

Summary
FAK is known as an integrin- and growth factor-associated tyrosine kinase promoting cell motility. Here, we show that during mouse development, FAK inactivation results in p53- and p21-dependent mesodermal cell growth arrest. Reconstitution of primary FAK-/-p21-/-fibroblasts revealed that FAK, in a kinase-independent manner, facilitates p53 turnover via enhanced Mdm2-dependent p53 ubiquitination. p53 inactivation by FAK required FAK FERM F1 lobe binding to p53, FERM F2 lobe-mediated nuclear localization, and FERM F3 lobe for connections to Mdm2 and proteasomal degradation. Staurosporine or loss of cell adhesion enhanced FERM-dependent FAK nuclear accumulation. In primary human cells, FAK knockdown raised p53-p21 levels, slowed cell proliferation, but did not cause apoptosis. Notably, FAK knockdown plus cisplatin triggered p53-dependent cell apoptosis which was rescued by either full length FAK or FAK FERM re-expression. These studies define a scaffolding role for nuclear FAK in facilitating cell survival through enhanced p53 degradation under conditions of cellular stress.
Introduction
Integrins are transmembrane receptors that mediate cell attachment to ECM and also cooperate with growth factor receptors to generate intracellular signals regulating growth and survival. Disruption of these signals can trigger cell cycle arrest or apoptosis (Gilmore, 2005). One of the executioners of these events, is the p53 tumor suppressor protein (Reddig and Juliano, 2005). Under steady-state growth conditions, p53 expression is maintained at low levels via poly-ubiquitination and proteasomal degradation (Vousden, 2002). Murine double minute-2 (Mdm2) is one of several ubiquitin E3 ligases that regulate p53 levels in cells (Iwakuma and Lozano, 2003). Activation of p53 in response to stress signals is associated with increased p53 stability (Harris and Levine, 2005) leading to the enhanced transcription of targets such as p21Cip/WAF1 (p21) cyclin-dependent kinase inhibitor (Oren, 2003). ECM-integrin signals can counter-act p53 activation (Stromblad et al., 1996) and enhance proteasomal degradation of p21 (Bao et al., 2002), but regulatory connections between integrins and p53 are not clear.
Focal adhesion kinase (FAK) and the related Pyk2 kinase are cytoplasmic tyrosine kinases activated by integrins and growth factor receptors (Parsons, 2003). FAK inactivation during development yields an embryonic lethal phenotype and the growth of FAK-/- fibroblasts and endothelial cells is facilitated by p53 inactivation (Ilic et al., 1998; Ilic et al., 1995; Ilic et al., 2003). Although integrin-stimulated FAK auto-phosphorylation at Y397, Src-family PTK binding to FAK pY397, and the formation of a FAK-Src signaling complex promote cell motility and cell survival, (Cox et al., 2006; Mitra and Schlaepfer, 2006), the connections between FAK and p53 regulation remain loosely defined.
Recent studies have elucidated the structure of the FAK N-terminal band 4.1, ezrin, radixin, moesin homology (FERM) domain which is comprised of 3 distinct lobes (F1, F2, and F3) (Lietha et al., 2007) and acts to regulate FAK kinase activity through an intra-molecular inhibitory mechanism (Cohen and Guan, 2005; Lietha et al., 2007). Interestingly, FAK can be modified by small ubiquitin-related modifier (SUMO) addition at lysine-152 within the FERM domain (Kadare et al., 2003), SUMOylated FAK is nuclear-enriched, and exogenous FAK FERM expression is nuclear-localized (Golubovskaya et al., 2005; Stewart et al., 2002). Despite reports of FAK in the nucleus, the mechanism(s) that promote or regulate FAK nuclear accumulation and the biological role of nuclear FAK remain unknown.
Here, we show that loss of FAK activates p53 during mouse embryogenesis and that FAK-/- embryo cell proliferation ex vivo is made possible by either p53 or p21 inactivation. Although FAK has been proposed to inhibit p53 transcriptional activity (Golubovskaya et al., 2005), we find that the MG132 proteosomal inhibitor blocks FAK regulation of p53. We show that FAK inactivates p53 in a kinase-independent manner via the FAK FERM domain acting as a scaffold to enhance Mdm2-dependent p53 ubiquitination. Although FERM domain SUMOylation is not required, p53 regulation is dependent on FAK nuclear translocation. Importantly, in FAK knock-down human diploid fibroblasts, DNA damage-associated cell apoptosis could be rescued by wild type but not nuclear-excluded FAK FERM re-expression. These results support a biological role for nuclear FAK in promoting cell proliferation and survival by facilitating p53 turnover.
Results
Mesoderm proliferation defects in E8.5 FAK-/- mouse embryos
The fak knockout results in an early embryonic lethal (E.8.5) phenotype (Ilic et al., 1995) and size comparisons between normal FAK+/+, FAK+/- and FAK-/- littermates at E8.0-8.5 show that both FAK+/- and FAK-/- embryos are smaller than FAK+/+ embryos (Fig. 1A). By E11.5, FAK+/- embryos become indistinguishable from FAK+/+ embryos whereas FAK-/- embryos do not survive beyond E8.5-9.0 (Ilic et al., 1995). To evaluate relative levels of cell proliferation during development, head-fold regions of FAK+/+ and FAK-/- littermate embryos were immuno-stained with antibodies to phosphorylated serine-10 histone H3 (pHis3) (Fig. 1B). Equal numbers of pHis3-stained head-fold ectoderm cells were detected in FAK+/+ and FAK-/- embryos (Fig. 1C). In contrast, few pHis3-stained mesoderm cells were detected in FAK-/- compared to FAK+/+ embryos (Fig. 1B and 1C) supporting the notion that a proliferation defect induced by fak loss may occur in vivo.
Figure 1.
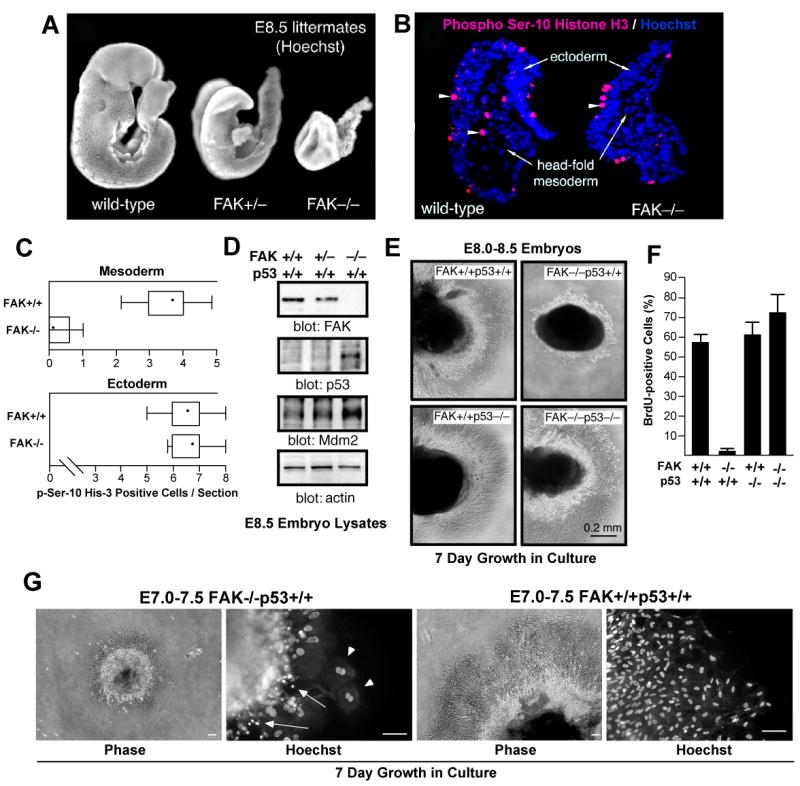
FAK-/- embryo mesoderm cell proliferation block is p53-dependent. (A) Hoechst DNA staining of wild-type (FAK+/+), FAK+/-, and FAK embryos at E8.5. (B) Mesoderm region of FAK-/- embryos lack mitotic cells. Saggital head-fold sections of E8.0-8.5 FAK+/+ and FAK-/- littermate embryos stained with phosphoserine-10 Histone H3 (red, arrowheads) and Hoechst (blue). (C) Quantitation of phosphoserine-10 Histone H3 staining. Number of mitotic cells (per headfold region section) in the ectoderm and mesoderm of FAK-/- and FAK+/+ littermates plotted as Box-and-whisker diagrams: dot (mean), box (25 to 75 percentile), and bars (minimum and maximum value). (D) Immunoblotting of embryo lysates shows increased p53 and Mdm2 protein expression from E.8.5 FAK-/- compared to FAK+/+ or FAK+/- littermates. (E) p53 prevents FAK-/- embryo proliferation ex vivo. The indicated E8.0-8.5 embryos were dissected and cultured in Matrigel for 7 days. Phase contrast images of Matrigel-embedded embryos (dark) and surrounding cells growing out from the embryo mass. (F) Rescue of FAK-/- cell proliferation defects by p53 deletion. Anti-BrdU staining (green) and Hoechst (blue) labeling of dissociated cells from embryo explants cells at 40X. Percentage of BrdU-positive cells for the indicated genotype. Data are mean +/- SEM from 3 independent experiments. (G) E7.0-7.5 time of embryo extraction does not alter FAK-/-p53+/+ proliferation defects ex vivo. Phase images of embryos in Matrigel culture for 7 days (scale bar is 100 μm). Hoechst staining shows cells with multiple (arrowheads) and fragmented (arrows) nuclei in FAK-/-p53+/+ but not FAK+/+p53+/+ cultures.
p53 accumulation prevents FAK-/- embryo proliferation ex vivo
To elucidate the potential role of p53 in FAK-/- embryo growth defects, we evaluated p53 protein levels in E8.5 embryo lysates (Fig. 1D). During development, p53 protein is rapidly turned over and not normally detected before E10.5-11.0 (Gottlieb et al., 1997). High levels of p53 and p53-responsive targets such as Mdm2 were detected in FAK-/- but not FAK+/+ embryos suggestive of p53 activation in the absence of FAK (Fig. 1D). To evaluate the contribution of p53 in FAK-/- embryo growth, a p53-/- mutation was introduced onto the FAK-/- background by mating FAK+/- and p53-/- mice. p53 inactivation did not rescue the E8.5 embryonic lethality of FAK-/- embryos (data not shown). However, loss of p53 enabled primary FAK-/- p53-/- embryo cell growth ex vivo whereas FAK-/-p53+/+ cells failed to proliferate (Fig. 1E and F). To quantify differences in cell growth, embryos were dissociated at day 6 ex vivo and cells cultured in the presence of bromodeoxyuridine (BrdU). Anti-BrdU staining revealed that FAK+/+p53+/+ and FAK+/+p53-/- cells showed a similar high rate of proliferation (50-60%) whereas <2% of cells in FAK-/-p53+/+ embryos were BrdU-positive (Fig. 1F). Importantly, p53 inactivation released this proliferation block resulting in ∼70% BrdU-positive cells from FAK-/-p53-/- embryos. This defect in FAK-/-p53+/+ embryo cell growth was not linked to the E8.0-8.5 time of extraction as cell proliferation defects and DNA aberrations were observed in cultured E7.0-7.5 FAK-/-p53+/+ but not FAK+/+p53+/+ embryos (Fig. 1G). Together, these results support the conclusion that p53 is active in FAK-/- embryos and that p53-associated signals block FAK-/- cell proliferation.
p53-associated proliferation block in FAK-/- cells is p21-dependent
To determine whether the absence of FAK results in changes in embryo protein expression, immunoblotting was performed on FAK+/+, FAK+/-, and FAK-/- E8.0-8.5 embryo lysates (Fig. 2A). The up-regulation of the FAK-related Pyk2 kinase was detected upon inactivation of one or both fak alleles. Comparisons of FAK+/+ and FAK-/- embryo lysates showed decreases in cyclin B and cyclin E, and increases in cyclin-dependent kinase inhibitors p57(Kip2), p27(Kip1), and p21(Cip1) in the absence of FAK (Fig. 2A). These changes may reflect reduced cell proliferation within FAK-/- embryos, however, other cell cycle regulatory proteins such as p16(Ink4a) remained unchanged.
Figure 2.
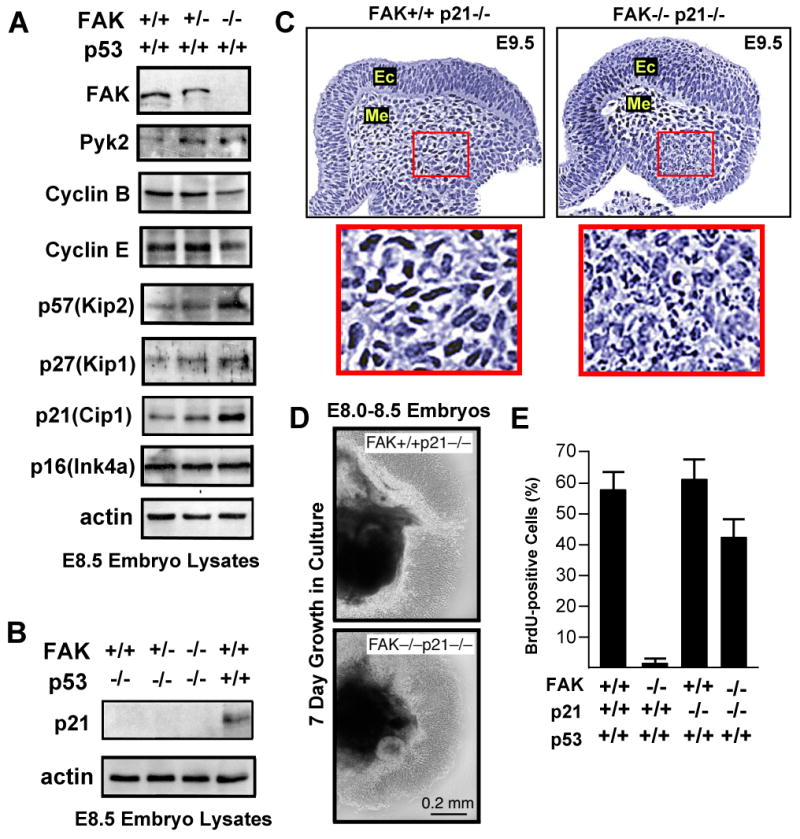
p53-mediated proliferation block of FAK-/- cells is p21-dependent. (A) Increased expression of p53, Pyk2, or cyclin-dependent kinase inhibitors and decreased expression of cyclins in FAK-/- compared to FAK+/+ littermates as determined by immunoblotting of E.8.5 embryo protein lysates. (B) Lack of detectable p21 expression in lysates from FAK-/- E8.5 embryos on a p53-/- background. (C) FAK-/-p21-/- embryos exhibit lethality at E9.5. H&E staining of saggital headfold sections. Ec=ectoderm and Me=mesoderm. Inset, fragmented nuclei observed in mesoderm region of FAK-/-p21-/- but not FAK+/+p21-/- embryos. (D) E8.0-8.5 FAK-/-p21-/- embryo cells proliferate equally to FAK+/+p21-/- cells in 7 day Matrigel culture ex vivo. Phase contrast images of embryo mass (dark) and surrounding cells. (E) p21 inactivation promotes FAK-/- embryo cell proliferation as determined by the percentage of BrdU-positive cells counted for the indicated genotype. Data are mean +/- SEM from 3 independent experiments.
In cell culture, p21 expression is enhanced by disruption of ECM-integrin linkages (Bao et al., 2002) and inhibited by FAK signaling (Bryant et al., 2006). During embryogenesis, elevated p21 levels in the absence of FAK were dependent upon p53, as p21 expression was not detected in FAK+/+, FAK+/-, or FAK-/- embryos on a p53-/- background (Fig. 2B). To test whether elevated p21 levels contribute to the p53-dependent block in FAK-/- embryo cell proliferation, a p21-/- mutation was introduced into a FAK-/-p53+/+ background by mating FAK+/- and p21-/- mice. FAK-/-p21-/- mice exhibited embryonic lethality at ∼E9.5 with increased mesoderm DNA fragmentation compared to FAK+/+p21-/- littermates (Fig. 2C). However, cells from E8.0-8.5 FAK-/-p21-/- embryos proliferated ex vivo at ∼75% the level of FAK+/+p21-/- cells (Fig. 2D and E). This release of the p53-dependent cell proliferation block was verified by BrdU incorporation into FAK-/-p21-/-p53+/+ cells (∼41%) but not FAK-/-p21+/+p53+/+ cells (<2%) (Fig. 2E). These studies support the notion that fak inactivation triggers a p53-p21-dependent cell proliferation block, yet release of this checkpoint is not sufficient to overcome an embryonic lethal FAK-null phenotype.
FAK promotes p53 turnover via FAK FERM domain-enhanced p53 ubiquitination
The inability of FAK-/-p53+/+ fibroblasts to grow in culture severely limits experimental analyses. To study the signaling linkage between FAK and p53, we generated FAK-/-p21-/- and FAK+/+p21-/- fibroblasts from E8.5 embryo littermates. In early passage FAK-/-p21-/-cells, steady state p53 levels were elevated and transient FAK re-expression resulted in decreased p53 levels (Supplemental Fig. S1A). This effect of FAK on p53 levels was associated with enhanced p53 turnover as determined by metabolic labeling of cells (Supplemental Fig. S1B-F).
To address which domains of FAK were required to promote p53 turnover, wild type (WT) FAK or various FAK mutants (Fig. 3A) were re-expressed via adenovirus (Ad) in low passage FAK-/-p21-/- fibroblasts and steady-state p53 levels were determined after 48 h by anti-p53 blotting (Fig. 3B). Surprisingly, WT FAK and various FAK signaling mutants functioned equally to reduce p53 levels by 60 to 90% compared to Mock-transduced cells (Fig. 3B). Over-expression of the FAK C-terminal domain (termed FAK-related non-kinase, FRNK) slightly elevated p53 levels whereas FAK residues 1-402 encompassing the FERM domain reduced p53 levels >80% (Fig. 3B). As N-terminal truncated and activated FAK (Δ1-100) did not affect p53 levels and kinase-dead FAK decreased p53 levels (Fig. 3B), our results support the notion that the FAK FERM domain plays a unique role in promoting p53 turnover in a kinase-independent manner.
Figure 3.

FAK FERM inhibits p53 activity through enhanced Mdm2-dependent ubiquitination and proteasomal degradation. (A) Epitope-tagged FAK construct schematic. Indicated is the band 4.1, ezrin, radixin, moesin (FERM) and central kinase domains. Filled regions indicate proline-rich motifs. Translation of FAK Δ1-100 starts at Met-101, Myc-tagged FERM encompasses residues 1-402, mutation of Y397 phosphorylation (F397), kinase-dead (KD, R454), Pro-null (Pro 712, 713, 872, 873, 876, and 877 mutated to Ala), and FRNK residues 691-1052 are indicated. (B) FAK FERM but not FAK kinase activity is required for the reduction of steady state p53 levels in FAK-/-p21-/- fibroblasts. Cells were transduced with Ad-Tet transactivator (TA, Mock), or Ad-TA plus the indicated Ad-FAK, Ad-FERM, or Ad-FRNK constructs. After 48 h, lysates were blotted with the indicated antibodies, p53 levels were quantified by densitometry, and mean values +/- SD are expressed as percentage of control (Mock) from 2 experiments. (C) Addition of MG132 (40 μM, 3h) prior to cell lysis prevents Ad-FAK and Ad-FERM-mediated p53 degradation in FAK-/-p21-/- fibroblasts. (D) FAK and FERM but not FRNK inhibit p53 activity as measured by a p21 promoter luciferase assay in FAK-/-p21-/- cells 48h after transfection. Values are means presented as percent of Mock control +/- SD from 3 experiments. (E) FAK and FERM but not FRNK promote enhanced p53 ubiquitination. HEK293 cells were co-transfected with flag-p53 and the indicated FAK constructs. MG132 was added 3h prior to lysis, p53 was isolated by IP, and analyzed by anti-ubiquitin and flag-tag blotting. (F) Mdm2 expression is required for FERM-enhanced p53 ubiquitination. Mdm2-/-p53-/- or Mdm2+/+p53-/- fibroblasts were transfected with flag-p53 and then transduced with Mock or Ad-FAK FERM. MG132 was added 3h prior to lysis, and p53 IPs were analyzed by anti-ubiquitin, anti-p53 (DO-1), and flag-tag blotting. Anti-Myc blotting was used to detect FAK FERM and anti-actin for loading control. Arrows indicate p53-shifted bands induced by FERM expression in Mdm2+/+ cells.
p53 levels in cells are regulated by a balance between protein expression and degradation. Addition of MG132 proteasome inhibitor for 3 h prior to cell lysis blocked FAK and FAK FERM effects on p53 (Fig. 3C) supporting the conclusion that FAK FERM facilitates p53 turnover. To determine whether changes in p53 expression affect p53 transcriptional activity, a 2.4 kb region of the p21 promoter coupled to luciferase was used as a p53 activity reporter in FAK-/-p21-/- cells (Fig. 3D). High p53 activity was detected in growing cells and this was inhibited >3-fold by WT FAK and FAK FERM but not FRNK expression. MG132 addition prevented FAK and FAK FERM inhibition of p21 promoter activity (data not shown) and accordingly, full-length FAK, FAK FERM but not FRNK increased p53 ubiquitination (Fig. 3E). To elucidate the ubiquitin E3 ligase for FAK FERM-mediated p53 modification, flag-p53 was transfected in Mdm2-/-p53-/- and Mdm2+/+p53-/- fibroblasts. Elevated levels of ubiquitinated p53 and p53 band shifts were detected upon Ad-FAK FERM over-expression only in Mdm2+/+ cells (Fig. 3F). These results show that FAK FERM promotes p53 turnover through enhanced Mdm2-associated p53 ubiquitination and degradation.
FAK FERM F2 lobe facilitates FAK nuclear localization
As activated p53 is primarily nuclear-localized and exogenous FAK FERM is nuclear-enriched (Golubovskaya et al., 2005; Stewart et al., 2002), analyses were undertaken to determine the determinants of FAK FERM nuclear targeting. Fusions of FAK residues (1-402) to green fluorescent protein (GFP) exhibit strong nuclear localization independent of Y397 phosphorylation (Supplemental Fig. S2A). The classic nuclear localization sequence typically contains clusters of basic amino acids. The avian FAK FERM domain is a three lobed (F1-F3) structure (Lietha et al., 2007). Primary sequence alignments of the murine FAK FERM F2 lobe region showed that FAK and Pyk2 contained patches of basic amino acids that are not conserved in other FERM-containing proteins such as ezrin, radixin, moesin, or merlin (Fig. 4A). Although these FAK F2 FERM domain basic residues are separated by primary sequence, they comprise surface-exposed clusters (Fig. 4B). The largest patch consists of residues lysine (K) 190, K191, K216, K218, arginine (R) R221, and K222 whereas a second smaller basic patch is comprised of R204, R205, and K209. Residues R177 and R178 are partially exposed within the FAK FERM F2 lobe structure (Fig. 4B).
Figure 4.
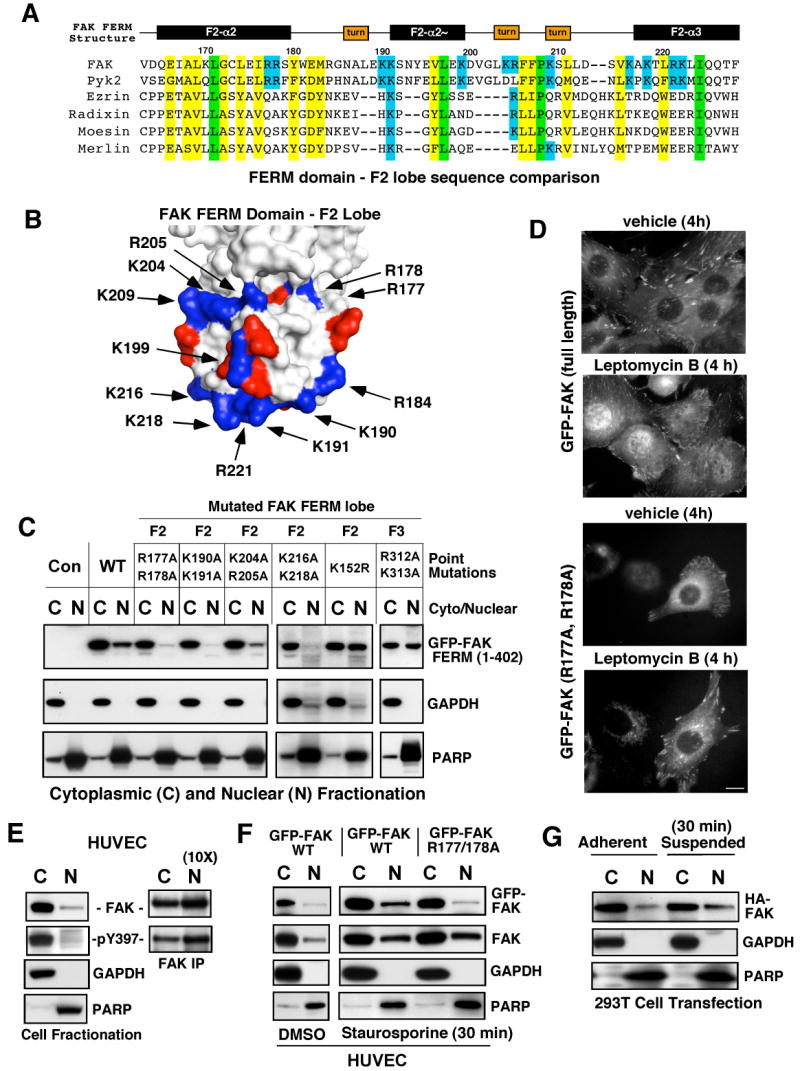
Determinants of FAK nuclear localization. (A) Structure-based alignment of FAK FERM F2 lobe residues (Lietha et al., 2007) with other FERM-containing proteins. Conserved basic residues within FAK and Pyk2 are highlighted in blue, total conserved FERM residues are highlighted in yellow and identical residues in green. (B) Localization of basic residue clusters on the surface of the FAK FERM F2 lobe. The FAK FERM domain (Lietha et al., 2007) F2 lobe was visualized using MacPyMOL. Basic residues (blue) are numbered according to the primary FAK sequence and acidic residues (red) are indicated. A putative nuclear targeting motif is comprised of residues at the tip of the F2 lobe (K190, K191, K216, K218, and R221). (C) FAK FERM domain analyzed by cellular fractionation. The indicated residues within GFP-FAK FERM (1-402) were mutated and constructs expressed in 293T cells. Cell lysates were separated into cytosolic (C) and nuclear (N) fractions, resolved by SDS-PAGE, and anti-GFP blotting was used to detect FAK FERM. Antibodies to gylceraldehyde-3-phosphate dehydrogenase (GAPDH) and poly ADP-ribose polymerase (PARP) were used to verify fractionation specificity, respectively. (D) Live cell imaging was used to follow GFP-FAK WT and GFP-FAK R177/178A distribution upon leptomycin B (10 ng/ml) or ethanol (vehicle) addition for 4 h. Scale bar is 10 μm. (E) FAK is partially nuclear-localized. HUVECs were separated into cytosolic and nuclear fractions, and blotted for FAK, GAPDH, and PARP. Ten-fold excess nuclear lysates was used to analyze FAK tyrosine phosphorylation by IP. (F) WT but not R177/178A FAK nuclear accumulation by HUVEC fractionation. HUVECs were treated with 1 μM staurosporine for 30 min, lysates separated into cytosolic or nuclear fractions, and immunoblotted with antibodies to GFP, FAK, GAPDH, and PARP. (G) 293T cells were transfected with HA-FAK and then fractionated into cytosolic and nuclear fractions under adherent and suspended conditions. Lysates blotted with anti-HA, GAPDH, and PARP.
Alanine substitutions were introduced within the FAK FERM F2 lobe and the effects on FAK FERM nuclear localization were monitored by fluorescence microscopy (data not shown) and cellular fractionation into nuclear (N) or cytosolic (C) extracts (Fig. 4C). Mutations in the largest basic patch (K190/191A or K216/218A) blocked whereas mutations in a second smaller patch (K204/R205A) slightly reduced FERM nuclear accumulation. R177/R178A mutations also prevented FERM nuclear localization whereas mutation of surface exposed basic residues in the FAK FERM F3 lobe (R312/K313A) or the SUMOylation site (K152R) did not affect FERM nuclear targeting (Fig. 4C). To prove the role of the FERM domain promoting FAK nuclear localization, GFP-FAK WT and GFP-FAK R177/R178A were expressed in FAK-/- fibroblasts or human umbilical endothelial cells (HUVECs) (data not shown) and GFP localization monitored by time-lapse imaging in the presence or absence of the nuclear export inhibitor leptomycin B (Fig. 4D). In vehicle-treated cells, both FAK constructs were detected in focal contacts and the cytoplasm and leptomycin B addition promoted WT but not FAK R177/R178A nuclear accumulation within 4 h.
To analyze whether endogenous FAK is present in the nucleus, nuclear (N) or cytosolic (C) HUVEC extracts were analyzed for FAK distribution (Fig. 4E). Approximately 5-10% of HUVEC FAK is nuclear-associated and tyrosine phosphorylated. To determine whether cellular stress may enhance FAK nuclear accumulation, HUVECs were treated with the protein kinase inhibitor staurosporine (Fig. 4F). Within 30 min, staurosporine enhanced endogenous FAK, GFP-FAK, but not FERM F2 lobe mutated GFP-FAK R177/178A nuclear accumulation. This was accompanied by FAK dephosphorylation, loss of FAK from focal contacts, and preceded staurosporine-induced HUVEC apoptosis occurring within 4-8 h (Supplemental Fig. S2B-F). Staurosporine caused a rapid redistribution of GFP-FAK R177/178A from focal contacts to punctate cytoplasmic clusters but not to the nucleus (Supplemental Fig. S2E). Loss of integrin-mediated cell adhesion is another cellular stressor, and this was also found to increase FAK nuclear accumulation within 30 min (Fig. 4G). Together, these results support the importance of the FAK FERM F2 lobe in nuclear targeting. Moreover, the distribution of FAK at focal contacts, in the cytoplasm, or the nucleus is affected by cellular stress.
FAK nuclear localization is important for p53 inhibition
Primary FAK-/-p21-/- fibroblasts are a unique model system to determine the role of FAK in p53 regulation. Increased expression of the FAK-related Pyk2 kinase was detected FAK-/-p21-/- fibroblasts and lentiviral-mediated Pyk2 shRNA was used to maintain low levels of Pyk2 for FAK re-expression studies (data not shown). To address whether FAK FERM nuclear localization is important for p53 regulation, stable pooled populations of GFP-FAK FERM (1-402) or GFP-FRNK-expressing FAK-/-p21-/- fibroblasts were established. WT, K152R SUMOylation mutant, and R312/K313A FAK FERM F3 lobe mutated proteins were nuclear-localized, F2 lobe FERM mutants (R177/R178A and K190/K191A) were cytoplasmic-distributed, and GFP-FRNK localized to focal contacts (Fig. 5A and Supplemental Table 2). Steady state p53 levels in FAK FERM-expressing cells were evaluated by immunoblotting and compared to parental FAK-/-p21-/- and GFP-expressing controls. Cells expressing WT FERM and the SUMOylation (K152R) FERM mutant exhibited reduced p53 levels (Fig. 5B). Importantly, mutations in the FERM F2 lobe (K190/K191A and R177/R178A) blocked FAK FERM effects on lowering p53 levels (Fig. 5B). However, mutations in the FAK FERM F3 lobe (R312/K313A) also blocked the ability of FAK FERM to reduce p53 levels (Fig. 5B) even though R312/K313A FAK FERM was nuclear-localized (Fig. 5A). As p53 transcriptional activity paralleled p53 expression levels in FAK FERM reconstituted FAK-/-p21-/- fibroblasts (data not shown), the combined results support the notion that FAK FERM nuclear localization is required but not sufficient for p53 regulation.
Figure 5.
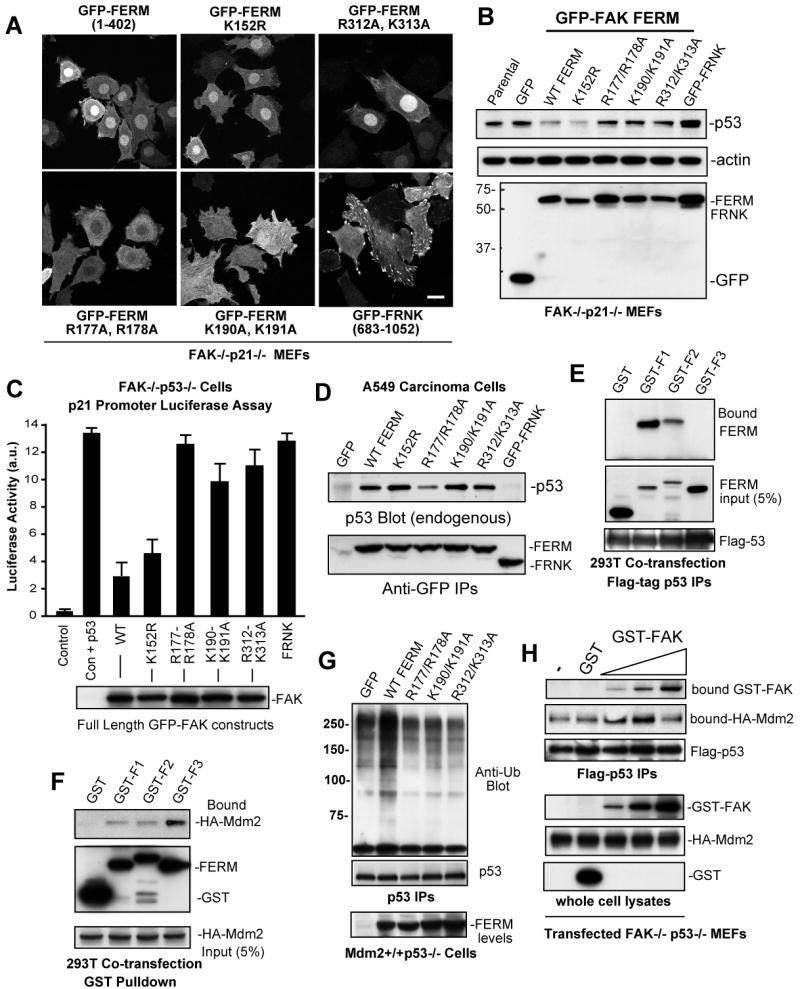
Separate FAK FERM lobes mediate p53 binding, nuclear localization, and Mdm2 association. (A) The indicated GFP-FAK FERM (1-402) constructs or GFP-FRNK were stably-expressed in FAK-/-p21-/- (Pyk2 shRNA) fibroblasts and intracellular distribution visualized by confocal microscopy. Scale bar is 20 μm. (B) Steady-state p53 expression is reduced by FAK FERM expression in FAK-/-p21-/- (Pyk2 shRNA) fibroblasts as detected by p53, actin, and GFP bloting of lysates. (C) FERM domain mutations disrupt full length FAK inhibition of p53 transcriptional activity. FAK-/-p53-/- fibroblasts were transiently-transfected with a 2.4 kb p21 promoter luciferase construct (Vector, V) or in combination with p53 (Vec+p53) and the indicated FAK constructs. Luciferase activity is arbitrary units (a.u.). Values are means of 2 experiments +/- SD. Blotting verified equal FAK construct expression (below). (D) F2 FERM mutations can weaken p53 association. Ad-FAK FERM or FRNK constructs were expressed in A549 cells and association with endogenous p53 analyzed by IP and blotting. (E) FAK FERM F1 lobe binds p53. 293T cells were co-transfected with flag-p53 and the indicated FAK F1, F2, or F3 FERM lobes as GST fusion proteins. Anti-GST blotting of Flag IPs was used to detect FERM lobe association with p53. (F) FAK FERM F3 lobe binds Mdm2. 293T cells were co-transfected with HA-Mdm2 and the indicated FAK F1, F2, or F3 FERM lobes as GST fusion proteins. Cells were treated with MG132 prior to cell lysis (40 μM, 3h), incubated with glutathione agarose, and anti-HA blotting detected bound Mdm2. (G) FAK FERM mutations disrupt FERM-enhanced p53 ubiquitination. Mdm2+/+p53-/- fibroblasts were transfected with flag-p53, transduced with the indicated Ad-FAK FERM constructs, and MG132 was added 3h prior to lysis. p53 IPs were analyzed by anti-ubiquitin, flag-tag, and GFP blotting. (H) Biphasic FAK effects on Mdm2-p53 complex formation. HA-Mdm2 and flag-p53 were transfected into FAK-/-p53-/- fibroblasts, MG132 was added 3h prior to lysis, and GST (500 ng) or increasing amounts of recombinant GST-FAK (10 ng-500 ng) were added prior to p53 isolation by IP. Bound Mdm2 and FAK within a p53 complex were detected by anti-HA and GST blotting.
To determine whether full-length FAK modulates p53 activity in a similar manner as the FAK FERM, co-transfection assays were performed in FAK-/-p53-/- fibroblasts (Fig. 5C). The p21 promoter-luciferase reporter was responsive to p53 re-expression resulting in ∼14-fold increased activity. Co-expression of WT or SUMOylation mutant K152R FAK resulted in >65% inhibition of p53 activity whereas equal expression of F2 lobe mutated R177/R178A FAK lowered p53 activity only 5-10% (Fig. 5C). F2 lobe mutated K190/K191A or F3 lobe mutated R312/K313A FAK only weakly inhibited p53 activity. Taken together, these results support the conclusion that FAK FERM F2 lobe-mediated nuclear translocation is important for p53 inhibition. However, the fact that FERM F3 lobe mutations (R312/K313A) did not affect nuclear translocation, yet disrupted the ability of FAK to inhibit p53, indicates that FAK-mediated regulation of p53 involves multiple FERM-associated mechanisms.
FAK FERM acts as a scaffold to facilitate p53 and Mdm2 association
Co-immunoprecipitation (co-IP) analyses have shown that FAK and p53 can form a complex (Golubovskaya et al., 2005), but the binding determinants for p53 within the FAK FERM domain remain unknown. GFP-FAK FERM or GFP-FRNK constructs were expressed in human A549 cells and evaluated for endogenous p53 association by co-IP analyses (Fig. 5D). p53 associated with FAK FERM but not FRNK and p53 binding to FAK FERM was weakened by mutations in the F2 (R177/R178A) but not F3 (R312/K313A) lobes (Fig. 5D). Similar results were obtained by GST-p53 pull down assays (Supplemental Fig. S3A). Interestingly, F2 lobe mutated K190/K191A FAK FERM can form a complex with p53 (Fig. 5D), but these FERM mutations disrupt nuclear localization (Fig. 5A) and p53 regulation (Fig. 5B and C). Additionally, F3 lobe mutated R312/K313A FAK FERM localizes to the nucleus (Fig. 5A), binds to p53 (Fig. 5D), but does not promote p53 turnover (Fig. 5B). Together, these results support the notion that FERM-mediated nuclear localization, p53 binding, and FERM-enhanced p53 turnover are separable events.
To determine whether individual FAK FERM lobes can associate with p53 or Mdm2 in cells, the F1, F2, or F3 regions were transiently-expressed as GST fusion proteins in 293T cells (Fig. 5E and F). The FAK FERM F1 lobe strongly associated with p53 in vivo whereas weak binding was detected with FAK FERM F2 lobe, and no p53 binding was detected with the FAK FERM F3 lobe (Fig. 5E). This result supports the notion that the FAK FERM p53 binding site likely spans the F1 and F2 FERM lobe regions. For Mdm2, FAK FERM F3 lobe bound the strongest with only weak interactions detected with F1 or F2 FAK FERM lobes (Fig. 5F). Interestingly, F3 lobe mutated R311/K312A FERM bound Mdm2 and surprisingly, F2 lobe mutated R177/R178A FERM exhibited enhanced Mdm2 binding (Supplemental Fig. S3B). Co-transfection studies revealed that both F2 and F3 lobe FERM mutations disrupted FAK FERM-enhanced p53 ubiquitination (Fig. 5G) and support the notion that FAK-enhanced p53 ubiquitination may be part of a nuclear-localized complex. Accordingly, although F2 lobe mutated K190/K191 FERM can bind p53, this mutant is cytoplasmic-distributed and fails to promote p53 ubiquitination. For F3 lobe mutated R312/K313A FERM, it is likely that this mutation disrupts the binding of another protein other than Mdm2 that is needed to facilitate p53 ubiquitination.
To support the notion that FAK may act as a scaffold to enhance the formation of a p53-Mdm2 complex, FAK-/-p53-/- fibroblasts were transfected with flag-p53, HA-Mdm2 and increasing amounts of recombinant GST or GST-FAK added to cell lysates. A p53-Mdm2 association was detected in the absence of FAK and this was not affected by purified GST addition (Fig. 5H). GST-FAK addition increased the amount of p53-bound Mdm2 through the formation of a p53-Mdm2-FAK complex in a dose-dependent manner. However, excess GST-FAK did not enhance p53-Mdm2 complex formation even though increased GST-FAK bound to p53 (Fig. 5H). This result is consistent with a biphasic scaffolding effect of FAK in facilitating p53 and Mdm2 interactions. Together with the FERM mutational results, these studies support the conclusion that nuclear-localized FAK functions as a FERM-associated scaffold for p53 and Mdm2 binding that facilitates p53 ubiquitination leading to p53 turnover.
FAK regulation of cell proliferation and p53-dependent apoptosis in human fibroblasts
As our studies support the importance of FAK in regulating p53 activation during development and in mouse fibroblasts, lentiviral-mediated FAK shRNA knockdown was used to test whether a FAK-p53 signaling axis exists in human fibroblasts and HUVECs. Within 72 h, >80% of cells showed strong lentiviral GFP expression and neither anti-FAK or scrambled (Scr) shRNA expression promoted cell apoptosis (Supplemental Fig. S4). However, >75% reduction in FAK expression was associated with ∼2-fold increased p53-p21 protein levels and reduced cell proliferation (Fig. 6A, 6B, and Supplemental Fig. S5). To determine whether decreased FAK and elevated p53 levels would sensitize cells to DNA damaging agents, Scr and FAK shRNA-expressing fibroblasts were treated with cisplatin. FAK shRNA cells showed ∼4-fold increased apoptosis compared to cisplatin-treated Scr shRNA cells (Fig. 6C and D) and the combination FAK shRNA plus cisplatin lead to elevated p53 protein levels compared to cisplatin-treated Scr shRNA fibroblasts (Fig. 6E).
Figure 6.
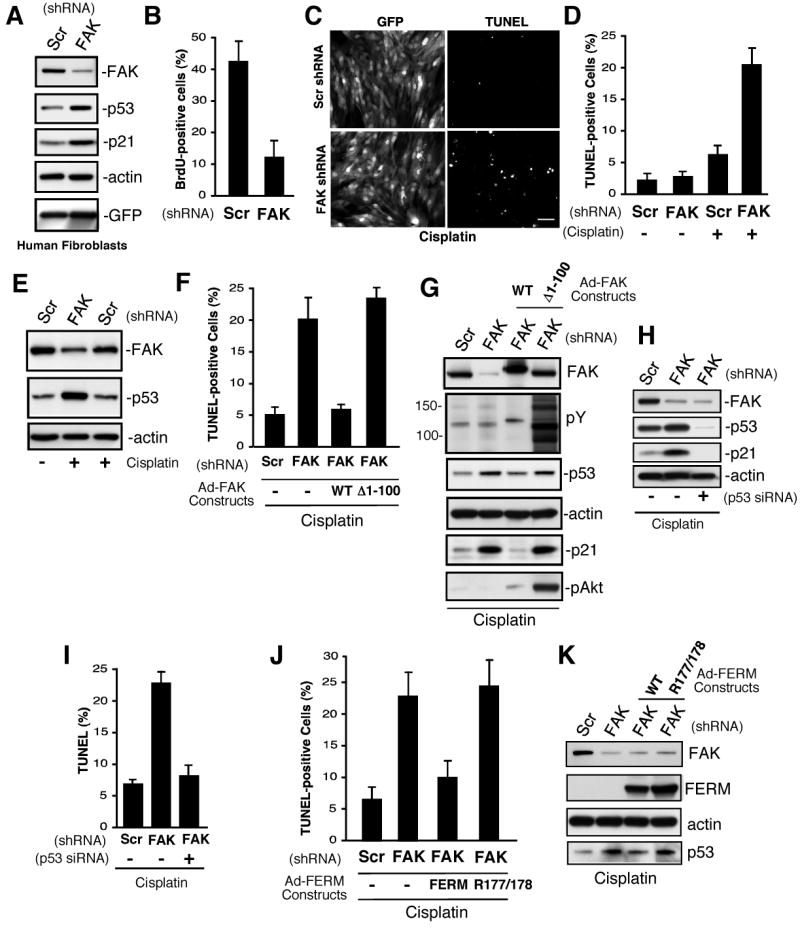
FAK controls human diploid fibroblast proliferation and p53-dependent apoptosis. (A) Lysates from scrambled (Scr) and FAK shRNA infected cells after 72 h were analyzed by anti-FAK, p53, p21, actin, and GFP blotting. (B) FAK shRNA inhibits human fibroblast proliferation. Cells were infected with the indicated lentivirus for 72 h, BrdU was added for 16h in growth media, and cells stained with anti-BrdU antibody. Mean values +/-SD are percent of total GFP-positive cells. (C, D) FAK shRNA sensitizes human fibroblasts to cisplatin-stimulated apoptosis. Cells were infected with the indicated lentivirus for 48 h, cisplatin (20 μg/ml) was added for 48 h, cells were fixed, and then analyzed by TUNEL staining. (C) Representative images of GFP-expressing and TUNEL-stained fibroblasts. Scale bar is 200 μm. (D) Mean values +/- SD for cisplatin-stimulated apoptosis were obtained by counting three TUNEL-stained 10X fields of cells from two coverslips. Only GFP-positive cells were counted and the data represents two independent experiments. (E) Elevated p53 levels in cisplatin-treated FAK shRNA-expressing fibroblasts as treated as in panel D and analyzed by anti-FAK, p53, and actin blotting. (F, G) FERM domain integrity is required for rescue of cisplatin-stimulated apoptosis. Cells were infected with Scr or FAK shRNA lentivirus (48 h), transducted with Ad-FAK or Ad-FAK (Δ1-100), and after 24h, cisplatin (20 μg/ml, 48 h) was added prior to analysis by TUNEL staining. (F) Mean values +/- SD for cisplatin-stimulated apoptosis were obtained as described for panel D. (G) FAK but not Δ1-100 FAK reverses cisplatin-stimulated increases in p53 and p21 expression as determined by blotting. Δ1-100 FAK activates Akt as determined by phospho-specific blotting. (H, I) FAK shRNA-enhanced cisplatin-stimulated apoptosis is p53 dependent. Fibroblasts were transfected with p53 siRNA, transduced with Scr or FAK shRNA lentivirus, and treated with cisplatin (20 μg/ml, 48 h). (H) Blotting for FAK, p53, and p21 blotting show changes in protein expression with actin blotting as control. (I) Mean values +/- SD for cisplatin-stimulated apoptosis were obtained as for panel d. (J, K) FAK FERM domain rescue of cisplatin-stimulated apoptosis. Cells were transduced with Scr or FAK shRNA lentivirus (48 h), infected with Ad-Myc-FERM WT or Ad-Myc-FERM R177/R178 (24 h), and then treated with cisplatin (20 μg/ml, 48h). (J) Cells were analyzed fro TUNEL staining as in panel D. (K) Blotted for FAK, Myc tag (FERM), actin, and p53 shows that FERM mutation blocks p53 regulation.
To verify that increased cisplatin-stimulated apoptosis was due to decreased FAK, human fibroblasts were treated with FAK shRNA and after 48 h, cells were transduced with murine Ad-FAK WT or Ad-FAK Δ1-100 containing a deletion in the FAK FERM domain (Fig. 6F and G). This truncation of FAK activates FAK kinase activity and facilitates FAK signaling (Mitra and Schlaepfer, 2006), but FAK Δ1-100 does not function to regulate p53 levels in mouse fibroblasts (Fig. 3B). WT FAK re-expression blocked cisplatin-stimulated apoptosis in FAK shRNA cells whereas Δ1-100 FAK had no effect (Fig. 6F). Importantly, Δ1-100 FAK exhibited high activity levels as determined by anti-phosphotyrosine blotting of fibroblast lysates and led to the robust activation of survival-associated signaling pathways such as Akt (Fig. 6G). However, only WT FAK reduced p53 levels and prevented p21 expression in cisplatin-treated cells (Fig. 6G). Importantly, cisplatin-stimulated apoptosis in FAK shRNA-expressing fibroblasts was dependent upon p53 as siRNA-mediated p53 reduction blocked both increased p21 expression and cell death (Fig. 6H and I).
To prove the importance of the FAK FERM domain function in preventing cisplatin-stimulated apoptosis, cell survival analyses were performed after Ad-FERM WT or Ad-FERM R177/R178A expression in FAK shRNA knockdown human fibroblasts (Fig. 6J and K). FAK FERM but not R177/178A FERM blocked p53 accumulation and only WT FAK FERM functioned to prevent apoptosis. Theses results are strong support for the biological importance of the FAK FERM domain in the regulation of p53-dependent cell survival.
Discussion
Null mutation of FAK results in an early embryonic lethal phenotype (Ilic et al., 1995). Here, we show that fak inactivation is associated with the cessation of mesodermal cell proliferation and p53 activation during development. Interestingly, FAK-null embryo ectoderm did not exhibit growth defects in vivo and this may be due to different regulatory signals in epithelial versus mesodermal cells. However, as FAK-null primary keratinocytes can proliferate in culture (Schober et al., 2007), cells of ectodermal origin may have different or compensatory survival mechanisms compared to mesodermal fibroblasts. Interestingly, genetic inactivation of p53 or p21 in FAK-null embryos did not rescue the embryonic lethal phenotype that remains associated with altered morphogenesis (Ilic et al., 1995). However, loss of p53 or p21 enabled FAK-/- embryo cell proliferation ex vivo. Thus, p53 activation induces cell cycle arrest through p21 when FAK expression is impaired.
Unlike FAK, loss of other integrin-associated signaling proteins such as Src, p130Cas, paxillin, talin, or vinculin do not result in p53 activation or the blockage of primary fibroblast proliferation. Thus, FAK regulation of p53 during development is unlikely to involve canonical integrin signaling connections (Fig. 7). Moreover, FAK connections to p53 are conserved in primary human cells as FAK shRNA knockdown promotes increased p53/p21 levels accompanied by decreased cell proliferation. Interestingly, conditional inactivation of FAK in endothelial (Braren et al., 2006; Shen et al., 2005) or Schwann cells (Grove et al., 2007) is linked to apoptosis or decreased cell proliferation, respectively. This may be associated with p53 activation as primary FAK-/- endothelial cell proliferation was facilitated by p53 inactivation (Ilic et al., 2003).
Figure 7.
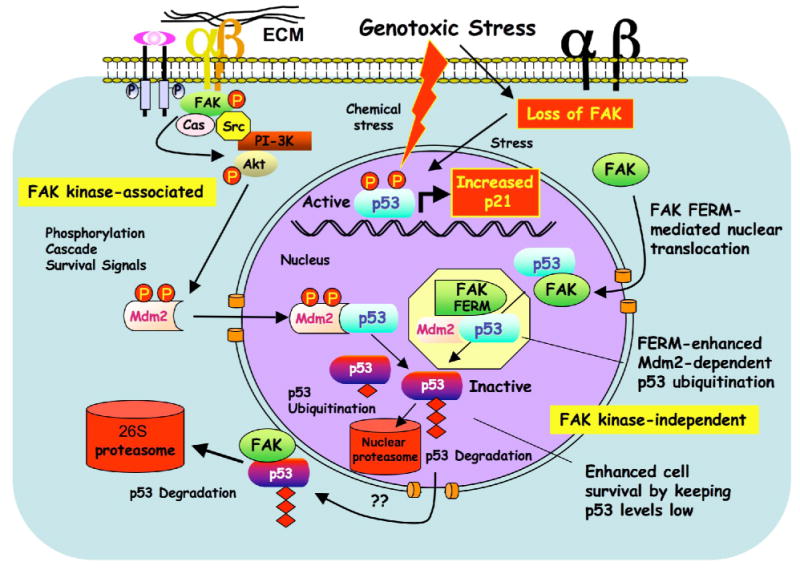
Model of FAK FERM-mediated p53 turnover and cell survival. FAK can function with integrins and growth factor receptors to promote cell survival through signaling cascades such as Akt that can activate ubiquitin E3-ligases such as Mdm2 to maintain low p53 levels. This canonical survival pathway involves FAK kinase activity (left). Under reduced integrin adhesion or conditions of cellular stress, FAK leaves focal contacts sites. This increases the cytoplasmic pool of FAK and enhances FAK nuclear accumulation via FAK-FERM-mediated targeting. Nuclear FAK acts as a scaffold to stabilize a p53-Mdm2 complex, leading to p53 polyubiquitination, and subsequent p53 degradation by nuclear or cytoplasmic proteasomes. This regulatory connection between FAK and p53 is dependent on the FAK FERM domain, but does not require FAK kinase activity (right).
FAK FERM survival pathway
Strikingly, we found that FAK catalytic activity was not required for FAK-mediated regulation of p53. Although FAK kinase-associated signals have been shown to repress p21 expression (Bryant et al., 2006; Ding et al., 2005), we found that kinase-inactive FAK functioned equally to WT FAK in repressing p53 expression using FAK-/-p21-/- MEFs. Moreover, under stress induced by FAK knockdown combined with cisplatin addition to trigger p53 activation, re-expression of WT but not truncated (Δ1-100) FAK repressed p53/p21 expression and prevented p53-mediated apoptosis in human fibroblasts. Notably, Δ1-100 FAK was highly active, facilitated signals leading to Akt phosphorylation, but did not function to promote survival. The inability of Δ1-100 FAK to counter-act p53 activation is associated with the disruption of FERM domain function as exogenous FAK FERM re-expression was sufficient to prevent cisplatin-stimulated apoptosis under FAK knockdown conditions.
To date, many studies have focused on the role of FAK kinase-associated cell survival pathways indirectly through dominant-negative type experiments (Reddig and Juliano, 2005). However, as a small molecule FAK catalytic inhibitor did not effect either normal or carcinoma cell proliferation or survival in culture (Slack-Davis et al., 2007), these over-expression studies may need to be re-evaluated. As FAK functions as both a scaffold and signaling kinase, it is also difficult to distinguish the survival and proliferation alterations caused by the loss of FAK expression. Using a strategy of FAK re-expression within FAK-/- or FAK knockdown fibroblasts, our studies have identified the FAK FERM domain as the key module of a kinase-independent FAK-p53 regulatory cell survival circuit. Previously, FAK FERM domain over-expression has been shown to suppress apoptotic stimuli via interactions with the death domain kinase receptor-interacting protein (Kurenova et al., 2004) and indirectly implicated in the transcriptional inhibition of p53 via binding to the p53 transactivation domain (Golubovskaya et al., 2005). Our studies support a different mode of p53 inhibition by FAK as proteasomal inhibitors prevent FAK inhibition of p53. We show that FAK inhibits p53 via FAK FERM nuclear translocation, FERM-mediated binding to p53, and FERM-enhanced Mdm2-dependent p53 ubiquitination (Fig. 7).
Nuclear FAK, p53 binding, and FERM-enhanced p53 degradation
The FAK FERM domain is comprised of three lobes (F1-F3) with similarity to other known FERM structures despite low primary sequence conservation (Lietha et al., 2007). Through mutagenesis, biochemical cell fractionation, and confocal microscopy, we identified a patch of basic residues as a nuclear localization motif within the FAK FERM F2 lobe (K190, K191, K216, K218, and R221). These basic residues are separated by primary sequence, conserved between FAK and Pyk2, and not present in other FERM domain containing proteins. As other F2 lobe (R177/R178A) but not F3 lobe (R312/K313A) FAK FERM mutations also disrupt FAK nuclear localization, correct F2 lobe FERM folding is also likely important for FAK nuclear targeting.
Co-immunoprecipitation analyses using the individual FAK FERM lobes revealed that the F1 lobe bound strongest to p53 whereas the F3 lobe bound to Mdm2 independent of p53 association. Overall, p53 inactivation by FAK required FAK FERM F1 lobe binding to p53, FERM F2 lobe-mediated nuclear localization, and FERM F3 lobe for connections to Mdm2 and proteasomal degradation. Importantly, mutations in the FERM F2 lobe (K190/K191A and R177/R178A) and within the FAK FERM F3 lobe (R312/K313A) blocked the ability of FAK FERM to reduce p53 levels. The mechanism(s) associated with these mutational effects are likely distinct. F2 lobe R177/R178A and K190/K191A FERM mutants can weakly bind p53, but they are cytoplasmically-distributed and do not promote p53 ubiquitination. Even though F3 lobe mutated R312/K313A FAK FERM was nuclear-localized and can bind Mdm2, this mutation also disrupts the ability of FAK FERM to facilitate p53 ubiquitination. As FAK forms a multi-protein scaffolding complex with p53 and Mdm2 in a biphasic manner, we speculate that the presence of FAK in the nucleus acts to stabilize a p53-Mdm2 complex, leading to p53 polyubiquitination, and subsequent p53 degradation by nuclear or cytoplasmic proteasomes.
What conditions may promote FAK translocation to the nucleus? The nuclear export inhibitor leptomycin B promoted FAK nuclear accumulation in hours whereas staurosporine treatment or loss of cell adhesion enhanced FAK nuclear distribution within 30 min. Nuclear FAK accumulation was also associated with a loss of FAK at focal contacts and this supports the notion that there is a cytoplasmic pool of FAK that is free to shuttle in and out of the nucleus. We propose a model whereby under conditions of cellular stress or reduced integrin signaling, the cytoplasmic pool of FAK is elevated, leading to increased FAK nuclear accumulation, which acts to enhance cell survival by facilitating p53 turnover. At present, the signals that regulate FAK cytoplasmic to nuclear movement remain undetermined. Although SUMOylated FAK was nuclear-enriched (Kadare et al., 2003), we found that K152 FAK SUMOylation was not essential for nuclear translocation. Interestingly, some transformed cell types can survive under suspension conditions and this associated with reduced p53 activation (Lewis et al., 2002). As FAK levels are elevated in many tumor cells (McLean et al., 2005), and we find that FAK can form a complex with p53 in both normal and tumor cells, future studies will be aimed at determining whether this FAK-p53 cell survival pathway may also play a role in promoting tumor progression.
Experimental Procedures
Mice
p53-/-, p21-/-, and heterozygous FAK mice were housed and bred according to AALAC-approved institutional guidelines. Genomic DNA from placental cone and yolk sac were used for genotyping by PCR as described (Deng et al., 1995; Furuta et al., 1995; Tsukada et al., 1993). Littermates from crossings of FAK+/- mice were dissected at E7.5 or E8.5, fixed briefly in 3.8% paraformaldehyde for 20 min and embedded in OCT. Sections (5 μm) were stained with anti-phosphoserine-10 Histone3 (Upstate Biotech) and Hoechst 33342 (10 μg/ml, Molecular Probes) to identify proliferating cells.
Embryo explant culture
E7.5-8.5-old embryos were dissected from yolk sac and fetal membranes, placed in a drop of Matrigel (BD Biosciences), and cultured for 7 days in 10% FBS DMEM supplemented with 10-4 M β-mercaptoethanol. To determine cell proliferation ex vivo, 10 μM BrdU was added to Day 6 embryo explants. After 24 h, embryos were disaggregated using trypsin and cells were plated on 10 μg/ml fibronectin-coated wells (Roche) in complete medium containing 10 μM BrdU for an additional 24 h. Cells were fixed, stained with FITC-labeled anti-BrdU antibody (Roche), and Hoechst 33342 (10 μg/ml) was added as a DNA counterstain. Data represents a minimum of five litters from mating of FAK-heterozygous mice on various genetic backgrounds.
Cells
FAK-/-p53-/- and mouse embryonic fibroblasts (MEFs) were generated as described (Ilic et al., 1995). FAK-/-p21-/- and FAK+/+p21-/- MEFs were from E8.0 embryos and maintained on dishes pre-coated with 0.1% gelatin up to 15 passages. Mdm2-/-p53-/- MEFs were provided by S. Jones (UMass Medical School). Human diploid foreskin BJ fibroblast and A549 lung carcinoma cells were obtained from ATCC. BJ fibroblast passage was limited to 50 doublings. Pyk2 shRNA-expressing FAK-/-p21-/- cells were generated by lentiviral infection and sorting for GFP. Lentiviral-associated GFP expression was removed by Ad-Cre transduction and verified by flow cytometry. Primary HUVECs were from Glycotech (Gaithersburg, MD).
Immunoblotting, antibodies, and chemicals
E8.5 embryos were dissected from yolk sac and fetal membranes and then snap frozen. 5-10 embryos from 3 or more pregnant females were pooled together for each analysis. Embryos or cell lysates were made using modified RIPA buffer as described (Mitra et al., 2006). Anti-FAK (4.47) and phosphotyrosine (4G10), Cyclin B1 (clone V152), and Cyclin E (07-687) were from Millipore. Mouse anti-HA (16B12), Myc-tag (9E10), and GFP (B34) were from Covance Research and rat anti-HA tag (clone 3F10) was from Roche. Anti-β-actin (AC-17) and Flag-tag (M4) were from Sigma. Anti-Mdm2 (SMP-14), p21 (F-5), p16Ink4a (F-12), p57Kip2 (H-91), and p53 (DO-1) were from Santa Cruz. Anti-Pyk2 (clone 11), PARP (clone 42), and p27Kip1 (clone 57) were from BD-Transduction. Anti-GAPDH (374, Chemicon), p53 (PAb240 and PAb122, Invitrogen), ubiquitin (FK-2, Biomol), pAkt (Ser-473, Cell Signaling Technology) and GST (30001, Pierce) were purchased. Anti-human p53 (CM1) and mouse p53 (CM5) were from Novocasta. Rabbit polyclonal anti-GFP was raised against recombinant 6-His-tagged-GFP produced from baculovirus culture. Affinity-purified anti-GFP antibodies were used for GFP-FAK FERM immunoprecipitation studies. Image J (1.36b) was used for densitometry and measurement of pixel intensity. Purified GST-FAK was from Active Motif. MG132, cisplatin, and staurosporine were from Calbiochem and Leptomycin B was from LC Laboratories.
FAK nuclear localization and subcellular fractionation
FAK-/-p21-/- MEFs expressing GFP-FAK FERM constructs were grown on glass coverslips coated with 0.1% gelatin, washed, fixed in 3.7% paraformaldehyde, and visualized by confocal microscopy (Biorad Radiance 2100). FAK-/-p53-/- MEFs expressing GFP-FAK WT or GFP-FAK R177/R178A mutant were grown on gelatin-coated glass coverslips in the presence or absence of leptomycin B (10 ng/ml) for 4 h, fixed in 3.7% paraformaldehyde, and visualized at 60X using an inverted microscope (Olympus). Cellular fractionation studies were performed on transfected 293T cells and HUVECs in the absence or the presence of 1 μM staurosporine. After 48 h, cells were washed in cold PBS, and lysed with Cyt Buffer (10 mM Tris pH 7.5, 0.05% NP-40, 3 mM MgCl2, 100 mM NaCl, 1 mM EGTA, 20 μg/ml aprotinin, 1 mM orthovanadate, and 10 μg/ml leupeptin). Cells were scrape-loaded into tubes, incubated for 5 min at 4°C, spun at 800 x g at 4°C (5 min), and cytosolic supernatants collected. Cells pellets were further washed with Cyto buffer twice to remove residual cytosolic proteins. Purified nuclei were resuspended in RIPA buffer, spun at 16,000 x g for 15 minutes, and the supernatant collected as the nuclear fraction. Samples were separated by SDS-PAGE and immunoblotted for gylceraldehyde-3-phosphate dehydrogenase (GAPDH) and poly ADP-ribose polymerase (PARP) as cytoplasmic and nuclear markers, respectively.
Supplementary Material
Acknowledgments
We thank P. Leder (Harvard) for p21-/- and S. Aizawa (RIKEN, Japan) for p53-/- mice. We are indebted to C. Kannemeier (Scripps), E. Choi (Scripps), and the Scripps Flow facility for technical help. We appreciate this assistance of V. Ossovskaya (UCSF) and P. Sun (Scripps) for discussions and C. Damsky (UCSF) for critical review. Part of these studies was conducted in a facility constructed with support from the Research Facilities Improvement Program (C06RR16490). Y. Lim was support in part by a Korean Research Foundation Grant (M01-2005-000-10071). D. Ilic was supported by grant CA87652. D. Schlaepfer is an American Heart Association Established Investigator (0540115N) and is supported by grants CA87038, CA75240 and CA102310 from the NIH. None of the authors have a financial interest related to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bao W, Thullberg M, Zhang H, Onischenko A, Stromblad S. Cell attachment to the extracellular matrix induces proteasomal degradation of p21(CIP1) via Cdc42/Rac1 signaling. Mol Cell Biol. 2002;22:4587–4597. doi: 10.1128/MCB.22.13.4587-4597.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol. 2006;172:151–162. doi: 10.1083/jcb.200506184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant P, Zheng Q, Pumiglia K. Focal adhesion kinase controls cellular levels of p27/Kip1 and p21/Cip1 through Skp2-dependent and -independent mechanisms. Mol Cell Biol. 2006;26:4201–4213. doi: 10.1128/MCB.01612-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LA, Guan JL. Residues within the first subdomain of the FERM-like domain in focal adhesion kinase are important in its regulation. J Biol Chem. 2005;280:8197–8207. doi: 10.1074/jbc.M412021200. [DOI] [PubMed] [Google Scholar]
- Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem. 2006;99:35–52. doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Ding Q, Grammer JR, Nelson MA, Guan JL, Stewart JE, Jr, Gladson CL. p27Kip1 and cyclin D1 are necessary for focal adhesion kinase regulation of cell cycle progression in glioblastoma cells propagated in vitro and in vivo in the scid mouse brain. J Biol Chem. 2005;280:6802–6815. doi: 10.1074/jbc.M409180200. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Ilic D, Kanazawa S, Takeda N, Yamamoto T, Aizawa S. Mesodermal defect in late phase of gastrulation by a targeted mutation of focal adhesion kinase, FAK. Oncogene. 1995;11:1989–1995. [PubMed] [Google Scholar]
- Gilmore AP. Anoikis. Cell Death Differ. 2005;12 2:1473–1477. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- Golubovskaya VM, Finch R, Cance WG. Direct Interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280:25008–25021. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- Gottlieb E, Haffner R, King A, Asher G, Gruss P, Lonai P, Oren M. Transgenic mouse model for studying the transcriptional activity of the p53 protein: age-and tissue-dependent changes in radiation-induced activation during embryogenesis. EMBO J. 1997;16:1381–1390. doi: 10.1093/emboj/16.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove M, Komiyama NH, Nave KA, Grant SG, Sherman DL, Brophy PJ. FAK is required for axonal sorting by Schwann cells. J Cell Biol. 2007;176:277–282. doi: 10.1083/jcb.200609021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- Ilic D, Almeida EA, Schlaepfer DD, Dazin P, Aizawa S, Damsky CH. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Ilic D, Kovacic B, McDonagh S, Jin F, Baumbusch C, Gardner DG, Damsky CH. Focal adhesion kinase is required for blood vessel morphogenesis. Circ Res. 2003;92:300–307. doi: 10.1161/01.res.0000055016.36679.23. [DOI] [PubMed] [Google Scholar]
- Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res. 2003;1:993–1000. [PubMed] [Google Scholar]
- Kadare G, Toutant M, Formstecher E, Corvol JC, Carnaud M, Boutterin MC, Girault JA. PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J Biol Chem. 2003:47434–47440. doi: 10.1074/jbc.M308562200. [DOI] [PubMed] [Google Scholar]
- Kurenova E, Xu LH, Yang X, Baldwin AS, Jr, Craven RJ, Hanks SK, Liu ZG, Cance WG. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol Cell Biol. 2004;24:4361–4371. doi: 10.1128/MCB.24.10.4361-4371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JM, Truong TN, Schwartz MA. Integrins regulate the apoptotic response to DNA damage through modulation of p53. Proc Natl Acad Sci U S A. 2002;99:3627–3632. doi: 10.1073/pnas.062698499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, Lim ST, Bernard-Trifilo JA, Ilic D, Stupack DG, et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–5984. doi: 10.1038/sj.onc.1209588. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005;24:425–439. doi: 10.1007/s10555-005-5134-3. [DOI] [PubMed] [Google Scholar]
- Schober M, Raghavan S, Nikolova M, Polak L, Pasolli HA, Beggs HE, Reichardt LF, Fuchs E. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J Cell Biol. 2007;176:667–680. doi: 10.1083/jcb.200608010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen TL, Park AYJ, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan JL. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. 2005;169:941–952. doi: 10.1083/jcb.200411155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG, Parsons JT. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–14852. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- Stewart A, Ham C, Zachary I. The focal adhesion kinase amino-terminal domain localises to nuclei and intercellular junctions in HEK 293 and MDCK cells independently of tyrosine 397 and the carboxy-terminal domain. Biochem Biophys Res Commun. 2002;299:62–73. doi: 10.1016/s0006-291x(02)02547-0. [DOI] [PubMed] [Google Scholar]
- Stromblad S, Becker JC, Yebra M, Brooks PC, Cheresh DA. Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin alphaVbeta3 during angiogenesis. J Clin Invest. 1996;98:426–433. doi: 10.1172/JCI118808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada T, Tomooka Y, Takai S, Ueda Y, Nishikawa S, Yagi T, Tokunaga T, Takeda N, Suda Y, Abe S, et al. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene. 1993;8:3313–3322. [PubMed] [Google Scholar]
- Vousden KH. Activation of the p53 tumor suppressor protein. Biochim Biophys Acta. 2002;1602:47–59. doi: 10.1016/s0304-419x(02)00035-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.