linker region: an intersubunit interaction between C-linkers of neighboring subunits, and an intrasubunit interaction between the C-linker and its CNBD. We show that breaking these salt bridges in both HCN2 and CNGA1 channels through mutation causes an increase in the favorability of channel opening. The wild-type behavior of both HCN2 and CNGA1 channels is rescued by switching the position of the positive and negative residues, thus restoring the salt bridges. These results suggest that the salt bridges seen in the HCN2 COOH-terminal crystal structure are also present in the intact HCN2 channel. Furthermore, the similar effects of the mutations on HCN2 and CNGA1 channels suggest that these salt bridge interactions are also present in the intact CNGA1 channel. As disrupting the interactions leads to channels with more favorable opening transitions, the salt bridges appear to stabilize a closed conformation in both the HCN2 and CNGA1 channels. These results suggest that the HCN2 COOH-terminal crystal structure contains the C-linker regions in the resting configuration even though the CNBD is ligand bound, and channel opening involves a rearrangement of the C-linkers and, thus, disruption of the salt bridges. Discovering that one portion of the COOH terminus, the CNBD, can be in the activated configuration while the other portion, the C-linker, is not activated has lead us to suggest a novel modular gating scheme for HCN and CNG channels.
Keywords: salt bridge, HCN2, CNGA1, COOH terminal, gating
INTRODUCTION
Cyclic nucleotide-gated (CNG) channels produce the primary electrical signal in photoreceptors upon absorption of a photon (Burns and Baylor, 2001). These channels also generate the electrical signal in olfactory receptors, and are expressed in several other sensory and nonsensory tissues (Zufall and Munger, 2001). CNG channels are voltage independent, nonselective cation channels that are gated by the direct binding of cyclic nucleotides (Fesenko et al., 1985; Kaupp and Seifert, 2002; Matulef and Zagotta, 2003). Hyperpolarization-activated cyclic nucleotide-modulated (HCN) channels are a related family of channels with a very different role. These channels have been found to control pacemaker activity in the heart and brain, resting potential, dendritic integration, and synaptic transmission (Accili et al., 2002; Biel et al., 2002; Robinson and Siegelbaum, 2003). HCN channels are cation channels that are weakly selective for conducting potassium ions and are activated by membrane hyperpolarization and modulated by the direct binding of cyclic nucleotides.
Despite their differences in physiological roles and voltage dependence, HCN and CNG channels likely have very similar structures. Both channels belong to the superfamily of voltage-gated potassium channels and are composed of four subunits, each with six transmembrane helices, arranged around a central pore (Jan and Jan, 1990). Furthermore, the intracellular COOH-terminal region of each CNG and HCN subunit contains a cyclic nucleotide-binding domain (CNBD) (Matulef and Zagotta, 2003; Robinson and Siegelbaum, 2003). Binding of cyclic nucleotides to these CNBDs produces an increase in open probability in both classes of channels. CNG channels exhibit a large increase in open probability across all voltages, whereas HCN channels exhibit a smaller increase in open probability, and only at hyperpolarized voltages.
There are several members of the CNG gene family: CNGA1, CNGA2, CNGA3, CNGA4, CNGB1, and CNGB3 (Matulef and Zagotta, 2003). These subunits combine to form heteromeric native channels; for example, native rod photoreceptors are composed of CNGA1 and CNGB1 subunits. CNGA1 subunits can form functional homomeric channels in exogenous expression systems. CNGA1 homomeric channels exhibit significant cyclic nucleotide selectivity. Saturating concentrations of cGMP (guanosine 3′,5′-cyclic monophosphate) produce a large increase in open probability while saturating cAMP (adenosine 3′,5′-cyclic monophosphate) produces only a small increase in open probability.
There are four known isoforms of HCN channels: HCN1, HCN2, HCN3, and HCN4 (Robinson and Siegelbaum, 2003). While the subunit composition of native channels is not known, each isoform can form functional homomeric channels, although with varying cyclic nucleotide dependence. HCN2 is one of the isoforms that is more responsive to cyclic nucleotides. For HCN2 homomeric channels, saturating concentrations of both cAMP and cGMP produce the same effects of increasing open probability at hyperpolarizing voltages, speeding activation kinetics and shifting the voltage dependence of activation to more depolarized voltages (DiFrancesco and Tortora, 1991; Ludwig et al., 1998; Santoro and Tibbs, 1999).
Recently, a crystal structure was solved for the intracellular COOH-terminal region of the HCN2 channel (Zagotta et al., 2003). This structure consists of a tetramer of the COOH-terminal region of HCN2 bound to either cAMP (Fig. 1) or cGMP. The COOH-terminal region includes the CNBD and the C-linker, the region that connects the CNBD to the end of the sixth transmembrane (S6) helix in the pore region. The C-linker comprises six α-helices (A'–F') and the CNBD comprises three α-helices (A–C) with eight β-strands (1–8) forming a β-roll between helices A and B. Virtually all of the intersubunit interactions in the COOH-terminal region occur between the C-linker domains. The interacting region has been likened to an “elbow on the shoulder.” where one subunit has its “elbow” on the “shoulder” of its neighbor. The A' and B' helices of the C-linker form the “elbow” and the C' and D' helices form the “shoulder” (Fig. 1, inset). The interaction between the “elbow” and “shoulder” is extensive and involves many hydrogen bonds, hydrophobic interactions, and salt bridge interactions.
Figure 1.

Structure of HCN2 C-linker and CNBD. Structure of the HCN2 COOH-terminal region (Zagotta et al., 2003), viewed from the side (left) and from the membrane (right). The structure is positioned below the membrane-spanning portion of the channel, as it is thought to be in vivo. The structure contains four subunits, two in dark gray and two in light gray, with the C-linkers making up the top half of the structure and the CNBDs the bottom half. cAMP (yellow) is bound in the CNBD of each subunit. Residues of the putative salt bridges are shown in CPK format: K472 (blue), E502 and D542 (both in red). Enlargement of the region with these salt bridges (inset). Only two subunits are shown here, with the A'–D' helices and β roll labeled, and the residues are now colored according to their elements (carbon, gray; nitrogen, blue; and oxygen, red). Plus sign within a square is K472, minus sign within a square is D542 from the same subunit, and minus sign within a circle is E502 from the neighboring subunit.
In this study, we examined these C-linker interactions by mutating a pair of salt bridges predicted by the HCN2 COOH-terminal crystal structure. We found in both HCN2 and CNGA1 channels that breaking the predicted salt bridges by mutation results in channels that favor channel opening more than wild-type channels and restoring the salt bridge by switching the positions of the positive and negative residues rescues wild-type behavior. Thus, the salt bridges seen in the HCN2 COOH-terminal crystal structure are present in both the intact HCN2 and CNGA1 channels. Our data also suggest that channel opening involves a rearrangement of the C-linkers and subsequent disruption of the salt bridges. Moreover, the finding that breaking the interactions of the C-linkers increases the favorability of opening in both channels suggests that the C-linkers in the HCN2 COOH-terminal crystal structure may be in the resting configuration despite the CNBDs being bound with ligand.
MATERIALS AND METHODS
Mutagenesis
In this paper, CNGA1cys-free is the cysteine-less version of the bovine CNGA1 channel (Matulef et al., 1999). All CNGA1 mutant channels were made in the CNGA1cys-free background, except for the D502R mutant, which was made in the cysteine-containing CNGA1 channel with a COOH-terminal enhanced cyan fluorescent protein (CNGA1-eCFP) to increase expression levels (Zheng et al., 2002). The CNGA1cys-free and CNGA1-eCFP channels gate similarly (the respective ΔGcGMP values are −2.50 ± 0.05 kcal/mol and −2.46 ± 0.13 kcal/mol, see below). Mouse HCN2 wild-type, K472E, E502K, and K472E+E502K channel DNAs were gifts from S.A. Siegelbaum (Columbia University, New York, NY). HCN2 mutant channels were made from the wild-type HCN2. Mutant complimentary DNAs were constructed by PCR methods previously described (Gordon and Zagotta, 1995a), verified by sequencing the PCR-amplified regions completely, and subcloned into the pGEMHE vector. cDNAs were linearized and cRNA was transcribed in vitro using the mMessage mMachine kit (Ambion).
Electrophysiology
Xenopus laevis oocytes were defolliculated and injected with cRNA as previously described (Zagotta et al., 1989). Recordings were made in the excised, inside-out patch configuration (Hamill et al., 1981) using an Axopatch 200A patch-clamp amplifier (Axon Instruments) and an RSC-100 rapid solution changer (Biologic) for internal solution application. Data were acquired with PULSE acquisition software (HEKA Elektronik). Patch pipettes were pulled from borosilicate glass and had resistances of 0.25–1 MΩ after fire polishing. The solutions for CNGA1 recordings were as follows: pipette (external) solution, 130 mM NaCl, 3 mM HEPES, 0.2 mM EDTA, pH 7.2, 500 μM Niflumic acid; bath (internal) solution, 130 mM NaCl, 3 mM HEPES, 0.2 mM EDTA, pH 7.2, with either no cyclic nucleotides or 16 mM cGMP, cIMP, or cAMP. The solutions for HCN2 recordings were as follows: pipette (external) solution, 130 mM KCl, 3 mM HEPES, 0.2 mM EDTA, pH 7.2; bath (internal) solution, 130 mM KCl, 3 mM HEPES, 0.2 mM EDTA, pH 7.2, with either no cyclic nucleotides or 100 μM cAMP.
Data Analysis
Calculation of L and ΔG Values
For CNGA1 channels, the opening transition for a channel that is fully bound by ligand can be described by a simple closed-to-open equilibrium with equilibrium constant L. The behavior for the different agonists can each be explained by ligand-specific L values.
In Fig. 2, ΔG (change in free energy) values are shown for all three cyclic nucleotides. ΔGcGMP is calculated from a published value of LcGMP = 72.4 ± 13.6 for CNGA1cys-free channels (Johnson and Zagotta, 2001) using Eq. 1:
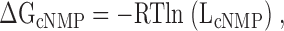 |
(1) |
where R is the gas constant, 1.987 cal·K−1·mol−1, and T is the temperature (298 K). From the LcGMP value, we could then calculate Imax, the current if the channel open probability was 1, using Eq. 2:
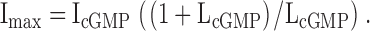 |
(2) |
Figure 2.
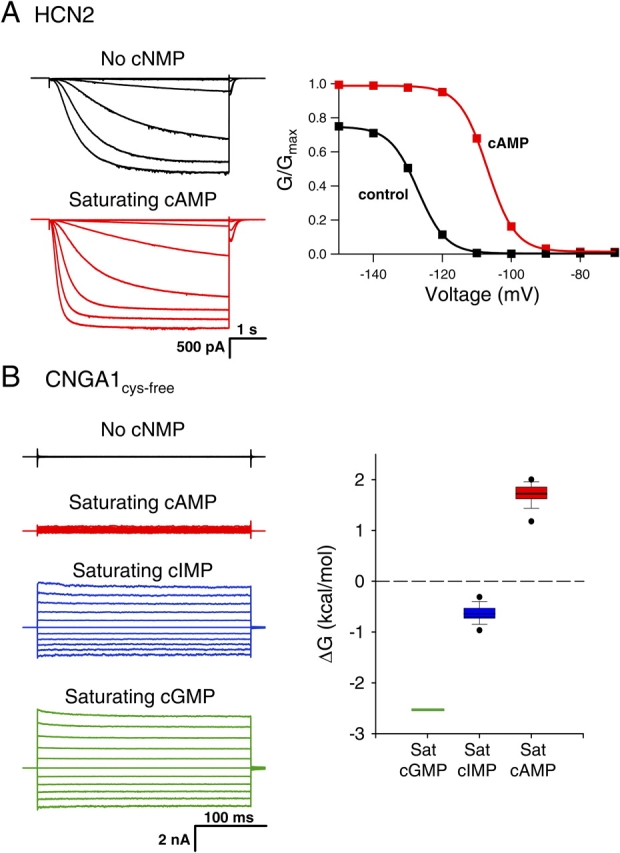
Behavior of wild-type HCN2 and CNGA1cys-free channels. (A) Behavior of wild-type HCN2 channels. Currents (left) and conductance–voltage relations (right) are shown in the absence (black) and presence of saturating cAMP (red). The currents were recorded in response to voltage pulses from a holding potential of 0 mV to test potentials between −70 and −150 mV, returning to a tail potential of −40 mV, and the conductance–voltage relations were obtained from normalized tail currents. (B) Behavior of CNGA1cys-free channels. Leak-subtracted currents (left) in response to voltage pulses from a holding potential of 0 mV to test potentials from −100 to +100 mV are shown in the absence (black, not leak subtracted) and presence of saturating cAMP (red), cIMP (blue), and cGMP (green). Box plot (right) of the ΔG for the opening transition with bound cGMP, cIMP, and cAMP. Center line is the median of the data, box is the 25th to 75th percentile of the data, and whiskers are the 5th and 95th percentile of the data. Outliers are shown as symbols.
Then, Eqs. 3 and 4 give L values:
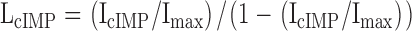 |
(3) |
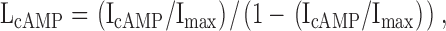 |
(4) |
where IcGMP, IcIMP, and IcAMP are the macroscopic currents with saturating cyclic nucleotide concentrations in response to voltage pulses to +60 mV. Using Eq. 1, we could then calculate the ΔGcAMP and ΔGcIMP values.
In Fig. 7, ΔGcGMP values are shown for all of the CNGA1 channels. For these calculations, we assumed that the ratios of LcAMP/LcGMP and LcIMP/LcGMP were constant for all of the mutants. Similar ratios were also found for a number of mutations in both the cysteine-containing CNGA1 and the CNGA1cys-free channels (Varnum et al., 1995; Johnson and Zagotta, 2001; Flynn and Zagotta, 2003). The LcAMP/LcGMP and LcIMP/LcGMP values were determined through the calculations performed for Fig. 2, and the mean ratio values, LcAMP/LcGMP = 0.00081 and LcIMP/LcGMP = 0.042, were held constant for fitting L values for all of the CNGA1 channels. Using these set ratios and the measured currents for IcGMP, IcIMP, and IcAMP, we determined LcGMP by fitting the currents with Eq. 5 using a least-squares algorithm.
Figure 7.

Thermodynamic mutant cycle analysis for CNGA1 channels. (A) Box plot of ΔGcGMP values for CNGA1cys-free and mutant CNGA1 channels. The dashed line indicates the median ΔGcGMP value for CNGA1cys-free. (B and C) Thermodynamic mutant cycles for the intersubunit salt bridge (B) and intrasubunit salt bridge (C). The letters are the amino acid abbreviations for the residues, the number indicates the residue number, and the charge of the residue is also shown. The lines indicate when a salt bridge is possible between residues with opposite charges. ΔΔΔG values were calculated from ΔG values (see materials and methods) and are shown as mean ΔΔΔG ± SEM of ΔΔΔG.
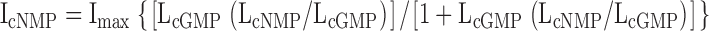 |
(5) |
Thermodynamic Mutant Cycle Analysis
The thermodynamic mutant cycle analysis was performed as previously described (Frisch et al., 1997; Vaughan et al., 2002). Eq. 6 shows the calculation for the intersubunit salt bridge and Eq. 7 shows the calculation for the intrasubunit salt bridge:
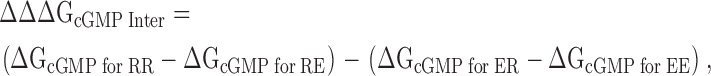 |
(6) |
where RE is the CNGA1cys-free channel, RR is the E462R mutant, EE is the R431E mutant, and ER is the double mutant R431E+E462R.
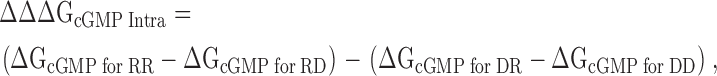 |
(7) |
where RD is the CNGA1cys-free channel, RR is the D502R mutant, DD is the R431D mutant, and DR is the double mutant R431D+D502R.
Conductance–Voltage Relations
To obtain GV curves for all HCN2 experiments, peak tail current amplitudes at −40 mV, either with or without saturating cAMP, were normalized by the largest peak tail current amplitude. These normalized data were then plotted against the test voltage potential and fit with Boltzmann curves:
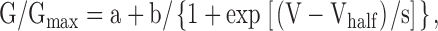 |
(8) |
where a = normalized leak current, b = maximal normalized tail current amplitude, V = test voltage (mV), Vhalf = midpoint activation voltage (mV), and s = slope of relation (mV).
Statistics
All numerical values are indicated as mean ± SEM. Statistical significance was estimated by Student's t test, unless otherwise indicated, and P values <0.05 were considered significant.
Modular Gating Modeling
Fig. 9 shows a modular gating model for HCN2 and CNGA1 channels. We calculated the probability of the channels being open by using the equilibrium constants and allosteric factors for each of these transitions. Using this model, we simulated conductance–voltage relations for wild-type and K472E HCN2 and fractional activations for wild-type and R431E CNGA1 channels. To account for the voltage dependence of the HCN2 channels, we added another module, the voltage sensor, which can be resting or activated (with equilibrium constant, J) and is coupled to the pore module (with allosteric factor, E). For the wild-type HCN2 simulation, the variables were set as follows: L0 = 2 × 10−4, C = 22, M = 0.01, F = 5, K0 = 0, KcAMP = 1000, J = 3.6 × 10−12, and E = 11500. For the K472E HCN2 simulations, the only difference from the wild-type values was setting M = 4. For the CNGA1cys-free simulation, the variables were set as follows: L0 = 10−5, C = 107, M = 10−6, FcGMP = 45, FcIMP = 13.5, FcAMP = 5, and K = 1000. For the R431E CNGA1 simulation, the only difference from the CNGA1cys-free values was setting M = 4 × 10−4.
Figure 9.

Model of modular gating scheme for HCN and CNG channels. (A) HCN and CNG gating model with three modules: pore, C-linker, and CNBD. Each module is represented by a boxed equilibrium, indicating that each module can be in one of two configurations. Pore can be closed (C) or open (O), C-linker can be resting (R) or activated (A), and CNBD can be unbound (U) or bound (B) with ligand. Equilibrium constants are shown for each module: L for pore, M for C-linker, and K for CNBD. The pore and the C-linker influence each other through allosteric factor C, and the C-linker and CNBD through allosteric factor F. (B) Simulated conductance–voltage (GV) relations for wild-type (top) and K472E (bottom) HCN2 channels in the absence (black) and presence of saturating cAMP (red). A voltage-dependent module that was coupled to the pore was added for these calculations. The values of the parameters used are shown in the materials and methods. (C) Simulated current traces in response to voltages for CNGA1cys-free (top) and R431E (bottom) channels in the presence of saturating cAMP (red), cIMP (blue), and cGMP (green). For the R431E simulations, the current values for cIMP are so similar to cGMP that the two traces overlay. Diagrams to the left show attractive electrostatic interactions by solid lines and repulsive electrostatic interactions by dotted lines with wild-type residues as open symbols and mutant residues as shaded symbols.
RESULTS
HCN2 channels are activated by both membrane hyperpolarization and cAMP binding. Homomeric wild-type HCN2 channels were expressed in Xenopus laevis oocytes, and currents were recorded using the patch-clamp technique in the inside-out configuration. Fig. 2 A shows HCN2 currents in response to voltage steps to potentials between −70 and −150 mV. In response to this hyperpolarization, wild-type HCN2 channels opened with a predominantly exponential time course after an initial lag, with a midpoint for the steady-state activation curve (Vhalf) of −125 ± 2.0 mV (Fig. 2 A, right; see Table II). Saturating concentrations (100 μM) of cAMP increased the rate of activation and the steady-state current levels at hyperpolarizing membrane potentials, resulting in a shift in the voltage dependence of activation to more depolarized potentials (Vhalf = −111 ± 1.7 mV) and an increase in the maximal current at hyperpolarized voltages (Fig. 2 A, red). Thus, cAMP stabilizes the closed-to-open equilibrium for HCN2 channels, enabling activation with less hyperpolarization and more complete activation at hyperpolarized voltages.
CNGA1 channels are also activated by cyclic nucleotide binding, although unlike HCN2 channels they are nearly voltage independent. Leak-subtracted CNGA1cys-free currents in response to voltage pulses to potentials between −100 and +100 mV are shown in Fig. 2 B. cGMP dramatically increased the steady-state currents over the control (no cNMP) currents, while saturating cIMP and cAMP produced less current than saturating cGMP. This behavior reflects the fact that the cyclic nucleotides stabilize the closed-to-open equilibrium of CNGA1, with cGMP having the most favorable closed-to-open transition and cAMP the least (Varnum et al., 1995; Sunderman and Zagotta, 1999). This difference in cyclic nucleotide behavior is exhibited in the changes in free energy (ΔG) between the closed and open states, which are calculated from the open probability (Fig. 2 B, right). Therefore, cGMP is a full agonist for CNGA1 channels while cIMP and cAMP are partial agonists.
The COOH-terminal regions of both HCN2 and CNGA1 channels, as well as those of other members of the HCN, CNG, and related channel families, exhibit high sequence similarity (Fig. 3). This region encompasses the C-linker and the CNBD. We have studied two salt bridge interactions in this region that are predicted by the HCN2 COOH-terminal crystal structure. The salt bridges involve the same positively charged residue and two different negatively charged residues, forming one intersubunit interaction and one intrasubunit interaction (Fig. 1). These residues are very well conserved across all of the HCN, CNG, and related channel families; the residue at the position of the positive charge is always a lysine or an arginine and the residues at the positions of the two negative charges are almost always glutamate or aspartate (Fig. 3). The positively charged residue is located on the B' helix of the C-linker region (Figs. 1 and 3, plus sign within a square). In the HCN2 COOH-terminal crystal structure, this residue forms an intersubunit “elbow on the shoulder” salt bridge interaction with a negatively charged residue on the D' helix of the neighboring C-linker (Figs. 1 and 3, minus sign within a circle). Additionally, in the HCN2 COOH-terminal crystal structure, the same positively charged residue forms an intrasubunit salt bridge with a negatively charged residue located in the β roll of the CNBD from the same subunit (Figs. 1 and 3, minus sign within a square). These symbols are used throughout the paper to indicate the charge and subunit location of the residues in the salt bridges.
Figure 3.

Sequence alignment of COOH-terminal regions from HCN, CNG, and related channels. Sequence alignment of the COOH-terminal region includes all members of the CNG channel family (bovine CNGA1, rat CNGA2, bovine CNGA3, rat CNGA4, bovine CNGB1, murine CNGB3) and HCN channel family (murine HCN2, mouse HCN1, rat HCN3, rat HCN4), as well as SPIH (the sea urchin sperm homologue of HCN), and the human ether-a-go-go related gene (HERG) and Drosophila Eag K+ channels. Above the sequence alignment, the tertiary structure elements are shown: α-helices (red rectangles), β-sheets (blue arrow), and uncoiled regions (line). Residues highlighted in blue are similar. Putative salt bridge residues studied are boxed and labeled.
Intersubunit Salt Bridge
To determine whether the intersubunit salt bridge in the HCN2 COOH-terminal crystal structure is present in intact HCN2 and CNGA1 channels, we mutated the positive and negative residues such that the salt bridge would be disrupted (Fig. 4). Replacing the positive residue at the HCN2 B' helix site with a negative residue (K472E) resulted in channels that activated slowly and were no longer modulated by cAMP; current traces with voltage steps to −130 mV in the presence of cAMP were very similar to currents in the absence of cAMP (Fig. 4 B). The ratio of the cAMP and control rates of activation for this mutant, 0.98 ± 0.04 (n = 4), is significantly decreased from that of wild type, 3.6 ± 0.48 (n = 7) (P < 0.05) (Table I). In addition, not only was cAMP no longer able to increase the current, it slightly decreased the maximum current level (Fig. 4 B). Substituting the negative residue at the HCN2 D' helix site with a positive residue (E502K) produced a channel that did not behave appreciably different from wild-type HCN2 channels; cAMP still augmented the rate of activation and the steady-state current (Fig. 4 C; Table I). However, making both mutations together, effectively switching the position of the positive and negative residues between the B' helix site and the D' helix site (K472E+E502K), rescued wild-type HCN2 behavior. cAMP had a similar effect on this double mutant as it did on the wild-type channel (Fig. 4 D; Table I). This functional rescue suggests that the intersubunit salt bridge was reformed and indicates that this salt bridge observed in the crystal structure of the HCN2 COOH-terminal fragment is present in intact channels.
Figure 4.
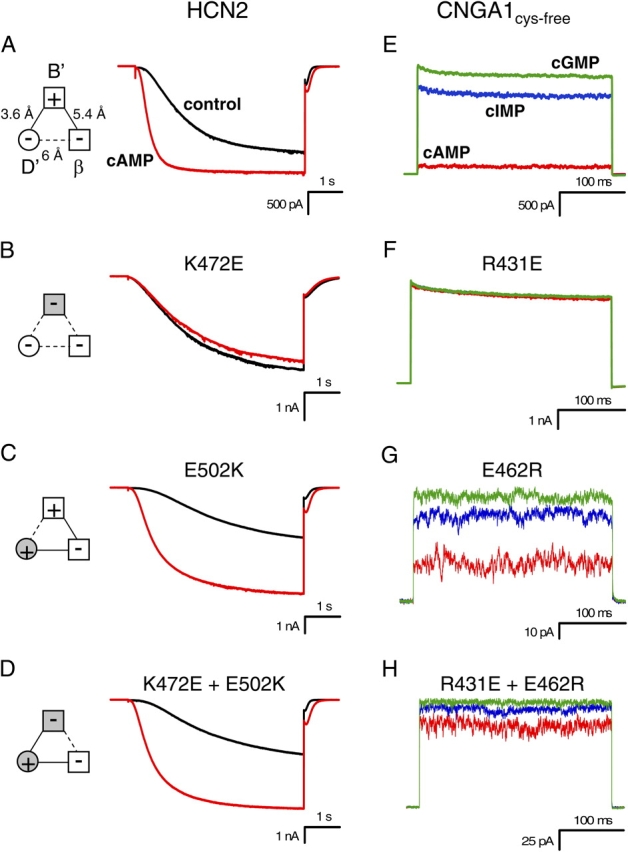
Mutations of intersubunit salt bridge. (A–D) Behavior of wild-type HCN2 (A), K472E (B), E502K (C), and K472E + E502K (D) channels. Currents in response to voltage pulses to −130 mV are shown in the absence (black) and presence of saturating cAMP (red). Diagrams to the left show attractive electrostatic interactions by solid lines and repulsive electrostatic interactions by dotted lines with wild-type residues as open symbols and mutant residues as shaded symbols. (E–H) Behavior of CNGA1cys-free (E), R431E (F), E462R (G), and R431E + E462R (H) channels. For R431E (F), the current values for cIMP are so similar to cGMP that the two traces almost overlay. Current in response to voltage pulses to +100 mV are shown in the presence of saturating cAMP (red), cIMP (blue), and cGMP (green).
TABLE I.
Rate Data for HCN2 Channels and Fractional Activation Data for CNGA1 Channels
| HCN2 channel | RatecAMP/RateControl | CNGA1 channel | Imax cAMP/Imax cGMP |
|---|---|---|---|
| Wild type (7) | 3.6 ± 0.48 | CNGA1cys-free (14) | 0.055 ± 0.01 |
| K472E (4) | 0.98 ± 0.04 | R431E (5) | 0.95 ± 0.01 |
| E502K (6) | 2.2 ± 0.37 | E462R (5) | 0.38 ± 0.05 |
| K472E + E502K (4) | 2.3 ± 0.52 | R431E + E462R (3) | 0.82 ± 0.04 |
| R431D (5) | 0.92 ± 0.03 | ||
| D542K (5) | 2.7 ± 0.99 | D502R (3) | 0.18 ± 0.03 |
| K472E + D542K (3) | 2.3 ± 1.0 | R431D + D502R (3) | 0.11 ± 0.03 |
| E502K + D542K (5) | 1.0 ± 0.05 |
All values are mean ± SEM. The n values are in parentheses after each channel construct. HCN2 channel rates (1/τ): time constants (τ) were calculated from single-exponential fits to currents in response to voltage pulses to −130 mV, either in the presence or absence of cAMP.
We created the equivalent mutations in CNGA1 channels in order to discern whether this salt bridge is present in CNGA1 channels as well (Fig. 4). Substituting the positive residue at the B' helix site with a negative residue (R431E) caused a significant increase in cAMP- and cIMP-induced currents relative to the cGMP-induced current as compared with CNGA1cys-free (P < 0.05) (Fig. 4, E and F; Table I). Thus, the effect of this mutation was to enable partial agonists to act as full agonists. The ability of cAMP and cIMP to induce the same amount of current as cGMP reflects a decrease in the ΔG of opening, corresponding to an increase in the favorability of opening, produced by disrupting the salt bridges. Mutating the negative residue at the D' helix site to a positive residue (E462R) also resulted in an increase in the fractional activation by cIMP and cAMP, although not as much as the positive-to-negative mutant (Fig. 4 G; Table I). Switching the position of the positive and negative residues between the B' helix site and the D' helix site (R431E+E462R) created a channel whose cAMP and cIMP fractional activation was less than the B' helix site positive-to-negative mutant, but not as low as CNGA1cys-free, producing a partial rescue of CNGA1cys-free behavior (Fig. 4 H; Table I). These results indicate that the intersubunit salt bridge seen in the HCN2 COOH-terminal crystal structure is present in intact CNGA1, as well as HCN2, channels.
Intrasubunit Salt Bridge
The intrasubunit salt bridge in the HCN2 COOH-terminal crystal structure involves the same positive residue as the intersubunit salt bridge, but a different negative residue. We also wanted to determine whether this salt bridge was present in intact channels. The same positive-to-negative HCN2 mutation at the B' helix site (K472E) is shown for this second salt bridge, which, as previously described, produced channels that were no longer modulated by cAMP (Fig. 5, A and B; Table I). The negative-to-positive mutation at the β roll site (D542K) for this intrasubunit salt bridge resulted in channels that were modulated by cAMP, although to a lesser degree than wild-type HCN2 (Fig. 5 C; Table I). Finally, switching the positive and negative residues between the B' helix site and the β roll site (K472E+D542K) rescued wild-type–like cAMP modulation (Fig. 5 D; Table I). Thus, the intrasubunit salt bridge seen in the HCN2 COOH-terminal crystal structure is present in HCN2 full-length channels as well.
Figure 5.
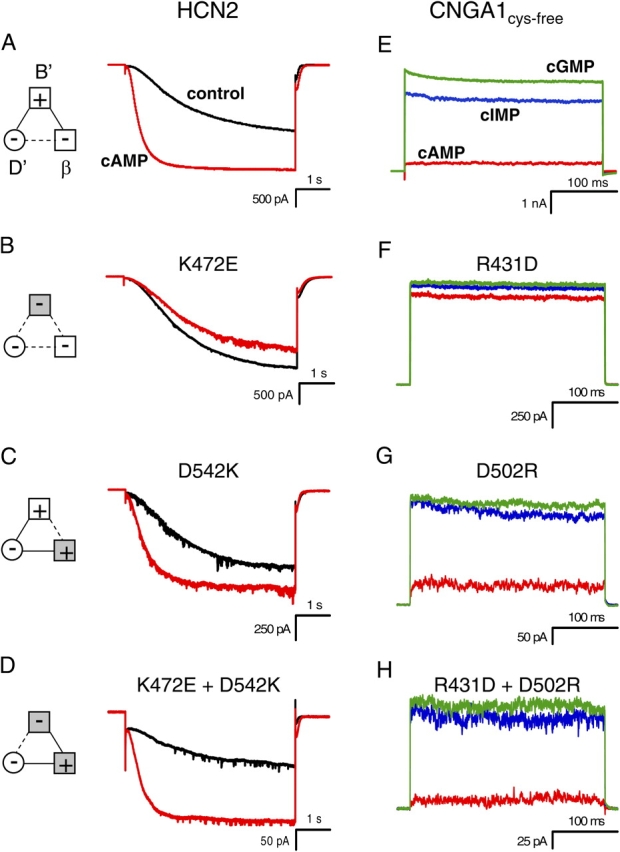
Mutations of intrasubunit salt bridge. (A–D) Behavior of wild-type HCN2 (A), K472E (B), D542K (C), and K472E + D542K (D) channels. Currents in response to voltage pulses to −130 mV are shown in the absence (black) and presence of saturating cAMP (red). Diagrams to the left show attractive electrostatic interactions by solid lines and repulsive electrostatic interactions by dotted lines with wild-type residues as open symbols and mutant residues as shaded symbols. (E–H) Behavior of CNGA1cys-free (E), R431D (F), D502R (G), and R431D + D502R (H) channels. Current in response to voltage pulses to +100 mV are shown in the presence of saturating cAMP (red), cIMP (blue), and cGMP (green).
Once again, the equivalent intrasubunit salt bridge mutations were made in CNGA1 channels. As before, substituting the positive residue at the B' helix site with a negative residue (R431D, this time an aspartate instead of a glutamate) created channels that had a fractional activation by cAMP and cIMP that was significantly increased from that of CNGA1cys-free (P < 0.05) (Fig. 5, E and F; Table I). This was the same phenotype as seen before and indicates that the ΔG of opening was decreased relative to CNGA1cys-free. Making a channel whose negative residue at the β roll site was replaced with a positive one (D502R) resulted in a channel whose fractional activation by cAMP and cIMP was slightly more than CNGA1cys-free (Fig. 5 G; Table I). However, reinstating the salt bridge by switching the positive and negative residues between the B' helix site and the β roll site (R431D+D502R) rescued CNGA1cys-free behavior such that the fractional activation of cIMP and cAMP was similar to that of CNGA1cys-free channels (Fig. 5 H; Table I). These results suggest that the intrasubunit salt bridge is present in intact CNGA1 and HCN2 channels, as predicted by the HCN2 COOH-terminal crystal structure.
Voltage Dependence of HCN2 Channels
Another way to quantify the effects of the intersubunit and intrasubunit salt bridge mutations on HCN2 channels is to record the changes in their voltage dependence. Conductance–voltage (GV) relations for wild-type and mutant HCN2 channels, in both the absence and presence of cAMP, are shown in Fig. 6 and summarized in Table II. From the GV relations, it was evident that the positive-to-negative mutation at the B' helix site (K472E) did more than prevent cAMP-induced increases in activation rate and steady-state current. The K472E mutation also significantly shifted the control voltage dependence to more depolarized voltages and significantly decreased the slope of both the control and cAMP GV curves (P < 0.05) (Fig. 6, A and B; Table II). The depolarizing shift suggests that this mutation increased the favorability of the regulatory conformational change in HCN2 channels such that the channels appeared to be regulated by cAMP even without cAMP present. Both of the negative-to-positive mutations from the inter- and intrasubunit salt bridges (E502K at the D' helix site and D542K at the β roll site, respectively) slightly decreased the cAMP-induced shift of the voltage dependence curves and slightly decreased the slope of those GV curves as well, although not as much as the positive-to-negative mutation in the B' helix site, K472E (Fig. 6, C and D; Table II). However, both channels in which the position of the positive and negative residues have been switched (either K472E+E502K between the B' helix site and the D' helix site for the intersubunit salt bridge or K472E+ D542K between the B' helix site and the β roll site for the intrasubunit salt bridge) had GV relations very similar to wild type (Fig. 6, E and F; Table II). Despite their slow kinetics of activation, our finding that the salt bridge mutations increase the favorability of the regulatory conformational change suggests that the salt bridges stabilize the resting conformation of the channel. This is true for the HCN2 channels as well as the CNGA1 channels, as the phenotypic effects of disrupting the salt bridges are qualitatively similar; both the rightward shift in the GV relation for mutant HCN2 and the increase in fractional activation of partial agonists for mutant CNGA1 reflect a favoring of channel opening.
Figure 6.

Conductance–voltage relations for HCN2 channels. (A–F) Conductance versus voltage plots for wild-type HCN2 (A), K472E (B), E502K (C), D542K (D), K472E+E502K (E), and K472E+D542K (F). Each plot indicates GV curves, obtained from normalized tail currents at −40 mV, in the absence (black) and presence of saturating cAMP (red). The data are fit with a Boltzmann relation. Diagrams on the left and the right show attractive electrostatic interactions by solid lines and repulsive electrostatic interactions by a dotted line with wild-type residues as open symbols and mutant residues as shaded symbols.
TABLE II.
Conductance–Voltage Data for HCN2 Channels
| Control | Saturating cAMP | ||||
|---|---|---|---|---|---|
| HCN2 channel | Vhalf (mV) | Slope (mV) | Vhalf (mV) | Slope (mV) | ΔVhalf (mV) |
| Wild type (7) | −125 ± 2.0 | 3.76 ± 0.30 | −111 ± 1.7 | 4.18 ± 0.24 | 14.3 ± 1.8 |
| K472E (4) | −113 ± 3.1 | 9.59 ± 0.89 | −112 ± 2.9 | 9.06 ± 0.91 | 0.75 ± 1.3 |
| E502K (6) | −128 ± 2.9 | 5.66 ± 0.97 | −116 ± 2.8 | 5.4 ± 1.1 | 12.3 ± 0.94 |
| K472E + E502K (4) | −125 ± 3.8 | 5.55 ± 0.82 | −110 ± 3.4 | 5.72 ± 0.41 | 14.9 ± 1.6 |
| D542K (5) | −124 ± 1.9 | 7.03 ± 0.61 | −114 ± 1.9 | 6.35 ± 0.75 | 10.1 ± 1.4 |
| K472E + D542K (3) | −133 ± 1.9 | 3.65 ± 0.57 | −119 ± 3.6 | 3.52 ± 0.31 | 14.2 ± 1.9 |
| E502K + D542K (4) | −118 ± 2.8 | 7.58 ± 1.4 | −122 ± 2.8 | 7.27 ± 1.1 | −3.9 ± 0.95 |
All values are mean ± SEM. The n values are in parentheses after each channel construct. ΔVhalf = Vhalf cAMP − Vhalf control.
Non-additive Effects of Salt Bridge Mutations
We have shown that disrupting these salt bridges through mutation changed the behavior of both HCN2 and CNGA1 channels and that switching the position of the positive and negative residues partially or fully rescued the wild-type HCN2 and CNGA1cys-free behavior. The nonadditive effects (coupling) of the individual mutations are indicative of an interaction between the residues, in this case the presence of salt bridges. To more quantitatively measure this coupling energy, we conducted a thermodynamic mutant cycle analysis. This analysis is possible in CNGA1 channels because, due to strong coupling between conformational changes of the C-linker and the pore, the closed-to-open transition of the channel can be reduced to a two state model, thus enabling us to calculate ΔGcGMP values from the open probabilities. Thermodynamic mutant cycle analysis is more difficult in HCN2 channels because the conformational changes in the C-linker are probably more weakly coupled to pore opening than in CNGA1 (see discussion).
The ΔGcGMP values for CNGA1 channels were calculated using the fractional activation by cIMP and cAMP (see materials and methods). For all of the mutant CNGA1 channels, the ΔGcGMP values are more negative than CNGA1cys-free (Fig. 7 A), indicating that opening is more favorable in the mutant than in the CNGA1cys-free channel. The ΔGcGMP values for all of the mutants, except the double mutant of the B' helix site and the β roll site, are statistically different from the CNGA1cys-free ΔGcGMP values (P < 0.05). Thermodynamic mutant cycle analysis then involves calculating the coupling energy (ΔΔΔG) from these ΔGcGMP values (see materials and methods) (Frisch et al., 1997). If the effects of mutations at the two residues are independent, the ΔΔΔG value is expected to be 0 kcal/mol. However, if the ΔΔΔG values are different from zero, this indicates coupling between the two mutations, suggestive of a molecular interaction (Frisch et al., 1997). The ΔΔΔG values for the intersubunit and intrasubunit salt bridges are −2.2 ± 0.3 kcal/mol and −3.8 ± 0.3 kcal/mol, respectively (Fig. 7, B and C). A z test indicated that both of these ΔΔΔG values are significantly less than zero (P < 0.05). Thus, the mutations within each salt bridge show coupling. Even in the intersubunit salt bridge, where switching the position of the B' helix positive residue and the D' helix negative residue only partially rescued CNGA1cys-free behavior, the coupling energy is large (Fig. 7 B). Therefore, the effects of the mutations are nonadditive, as expected for a salt bridge.
Thus far, we have considered the two negatively charged residues separately, focusing on how each interacts with the positively charged B' helix site within its own salt bridge. But is there coupling between mutations at the D' helix site and mutations at the β roll site? We were not able to perform thermodynamic mutant cycle analysis for these two residues in CNGA1, due to lack of functional expression of the double mutant channel. However, we were able to express this double mutation in the HCN2 channel. Although in HCN2 channels we cannot calculate the energetic effects of these mutants, we can compare the magnitude of the effects of each single negative-to-positive mutation at the D' helix site and the β roll site with the effects of mutating both negative residues to positive residues. The channel with both negative-to-positive mutations at the D' helix site and the β roll site shows a much greater (nonadditive) change from wild type than either of the single negative-to-positive mutant channels (compare Figs. 6, C and D, with Fig. 8 B). In fact, the behavior of the double negative-to-positive mutant channel (E502K+D542K) resembles the dramatic phenotype of the positive-to-negative mutation of the B' helix site (K472E) (Fig. 8). Both of these channels showed many of the same changes from wild-type behavior: a lack of cAMP-induced changes in activation kinetics and shift in voltage dependence, slight inhibition of steady-state currents instead of augmentation, and reduction of GV slope (Fig. 8; Table II). One difference between the behaviors of the two mutants was that E502K+D542K did not show the same shift in control Vhalf that was seen with K472E. Overall, it appears that in addition to an interaction between the residues within a salt bridge, there is also an interaction, exhibited by the nonadditive effect of the double negative-to-positive mutant, between the negatively charged residues in different salt bridges.
Figure 8.

Double negative-to-positive HCN2 mutant has same phenotype as B' helix site positive-to-negative mutant. Behavior of K472E (A) and E502K+D542K (B) HCN2 channels. Currents in response to voltage pulses to −130 mV, as well as conductance–voltage relations of the normalized conductance from tail currents at −40 mV, are shown in the absence (black) and presence of saturating cAMP (red). Diagrams at the top of each column show attractive electrostatic interactions by solid lines and repulsive electrostatic interactions by dotted lines with wild-type residues as open symbols and mutant residues as shaded symbols. The GV curves are fit with a Boltzmann relation.
DISCUSSION
We studied two salt bridges present in the C-linker region of the HCN2 COOH-terminal crystal structure, one intersubunit and one intrasubunit, which involve the same positively charged residue at the B' helix site and two different negatively charged residues at the D' helix site and the β roll site, respectively. Through mutation experiments, we found that both salt bridges are present in intact functional HCN2 and CNGA1 channels. The mutation that had the largest effect on channel behavior was the positive-to-negative mutation at the B' helix site predicted to disrupt both salt bridges. For both channels, this mutation increased the favorability of the regulatory conformational change, thus increasing the favorability of channel opening. Smaller effects were seen for both single negative-to-positive mutants (at the D' helix site and the β roll site), predicted to disrupt the inter- and intrasubunit salt bridges, respectively. They too, however, produced channels that favored channel opening more than wild-type HCN2 and CNGA1cys-free. The channels with the position of the positive and negative residues switched (either between the B' helix site and the D' helix site or between the B' helix site and the β roll site) partially or completely rescued wild-type HCN2 and CNGA1cys-free behavior. All mutations resulted in qualitatively similar phenotypes for both HCN2 and CNGA1 channels. These findings are expected to apply also to wild-type (cysteine-containing) CNGA1 and heteromeric CNG channels, which exhibit a high degree of sequence and functional similarity to CNGA1cys-free channels. Therefore the HCN2 COOH-terminal crystal structure, at least at the level of these residue interactions, applies to both HCN2 and CNGA1 channels.
The most surprising aspect of our results is that disrupting the salt bridges through mutation leads to channels whose favorability of channel opening is increased over wild type. The HCN2 COOH-terminal crystal structure was solved in the presence of cAMP (Zagotta et al., 2003). Hence, it was presumed that the entire COOH terminus was captured in the active (favors the open channel) configuration. Therefore, the disruption of any state-dependent interaction in the crystal structure was expected to destabilize the open configuration. However, all of the single mutations we made in both HCN2 and CNGA1 channels increased the favorability of channel opening. This could result if disrupting the salt bridges destabilized the closed state and would thus suggest that the HCN2 COOH-terminal crystal structure, at least at the level of these residue interactions, is in the resting (favors the closed channel) configuration. Alternatively, the HCN2 COOH-terminal crystal structure may be in an activated conformation, but the salt bridges are stronger in the resting conformation. Finally, it is possible that mutation of these salt bridges produced a large structural change in the COOH-terminal region that altered other state-dependent interactions in the channel.
How, in the HCN2 COOH-terminal crystal structure, can the CNBD be bound by ligand but the C-linker be in its resting conformation? Gating in ion channels can be thought of as a series of coupled conformational changes in separate domains of the channel (Horrigan and Aldrich, 2002). For example, the cyclic nucleotide-dependent activation of HCN2 and CNGA1 channels can be described through interactions between three domains: the pore, the C-linker, and the CNBD (Fig. 9 A). Each domain, or module, is in equilibrium between two possible conformations. The pore can be closed or open, the C-linker can be resting or activated, and the CNBD can be unbound or bound with ligand. The modules are then coupled to each other, indicating that the conformation of one module affects the conformation of another module. For example, the opening of the channel is C times more favorable when the C-linker is in its activated state and the activation of the C-linker is F times more favorable for each cyclic nucleotide bound. While the parameters of the model are not uniquely determined by our experimental data, we were able to simulate channel behavior for both HCN2 and CNGA1 (Fig. 9, B and C). For HCN2, an additional voltage sensor module was coupled to channel opening (see materials and methods). By only changing the equilibrium constant, M, for the C-linker module, we could convert wild-type behavior into the behavior of the positive-to-negative B' helix mutation. This model also explains how, in the HCN2 COOH-terminal crystal structure, the CNBD can be bound by ligand but the C-linker can be in its resting conformation. Finally, it explains how cAMP and cGMP can both appear to be full agonists on the HCN2 channel yet both only shift the voltage dependence of opening by 15 mV. Both cAMP and cGMP can exert a saturating effect on the C-linker module, that then exerts a fixed effect (e.g., 15 mV shift) on pore opening, determined by the allosteric factor C.
Why would the C-linker in the HCN2 COOH-terminal crystal structure be in a resting conformation if the CNBD is bound with ligand? One possible answer is suggested by the modular gating scheme in Fig. 9 A. If the resting state of the C-linker normally has an inhibitory effect on pore opening, then removing the pore (as done for crystallizing the HCN2 COOH-terminal fragment) should promote the resting state of the C-linker. Therefore, even in the presence of ligand, the unloaded (isolated from the pore) C-linker might remain in its resting state. In fact, inhibition by the COOH-terminal was proposed by Wainger et al. (2001) in their study with COOH-terminal deletion mutants of HCN1 and HCN2 channels. These authors concluded that the CNBD exerts an inhibitory effect on channel activation by shifting the Vhalf of channel activation to more hyperpolarized potentials. The CNBD deletion has a phenotype very similar to the HCN2 positive-to-negative mutation at the B' helix site, with a control Vhalf that is similar to the cAMP Vhalf for wild type. Noticeably, their deletion of the CNBD also removes the β roll salt bridge site, and consequently will disrupt the salt bridge interactions. Therefore, it is possible that it is not the entire CNBD that is inhibiting pore opening, rather it is the presence of these salt bridges.
For the channels with mutations at the D' helix site and the β roll site, why does the channel with both negative-to-positive mutations show a much greater (nonadditive) change from wild type than either of the single negative-to-positive mutant channels? In the single negative-to-positive mutant channels, it is possible that there is a compensatory mechanism that preserves some of the wild-type structural stability in this region. We propose two possible compensatory mechanisms that may preserve structural stability. The first is that when one salt bridge is disrupted through mutation, the other salt bridge strengthens, preserving the stability of the region. For example, when the negatively charged residue at the D' helix site is mutated to a positive residue, the intersubunit salt bridge is disrupted, but the intrasubunit salt bridge may compensate by strengthening and thus may stabilize this region of the channel.
The second compensatory mechanism proposes that when a salt bridge is disrupted by mutation, a new attractive electrostatic interaction and a new repulsive interaction are formed between the three salt bridge residues, thus maintaining the same types of interactions seen in wild-type channels. This mechanism is based on the charges and relative positioning of the salt bridge residues in the HCN2 COOH-terminal crystal structure. These three residues can be thought of as corners of a triangle with sides of 3.6, 5.4, and 6.0 Å (Fig. 4 A diagram). In the HCN2 COOH-terminal crystal structure, there are two attractive electrostatic interactions and one repulsive interaction. For the negative-to-positive mutation at the D' helix site, the intersubunit salt bridge is disrupted, and this side of the triangle becomes a repulsive interaction instead of an attractive interaction (Fig. 4 C). To compensate, the repulsive interaction between the D' helix site and the β roll site becomes an attractive interaction, perhaps forming a new salt bridge. Overall, there are the same numbers of attractive and repulsive interactions as in wild-type channels, which might compensate for the changes wrought by mutating the residues. It is also possible that both compensatory mechanisms might arise. Either of these compensatory mechanisms allows for an increase in structural stability despite the disruption of a salt bridge, thus accounting for a phenotype that is practically wild type. However, these compensatory mechanisms are not possible for the positive-to-negative B' helix site mutation or the negative-to-positive double mutation (Fig. 8). Another possible compensatory mechanism would be that the mutations caused the formation of a new salt bridge with an amino acid other than those residues discussed here. This does not seem likely, as in the HCN2 COOH-terminal crystal structure there are no possible salt bridge partners for the B' helix residue, and no possible salt bridge partners within 8 Å for the D' helix and β roll residues.
If the HCN2 COOH-terminal crystal structure represents the resting configuration of the C-linker, what then is the structure of the activated state? Previously, it has been shown that several histidine residues in the CNGA1 A' helix, for example, H420, are able to coordinate Ni+2 between the same residue on neighboring subunits in the open state of the channel (Gordon and Zagotta, 1995b,c; Johnson and Zagotta, 2001). But this result is in contrast to the HCN2 COOH-terminal crystal structure, where H420 residues are very distant from one another. Together with the HCN2 COOH-terminal crystal structure and the results presented in this paper, these results lead us to propose the following model for the resting to activated transition of the C-linker (Fig. 10). In the absence of ligand, the C-linker is in a compact, tense quaternary configuration and the intersubunit “elbow on the shoulder” interactions, as seen in the HCN2 COOH-terminal crystal structure, are present. This conformation inhibits pore opening. Upon cyclic nucleotide binding to the CNBDs, there is a quaternary rearrangement of the C-linkers to a more relaxed structure, where the COOH-terminal portion of the A' helices move closer together, toward the central axis of the channel. This rearrangement would result in the residues of the salt bridges becoming more distant from each other, thus disrupting the salt bridge interactions.
Figure 10.

Model for COOH-terminal region in the resting and activating configurations. Cartoons of HCN2 COOH-terminal region from the side view, both in the resting configuration (left) and the active configuration (right). COOH-terminal regions of two subunits (dark and light gray) are shown below the membrane-spanning portion of the channel. α helices are shown with narrow cylinders, and the β rolls of the CNBDs are shown with wide cylinders. Salt bridge residues are shown: the positive B' helix residue (plus sign within a square) and the two negative residues on the D' helix and the β roll (minus signs within a circle or square, respectively).
(SCHEME 1).

Acknowledgments
We would like to thank S.A. Siegelbaum for HCN2 clones and E.C. Young for preliminary work on HCN2 mutants. We thank K.D. Black, S. Cunnington, G. Sheridan, and H. Utsugi for technical assistance, G.E. Flynn, J.P. Johnson Jr., L.D. Islas, N.G. Shuart, M.C. Trudeau, and J. Zheng for thoughtful discussions, and J.P. Johnson Jr., M.C. Trudeau, and T.I. Brelidze for comments on the manuscript.
This study was supported by the Howard Hughes Medical Institute, the National Institutes of Health grant R01 EY010329 (awarded to W.N. Zagotta), and the University of Washington Neurobiology training grant T32 GM07108 (awarded to K.B. Craven).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: cAMP, adenosine 3′,5′-cyclic monophosphate; cGMP, guanosine 3′,5′-cyclic monophosphate; CNBD, cyclic nucleotide-binding domain; CNG, cyclic nucleotide-gated; HCN, hyperpolarization-activated cyclic nucleotide-modulated.
References
- Accili, E.A., C. Proenza, M. Baruscotti, and D. DiFrancesco. 2002. From funny current to HCN channels: 20 years of excitation. News Physiol. Sci. 17:32–37. [DOI] [PubMed] [Google Scholar]
- Biel, M., A. Schneider, and C. Wahl. 2002. Cardiac HCN channels: structure, function, and modulation. Trends Cardiovasc. Med. 12:206–212. [DOI] [PubMed] [Google Scholar]
- Burns, M.E., and D.A. Baylor. 2001. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu. Rev. Neurosci. 24:779–805. [DOI] [PubMed] [Google Scholar]
- DiFrancesco, D., and P. Tortora. 1991. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 351:145–147. [DOI] [PubMed] [Google Scholar]
- Fesenko, E.E., S.S. Kolesnikov, and A.L. Lyubarsky. 1985. Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature. 313:310–313. [DOI] [PubMed] [Google Scholar]
- Flynn, G.E., and W.N. Zagotta. 2003. A cysteine scan of the inner vestibule of cyclic nucleotide-gated channels reveals architecture and rearrangement of the pore. J. Gen. Physiol. 121:563–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch, C., G. Schreiber, C.M. Johnson, and A.R. Fersht. 1997. Thermodynamics of the interaction of barnase and barstar: changes in free energy versus changes in enthalpy on mutation. J. Mol. Biol. 267:696–706. [DOI] [PubMed] [Google Scholar]
- Gordon, S.E., and W.N. Zagotta. 1995. a. Localization of regions affecting an allosteric transition in cyclic nucleotide-activated channels. Neuron. 14:857–864. [DOI] [PubMed] [Google Scholar]
- Gordon, S.E., and W.N. Zagotta. 1995. b. A histidine residue associated with the gate of the cyclic nucleotide-activated channels in rod photoreceptors. Neuron. 14:177–183. [DOI] [PubMed] [Google Scholar]
- Gordon, S.E., and W.N. Zagotta. 1995. c. Subunit interactions in coordination of Ni2+ in cyclic nucleotide-gated channels. Proc. Natl. Acad. Sci. USA. 92:10222–10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Horrigan, F.T., and R.W. Aldrich. 2002. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 120:267–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan, L.Y., and Y.N. Jan. 1990. A superfamily of ion channels. Nature. 345:672. [DOI] [PubMed] [Google Scholar]
- Johnson, J.P., Jr., and W.N. Zagotta. 2001. Rotational movement during cyclic nucleotide-gated channel opening. Nature. 412:917–921. [DOI] [PubMed] [Google Scholar]
- Kaupp, U.B., and R. Seifert. 2002. Cyclic nucleotide-gated ion channels. Physiol. Rev. 82:769–824. [DOI] [PubMed] [Google Scholar]
- Ludwig, A., X. Zong, M. Jeglitsch, F. Hofmann, and M. Biel. 1998. A family of hyperpolarization-activated mammalian cation channels. Nature. 393:587–591. [DOI] [PubMed] [Google Scholar]
- Matulef, K., and W.N. Zagotta. 2003. Cyclic nucleotide-gated ion channels. Annu. Rev. Cell Dev. Biol. 19:23–44. [DOI] [PubMed] [Google Scholar]
- Matulef, K., G.E. Flynn, and W.N. Zagotta. 1999. Molecular rearrangements in the ligand-binding domain of cyclic nucleotide-gated channels. Neuron. 24:443–452. [DOI] [PubMed] [Google Scholar]
- Robinson, R.B., and S.A. Siegelbaum. 2003. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu. Rev. Physiol. 65:453–480. [DOI] [PubMed] [Google Scholar]
- Santoro, B., and G.R. Tibbs. 1999. The HCN gene family: molecular basis of the hyperpolarization-activated pacemaker channels. Ann. NY Acad. Sci. 868:741–764. [DOI] [PubMed] [Google Scholar]
- Sunderman, E.R., and W.N. Zagotta. 1999. Mechanism of allosteric modulation of rod cyclic nucleotide-gated channels. J. Gen. Physiol. 113:601–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum, M.D., K.D. Black, and W.N. Zagotta. 1995. Molecular mechanism for ligand discrimination of cyclic nucleotide-gated channels. Neuron. 15:619–625. [DOI] [PubMed] [Google Scholar]
- Vaughan, C.K., P. Harryson, A.M. Buckle, and A.R. Fersht. 2002. A structural double-mutant cycle: estimating the strength of a buried salt bridge in barnase. Acta Crystallogr. D Biol. Crystallogr. 58:591–600. [DOI] [PubMed] [Google Scholar]
- Wainger, B.J., M. DeGennaro, B. Santoro, S.A. Siegelbaum, and G.R. Tibbs. 2001. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 411:805–810. [DOI] [PubMed] [Google Scholar]
- Zagotta, W.N., T. Hoshi, and R.W. Aldrich. 1989. Gating of single Shaker potassium channels in Drosophila muscle and in Xenopus oocytes injected with Shaker mRNA. Proc. Natl. Acad. Sci. USA. 86:7243–7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta, W.N., N.B. Olivier, K.D. Black, E.C. Young, R. Olson, and E. Gouaux. 2003. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 425:200–205. [DOI] [PubMed] [Google Scholar]
- Zheng, J., M.C. Trudeau, and W.N. Zagotta. 2002. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron. 36:891–896. [DOI] [PubMed] [Google Scholar]
- Zufall, F., and S.D. Munger. 2001. From odor and pheromone transduction to the organization of the sense of smell. Trends Neurosci. 24:191–193. [DOI] [PubMed] [Google Scholar]