

Abstract
Patients who survive sepsis have significant deficiencies in their immune responses caused by poorly understood mechanisms. We have explored this phenomenon by studying dendritic cells (DCs) recovered from animals surviving severe peritonitis-induced sepsis, using the well-established cecal ligation and puncture (CLP) model. Immediately after the initiation of sepsis there is a depletion in DCs from the lung and spleen, which is followed by repopulation of these cells back to the respective organs. DCs recovered from surviving animals exhibited a significant and chronic suppression of interleukin-12 (IL-12), a key host defense cytokine. The suppression of DC-derived IL-12 persisted for at least 6 weeks after CLP and was not due to immunoregulatory cytokines, such as IL-10. Using chromatin immunoprecipitation (ChIP) techniques, we have shown that the deficiency in DC-derived IL-12 was due to epigenetic alterations. Specifically, IL-12 expression was regulated by stable reciprocal changes in histone H3 lysine-4 trimethylation (H3K4me3) and histone H3 lysine-27 dimethylation (H3K27me2), as well as changes in cognate histone methyltransferase (HMT) complexes on the Il12p35 and Il12p40 promoters. These data implicate histone modification enzymes in suppressing DC-derived IL-12, which may provide one of the mechanisms of long-term immunosuppression subsequent to the septic response.
Introduction
The clinical manifestations of sepsis are often due to a dysregulated host immune response to either a known or uncharacterized insult.1,2 Despite advances in intensive care technology and mechanical ventilator support, efficacious pharmacologic options are limited, which reflect our insufficient understanding of the host-dependent mechanisms that underlie this pathophysiologic disorder.3 Many clinical studies have concentrated on the short-term outcomes of septic patients and have reported intensive care mortalities of 30% to 50%, irrespective of whether an infectious agent was identified.4,5 These studies have established that sepsis is an acute life-threatening disorder.
However, there is an additional insidious clinical aspect of patients who survive severe sepsis, because epidemiologic studies have shown that these survivors have a statistically significant risk of dying for 5 years after recovering from sepsis that exceeded normal predictions.6 Clinical records have documented that these patients who survive severe sepsis are more susceptible to disorders normally controlled by a fully functional host response long after being discharged from the intensive care unit,6,7 suggesting that profound alterations have occurred in the innate or adaptive immune response or both. Although immunosuppression is responsible at least in part for the acute rate of morbidity and mortality associated with severe sepsis, the mechanistic contributions that support the long-term consequences of aberrant immunoregulation in this disorder are not fully known.
A recent study of immunoregulation after severe sepsis has identified alterations in dendritic cell (DC) number and function as potential mechanisms for the long-term alteration in the host's immune response.8 Furthermore, we have now characterized DC-derived IL-12 as a key host defense cytokine that is significantly down-regulated even 6 weeks after the original septic event. This sepsis-dependent alteration in DC-derived IL-12 has been shown to render animals susceptible to a secondary microbial challenge and to induce an exacerbated Th2 cytokine response to an acquired immune event,9,10 but the mechanism underlying the long-term suppression of DC-derived IL-12 is not known.
A growing body of evidence has shown that epigenetic mechanisms are involved in the regulation of various genes expressed during normal embryonic development and cancer.11,12 In addition, the inappropriate regulation of histone-modifying enzymes has been documented in both autoimmune diseases and cancer.13,14 Among various histone modifications, methylation of histone H3 at lysine-4 (H3K4) and at lysine-27 (H3K27), each mediated by distinct histone methyltransferase (HMT) complexes, are highly correlated with transcriptional activation15,16 and repression,17 respectively. Interestingly, histone methylation has been implicated in the development of the acquired immune responses, especially in the differentiation and maintenance of T helper type 1/T helper type 2 (Th1/Th2) memory cells.18 However, most of this evidence has been derived from in vitro studies of epigenetic changes associated with T-cell activity. In the present study we have focused on the mechanisms responsible for the long-term regulation of IL-12 expression from DCs recovered from animals surviving severe experimental sepsis. We provide evidence for stable and long-term changes in histone methylation and characterize some of the responsible HMT complexes associated with the regulation of DC-derived IL-12 in postseptic animals. These epigenetic changes are potentially important for facilitating stable alterations in cytokine gene expression, which mechanistically contribute to the long-term suppression of the host's immune system after severe sepsis.
Methods
Mice
Female C57BL/6 mice (6–8 weeks; Taconic Farms, Germantown, NY) and OVA-specific TCR-transgenic OT II mice (The Jackson Laboratory, Bar Harbor, ME) were housed under specific pathogen-free conditions at the Unit for Laboratory Animal Medicine of the University of Michigan and treated in accordance with the guidelines of the animal ethical committee.
Cecal ligation and puncture
Cecal ligation and puncture (CLP) surgery was performed on mice as previously described.10 In brief, mice were anesthetized with an intraperitoneal injection of 2.25 mg ketamine HCL (Abbott Laboratories, Abbott Park, IL) and 150 μg xylazine (Lloyd Laboratories, Shenandoah, IA). Under sterile surgical conditions, a 1-cm midline incision was made to the ventral surface of the abdomen, and the cecum was exposed. The cecum was partially ligated at its base with a 3.0 silk suture and punctured 9 times with 21-gauge needle. The cecum was returned to the peritoneal cavity, and the abdominal incision was closed using surgical staples. Sham-operated mice underwent identical operation except for cecal ligation and puncture and served as controls. Mice in both groups were injected intraperitoneally with the antibiotic ertapenem (Invanz) at 75 mg/kg (Merck, Whitehouse Station, NJ) beginning at 6 hours after surgery and reinjected every 24 hours until day 3 after surgery.
Flow cytometric analysis
Whole lungs and spleens were dispersed in 0.2% collagenase type IV (Sigma-Aldrich, St Louis, MO) in RPMI 1640 (Mediateck, Herndon, VA) plus 5% fetal bovine serum (Atlas Biologicals, Fort Collins, CO) at 37°C for 30 minutes. After lysing red blood cells (RBCs) with ammonium chloride buffer (4.01 g NH4Cl, 0.42 g NaHCO3, and 0.185 g tetrasodium EDTA in 500 mL dH2O), Fc binding was blocked by a 10-minute incubation with purified rat anti–mouse CD16/CD32 (FcγIII/II receptor). Then the cells were stained with FITC-labeled anti-CD11c (HL3), PerCP-Cy5.5–anti-CD11b (M1/70) in combination with each of the following antibodies: PE–anti-I-Ab (AF6-120.1), PE–anti-CD40 (HM40-3), PE–anti-CD80 (16-10A1), or PE–anti-CD86 (GL1) in Dulbecco PBS plus 0.2% BSA plus 0.1% NaN3 for 30 minutes at 4°C in the dark. The appropriate IgG isotypes were used as controls. All antibodies and IgG isotypes were purchased from BD Biosciences PharMingen (San Diego, CA). The cells were fixed in 1% paraformaldehyde and kept in the dark at 4°C until analysis with a FACSCaliber (CELLQuest software; Becton Dickinson, Franklin Lakes, NJ).
Isolation of splenic DCs and lung DCs
Whole lungs and spleens were dispersed as described in “Flow cytometric analysis.” After lysing RBCs, cell suspensions were enriched with anti-CD11c magnetic beads (Miltenyi Biotec, Auburn, CA). Briefly, the cells were resuspended in 400 μL buffer (PBS/0.5% BSA) containing 100 μL CD11c microbeads. After a 15-minute incubation at 4°C, free beads were washed away, and the cells conjugated with beads were passed through MS+ columns for positive selection. Especially for lung DC isolation, total dispersed lung cells were incubated in 100-mm cell culture dish for 1 hour at 37°C to remove the adherent CD11c+ macrophages before positive selection with anti-CD11c magnetic beads. Purified DCs were counted on a hemocytometer and subsequently diluted at 2 × 106/mL. The aliquots of 200 μL containing these cells were added to 96-well plates and stimulated with a group of TLR agonists: 2.5 μg/mL Pam3cys (EMC Microcollections, Tubingen, Germany), 1 μg/mL lipopolysaccharide (LPS from Escherichia coli 0111:B4; Sigma-Aldrich), or 2μM mouse CpG-DNA (HyCult Biotechnology, Canton, MA). After 6 hours of stimulation, total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA), and quantitative real-time polymerase chain reaction (PCR; Taqman) was performed to measure gene expression of inflammatory cytokines (Applied Biosystems, Foster City, CA). After 24 hours of stimulation, cell-free supernatant from each sample was subject to enzyme-linked immunoabsorbent assay (ELISA) to measure protein levels of inflammatory cytokines. In the experiments neutralizing IL-10, 1 or 10 μg/mL polyclonal neutralizing anti-10 antibody (R&D Systems, Rochester, MN) was used with goat IgG as control.
Quantitative real-time PCR
Total RNA was isolated from splenic DCs using Trizol according to the manufacturer's directions. Reverse transcription was performed to yield cDNA in a 25-μL reaction mixture containing 1X first strand (Invitrogen), 250 ng of oligo (dT)12–18 primer, 1.6mM dNTPs (Invitrogen), 5 U of RNase inhibitor (Invitrogen), and 100 U of Moloney murine leukemia virus reverse transcriptase (Invitrogen). Messenger RNA levels of IL-12p35 and IL-12p40 were then analyzed by reverse transcription–PCR with Taqman gene expression assay (Applied Biosystems) using a Taqman 7500 sequence detection system. GAPDH was analyzed as an internal control.
Measurement of cytokine protein levels
Concentrations of IL-10 and IL-12p70 were measured in isolated splenic DC culture supernatants using a Luminex Bio-Plex 200 system (Bio-Rad, Hercules, CA), according to the manufacturer's protocol. Briefly, 96-well multiplex assay plate was coated with anti–mouse cytokine 2-plex conjugated beads (IL-10 and IL-12p70). Plates were rinsed 2 times with wash buffer A, and cell-free supernatants, as well as series diluted standard cytokine, were loaded and incubated for 30 minutes at room temperature with gentle vortex. After 3 washings, the biotinylated mouse cytokine 2-plex detection antibody was added for 30 minutes at room temperature with gentle vortex. The plates were washed again, and PE-conjugated streptavidin was added for 10 minutes at room temperature with gentle vortex. Plates were washed and read using Luminex Bio-Plex 200 system plate reader. Murine stock cytokines of known concentrations, which came together with the kit, were used to generate the standard curves from which the cytokine concentrations present in the samples were calculated. The threshold of each cytokine is 5 pg/mL.
Th1/Th2-polarizing capacity assay
To determine the effect of DCs recovered from septic mice in driving the cytokine phenotype from T cells, we used the following protocol: splenic DCs isolated from sham or CLP mice on day 11 after surgery were pulsed with 5 μg/mL OVA323–339 peptides (Peptides International, Louisville, KY) for 4 hours. After removing soluble OVA peptide in the medium, CD4+ T cells isolated from spleens of naive OT II mice with anti-CD4 magnetic beads (Miltenyi Biotec) were added to the DC culture. After 24 hours, supernatants were collected and stored at −80°C until measurement of protein levels of IL-4, IL-5, IL-13, and IFN-γ by ELISA.
Chromatin immunoprecipitation
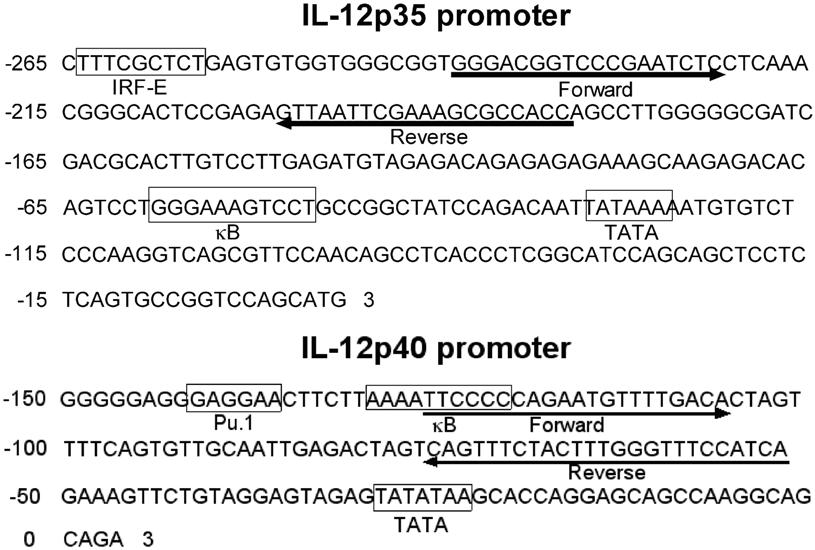
The chromatin immunoprecipitation (ChIP) procedure was performed using an assay kit (Upstate Biotechnology, Charlottesville, VA) according to the manufacturer's instructions. Briefly, 1 × 106 isolated splenic DCs were stimulated with 1 μg/mL LPS for 6 hours. DNA-protein structure was then cross-linked by 1% formaldehyde for 10 minutes at 37°C. Cells were collected and lysed in 400 μL SDS lysis buffer. The resulting lysates were sonicated to obtain DNA fragments ranging from 200 to 1000 bp (base pairs) using a Branson Sonifier 450 (VWR, West Chester, PA) under the following condition: 6 times for periods of 15 seconds each. After centrifuging, the supernatant containing chromatin was diluted, and an aliquot (2% volume) was saved to indicate the input DNA in each sample. The remaining chromatin fractions were precleared with salmon sperm DNA/protein A agarose beads followed by immunoprecipitation with the following antibodies: anti–acetyl histone H3 (06-599; Upstate Biotechnology), anti–H3K4me3 (ab8580; Abcam, Cambridge, MA), anti–H3K27me2 (07-452; Upstate Biotechnology), anti-MLL, anti-WDR5, and anti-EED (Dr Yali Dou),16 anti-RbBP5 (A300-109A; Bethyl Laboratories, Montgomery, TX), or anti-SUZ12 (ab12073; Abcam) overnight at 4°C with gentle rotation. Cross-linking was reversed for 4 hours at 65°C and was followed by proteinase K digestion. DNA was purified by standard phenol/chloroform and ethanol precipitation and was subjected to real-time PCR. Primers for mouse Il12p35 promoters were as follows: forward, 5′-GGGACGGTCCCGAATCTC; reverse, 5′-GGTGGCGCTTTCGAATTAAC. Primers for mouse Il12p40 promoter were as follows: forward, 5′-TTCCCCCAGAATGTTTTGACA; reverse, 3′-TGATGGAAACCCAAAGTAGAAACTG.
Statistics
Results were expressed as means plus or minus SEMs. Student t test and one-way ANOVA were used to detect statistical significance.
Results
Tissue myeloid DCs undergo rapid depletion followed by reconstitution after severe sepsis
A well-documented cellular event after both clinical and experimental sepsis is the rapid depletion of different leukocyte populations, which occurs mainly by an apoptotic event.19 We have profiled the initial depletion and repopulation kinetics of lung and splenic myeloid (DCs; CD11C+CD11B+MHCIIhi) in a model of experimental sepsis in which approximately 60% of the animals are long-term survivors (Figure 1A). In this model there was a significant loss of lung and spleen mDCs during the first 3 days after the initiation of experimental sepsis, which was followed by a gradual reconstitution during the next 3 weeks in the spleen and 5 to 6 weeks in the lungs. In addition, there was a profound loss of DCs in the bone marrow during the first 7 days, such that no DCs could be cultured and expanded from the bone marrow during this time frame (data now shown).
Figure 1.

Depletion of tissue mDCs followed by reconstitution after experimental peritonitis. (A) At different time points after CLP procedure, spleen and lung were collected and dispersed. The cells were stained with FITC–anti-CD11c, PerCP-Cy5.5–anti-CD11b and PE–anti-I-Ab. Myeloid dendritic cells (mDCs) were characterized as CD11c+CD11b+MHCIIhi. *P ≤ .05 compared with mDC number observed in spleen or lung from sham mice. Error bars represent SEM. (B) At day 11 after CLP procedure, spleens were collected and dispersed. Expression of CD40, CD80, CD86, and MHCII were investigated on the cell surface of CD11c+CD11b+ mDCs. Black line indicates isotype control; dashed line, sham group; gray line, CLP group.
In addition to assess DC numbers, we also determined whether severe sepsis led to any alteration in the expression of MHCII and the costimulatory molecules CD40, CD80, and CD86, each of which might influence DC functions.20 At day 11 after CLP, there was no difference in the expression of any of the above molecules (Figure 1B). Similar observations were made when we measured the expression levels of MHCII and costimulatory molecules on DC surfaces at 6 weeks after CLP (data now shown).
Deficiency in IL-12 production by postseptic splenic DCs
DCs serve as sentinels at portals of potential pathogen entry and orchestrate host innate and adaptive immunity against invading pathogens by the production of inflammatory cytokines and the subsequent presentation of antigen to T cells in the draining lymph nodes.21 One of the key cytokines that facilitates these important immune responses is IL-12, which is a heterodimeric inflammatory cytokine composed of an IL-12p35 and IL-12p40 subunit.22 BecauseIL-12 generated by DCs is the key molecule to direct a type-1 immune response,23 we sought to determine whether severe sepsis altered the capacity of DCs to produce IL-12. At day 11 after the initiation of experimental sepsis, a time point when DCs were repopulating the spleen, these cells were recovered and purified from sham or CLP mice and stimulated with a series of toll-like receptor (TLR) agonists (Pam3Cys [TLR2], LPS [TLR4], or CpG-DNA [TLR9]) and assessed for the expression of IL-12 (Figure 2). Real-time PCR showed that after sepsis splenic DCs had significantly lower mRNA levels of both Il12p35 and Il12p40 but a higher mRNA level of regulatory cytokine Il10 compared with similarly treated sham splenic DCs (Figure 2A). Consistently, postseptic DCs, recovered at day 11, generated a significantly lower protein level of IL-12p7, but a higher protein level of IL-10 in response to the stimulation with TLR agonists in in vitro culture systems (Figure 2B). We believe that severe sepsis results in a systemic defect in DC-derived IL-12 production, because lung DCs isolated at day 11 after CLP also showed a deficiency in IL-12 production in response to TLR agonists, compared with similarly treated lung DCs from sham mice (Figure 2C).
Figure 2.

Impaired IL-12 production by splenic DCs and lung DCs day 11 after experimental peritonitis. (A) Purified splenic DCs from septic or control mice on day 11 after surgery were stimulated by a series of TLR agonists. Splenic DC mRNA levels of the 2 IL-12 subunits and IL-10 were determined by Taqman. (B) Splenic DC protein levels of IL-12p70 and IL-10 were measured by Bio-Plex. (C) Purified lung DCs from septic or sham mice on day 11 after surgery were stimulated by a series of TLR agonists, and protein levels of IL-12p70 and IL-10 were measured by Bio-Plex. Results are representative of 3 to 5 independent experiments and are expressed as mean plus or minus SEM. *P ≤ .05 compared with cytokine levels measured in splenic DCs from sham mice.
Down-regulation of IL-12 in postseptic DCs is not due to excessive IL-10 production
As a regulatory cytokine, IL-10 has been reported to directly inhibit IL-12 production both in vivo and in vitro.24,25 To test the hypothesis that the deficiency in IL-12 production by postseptic DCs was due to excessive IL-10 production in culture system, 2 concentrations of specific, high titer anti–IL-10 antibody (1 and 10 μg/mL) were used to block IL-10 production by both sham and postseptic splenic DCs in response to LPS stimulation (Figure 3). DCs from sham mice showed increased LPS-induced IL-12 production in the presence of anti–IL-10 in a dose-dependent manner, indicating that the anti–IL-10 antibody was active and that IL-10 does have an inhibitory effect on IL-12 production in control DCs. However, IL-12 production by postseptic DCs was not rescued by anti–IL-10, which suggested that the deficiency in IL-12 production by postseptic DCs was probably due to a mechanism independent of excessive IL-10 production.
Figure 3.

Deficient IL-12 production in postseptic DCs was not due to excessive IL-10 or other extrinsic effector molecules. Purified splenic DCs from septic or control mice on day 11 after surgery were stimulated by LPS, in the absence or presence of 1 or 10 μg/mL anti-IL-10 antibody. Protein levels of IL-12p70 and IL-10 were determined by Bio-Plex. Results are representative of 3 independent experiments. *P ≤ .05 compared with cytokine levels measured in splenic DCs from sham mice.
DC IL-12 suppression is a long-term consequence of severe experimental sepsis
We next determined whether the deficient phenotype in IL-12 production exhibited by postseptic DCs was preserved over time in animals surviving experimental sepsis. In this set of studies, cytokine production by splenic DCs was determined 6 weeks after sham and CLP procedures. No difference in weight or behavior was observed between the 2 groups of mice at this time point. However, splenic DCs recovered from postseptic mice showed significantly lower mRNA levels of Il12p35 and Il12p40 and produced lower IL-12p70 protein level in response to TLR agonists compared with similarly treated control splenic DCs (Figure 4). No difference in IL-10 production was observed between sham and postseptic splenic DCs, which suggested that the deficiency in IL-12 production by postseptic DCs, at this time point, was again not due to regulatory effects of IL-10. Collectively, severe sepsis results in a systemic decrease in DC-derived IL-12, which seems to be long-term maintained and mediated by intrinsic mechanisms.
Figure 4.

DC IL-12 suppression is a long-term consequence of severe experimental sepsis. Purified splenic DCs from septic or control mice 6 weeks after surgery were stimulated with a number of TLR agonists. IL-12 and IL-10 expression was determined at the mRNA level (A) and protein level (B). Results represent 3 independent experiments. *P ≤ .05 compared with cytokine levels measured in splenic DCs from sham mice. Error bars represent SEM.
Th2-polarized capacity of postseptic splenic DCs
Our previous studies showed that postseptic mice presented with an antigen challenge induced an enhanced Th2 cytokine profile and an exacerbated type-2–mediated granulomatous response.10 Therefore, we hypothesized that after severe sepsis, Ag-loaded DCs direct naive T cells toward a Th2-biased response. To test this notion, splenic DCs recovered from day 11 postseptic mice were pulsed with OVA peptide and cocultured with OVA peptide-specific CD4+ T cells isolated from TCR transgenic OT II mice. These T cells generated significantly higher levels of Th2 cytokines such as IL-4, IL-5, and IL-13 but a lower level of Th1 cytokine IFN-γ, compared with similar coculture experiments using splenic DCs from sham mice (Figure 5). These data suggest that postseptic DCs facilitate a Th2-polarized effect on T cells, which cause an altered cytokine environment in which the host immune system must operate.
Figure 5.

Th2-polarized capacity of postseptic splenic DCs. Purified splenic DCs from septic or control mice on day 11 after surgery were pulsed with 5 μg/mL OVA323–339 peptide and cocultured with splenic CD4+ T cells isolated from naive TCR transgenic OT II mice. Twenty-four hours later, the supernatants were collected to measure cytokine protein levels by Bio-Plex. Results represent 3 independent experiments and are expressed as means plus or minus SEM. *P ≤ .05 compared with cytokine protein levels in the supernatants of coculture between CD4+ T cells and sham splenic DCs.
Altered histone modifications on Il12 promoters in postseptic splenic DCs
A number of investigations underscore the importance of epigenetic changes in preserving long-term alterations in the expression profile of specific genes.11 This is especially true of advances made in the field of cancer and developmental biology. Yet the mechanistic role of epigenetic changes that may influence important DC-derived genes has not been explored. It is well documented that histone H3K4 and K27 methylations are involved in the regulation of specific genes.26,27 Specifically, methylation of H3K4, mediated by mixed-lineage leukemia (MLL) family HMTs, is correlated with transcription activation, whereas methylation of H3K27, mediated by enhancer of Zeste 2 (EZH2), is correlated with gene silencing.18 The long-term maintenance of the deficient IL-12 production in postseptic but not sham DCs prompted us to hypothesize that an epigenetic mechanism might be involved in this process. We tested this hypothesis by ChIP assay using anti-H3K4 trimethylation (H3K4me3) and anti-H3K27 dimethylation (H3K27me2) antibodies. Taqman primers were designed at the promoter regions of Il12p35 and Il12p40 around the essential binding sites for several key transcriptional factors, such as Pu.1, IFN regulatory factor, and κB (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). We found that splenic DCs from control mice showed a relative high H3K4me3 and low H3K27me2 at both Il12p35 and Il12p40 promoters, indicating that both of these genes are surrounded by permissive chromatin structure and are poised for expression on exposure to stimulus. Conversely, splenic DCs at day 11 after CLP showed significantly decreased H3K4me3 and increased H3K27me2 at Il12p35 and Il12p40 promoters, suggesting that chromatin structure was skewed toward a more repressive status after the CLP procedure (Figure 6A). This is a long-term effect because the repressive chromatin environment is present at 6 weeks after CLP; significantly decreased H3K4me3 and increased H3K27me2 at both Il12p35 and Il12p40 promoters were observed in postseptic splenic DCs (Figure 6B). Thus, the altered status of H3K4 and H3K27 methylations at Il12p35 and Il12p40 promoters correlated well with the decreased gene expression.
Figure 6.

Alterations in histone H3 methylation on Il12 promoters in postseptic splenic DCs. Splenic DCs were isolated from sham and CLP mice at either day 11 (A) or 6 weeks (B) after surgery. ChIP assay was performed to determine the histone H3 methylation status at the promoter regions of Il12p35 and Il12p40, respectively. The results shown are representative of 2 to 3 independent experiments and are expressed as means plus or minus SEM. *P ≤ .05 compared with splenic DCs from sham mice.
Differential recruitment of core components of HMT complexes on Il12 promoters
We next explored the molecular mechanism whereby postseptic splenic DCs exhibited decreased H3K4me3 and increased H3K27me2 on Il12 promoters. Methylations at H3K4 and H3K27 sites are mainly mediated by MLL family H3K4 methyltransferases and polycomb repressive complex 2 (PRC2) complex, respectively.15,17 MLL family HMTs share common structures, which include the catalytic subunit, as well as several structural proteins, including the WD40-repeat protein WDR5, RbBP5, and Ash2L.28,29 PRC2 complex consists of the core components EZH2, suppressor of Zeste 12 (SUZ12), and embryonic ectoderm development (EED).17 The activities of both MLL family HMTs and PRC2 require the interaction of the catalytic subunits with other components. To assess whether the alterations in histone methylation were the result of the altered recruitment or the complex integrity of MLL complex or PRC2 complex or both complexes at Il12 promoters, respectively, we performed ChIP assays using a series of antibodies directed against the core components of these2 complexes. Although there was no difference in MLL1 binding on H3K4 at Il12 promoters between postseptic splenic DCs and control DCs, the recruitment of WDR5 and RbBP5 on Il12 promoters was significantly decreased in postseptic DCs compared with sham DCs (Figure 6A). In contrast, the recruitment of EED and SUZ12 was significantly increased in postseptic DCs compared with sham DCs (Figure 7A). EZH2 binding was not investigated, because of the lack of ChIP-grade antibody. This same pattern was shown to be present 6 weeks after the initiation of the original septic event (Figure 7B). Thus, our data suggest that alterations in the levels of H3K4me3 and H3K27me2 are related to the corresponding HMT complexes on the Il12 promoters. These data further support the concept that epigenetic alterations skew IL-12 expression toward a more repressive status after the induction of severe sepsis.
Figure 7.

Altered recruitment of HMT complexes at Il12 promoters in postseptic splenic DCs. Splenic DCs were isolated from sham and CLP mice at either day 11 (A) or 6 weeks (B) after surgery. Using appropriate antibodies directed against the core components of the MLL and PRC2 complexes, recruitment of the core components to the promoter regions of Il12p35 and Il12p40 was determined by ChIP assay. The results shown are representative of 2 to 3 independent experiments and are expressed as means plus or minus SEM. *, α P ≤ .05 compared with splenic DCs from sham mice.
Discussion
The initiation and maintenance of the pathology observed after sepsis depends on ill-understood cellular and molecular mechanisms. These unknown processes contribute to the clinical challenge of treating the acute phase of sepsis, as well as a subsequent sustained immunosuppressive state found in patients who survive severe sepsis. The “immunoparalysis” in septic patients is often manifested by an inability to eradicate a primary infection or the development of new secondary infections.5 Thus, the initial episode of severe sepsis, characterized by a dysregulated inflammatory response, may lead to long-lasting complications about how the host responds to and deals with subsequent microbial challenges. The depletion of immune cells, such as DCs and lymphocytes, because of extensive apoptosis has been related to long-term immunosuppression.19 However, it seems unlikely that the decrease in immune cell numbers is the only mechanism whereby the septic host fails to mount an appropriate immune response, because we have observed a gradual restoration of immune cells after the initial apoptosis-induced immune cell depletion. Therefore, the long-term maintenance of abnormal expression patterns of key cytokine genes by immune cells is a potential mechanism worthy of investigation.
For a number of years, epigenetic mechanisms have been shown to play an essential role in the maintenance of gene expression patterns during embryogenesis and cancer.12,14 In the latter case, specific alterations in the enzymatic machinery involved in chromatin remodeling have been mechanistically associated with the development of various cancers. The combination of individual histone modifications, catalyzed by several families of histone-modifying enzymes, form a “histone code” that constitutes an important epigenetic determinant of the transcriptional state.11,30 It has been recently proposed that histone modifications regulate gene expression pattern in the immune system18; however, most of these studies have focused primarily on lymphocyte-derived cytokines. Chromatin remodeling events have been documented on the IFNγ promoter, Il4 locus (including Il5, Rad50, Il13, and Il4), and Il17 promoter during the differentiation of Th1, Th2, and Th17 memory T cells.18,31 MLL, an HMT enzyme specific for H3K4 methylation has been reported to be essential for the maintenance of memory Th2 cell response,32 whereas EZH2, a core component of PRC2, has been identified as an important molecule for the continuance of a Th1 cytokine phenotype by silencing of the cytokines Il4 and Il13.18,33 One of the many unanswered questions about the enzymatic machinery that directs the methylation of chromatin is what are the environmental factors that regulate the expression or activation of these enzyme complexes. In the setting of inflammation and immune reactivity, it is likely that specific cytokines themselves may play an instrumental role in regulating the enzymes that remodel the chromatin. Thus, the rather long list of cytokines that have a known regulatory influence on the expression of other cytokines could manifest this effect by controlling the chromatin-remodeling enzyme complexes.
Although epigenetic alterations have been shown to alter the expression of lymphocyte-derived cytokines, the signaling pathways that lead to the epigenetic changes in other parts of the immune system are just beginning to be examined. In a recent study, LPS-stimulated macrophages may shed some light on these phenomena.34 Although this study34 showed that TLR signaling could induce epigenetic changes, it also showed that signaling through TLR4 alone was not sufficient to build a long-term immune cell memory for the expression of certain genes. In memory T cells the candidate pathways include T-cell receptor and cytokine receptor signaling pathways. Because severe sepsis results in an overwhelmed innate immune response and a classic cytokine storm, it is reasonable to hypothesize that TLR signaling and cytokine/cytokine receptor signaling pathways are potential mechanistic contributors to the epigenetic changes found in DC-derived IL-12 expression. It is also highly possible that synergy between individual TLR signaling pathways exists that results in downstream epigenetic changes, because synergy between different TLR signaling pathways has been shown to promote the Th1-polarizing function of DCs.35 Investigations of the role of the epigenetic alterations, including DNA methylation and histone modifications, in long-term DC dysfunction and immunosuppression after a severe acute inflammatory response will probably provide mechanistic insight into the longer-term sequela of survivors of sepsis and acute lung injury. These future studies will be important in laying the groundwork for the development of potentially novel cell and mediator-based therapeutic interventions.
The chronic consequences linked to severe acute inflammation have been found in many severe acute human diseases, including the acute ischemia/reperfusion injury after organ transplantation,36 severe respiratory syncytial viral infection in neonates,37,38 and severe burn and trauma injury.39,40 Many of these clinical cases appear to be governed by severe, life-threatening inflammatory events, followed by a long-lasting improper or immunosuppressed immune response. One of the best examples of the chronic effects of severe acute inflammation is severe sepsis, both in patients and in experimental animal models. After the initial hyperinflammatory response, septic patients develop a sustained anti-inflammatory or immunosuppressive state that has been termed “immunoparalysis,” which is manifested by an inability to eradicate the primary infection or the development of new secondary infections.1,5,41–44 Thus, the initial episode of severe sepsis, characterized by a dysregulated inflammatory response, appears to lead to long-lasting complications about the manner in which the host responds to and deals with subsequent challenges. Our data address a lingering conundrum of how certain immune/inflammatory cells possess a long-term cytokine expression profile by “remembering” whether they should be actively transcribing specific genes. We believe that epigenetic alterations that dictate the expression of key cytokine genes are an important and fundamental mechanism that leads to the chronicity of many human diseases.
In the present study, we report for the first time that the chronic regulation of Il12 gene expression in postseptic mice is associated with an epigenetic mechanism of gene regulation by histone methylation in DCs. We have observed both H3K4me3 and H3K27me2 at Il12 promoters, which lead to gene activation and silencing, respectively. The existence of bivalent domains of H3K4me3 and H3K27me2 has recently been reported in embryonic stem cells45 and in terminally differentiated T cells,46 and these published data suggest that the relative ratio between these 2 markers determines transcription outcomes. We observed a decreased ratio between H3K4me3 and H3K27me2 on the promoter regions of Il12p35 and Il12p40, which was associated with down-regulation of IL-12 gene expression in postseptic splenic DCs. In addition, this decreased ratio of H3K4me3 and H3K27me2 was a result of altered binding of cognate HMT complexes. Thus, our study indicates that the alterations in histone methylation are involved in long-term aberrant gene expression pattern in DCs and that these contribute to the immunosuppression observed in postseptic animals and potentially in patients with from severe sepsis.
Given that transplantation of DCs treated in vitro with tumor antigen has provided a novel therapeutic approach to treat cancer,47–49 transplantation of immunocompetent DCs to patients who survive severe sepsis is a potential therapeutic strategy to rescue sepsis-associated immunosuppression. The broader window of treatment timing of this approach may bestow vast clinical benefits compared with treatments aimed at the acute phase of sepsis, during which a cytokine storm occurs so rapidly that treatment strategies often fail.2,3 Although chemical manipulation of histone modifications is in an early stage of development, this approach may present a future therapeutic strategy to reverse the long-term epigenetic changes induced by severe sepsis. Collectively, our studies assessing novel mechanisms that dictate the long-lived immunosuppression associated with severe sepsis survivors provide insight into the development of new therapeutic strategies for an understudied syndrome with significant clinical and financial implications to our health care system.
Supplementary Material
Acknowledgments
We thank Holly Evanoff and Pam Lincoln for their technical assistance. We also think Robin Kunkel and Judith Connett for editorial assistance.
This work was supported by National Institutes of Health grants HL31237, HL74024, and HL31963 (S.L.K.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.D., C.M.H., and S.L.K. designed the experiments; H.W. performed the experiments and collected and analyzed data; Y.D. contributed vital new antibodies; H.W., Y.D., C.M.H., and S.L.K. contributed to manuscript preparation; S.L.K. supervised the project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Steven L. Kunkel, Department of Pathology, University of Michigan Medical School, 4071 BSRB, 109 Zina Pitcher Pl, Ann Arbor, MI 48109-2200; e-mail: slkunkel@umich.edu.
References
- 1.Angele MK, Faist E. Clinical review: immunodepression in the surgical patient and increased susceptibility to infection. Crit Care. 2002;6:298–305. doi: 10.1186/cc1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Invest. 2003;112:460–467. doi: 10.1172/JCI19523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med. 2003;9:517–524. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 4.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 5.Reddy RC, Chen GH, Tekchandani PK, Standiford TJ. Sepsis-induced immunosuppression: from bad to worse. Immunol Res. 2001;24:273–287. doi: 10.1385/IR:24:3:273. [DOI] [PubMed] [Google Scholar]
- 6.Quartin AA, Schein RM, Kett DH, Peduzzi PN. Magnitude and duration of the effect of sepsis on survival. Department of Veterans Affairs Systemic Sepsis Cooperative Studies Group. JAMA. 1997;277:1058–1063. [PubMed] [Google Scholar]
- 7.Perl TM, Dvorak L, Hwang T, Wenzel RP. Long-term survival and function after suspected gram-negative sepsis. JAMA. 1995;274:338–345. [PubMed] [Google Scholar]
- 8.Flohe SB, Agrawal H, Schmitz D, Gertz M, Flohe S, Schade FU. Dendritic cells during polymicrobial sepsis rapidly mature but fail to initiate a protective Th1-type immune response. J Leukoc Biol. 2006;79:473–481. doi: 10.1189/jlb.0705413. [DOI] [PubMed] [Google Scholar]
- 9.Benjamim CF, Lundy SK, Lukacs NW, Hogaboam CM, Kunkel SL. Reversal of long-term sepsis-induced immunosuppression by dendritic cells. Blood. 2005;105:3588–3595. doi: 10.1182/blood-2004-08-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen H, Hogaboam CM, Gauldie J, Kunkel SL. Severe sepsis exacerbates cell-mediated immunity in the lung due to an altered dendritic cell cytokine profile. Am J Pathol. 2006;168:1940–1950. doi: 10.2353/ajpath.2006.051155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005;15:163–176. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 13.Ballestar E, Esteller M, Richardson BC. The epigenetic face of systemic lupus erythematosus. J Immunol. 2006;176:7143–7147. doi: 10.4049/jimmunol.176.12.7143. [DOI] [PubMed] [Google Scholar]
- 14.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 16.Dou Y, Milne TA, Tackett AJ, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 17.Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev. 2004;14:155–164. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol. 2006;24:607–656. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- 19.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813–822. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Zhao M, Han SM, Li ZM, Zhu GJ. [Testing and analyzing the lung functions in the normal population in Hebei province]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2004;26:463–466. [PubMed] [Google Scholar]
- 21.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 22.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 24.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 25.Zhou L, Nazarian AA, Smale ST. Interleukin-10 inhibits interleukin-12 p40 gene transcription by targeting a late event in the activation pathway. Mol Cell Biol. 2004;24:2385–2396. doi: 10.1128/MCB.24.6.2385-2396.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 27.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 28.Wysocka J, Swigut T, Milne TA, et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121:859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 29.Dou Y, Milne TA, Ruthenburg AJ, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 30.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem. 2007;282:5969–5972. doi: 10.1074/jbc.C600322200. [DOI] [PubMed] [Google Scholar]
- 32.Yamashita M, Hirahara K, Shinnakasu R, et al. Crucial role of MLL for the maintenance of memory T helper type 2 cell responses. Immunity. 2006;24:611–622. doi: 10.1016/j.immuni.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 33.Koyanagi M, Baguet A, Martens J, Margueron R, Jenuwein T, Bix M. EZH2 and histone 3 trimethyl lysine 27 associated with Il4 and Il13 gene silencing in Th1 cells. J Biol Chem. 2005;280:31470–31477. doi: 10.1074/jbc.M504766200. [DOI] [PubMed] [Google Scholar]
- 34.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 35.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagano H, Tilney NL. Chronic allograft failure: the clinical problem. Am J Med Sci. 1997;313:305–309. doi: 10.1097/00000441-199705000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Lemanske RF., Jr The childhood origins of asthma (COAST) study. Pediatr Allergy Immunol. 2002;13(Suppl 15):38–43. doi: 10.1034/j.1399-3038.13.s.15.8.x. [DOI] [PubMed] [Google Scholar]
- 38.Openshaw PJ, Dean GS, Culley FJ. Links between respiratory syncytial virus bronchiolitis and childhood asthma: clinical and research approaches. Pediatr Infect Dis J. 2003;22:S58–S64. doi: 10.1097/01.inf.0000053887.26571.eb. discussion S64–S65. [DOI] [PubMed] [Google Scholar]
- 39.Rodgers GL, Mortensen J, Fisher MC, Lo A, Cresswell A, Long SS. Predictors of infectious complications after burn injuries in children. Pediatr Infect Dis J. 2000;19:990–995. doi: 10.1097/00006454-200010000-00010. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi M, Takahashi H, Sanford AP, Herndon DN, Pollard RB, Suzuki F. An increase in the susceptibility of burned patients to infectious complications due to impaired production of macrophage inflammatory protein 1 alpha. J Immunol. 2002;169:4460–4466. doi: 10.4049/jimmunol.169.8.4460. [DOI] [PubMed] [Google Scholar]
- 41.Ertel W, Kremer JP, Kenney J, et al. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood. 1995;85:1341–1347. [PubMed] [Google Scholar]
- 42.Docke WD, Randow F, Syrbe U, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997;3:678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 43.Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. 2001;16:83–96. doi: 10.1097/00024382-200116020-00001. [DOI] [PubMed] [Google Scholar]
- 44.Benjamim CF, Hogaboam CM, Kunkel SL. The chronic consequences of severe sepsis. J Leukoc Biol. 2004;75:408–412. doi: 10.1189/jlb.0503214. [DOI] [PubMed] [Google Scholar]
- 45.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 46.Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci U S A. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang DH, Osman K, Connolly J, et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med. 2005;201:1503–1517. doi: 10.1084/jem.20042592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banchereau J, Ueno H, Dhodapkar M, et al. Immune and clinical outcomes in patients with stage IV melanoma vaccinated with peptide-pulsed dendritic cells derived from CD34+ progenitors and activated with type I interferon. J Immunother (1997) 2005;28:505–516. doi: 10.1097/01.cji.0000171292.79663.cb. [DOI] [PubMed] [Google Scholar]
- 49.Fay JW, Palucka AK, Paczesny S, et al. Long-term outcomes in patients with metastatic melanoma vaccinated with melanoma peptide-pulsed CD34(+) progenitor-derived dendritic cells. Cancer Immunol Immunother. 2006;55:1209–1218. doi: 10.1007/s00262-005-0106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}