

Abstract
Increased resistance of airways or blood vessels within the lung is associated with asthma or pulmonary hypertension and results from contraction of smooth muscle cells (SMCs). To study the mechanisms regulating these contractions, we developed a mouse lung slice preparation containing bronchioles and arterioles and used phase-contrast and confocal microscopy to correlate the contractile responses with changes in [Ca2+]i of the SMCs. The airways are the focus of this study. The agonists, 5-hydroxytrypamine (5-HT) and acetylcholine (ACH) induced a concentration-dependent contraction of the airways. High concentrations of KCl induced twitching of the airway SMCs but had little effect on airway size. 5-HT and ACH induced asynchronous oscillations in [Ca2+]i that propagated as Ca2+ waves within the airway SMCs. The frequency of the Ca2+ oscillations was dependent on the agonist concentration and correlated with the extent of sustained airway contraction. In the absence of extracellular Ca2+ or in the presence of Ni2+, the frequency of the Ca2+ oscillations declined and the airway relaxed. By contrast, KCl induced low frequency Ca2+ oscillations that were associated with SMC twitching. Each KCl-induced Ca2+ oscillation consisted of a large Ca2+ wave that was preceded by multiple localized Ca2+ transients. KCl-induced responses were resistant to neurotransmitter blockers but were abolished by Ni2+ or nifedipine and the absence of extracellular Ca2+. Caffeine abolished the contractile effects of 5-HT, ACH, and KCl. These results indicate that (a) 5-HT and ACH induce airway SMC contraction by initiating Ca2+ oscillations, (b) KCl induces Ca2+ transients and twitching by overloading and releasing Ca2+ from intracellular stores, (c) a sustained, Ni2+-sensitive, influx of Ca2+ mediates the refilling of stores to maintain Ca2+ oscillations and, in turn, SMC contraction, and (d) the magnitude of sustained airway SMC contraction is regulated by the frequency of Ca2+ oscillations.
Keywords: laser scanning confocal microscopy, asthma, airways, arterioles, mouse, lung slices
INTRODUCTION
Gas exchange in the lungs requires an appropriate matching of ventilation to blood perfusion and this is influenced by the caliber of the airways and blood vessels. Consequently, an understanding of the mechanisms that control the size of airways and arterioles is required to understand lung physiology and the development of obstructive lung disease and pulmonary hypertension. In general, the mechanisms that control the caliber of intrapulmonary airways or arterioles have been investigated in either whole lungs or isolated smooth muscle cells (SMCs). While these approaches provide valuable data, it is difficult to determine the site or size of airway or arteriole contraction by measurements of air flow in whole lungs or blood pressure in the pulmonary artery. Similarly, it is difficult to relate changes in intracellular Ca2+ concentration ([Ca2+]i) of isolated SMCs to the contractile responses of intact airways and arterioles.
Our solution to investigate how cellular physiology regulates the contraction of the small airways or arterioles was to examine living lung slices that retain many structural and functional properties of the lung. Relatively thick lung slices have been used to study the contractile response of airways (Dandurand et al., 1993; Martin et al., 1996; Minshall et al., 1997; Adler et al., 1998; Duguet et al., 2001; Martin et al., 2001; Wohlsen et al., 2001), but thinner lung slices, combined with confocal microscopy, provided us with the ability to study changes in [Ca2+]i of SMCs that underlie airway contraction (Bergner and Sanderson, 2002a,b, 2003). A subsequent study examined [Ca2+]i in strips of tracheal muscle but these do not preserve the lung architecture or address the physiology of small airways (Kuo et al., 2003). To also examine the equally important role of blood vessels in lung physiology, we modified the lung slice preparation to preserve the structure and function of the arterioles. Although we studied the contraction and relaxation of both the airways and arterioles simultaneously, we focus here, for clarity, on the responses of the airways. We compare the accompanying responses of the arterioles in a second paper (Perez and Sanderson, 2005).
Although 5-hydroxytryptamine (5-HT) is released by neuroendocrine cells in the airway mucosa of animals and humans (Lauweryns et al., 1973; Lommel, 2001; Adriaensen et al., 2003) the role of 5-HT in airway tone is not well understood. In mice, rats, and guinea pigs, 5-HT induced airway contraction, either in vivo, in isolated lungs, or in isolated trachea rings (Levitt and Mitzner, 1989; Eum et al., 1999; Fernandez et al., 1999; Held et al., 1999; Moffatt et al., 2004). However, this contraction seemed sensitive to atropine and 5-HT was thought to act via a muscarinic pathway. In humans, inhaled 5-HT did not produce bronchoconstriction (Raffestin et al., 1985; Cushley et al., 1986) but could facilitate cholinergic-induced bronchoconstriction (Dupont et al., 1999). In asthmatics, plasma levels of 5-HT are elevated and correlate with clinical severity (Lechin et al., 1996), but its role in asthma remains unknown.
Another unresolved aspect of airway SMC contractility is the role of membrane depolarization and the influx of extracellular Ca2+ (Janssen, 2002). Because contraction induced by either KCl or agonists appears to require extracellular Ca2+ (Li et al., 2003), it is believed that a sustained Ca2+ influx via voltage-, store-, or receptor-operated channels mediates contraction. However, sustained elevations of Ca2+ are often toxic to many cell types including SMCs. Consequently, we reexamined the mechanisms of agonist-induced (5-HT or acetylcholine [ACH]) contraction and voltage-dependent (KCl) contraction. We found that 5-HT and ACH induced Ca2+ oscillations in airway SMCs with a frequency that determines the size of the airway contraction. By contrast, KCl induced low frequency Ca2+ oscillations that induced twitching in airway SMCs. Although both mechanisms required an influx of Ca2+, this was found to be primarily required to refill internal stores to maintain the Ca2+ oscillations that mediated contraction.
MATERIALS AND METHODS
Materials
Cell culture reagents were obtained from Invitrogen and GIBCO BRL. Other reagents were obtained from Sigma-Aldrich. Hanks' balanced salt solution (GIBCO BRL) was supplemented with 20 mM HEPES buffer (sHBSS) and adjusted to pH 7.4. K+-sHBSS was prepared by replacing the sodium salts of sHBSS with potassium counterparts. High KCl isotonic solutions were prepared by mixing K+-sHBSS with normal sHBSS to obtain the desired concentration in K+. Hanks' “0” Ca2+ solution was prepared by supplementing sHBSS without Ca2+ and Mg2+ with 0.9 mM MgSO4 and 1 mM of Na2H2-EGTA.
Lung Slices
To preserve the normal morphology and study the physiological response of intrapulmonary airways and blood vessels, we modified the preparation of lung slices (Bergner and Sanderson, 2002a). Male BALB/C inbred mice (Charles River Breeding Labs, Needham, MA), between 7 and 9 wk old, were killed by intraperitoneal injection of 0.3 ml of pentobarbital sodium (Nembutal) as approved by the IACUC of the University of Massachusetts Medical School. The trachea was cannulated with an intravenous (IV) catheter tube with two input ports (20G Intima; Becton Dickinson) and secured with suture thread (Dexon II, 4–0; Davis and Geck) to ensure a good seal. A syringe filled with 3 ml of air was attached to one port while the other port was closed. The chest cavity was opened by cutting along the sternum and the ribs adjacent to the diaphragm. To reduce the intrapulmonary blood vessel resistance and facilitate vessel perfusion with gelatin, the collapsed lungs were gently reinflated to approximate their total lung capacity by injecting ∼1.5 ml of air. A warm (37°C) solution of gelatin (type A, porcine skin, 300 bloom, 6% in sHBSS) was perfused through the intrapulmonary blood vessels, via the pulmonary artery, by inserting the hypodermic needle of an infusion set (SV x S25BL; Terumo Corporation) into the right ventricle of the heart and slowly injecting ∼1 ml of gelatin solution. A small cotton–wool swab soaked in ice-cold sHBSS was placed only on the heart to solidify the gelatin before the perfusion needle was removed. The lungs were deflated by releasing the positive air pressure. A syringe filled with a warm (37°C) solution of 2% agarose (type VII or VII-A: low gelling temperature) in sHBSS was attached to the second port of the catheter. The IV tube was clamped proximal to the trachea and purged of air with the agarose solution by allowing the trapped air to escape via a 27G needle inserted into the IV tube proximal to the clamp. The IV clamp was removed and the lungs were reinflated by injecting ∼1.3 ml of agarose-sHBSS. Subsequently, ∼0.2 ml of air was injected into the airways to flush the agarose-sHBSS out of the airways and into the distal alveolar space. Immediately after agarose inflation, the lungs were washed with ice-cold sHBSS, and the animal was placed at 4°C for 15 min. The lung and heart were removed and placed in sHBSS (4°C) and cooled for an additional 30 min to ensure the complete gelling of the gelatin and agarose.
To cut thin lung slices, a single lung lobe was removed from the respiratory tree by cutting the main bronchus. The lung lobe was trimmed near the bronchus to produce a flat surface that was adhered to the mounting block of a vibratome (model EMS-4000; Electron Microscope Sciences) using cyanoacrylate glue (Krazy glue, Aron α; Electron Microscope Sciences). The mounted lobe was submerged in a bath of sHBSS maintained at ∼4°C. The lung lobe was sectioned into slices ∼130 μm thick starting at the lung periphery. The initial slices consisted mainly of agarose-filled alveoli and respiratory bronchioles, but as slicing advanced deeper into the lobe, small arterioles and airways start to appear. Airways were readily identified by their lining of epithelial cells with beating cilia. Serial sections with these features were collected and transferred individually to wells of a 24-well plate containing DMEM supplemented with antibiotics and anti-mycotics and NaHCO3. Fetal serum was not added because it can contain 5-HT (3–5 μM when used at 10%) that can stimulate contraction of SMCs (Abdullah et al., 1994). Slices were kept in culture media at 37°C and 10% CO2 for up to 3 d. At 37°C, the gelatin in the blood vessel lumen dissolved, leaving the blood vessel lumen empty.
Measurement of the Contractile Response of Airways and Arterioles Induced by Agonists
Lung slices containing at least one airway and an accompanying arteriole that were cut to reveal a transverse section were selected. Care was taken to ensure that the airways were lined with epithelial cells showing ciliary activity and that the blood vessel wall was intact and not collapsed. It was not uncommon to find blood vessels with elements of their walls separated from the lung parenchyma; these vessels were not used. Slices were transferred to a custom-made perfusion chamber consisting of a Plexiglas support for a 45 × 50 mm coverglass. A lung slice was placed in the center of the coverglass and held in place with a small sheet of nylon mesh (210 μm opening; Small Parts Inc.). To ensure the nylon mesh did not influence the contractile responses of the airways and arterioles or interfere with the phase-contrast optics, a small hole was cut in the mesh, and this was centered over the selected airway and arteriole. A second custom-cut (11 × 40 mm) coverglass edged with silicone grease (Valve Sealant; Dow Corning Co.) was placed over the slice and nylon mesh.
Perfusion of the lung slice in the chamber was performed by applying a gravity-fed flow of solution at one end of the glass chamber and suction at the other end of the chamber. The volume of the chamber was ∼100 μl with a perfusion rate of 800 μl/min. The application of different solutions was controlled by using a custom-built perfusion system consisting of eight solution reservoirs (30 ml) connected to a manifold with a single output tube (Warner Instruments Inc.). The flow from each reservoir was regulated by a valve (LFVA; Lee Company) under TTL control generated by an electronic interface and the image acquisition software.
For phase-contrast microscopy, lung slices were observed on an inverted microscope (Diaphot 300; Nikon) with a 20× objective, a zoom adaptor, and a reducing lens to give a magnification of 0.6–1.0 μm/pixel of the CCD camera (model TM-6710; Pulnix) depending on the zoom setting. Images were recorded from the CCD camera using a computer interface (Road Runner; BitFlow, Inc.) and image acquisition software (Video Savant; IO industries, Inc.). Digital images (648 × 484 pixels) were recorded in time lapse (0.5 Hz) and stored directly on a hard drive. Images were converted to stacks of TIFF images and analyzed with the PC software NIH Image/Scion image (Scion Corporation; download: www.scioncorp.com). After image calibration and the selection of an appropriate grayscale threshold to distinguish the lumen from the surrounding tissue, the area of the airway and arteriole lumen was calculated, with respect to time, by pixel summing. Values were normalized to the prestimulation luminal area (initial area). All measurements were performed at room temperature.
Measurements of Intracellular Ca2+
A stock solution of 2 mM Oregon green 488 BAPTA-AM (Molecular Probes) was prepared with 20 μl anhydrous DMSO and 50 μg Oregon green-AM. An equal volume (20 μl) of 20% Pluronic F-127 (Molecular Probes) in anhydrous DMSO was mixed with the stock solution and diluted in 2 ml of sHBSS containing 100 μM sulfobromophthalein (an inhibitor used to prevent dye extrusion through anion exchangers) to produce a final loading solution of 20 μM Oregon green-AM, 0.2% pluronic, and 100 μM sulfobromophthalein. Slices were incubated for 30 min at 30°C followed by 30 min at room temperature in the loading solution, followed by an additional hour at room temperature in sHBSS containing 100 μM sulfobromophthalein to allow for the deesterification of the dye. Lung slices were mounted in the perfusion chamber as described above.
Fluorescence imaging was performed using a custom-built video-rate confocal microscope (Sanderson and Parker, 2003) modified to capture four channels simultaneously (Sanderson, 2004). In brief, the 488-nm line of an argon or diode laser was scanned across the specimen with two oscillating mirrors (for X- and Y-scan) in an inverted microscope. The resultant fluorescence (>510 nm) was detected by a photomultiplier tube (PMT). A video frame capture board (Raven; Bit Flow, Inc.) digitizes the PMT signal (1 PMT per channel) to form an image (480 pixels × 400 lines) that was recorded using the software Video Savant to a hard disk (a stripped SCSI volume). For time lapse, a recording rate of 2 Hz was used, otherwise video rate (30 Hz) was used. For the higher speed recordings of Ca2+ waves and elemental Ca2+ events, images were acquired at 60 Hz (480 pixels × 170 lines). A line-scan analysis of these images was performed by extracting a row of pixels from each image and placing them sequentially, as a time sequence, in a single image. Alternatively, changes in fluorescence intensity within the images were analyzed by selecting regions of interest (ROI) of ∼5 × 5 pixels in a single SMC. Average fluorescence intensities of an ROI were obtained, frame-by-frame, using the Scion Image software with custom written macros that allow tracking of the ROI within an SMC as it moved with contraction. When necessary, a bleach correction was calculated from the bleaching rate observed during a period of 30 s before the perfusion of drugs. Final fluorescence values were expressed as a fluorescence ratio (Ft/F0) normalized to the initial fluorescence (F0).
Immunocytochemistry
Lung slices were washed in sHBSS, fixed with cold 100% acetone (−20°C) for 20 min, and washed in sHBSS containing 1 mg/ml BSA (sHBSS-BSA). Antibodies were diluted 1:250 in sHBSS-BSA. The slices were incubated for 1 h at room temperature with a FITC-conjugated mouse monoclonal antibody against α-smooth muscle actin (Sigma-Aldrich). Slices were washed three times in sHBSS-BSA, and fluorescence was recorded with the confocal microscope using 488 nm excitation light and a bandwidth >510 nm for emission. Phase-contrast images were recorded simultaneously with the fluorescence images by using a second PMT to detect the laser light transmitted by the specimen.
Statistics
A paired Student's t test was used to test for significant differences between means. All statistical values are expressed as mean ± SEM.
Online Supplemental Material
Videos consisting of sequences of phase-contrast or fluorescence images were produced in Video Savant by exporting the images as “mpeg” files. To show the time of addition of agonists, labels were added to each frame using a custom written script file. Videos are presented in time lapse and the playback speed is indicated for each video. Videos 1–4 are available at http://www.jgp.org/cgi/content/full/jgp.200409216/DC1.
RESULTS
Characteristics and Morphology of Lung Slices
Throughout the lungs, the intrapulmonary airways and arteries have a close anatomical association that follows a parallel course. As a result, an airway and an accompanying arteriole (a bronchiole–arteriole pair) was easily identified and visualized in a single microscopic field of view (Fig. 1, A and B). This also makes the identification of pulmonary veins (not shown) easier because they are found as individual structures at some distance away from the bronchiole–arteriole pair. This anatomical separation precludes a direct comparison of the arteriole and vein responses in the same experiment. Each bronchiole–arteriole pair, when observed in transverse section, usually consists of a larger airway and a smaller arteriole. The airway is characterized by a lining of cuboidal epithelial cells with actively beating cilia. The arteriole lumen is lined with a low profile, squamous endothelium. Both structures are surrounded by a dense layer of tissue that often has a fibrous appearance. More distally, the airway and arteriole are surrounded by the alveolar parenchyma consisting of thin-walled sacs (Fig. 1, A and B). Specific antibody staining for SMC α-actin (Fig. 1 A) reveals that the SMCs are located in the surrounding fibrous layer, directly below the epithelium or endothelium. It is important to note that in the lung slices used, only the alveoli remain filled with agarose. Before gelling, the agarose is flushed out of the airways with air. The gelatin is absent from the arterioles because it dissolves during incubation at 37°C. Consequently, the luminal compartments do not offer resistance to contraction. Agarose does not dissolve at 37°C but remains in the alveoli to keep the alveoli inflated and airways open.
Figure 1.
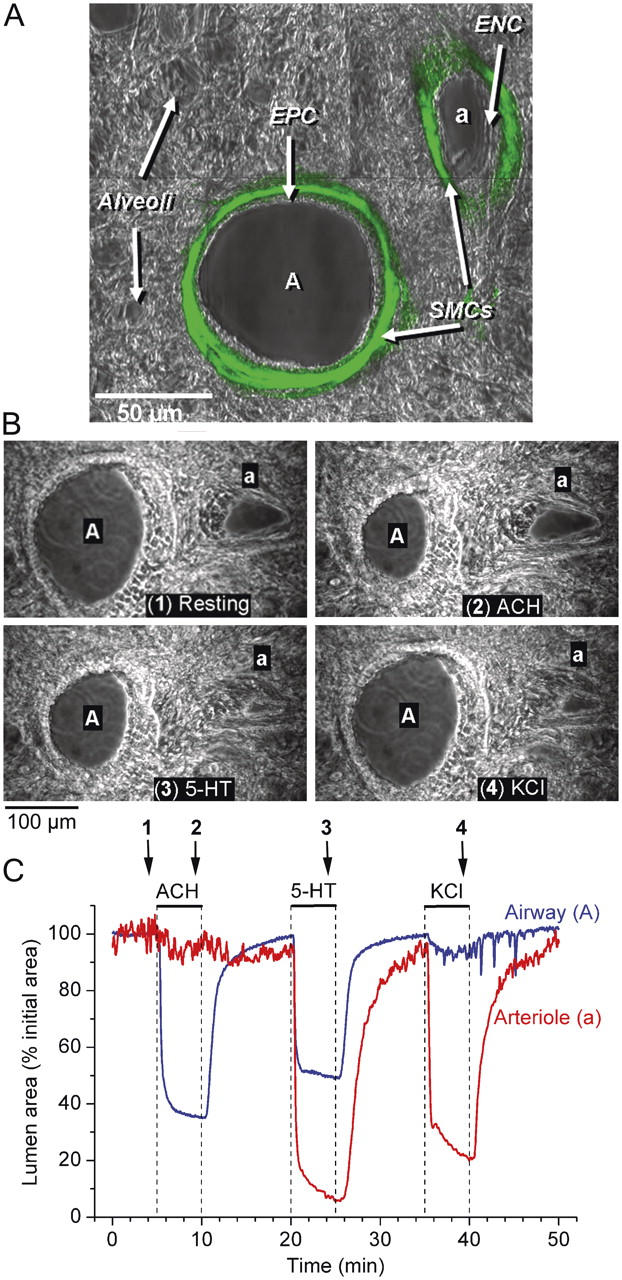
The localization of SMCs and the contractile responses of an airway and arteriole in a lung slice induced by ACH, 5-HT, and KCl. (A) A lung slice was fixed and stained with FITC-conjugated antibodies against smooth muscle α-actin. Fluorescence and phase-contrast images were recorded with a confocal microscope. The fluorescence image was assigned a green pseudo-color and superimposed on the phase-contrast image to show colocalization. The final image consists of a montage of six different images. The bronchiole airway (A) and the accompanying arteriole (a) are surrounded by alveolar parenchyma. A prominent layer of SMCs (green) is located below the epithelial (EPC) and endothelial (ENC) cells of the bronchiole and arteriole, respectively. (B) A series of phase-contrast images, recorded at different times (indicated by arrows in C) showing the appearance of an airway and arteriole before (1) and after stimulation with 1 μM ACH (2), 1 μM 5-HT (3), and 100 mM KCl (4). (C) The cross-sectional area of the lumen of an airway (blue line) and arteriole (red line) with respect to time in response to ACH, 5-HT, and KCl (top bars). ACH induced a contraction of the airway but not of the arteriole. 5-HT induced a greater contraction in the arteriole than in the airway. KCl induced twitching in both the airway and arteriole and a sustained contraction of the arteriole. Upon washout of agonists or KCl, the airway and arteriole relaxed. A movie of these data is shown in Video 1 (available at http://www.jgp.org/cgi/content/full/jgp.200409216/DC1). Representative experiment of six different slices from three mice.
The Contractile Response of Bronchiole–Arteriole Pairs to ACH, 5-HT, and KCl
To establish the contractile sensitivity of bronchiole–arteriole pairs to ACH, 5-HT, and high KCl, we measured changes in the luminal area with respect to time. In response to 1 μM ACH, the airway contracted and reduced its lumen area by ∼60% within 2 min (Fig. 1, B and C, blue line; Video 1, available at http://www.jgp.org/cgi/content/full/jgp.200409216/DC1) and after 5 min, the average airway luminal area had decreased to 57 ± 7% (n = 8 slices from 3 mice). Upon washout of ACH, the airway quickly relaxed, attaining 80–90% of its initial size within 3 min, but continued to slowly relax over the next 5–7 min. In contrast to the airways, the arterioles had no contractile response to ACH (Fig. 1 C, red line).
The exposure of the same bronchiole–arteriole pair to 1 μM 5-HT induced the contraction of both the airway and arteriole (Fig. 1, B and C). In the airway, 5-HT induced a contraction at a similar rate compared with that induced by ACH and reduced the luminal area by 41 ± 5% after 5 min (n = 9 slices from 3 mice). In the arteriole, 5-HT induced an initial fast contraction that reduced the lumen by up to 80% within 2 min. This was followed by a second phase of slower contraction. The average arteriole luminal area was reduced by 75 ± 6% after 5 min (n = 6 slices from 3 mice). Upon 5-HT washout, both the airway and arteriole relaxed; however, it is important to note that the airway relaxed quickly, whereas the arteriole relaxation had a delayed onset and occurred more slowly (Fig. 1 C).
The same bronchiole–arteriole pair responded to isotonic sHBSS containing 100 mM KCl with a large reduction of the arterial lumen but only a small reduction of the airway lumen (Fig. 1, B and C). However, unlike the sustained contraction induced by ACH and 5-HT, the contraction induced by KCl was irregular or spasmodic (Video 1). This was especially evident in the airway where uncoordinated transient contractions (or twitching) of individual airway SMCs only resulted in a small decrease in luminal area. After 5 min, KCl had induced a luminal reduction in airways of 14 ± 3% (n = 7 slices from 3 mice) and arterioles of 64 ± 8% (n = 6 slices from 3 mice). Removal of the KCl resulted in arteriole and airway relaxation although some airways continued to display transient contractions (four out of seven).
These results indicate that the SMCs of airways and arterioles respond very differently to different agonists. Consequently, in this paper, we have focused on characterizing and correlating the contractile responses and the underlining Ca2+ signaling of the airway SMCs to 5-HT, ACH, and KCl. The responses of the arteriole SMCs to these agonists are addressed in a separate study (Perez and Sanderson, 2005).
Concentration Dependence of Airway Contraction to 5-HT, ACH, and KCl
To determine the relative sensitivity of the contraction of the airways to 5-HT, ACH, and KCl, we measured the changes in luminal area in response to sequentially increasing concentrations of agonist (Fig. 2). Increasing concentrations of 5-HT or ACH (from 0.01 to 0.5 μM) induced an increasing reduction in luminal size (Fig. 2, A and C). At low concentrations of 5-HT or ACH (0.01 μM), the airway contraction was small and consisted of uncoordinated twitching of the airway wall. At higher agonist concentrations, the airway contraction proceeded quickly and smoothly without twitching. While a maximal luminal reduction was induced by concentrations of ≥1 μM for each agonist, the maximal response to 5-HT was less than that induced by the maximal concentration of ACH (Fig. 2 C). This relative sensitivity was verified in each lung slice by the addition of the complementary agonist at the end of each experiment (Fig. 2 A). The addition of 10 μM ACH, after 10 μM 5-HT, resulted in a larger contraction (69% and 59%, respectively; Fig. 2 A). Conversely, the addition of 10 μM 5-HT, after the addition of 10 μM ACH, resulted in a smaller airway contraction (38% and 49%, respectively). 5-HT induced a smaller airway contraction than ACH at all concentrations (Fig. 2 C). An average maximal contraction of 48 ± 4% (n = 5 from 3 mice) and 59 ± 2% (n = 6 from 3 mice) was induced by 10 μM 5-HT and ACH, respectively. The half maximal concentration for airway contraction (EC50) was 31 and 30 nM for 5-HT and ACH, respectively.
Figure 2.

The effect of concentration on 5-HT–, ACH-, and KCl-induced airway contraction. Lung slices with airways of similar sizes were stimulated with increasing concentrations of 5-HT, ACH, or KCl (perfusion time of 8 min). Relaxation of the airways between exposure to agonists or KCl was achieved by washing with sHBSS for 10 min. An image was recorded every 2 s and the airway lumen cross-sectional area was calculated and plotted with respect to time. (A) 5-HT induced a concentration-dependent increase in the contraction of airways in the range of 0.01–0.5 μM. Stimulation with 10 μM ACH induced a larger contraction as compared with 10 μM 5-HT. (B) A representative experiment showing the effect of increasing isotonic concentrations of KCl on the contraction of airways. (C and D) Summary of the concentration-dependent contractility of the airways to (C) ACH (open squares) and 5-HT (closed circles) or (D) KCl, calculated after 5 min of agonist or KCl exposure. Each point represents the mean ± SEM from at least three different experiments on different slices from at least two mice. For each agonist, the contractility data were fitted with a logistic function curve.
Exposure of the airway to a range of isotonic KCl concentrations between 25 and 150 mM commonly induced a transient contraction that subsided into a smaller sustained contraction of the lumen that varied little over the range of KCl concentrations tested (Fig. 2, B and D). It is worth noting that, at all KCl concentrations, the small overall contraction was very similar to that induced by a very low concentration of agonist and was achieved by uncoordinated twitching. However, the maximum response to KCl was small, consisting of a contraction of only 11 ± 1% in response to 100 mM KCl (n = 4 from 3 mice). The order of the sensitivity of the contractile response of airways was ACH ≥ 5-HT >>> KCl.
Ca2+ Oscillations in Airway SMCs Induced by 5-HT and ACH
In response to 1 μM 5-HT, the SMCs of the airway responded with an initial increase in [Ca2+]i that was quickly followed by the occurrence of oscillations in [Ca2+]i (Fig. 3). These Ca2+ oscillations occurred asynchronously, with each cell displaying repetitive increases in [Ca2+]i at different times with respect to neighboring SMCs (Fig. 3; Video 2, available at http://www.jgp.org/cgi/content/full/jgp.200409216/DC1). Individual Ca2+ oscillations occurred as Ca2+ waves propagating throughout the length of the cell (Video 3; also shown in Fig. 13), but no Ca2+ waves were observed to spread from one SMC to an adjacent SMC. The initiation of the Ca2+ oscillations in the SMCs was accompanied by a sustained contraction of the airway; the SMCs shortened and were displaced toward the airway lumen (Video 2).
Figure 3.
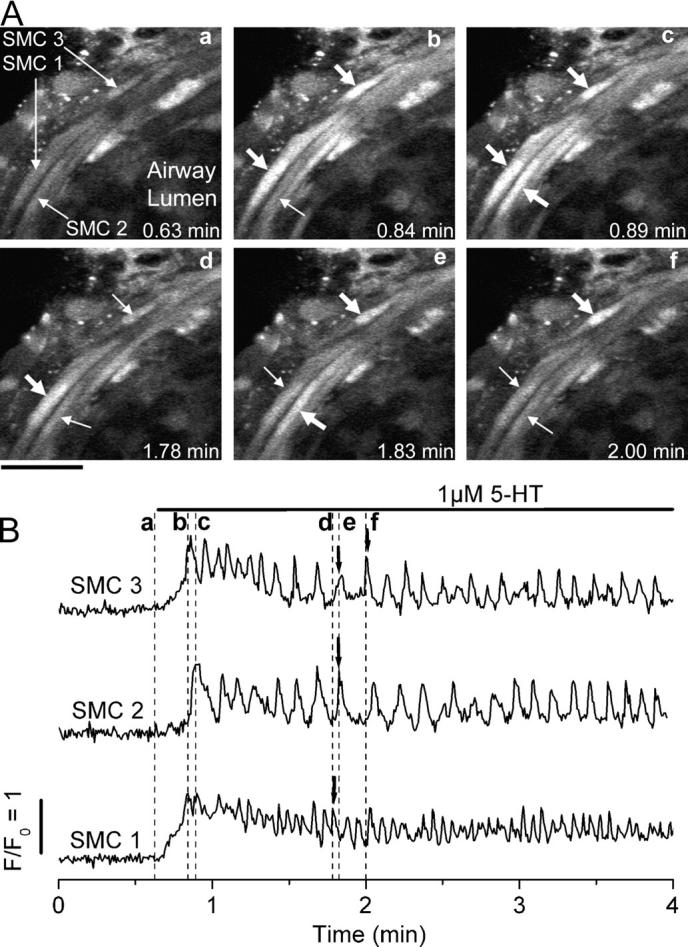
Ca2+ signaling induced by 5-HT in airway SMCs in lung slices. (A) Selected images recorded at times indicated in each panel (a–f) (and dashed lines in B) during the exposure to 5-HT. Bar, 20 μm. The responses of three different SMCs (indicated by arrows in a) are highlighted. After stimulation with 1 μM 5-HT, the fluorescence in each SMC increases and begins to oscillate asynchronously (thick arrows, b–f) above the basal level (thin arrows). Due to airway contraction, the cells are displaced toward the bottom right. (B) In all three airway SMCs, the Ca2+ signaling induced by 5-HT was characterized by an initial increase in [Ca2+]i followed by Ca2+ oscillations. Fluorescence images were recorded at 2 Hz. The fluorescence changes from small ROIs (∼5 × 5 pixels), as defined in the SMCs as indicated in A, a, were plotted as a ratio (Ft/F0) with respect to time. A representative experiment of at least five trials from different slices from three mice is shown. A movie showing the effect of 5-HT on Ca2+ signaling in airway SMCs is shown in Video 2 (available at http://www.jgp.org/cgi/content/full/jgp.200409216/DC1).
Figure 13.

Ca2+ waves and elemental Ca2+ events in airway SMCs during the stimulation with 5-HT, ACH, and KCl. Line scans from the longitudinal axes of single airway SMCs (inset, a) from high speed recordings (60 fps) of changes in Ca2+ during continuous perfusion with 1 μM 5-HT, 1 μM ACH, and 50 mM KCl and after removal of KCl by washing with HBSS, as indicated. The slopes of the white lines indicate the velocity (μm/s) and direction of the Ca2+ waves in the SMCs. KCl induce small Ca2+ events (arrows) preceding the Ca2+ wave. (Inset, b) An expanded view of a single Ca2+ event (image and trace) induced by KCl. Representative traces of at least four experiments from different slices of two mice.
With the exception of differences in frequency, the basic characteristics of the Ca2+ signals induced by ACH appeared similar to those induced by 5-HT (Fig. 4). In general, the pattern of Ca2+ oscillations consisted of two phases. The initial phase consisted of low amplitude and high frequency Ca2+ oscillations superimposed on an elevated [Ca2+]i that lasted ∼1 min (Fig. 4, A and C). During the second phase, the Ca2+ oscillations occurred with a stable frequency and similar amplitude superimposed on a decreasing baseline of [Ca2+]i (Fig. 4, B and D, and Fig. 8). This second phase persisted for the remaining time while the agonist was present (see Fig. 8, C and D). Each individual Ca2+ oscillation consisted of an initial rapid Ca2+ transient followed by a slower phase of declining [Ca2+]i. The mean duration of the Ca2+ oscillation in 1 μM 5-HT or ACH was 0.7 ± 0.2 s or 0.8 ± 0.2 s, respectively. Although the magnitude of the increase of the fluorescence (Ft/F0) during the first and subsequent Ca2+ oscillations fluctuated between values of 1.5 and 3.0, no major differences between agonists were identified.
Figure 4.
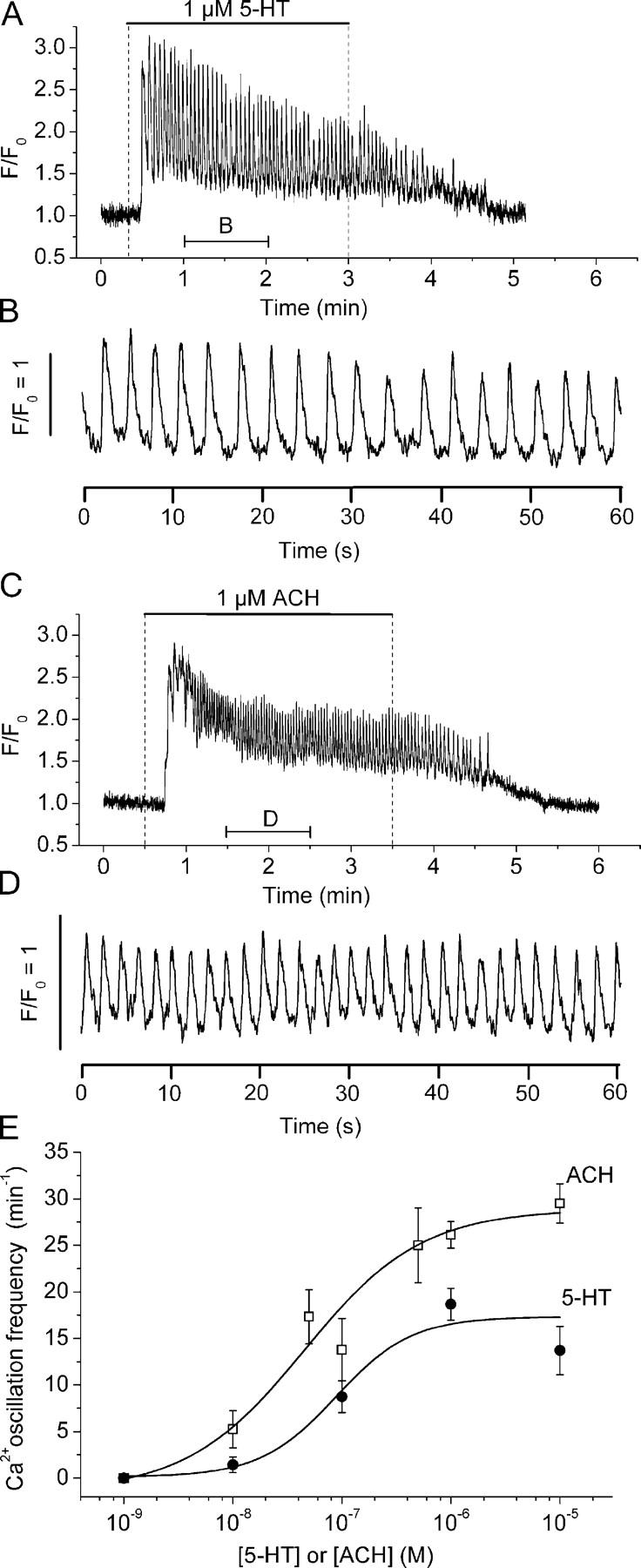
Comparison of Ca2+ oscillations induced by 5-HT and ACH in airway SMCs. Airway SMCs of lung slices stimulated with (A) 1 μM 5-HT or (C) 1 μM ACH. The Ca2+ responses to these agonists were characterized by an increase in [Ca2+]i followed by Ca2+ oscillations that persisted until the removal of the agonist. (B and D) An expanded region of 1 min, indicated by the lower bar in A and C, to show the details of the Ca2+ oscillations induced by each agonist. Representative traces of at least three experiments from different slices from two mice. A movie of the first 1 min after stimulation with 5-HT is shown in Video 3 (available at http://www.jgp.org/cgi/content/full/jgp.200409216/DC1). (E) Concentration–response curves for the frequency of Ca2+ oscillations induced by 5-HT (solid circles) and ACH (open squares). Changes in [Ca2+]i were determined in single SMCs for each agonist concentration in separate experiments. Each point represents the mean ± SEM from at least five different cells from different slices from at least two mice. Data points of 5-HT and ACH were each fitted with a logistic function curve.
Figure 8.
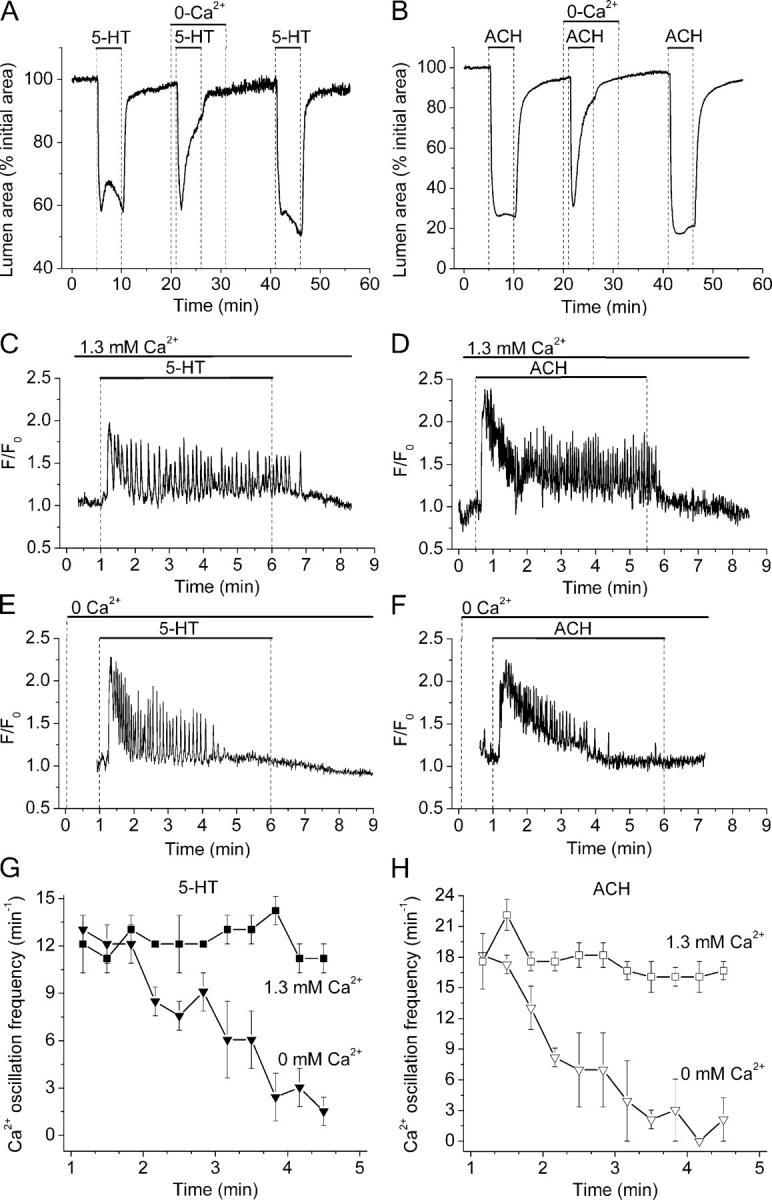
Role of extracellular Ca2+ during the contraction and the Ca2+ signaling of airway SMCs induced by 5-HT and ACH. (A and B) Contractile responses of airways in different lung slices sequentially exposed (top bars) to 1 μM 5-HT and 1 μM ACH in the presence or absence of extracellular Ca2+. Representative traces of five airways from three mice. (C–H) The changes in [Ca2+]i in small ROIs of single SMCs were determined in lung slices stimulated with (C, E, and G) 1 μM 5-HT or (D, F, and H) 1 μM ACH in the (C and D) presence or (E and F) absence of extracellular Ca2+ as indicated by top bars. In the absence or presence of extracellular Ca2+, both agonists induced an increase in [Ca2+]i followed by Ca2+ oscillations. Although Ca2+ oscillations persisted in the presence of extracellular Ca2+, they progressively reduce their frequency and stopped in the absence of extracellular Ca2+. (G and H) The frequency of agonist-induced Ca2+ oscillations, calculated every 20 s, in the presence (squares) or absence of Ca2+ (triangles) with respect to time. Symbols and bars are the mean ± SEM of five to eight SMCs in five paired experiments from different lung slices from three mice.
In the range from 10−8 to 10−6 M, both 5-HT and ACH induced a concentration-dependent increase in the frequency of the Ca2+ oscillations calculated during the second phase (Fig. 4 E). At all concentrations, 5-HT induced a Ca2+ oscillation frequency that was slower than that induced by ACH. For example, 1 μM 5-HT induced a frequency of 18 ± 2 cycles min−1 (n = 5 slices of 3 animals) while 1 μM ACH induced a frequency of 26 ± 2 cycles min−1 (n = 6 slices of 3 animals, P = 0.018, t test.). The half maximum concentration was 88 and 45 nM for 5-HT and ACH, respectively.
Ca2+ Signaling in Airway SMCs Induced by KCl
High concentrations of KCl also induced increases in [Ca2+]i and Ca2+ oscillations (Fig. 5; Video 4). However, the KCl-induced Ca2+ oscillations occurred more slowly and an initial phase of high frequency Ca2+ oscillations was absent. The frequency of the Ca2+ oscillations induced by KCl was low (one to three per minute) at all concentrations tested (25–100 mM; Fig. 5 D). An average frequency of 2.8 ± 1.4 cycles min−1; (n = 4 slices of 3 animals) occurred at 50 mM KCl but this was not significantly different from the frequencies recorded at higher or lower KCl concentrations. Each Ca2+ transient or oscillation had a long duration (11.8 ± 2.6 s) (Fig. 5 B). In some cases, the Ca2+ oscillations of neighboring cells had similar frequencies and appeared to be synchronized (four airways, Fig. 5). However, in other cases, neighboring cells showed asynchronous Ca2+ oscillations with different frequencies (four airways). In each case, the Ca2+ oscillations were accompanied by local transient contractions of either multiple or individual cells (Fig. 5 C; Video 4, available at http://www.jgp.org/cgi/content/full/jgp.200409216/DC1).
Figure 5.
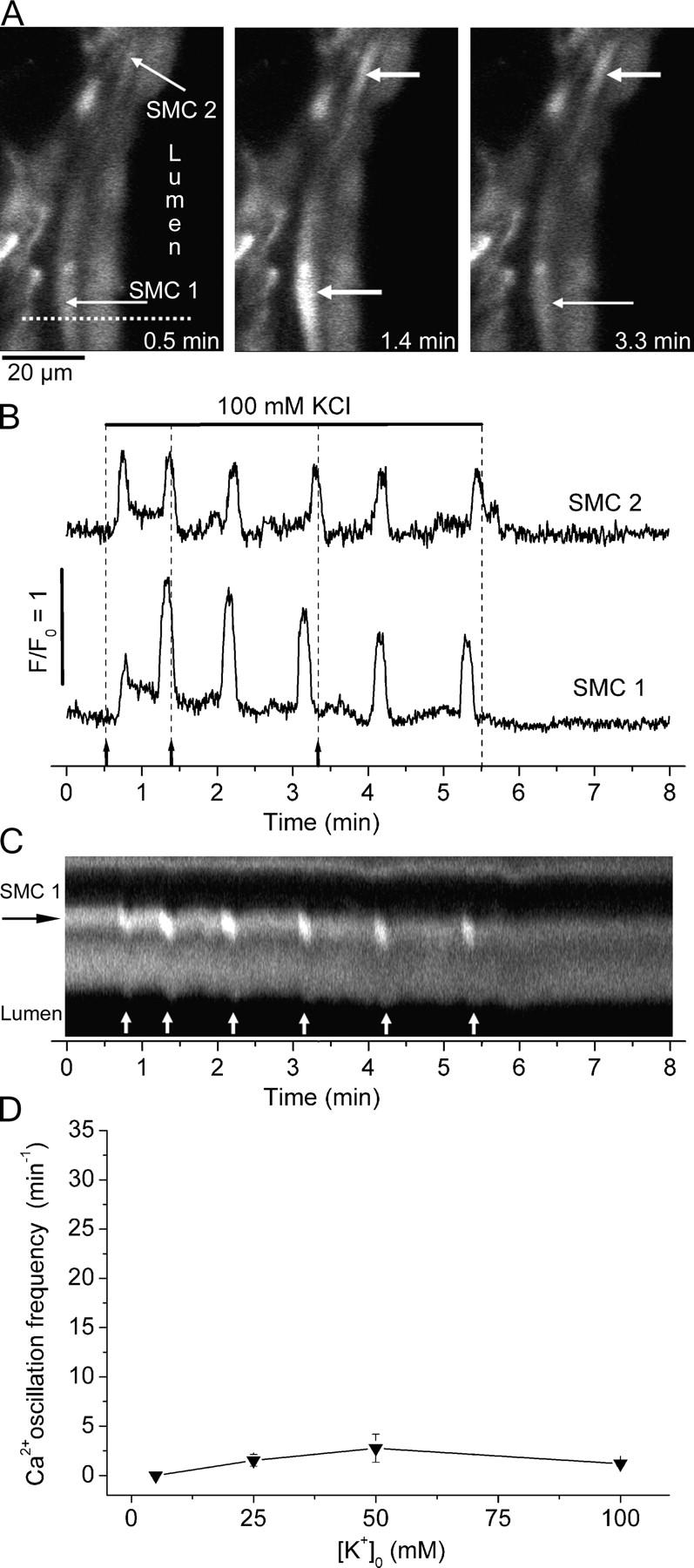
Ca2+ signaling induced by KCl in airway SMCs. (A) Selected images taken at different times (indicated in B by arrows and dashed lines) during the exposure to 100 mM KCl (bar) showing two cells responding with transient increases in [Ca2+]i (thick arrows) over a basal level (thin arrows). (B) Fluorescence changes acquired from a 5 × 5 pixel ROI within each SMC indicated in A showing the Ca2+ oscillations induced by KCl. (C) A line-scan plot, from the dotted line indicated in A, showing the Ca2+ oscillations induced by KCl in SMC 1 (white lines) and the accompanying transient contraction (downward deflections, arrows) toward the lumen (lower black area). (D) Concentration–response curve for the frequency of Ca2+ oscillations induced by KCl. Each point represents the mean ± SEM from six different cells from three different slices from three mice. Data points were joined with a straight line. A movie of the effect of KCl is shown in Video 4 (available at http://www.jgp.org/cgi/content/full/jgp.200409216/DC1).
Correlation of Airway Contraction with Ca2+ Oscillation Frequency
The correlation between the increasing frequency of the Ca2+ oscillations and the increasing airway contraction with increasing concentrations of 5-HT and ACH suggests that frequency of the Ca2+ oscillations determines the extent of airway contraction (Fig. 6). By contrast, an increasing correlation between airway contraction and the frequency of Ca2+ oscillations induced by KCl did not exist because the frequencies of the Ca2+ oscillations induced by a range of KCl concentrations were all very low and similar (Fig. 6). However, the contraction induced by these low frequency Ca2+ oscillations in response to high KCl matched the contraction induced by low frequency Ca2+ oscillations induced by low doses of agonists.
Figure 6.

Relationship between Ca2+ oscillation frequency and airway contraction induced by 5-HT, ACH, and KCl. Data from concentration–response curves of the frequency of the Ca2+ oscillations and the contractility for each agonist were replotted. Data were fitted with a sigmoidal curve (Boltzmann). For ACH (open squares) and 5-HT (filled circles), the airway contraction increases as a saturating function of the frequency of the Ca2+ oscillations. The low frequency Ca2+ oscillations induced by KCl (filled triangles) induced only a small contraction.
Type of 5-HT Receptor Involved in Airway Contraction
Because 5-HT2 and 5-HT3 receptors are known to produce increases in [Ca2+]i, 5-HT–specific antagonists and agonists were used to determine which receptor type mediates airway contraction. Ketanserin, a 5-HT2–specific antagonist, at 10 nM blocked the contraction induced by 1 μM 5-HT (Fig. 7 A). In addition, sequential stimulation with 10−8, 10−7, and 10−6 M DOI (2, 5-dimethoxy-4-iodoamphetamine hydrochloride), a 5-HT2–specific agonist, induced reversible airway contraction (three airways from three mice). On the other hand, SR 57227 (4-amino-1-(6-chloro-2-pyridyl)-piperidine hydrochloride), a 5-HT3 agonist, did not induce a substantial contraction at 10−7 or 10−6 M (the decrease in airway lumen area was <10%, three airways from three mice). These results suggest that the 5-HT2 receptor is responsible for mediating 5-HT responses.
Figure 7.

Type of receptor involved in the contraction of airways induced by 5-HT. (A) An airway sequentially stimulated with 1 μM 5-HT in the absence or presence of 10−8 M ketanserin (a specific 5-HT2 antagonist) as indicated by bars. (B) An airway sequentially stimulated with 1 μM 5-HT and 1 μM ACH in the absence or presence of 1 μM atropine as indicated by top bars. Atropine has no effect on 5-HT–induced airway contraction but completely blocked or relaxed ACH-induced contraction. Representative traces of at least three different slices from three mice.
Alternative Mechanisms of Action for 5-HT and KCl
Because a cholinergic pathway has been proposed as a mechanism by which 5-HT induced airway contraction in trachea and isolated lungs (Levitt and Mitzner, 1989; Eum et al., 1999; Fernandez et al., 1999; Held et al., 1999; Moffatt et al., 2004), we examined the effect of atropine on the 5-HT responses of lung slices to determine if any 5-HT effects occurred indirectly. Atropine (1 μM) had no effect on the airway contractile response (Fig. 7 B) when added, either before or after exposure to 1 μM 5-HT. By contrast, 1 μM atropine totally abolished the airway contractile response induced by 1 μM ACH. Similarly, 1 μM atropine induced a full relaxation of an airway that was precontracted with ACH (Fig. 7 B). These results indicate that 5-HT does not act via muscarinic receptors in the small airways of mice.
Although we believe that KCl is acting via membrane depolarization of the SMCs, it is possible that KCl may also act via membrane depolarization of neural terminals within the lung slice to locally release neurotransmitters. However, we found that KCl induced contraction of the airways in the presence of atropine and ketanserin, receptor antagonists that fully blocked the effect of ACH and 5-HT, respectively. Similarly, the possibility that KCl acts via the release of an α1-agonist was ruled out by the fact that the airways were totally unresponsive to phenylephrine. Because ATP (but not AMP or adenosine) can stimulate airway contraction (Bergner and Sanderson, 2002b) and may be coreleased from nerve terminals, we added 10 U/ml apyrase, a potent ATPase, during KCl stimulation. Under these conditions, a twitching response of the airways similar to that induced by KCl alone was observed. In addition, it should be noted that during agonist or KCl exposure, the lung slices were under constant perfusion, a condition that would be expected to quickly wash away any small concentrations of agonists released from nerves. However, the twitching response induced by KCl persisted with an approximately constant frequency during several minutes of perfusion with KCl. Furthermore, this response was elicited by several sequential stimulations with KCl (Fig. 2 B, Fig. 5 B, and Fig. 12), which would have been expected to run-down the release of agonists from isolated nerve terminals.
Figure 12.

Effect of caffeine and CPA on airway contraction induced by KCl. (A) Airways were sequentially stimulated with 50 mM KCl in the absence or presence of 20 mM caffeine (top bars). (B) A line-scan obtained from phase-contrast images in a region similar to that shown by the dotted line in Fig. 5 A during sequential stimulation with KCl in the absence or presence of 10 μM CPA (top bars). Twitches induced by KCl are observed as transient signals toward the lumen of the airway and represent the local displacement of cells produced by the contraction of one or a few SMCs. A few spontaneous twitches were observed before exposure to KCl. Representative data of at least four experiments from different slices from three mice.
Role of Extracellular Ca2+ in SMC Contraction and [Ca2+]i Signaling
In the presence of extracellular Ca2+, 5-HT or ACH stimulated a sustained contraction of the airway (Fig. 8, A and B). In the absence of Ca2+, 5-HT or ACH induced a similar initial contraction but this was followed by relaxation (Fig. 8, A and B). In Ca2+-free conditions, the contraction induced by ACH was 83% at 1 min but only 11% after 5 min. Similar relative changes were obtained with 5-HT (Fig. 8 A). These results indicate that the extracellular Ca2+ was not necessary to trigger the contraction but that Ca2+ was required to sustain airway contraction.
The basis for these two different responses was found by examining the changes in intracellular Ca2+ concentration ([Ca2+]i). In the presence of extracellular Ca2+, both 5-HT and ACH induced an initial increase in [Ca2+]i followed by Ca2+ oscillations (Fig. 8, C and D). After the initial settling period, the frequency of the Ca2+ oscillations remained steady during the presence of the agonist (Fig. 8, G and H). In the absence of extracellular Ca2+, the initial increase in [Ca2+]i and stimulation of Ca2+ oscillations was observed, but the frequency of the Ca2+ oscillations began to decline after ∼1.5 min, and the Ca2+ oscillations ceased ∼2–3 min later (Fig. 8, E–H). This progressive decrease in the frequency of the Ca2+ oscillations correlated with the progressive relaxation of the airway in the absence of Ca2+ (Fig. 8, A and B). By contrast, in the absence of extracellular Ca2+, high KCl did not stimulate a change in the [Ca2+]i, a result that correlates with the inability of KCl to induce airway contraction under these conditions (four airways from three mice).
Effect of Ni2+ and Nifedipine on Airway Contraction
The extent of the Ca2+ influx during agonist- or KCl-induced contraction was investigated with Ca2+ channel blockers. Stimulation with 1 μM 5-HT in the presence of 1 mM Ni2+ induced an airway contraction that was slightly smaller in magnitude than the control (Fig. 9 A). Similarly, stimulation with 1 μM ACH in the presence of 1 mM Ni2+ induced airway contraction, although the airway subsequently showed a slow relaxation (Fig. 9 B). Airway contraction induced by 5-HT or ACH was not affected by 10 μM nifedipine. By contrast, both nifedipine and Ni2+ completely blocked airway contraction stimulated by high KCl. These results indicate that while L-type Ca2+ channels may have a significant role in KCl-induced contraction, they have little influence in agonist-induced contraction and suggest that other Ca2+ channels serve to sustain contraction.
Figure 9.

Effect of Ni2+ on Ca2+ signaling and contraction induced by 5-HT and ACH. (A and B) Airways were sequentially stimulated with (A) 1 μM 5-HT or (B) 1 μM ACH in the absence or presence of 1 mM NiCl2 (top bars). Representative traces of at least four experiments from different slices from three mice. (C) Simultaneous recordings of changes in [Ca2+]i (top trace) in single SMCs and contraction of the airway lumen (bottom black trace) during the stimulation with ACH in the presence or absence of 1 mM NiCl2 as indicated by top bars. The frequency of the agonist-induced Ca2+ oscillations (gray trace) was calculated every 20 s. Symbols and bars are the mean ± SEM of six SMCs in five experiments from different lung slices from two mice.
To investigate the cause of the slow relaxation of the airway observed during the stimulation with ACH in the continued presence of Ni2+, we simultaneously measured the changes in SMC [Ca2+]i and contraction of the airway (Fig. 9 C). After the initial increase in [Ca2+]i and the onset of Ca2+ oscillations, the frequency of Ca2+ oscillations progressively decreased and this was accompanied by the relaxation of the airway. After removal of Ni2+, but still in the presence of ACH, the frequency of the Ca2+ oscillations increased again and the airway recontracted. These results suggest that the Ni2+ blocked Ca2+ channels that contribute to the maintenance of the Ca2+ oscillations and perhaps serve to refill internal Ca2+ stores.
The Contribution of Ca2+ Influx to Airway Contraction
To further investigate the mechanisms and Ca2+ source used during agonist and KCl-induced contractions and Ca2+ oscillations, lung slices were exposed to caffeine to empty internal stores by opening ryanodine receptors (RyRs). In response to sustained exposure of caffeine, the airways displayed a single transient contraction (Fig. 10 A). The maximal contraction (38 ± 6%, n = 12 slices from 4 mice) was attained within 8–10 s, and after 1 min, the airways were almost completely relaxed (5 ± 3%, n = 12 slices from 4 mice).
Figure 10.

Ca2+ signaling and contraction induced by caffeine in airway SMCs. (A) Repetitive contractile responses of an airway to sequential stimulation with 20 mM caffeine as indicated by top bars. (B, C, D, and E) Simultaneous recordings of changes in [Ca2+]i in single SMCs and contraction of the airway lumen during the stimulation with 20 mM caffeine in the presence or absence of extracellular Ca2+ or 1 mM NiCl2 as indicated by top bars. Representative traces of at least four experiments from different slices of two mice.
Under similar conditions, caffeine induced an initial transient increase in [Ca2+]i that was followed by a sustained plateau of increased [Ca2+]i (Fig. 10 B). The average peak and plateau ratio values were 2.8 ± 0.4 and 1.7 ± 0.3, respectively (n = 6 slices from 2 animals). Simultaneous recording of the changes in airway lumen area showed that the transient contraction accompanied the transient increase in Ca2+. However, immediately following the Ca2+ transient, the airway relaxed and remained relaxed during the decline in Ca2+ and during the extended period of elevated Ca2+. The caffeine-induced plateau of elevated Ca2+ was quickly abolished when extracellular Ca2+ was withdrawn (Fig. 10 C). Similarly, the elevated Ca2+ plateau was reduced when extracellular Ni2+ was added (Fig. 10 D). These manipulations did not alter the relaxed state of the airway. In the absence of extracellular Ca2+, caffeine was still capable of inducing a transient Ca2+ increase and contraction, but a sustained increase in Ca2+ did not occur (Fig. 10 E). However, the replacement of extracellular Ca2+ resulted in a rapid and sustained increase in Ca2+ that was accompanied by a very small contraction of the airway (3.3 ± 1.9%, n = 5 slices from 2 mice). These results indicate that caffeine initiated the emptying of the internal Ca2+ stores and that this induced an influx of extracellular Ca2+. Interestingly, the transient of Ca2+ only induced a transient airway contraction while the sustained Ca2+ influx did not maintain sustained contraction.
Role of Intracellular Ca2+ Stores During Stimulation with 5-HT, ACH, and KCl
With this basic information on how caffeine acts on lung slices, we examined the effect of caffeine on agonist- and KCl-induced contraction. Consistent with the previous experimental results, caffeine completely, but reversibly, blocked airway contraction and the generation of Ca2+ oscillations (n = 3 slices from 2 mice) induced by 5-HT and ACH (Fig. 11, A and B). These results further suggest that contraction and Ca2+ oscillations are dependent on internal Ca2+ stores.
Figure 11.
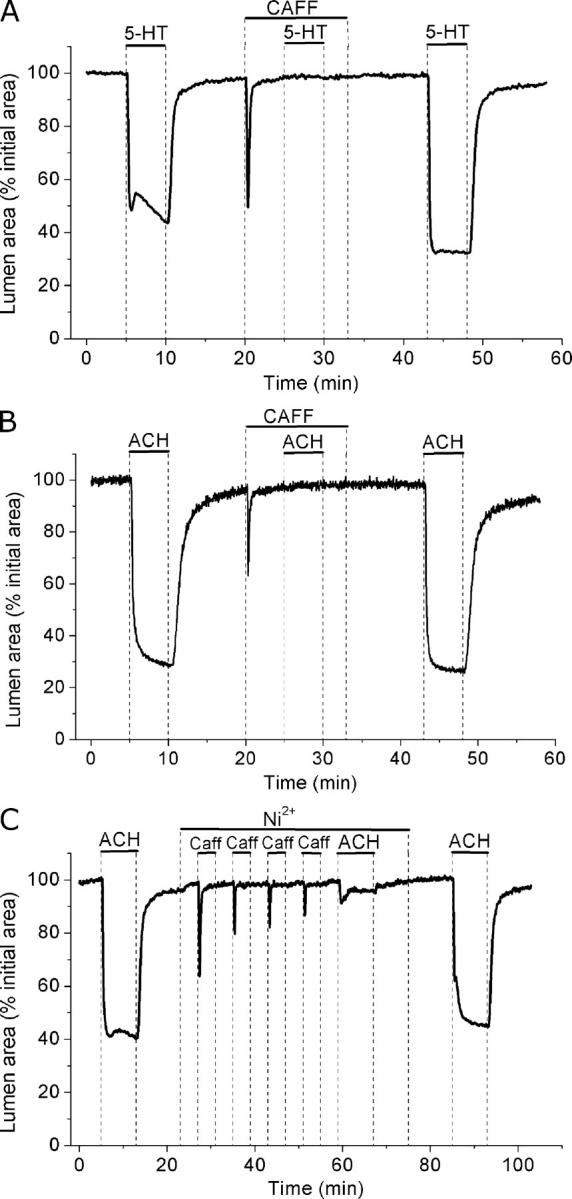
Effect of caffeine on airway contraction induced by 5-HT and ACH. Airways in lung slices were sequentially stimulated with (A) 1 μM 5-HT or (B) 1 μM ACH in the absence or presence of 20 mM caffeine (top bars). (C) The effect of depletion of internal stores on the response to 1 μM ACH induced by successive stimulation with 20 mM caffeine in the presence of 1 mM Ni2+. Representative traces of at least three experiments from different slices from two mice.
To further test if refilling of Ca2+ stores depends on a Ni2+-sensitive Ca2+ influx, we examined the effect of caffeine and Ni2+ on the response to ACH (Fig. 11 C). In the presence of Ni2+, the contractile response of an airway to successive exposures of caffeine progressively declined (Fig. 11 C). This indicated that the internal Ca2+ stores were progressively depleted. The subsequent stimulation with ACH only induced a small contraction (19.6 ± 4.6% relative to the control contraction) that relaxed (to 6.5 ± 0.6%) after 8 min (3 slices from 2 mice). Upon removal of Ni2+, ACH once again induced a strong contraction (86.9 ± 6.0% and 99.6 ± 10% after 8 min). In control experiments, in the absence of Ni2+, airway contraction did not decline in response to a similar series of caffeine exposures and this treatment did not affect the subsequent ACH-induced contraction (2 airways from 2 mice). These results indicate the presence of a Ni2+-sensitive Ca2+ influx to refill Ca2+ stores to sustain contraction.
Surprisingly, caffeine also completely blocked airway contraction or twitching induced by KCl (Fig. 12 A). The twitching response to KCl was also blocked with cyclopiazonic acid (CPA), an inhibitor of SR Ca2+ pump (SERCA) (Fig. 12 B). These results suggest that KCl-induced twitching as well as 5-HT– and ACH-induced contractions were dependent on cycles of release and reloading of the Ca2+ stores.
Ca2+ Waves and Elemental Ca2+ Events in Airway SMCs
To explore the mechanisms mediating fast Ca2+ oscillations induced by agonists and slow Ca2+ oscillations induced by KCl from internal stores, we compared the spatio-temporal properties of the Ca2+ oscillations induced by 5-HT, ACH, and KCl in airway SMCs using high speed (60 fps) recordings of the changes in [Ca2+]i. Line-scan plots from the longitudinal axis of single SMCs showed that Ca2+ oscillations induced by 5-HT and ACH consisted of Ca2+ waves that propagated along the entire longitudinal axis of the cell (Fig. 13). The average velocity of the Ca2+ waves induced by 5-HT and ACH was 27 ± 2 μm/s and 43 ± 4 μm/s, respectively. In general, as indicated by the consistent pattern of the line-scan analysis, the Ca2+ waves appeared to be initiated at specific locations within the cell. However, successive waves could change their initiation site and propagate in the opposite direction (Fig. 13).
In contrast to the regular patterns of agonist-induced Ca2+ waves, KCl-induced waves were preceded by multiple small, localized, and transient Ca2+ increases (or “elemental Ca2+ events”) that did not propagate through the whole cell (Fig. 13, arrows). These elemental Ca2+ events were also initiated from specific locations within a cell at an average frequency of 7 ± 4 per min (8 cells from 5 slices from 2 mice). However, their frequency increased with time, and was fastest just before a Ca2+ wave was initiated. The Ca2+ waves induced by KCl had slower propagating velocities (18 ± 2 μm/s) and a longer duration than those induced by 5-HT or ACH (Figs. 4, 5, and 13). After each Ca2+ wave had subsided, a refractory period elapsed before the elemental Ca2+ events started to occur again. KCl-induced Ca2+ waves were usually initiated from one location that showed the highest frequency of elemental signaling. Each elemental Ca2+ event occurred with a diameter of 4 ± 2 μm, had a rise-to-peak time of 80 ± 30 ms and a first order exponential decay of 420 ± 155 ms (Fig. 13, inset, n = 4 cells from 4 slices from 2 mice). Before the addition of KCl, the elemental Ca2+ events were not observed; after the removal of KCl, the elemental Ca2+ events were virtually abolished (Fig. 13, HBSS line). Ca2+ waves and elemental Ca2+ events were not observed during KCl stimulation in the presence of caffeine or CPA.
DISCUSSION
We further developed the lung slice preparation to compare the contractile responses of intrapulmonary airways and arterioles and the underlying changes in [Ca2+]i in the associated SMCs. Lung slices are well suited for these studies because the airways and blood vessels can be easily identified at regions which are considered important in the development of asthma and pulmonary hypertension and a link between the responses of the SMCs and the physiology of airways and blood vessels can be made. In addition, the robust responses of the airways and arterioles allow a quantitative evaluation of the contractile response to different agonists or concentrations of the same agonist. Other important qualities of lung slices include the ability to simultaneously study the response of two types of SMCs to the same conditions, a viability of several days and the use of serial sections and transgenic animals (Kotlikoff et al., 2004).
Airway or arteriole contraction is limited by the balance between the contractile forces produced by the SMCs and the recoil forces generated by surrounding alveolar tissue. In the animal, the recoil forces are established by lung inflation with air (Ding et al., 1987). In lung slices, the recoil forces are established by inflation with agarose. Because overinflation will reduce the contractile response, it is important to adjust the inflation volume of agarose to be about equal (but not greater than) the volume of the chest cavity (1–1.4 ml). At this volume, a reduction in the airway area of ∼50–60% in response to a maximal dose of 5-HT or ACH was consistently observed. While differences in contractile responses could be attributed to differences in the inflation volume, this possibility is minimized by using the same lung slice to evaluate multiple drugs. In addition, the similarity of the responses of slices to agonists from different mice suggests that the lung inflation was similar between each preparation.
The airways and arterioles of mouse lung slices responded differently to agonists and KCl. In response to 5-HT, the airways and arterioles displayed a sustained contraction but in response to ACH only the airways displayed contraction. This contrasts with arterial perfusion of isolated lungs where 5-HT only induced a small increase in airway resistance and pulmonary artery pressure (Held et al., 1999); this difference might be explained by the fact that 5-HT was not in contact with the SMCs in perfused lungs. In response to KCl, the airways of lung slices displayed SMC twitching while the arterioles displayed contraction and twitching. To understand the mechanisms underlying these responses, it was necessary to characterize the Ca2+ responses of the SMCs. However, for clarity, we only report here the contractility and Ca2+ signaling of airway SMCs induced by agonists and KCl. The responses of arteriole SMCs are the subject of a second study where we develop a hypothesis to propose how different frequencies of Ca2+ oscillations regulate contraction of airway and arteriole SMCs (Perez and Sanderson, 2005).
We have found that 5-HT and ACH induced airway contraction in a similar manner although the contraction induced by ACH was greater than that induced by 5-HT. A greater contractile response to MCH, as compared with 5-HT, was also observed in isolated mouse lungs (Held et al., 1999) and trachea rings (Moffatt et al., 2004). However, 5-HT was also believed to act via a cholinergic pathway. In our lung slices, atropine had no effect on 5-HT–induced contraction of airways even though atropine completely blocked airway contraction induced by ACH. These results suggest that 5-HT does not act via a cholinergic pathway and that 5-HT and ACH may be putative regulators of the caliber of the small airways of the mouse.
Agonist-induced (5-HT or ACH) contraction of the airways was generally sustained, whereas the KCl-induced contraction was characterized by SMC twitching. This difference indicates that agonist-induced contraction does not require membrane depolarization (Bolton et al., 1999a), a hypothesis supported by the fact that 5-HT and ACH can act via G protein–coupled receptors. 5-HT and ACH, but not KCl, also initiated contraction in the absence of extracellular Ca2+ or in the presence of nifedipine, an antagonist of L-type Ca2+ channels. A similar finding that nifedipine only slightly relaxed trachea SMCs contracted with ACH was reported by Kuo et al., (2003). However, both 5-HT and ACH only induced a transient contraction in the absence of extracellular Ca2+ or in the presence of Ni2+; results that indicate that external Ca2+ entry is necessary to maintain the sustained contraction. The persistent contraction of the airway in the presence of nifedipine suggests that L-type Ca2+ channels are not the route of Ca2+ entry. However, voltage-sensitive T-type Ca2+ currents have been found in bronchial SMCs and may play a role in excitation–contraction and the refilling of Ca2+ stores (Janssen, 1997; Yamakage et al., 2001). T-type Ca2+ channels have little sensitivity to nifedipine but are blocked by Ni2+; consequently, we cannot rule out the possibility that T-type Ca2+ channels are involved in refilling the Ca2+ stores. Because of the less-specific effects of Ni2+, it is also likely that other membrane channels, such as store-operated or noncapacitative, receptor-operated Ca2+ channels (transient receptor potential channels), contribute Ca2+ entry in airway SMCs (Berridge et al., 2003; Li et al., 2003; Ay et al., 2004). The findings that L-type Ca2+ channel blockers are ineffective as therapeutic agents for asthma and that agonist-induced changes in membrane potential are inadequate to activate L-type Ca2+ channels in airway SMCs further de-emphasizes the role suggested for L-type voltage-gated Ca2+ channels in airway contraction (Janssen, 2002).
Previous investigations of Ca2+ signaling in rat (Tolloczko et al., 1995, 1997) or dog (Yang et al., 1997; Yang, 1998) tracheal SMCs used low-speed sampling systems to report that 5-HT induced an initial transient followed by a sustained elevation in [Ca2+]i. By contrast, we found with video-rate confocal microscopy that 5-HT induces repetitive transients in [Ca2+]i or Ca2+ oscillations in airway SMCs. In most respects, the 5-HT–induced Ca2+ oscillations were similar to those induced by ACH, both in this and previous studies with lung slices (Bergner and Sanderson, 2002a, 2003) or isolated tracheal airway preparations (Prakash et al., 1997, 2000; Roux et al., 1997; Kuo et al., 2003). The Ca2+ oscillations persisted with a steady frequency and usually originated at one end of the cell and spread toward the other end as a Ca2+ wave, although the direction of the Ca2+ waves could be reversed. The Ca2+ waves were unsynchronized between neighboring cells and did not propagate to adjacent cells, suggesting that each wave originated within each cell. It is important to note that each Ca2+ oscillation did not generate a twitch of contraction but that the SMCs maintained a steady contractile state.
A significant characteristic of agonist-induced Ca2+ oscillations was the fact that the frequency of the Ca2+ oscillations increased with the concentrations of 5-HT or ACH. Similarly, the extent of the airway contraction was also concentration dependent over the same range. This relationship between contraction and Ca2+ oscillation frequency suggests that the size of the airway contraction is regulated by frequency modulation of the changes in [Ca2+]i (Berridge et al., 2003). Consistent with this idea is the fact that we could not establish a relationship between airway contraction and the magnitude or duration of the Ca2+ oscillations. Agonist-induced Ca2+ oscillations could initially have large amplitudes, and although this amplitude could decline with time, it did not alter the level of contraction. Similarly, slowing the Ca2+ oscillation frequency (e.g., with 0 extracellular Ca2+ or Ni2+) induced relaxation but did not alter the oscillation amplitude. Slow KCl-induced Ca2+ oscillations had a similar magnitude but a much longer duration (≥10 times) than the oscillations induced by agonists, yet the contraction induced by KCl was smaller. A similar relationship between the Ca2+ oscillatory frequency and contractility was reported for ACH (Bergner and Sanderson, 2002a) and ATP (Bergner and Sanderson, 2002b) in lung slices and for ACH in SMCs bundles isolated from porcine trachea (Kuo et al., 2003).
The most likely mechanism for the generation of 5-HT– or ACH-induced Ca2+ oscillations is agonist activation of PLC via a G protein–coupled receptor to generate inositol 1,4,5-trisphosphate (IP3) that, in turn, initiates repetitive cycles of Ca2+ release and uptake from the SR via the IP3 receptor (IP3R) (Roux et al., 1997; Pabelick et al., 2001; Bergner and Sanderson, 2002a; Janssen, 2002). However, caffeine inhibits the contractile response to ACH and 5-HT, a result suggesting that internal stores with RyRs contribute to the Ca2+ fluxes occurring during Ca2+ oscillations. While previous studies indicate that ACH binds to an M3 muscarinic receptor, 5-HT appears to act via the 5-HT2, Gq protein–coupled, receptor (Hoyer et al., 2002) because airway contraction was inhibited by ketanserin, a 5-HT2 receptor blocker, and stimulated by DOI, a 5-HT2 receptor agonist. Although 5-HT3 receptors can elevate [Ca2+]i, via a Ca2+ influx (Reeves and Lummis, 2002), its role appears minimal because 5-HT–induced increases in [Ca2+]i in the absence of extracellular Ca2+ and the selective 5-HT3 receptor agonist SR-57227 did not stimulate the airway contraction.
The IP3Rs, RyRs, and SERCA all appear to participate in the generation of Ca2+ oscillations, but the mechanisms that regulate the Ca2+ oscillation frequency are unclear. One idea is that the Ca2+ oscillation rate is influenced by the basal [Ca2+]i, which increases the sensitivity of the RyR to CICR and SERCA pump activity (Prakash et al., 2000; Pabelick et al., 2001). An alternative idea is that the frequency is regulated by the intracellular concentration of IP3 acting on the IP3R (Berridge et al., 2003) and that the elevated [Ca2+]i is a consequence of increases in Ca2+ oscillation frequency. Because CICR via the IP3R may also be influenced by [Ca2+]i, it may take a short period to establish a steady state for the Ca2+ oscillations after an initial surge of IP3 associated with agonist stimulation. We have observed an initial increase in the basal [Ca2+]i with high frequency Ca2+ oscillation, but it is interesting to note that the airway contraction remains steady, if not increases, during this stabilization period. Differences in the frequency rate between cells could be the result of heterogeneities in internal and external receptor expression.
Before exploring the hypothesis that KCl-induced contraction is mediated by the depolarization of SMCs, we ruled out the alternative hypothesis that KCl acts by stimulating the local release of neurotransmitters for several reasons. First, KCl stimulated SMC twitching in the presence of ketanserin or atropine, 5-HT and ACH receptor antagonists, and apyrase, an ATPase. Second, the airways did not respond to phenylephrine, an α1-agonist. These results indicate that a release of ACH, 5-HT, ATP, or noradrenaline from nerve terminals does not explain the contraction stimulated by KCl. Third, because our experiments were performed with constant perfusion, any neurotransmitter released would be quickly washed away and could not generate a sustained response. And, finally, the Ca2+ responses and contraction of the SMCs to KCl were very different to those induced by agonists.
The hypothesis for the mechanism by which KCl triggers Ca2+ oscillations is also constructed from several lines of evidence. First, while the KCl-induced Ca2+ oscillations were characterized as long-lasting Ca2+ waves that occurred at low frequencies, these Ca2+ waves were preceded by multiple, transient Ca2+ increases or “elemental Ca2+ events” that were similar to Ca2+ sparks or Ca2+ puffs observed in other cells. Second, these KCl-induced Ca2+ oscillation and events were abolished by caffeine and CPA, which suggests that these elemental Ca2+ signals involve RyRs and the release of intracellular Ca2+. The prolonged period of the KCl-induced Ca2+ wave suggests an extensive emptying of the internal Ca2+ store, and this is consistent with the development of a refractory period immediately following the Ca2+ waves in which no Ca2+ events were observed. Third, the propagation velocity of the KCl-induced Ca2+ waves is slow and similar to that induced by agonists, which supports the idea that the transient Ca2+ oscillation results from CICR from internal stores rather than from a Ca2+ influx initiated by fast propagating changes in membrane potential. Fourth, in the presence of extracellular KCl, oscillations in membrane potential would not be expected, but if they did occur, they would be expected to propagate to adjacent SMCs; however, Ca2+ waves induced by KCl occurred asynchronously between adjacent cells. And, finally, the KCl-induced Ca2+ oscillations required extracellular Ca2+ influx and were sensitive to nifedipine and Ni2+.
From this data we hypothesize that the KCl-induced Ca2+ oscillations are the result of the following events. Initially, KCl induces membrane depolarization and initiates an influx of Ca2+ via L-type and/or T-type Ca2+ channels. The cell compensates for this rise in [Ca2+]i by transporting the extra Ca2+ into the SR via SERCA pumps. Because of their limited capacity, the stores quickly overload as indicated by the increasing frequency of the elemental Ca2+ events, which reflect sensitized RyRs. Upon reaching a critical Ca2+ load, the elemental Ca2+ events trigger an extended phase of CICR via sensitized RyR to empty the store and generate a Ca2+ wave with a transient SMC contraction before initiating the cycle again.
It is unknown if the elemental Ca2+ events arise from pure clusters of RyRs or mixtures of RyR and IP3Rs. Ca2+ sparks, observed in other SMCs, occurred as localized and transient Ca2+ increases and had rise times of ∼20 ms and initial decay constants of ∼50 ms (Wellman and Nelson, 2003). The elemental Ca2+ events recorded here had longer rise times and decay constants. However, under conditions of Ca2+ overload, a Ca2+ spark can activate CICR of a neighboring cluster of RyRs to form a compound Ca2+ spark (Wang et al., 2004). In cardiomyocytes, compound Ca2+ sparks can evolve into propagating Ca2+ waves (Cheng et al., 1993, 1996), and we consistently observed that KCl-induced Ca2+ waves were initiated from sites where localized Ca2+ events were occurring.
The KCl-induced Ca2+ waves occur at low frequencies and result in twitching in the airway SMCs; yet, under these conditions, there appears to be a steady influx of Ca2+ but this is incapable of maintaining a sustained contraction. A steady influx of Ca2+ can also be invoked by emptying the internal Ca2+ stores with caffeine, but the SMCs remain relaxed. This is consistent with the idea that the contraction of airway SMCs is mediated by the frequency of the Ca2+ oscillations instead of a sustained elevation of Ca2+. An alternative explanation for the lack of contraction is that caffeine acts as a phosphodiesterase inhibitor to reduce the Ca2+ sensitivity of the SMC (Hall, 2004). However, the inhibition of the KCl-induced Ca2+ oscillations and twitching by caffeine and CPA suggests a direct action of caffeine on the Ca2+ stores.
The fact that a steady Ca2+ influx, initially driven by a change in membrane potential, has little effect on the regulation of airway SMC contraction suggests that Ca2+ sparks (Bolton et al., 1999b; Zhuge et al., 2004) do not serve as a relaxation mechanism for bronchiole SMCs. These Ca2+ events may counteract the overloading of the internal stores by membrane hyperpolarization when coupled to Ca2+-activated K+ channels (BK channels) but it appears that this process only occurs in airways exposed to KCl. As a result, cyclic Ca2+ release from internal stores seems to be the most important signal to sustain airway SMC contraction, while Ca2+ influx through nifedipine-resistant, Ni2+-sensitive channels is necessary to replenish the Ca2+ stores and maintain the frequency of the Ca2+ oscillations.
The sources of 5-HT in the lungs include pulmonary neuroendocrine cells that form neuroepithelial bodies in the airways of animals, including humans (Lauweryns et al., 1973; Junod, 1975; Gonmori et al., 1986; Prasada Rao and Mehendale, 1987; Ben-Harari et al., 1990) and mast cells (Wasserman, 1994). Although substantial amounts of 5-HT are also synthesized in the gut and stored in blood platelets, circulating 5-HT had a minor effect on airways (Held et al., 1999). Consequently, 5-HT that is released to the basolateral space in response to hypoxia or neural activity (Cutz et al., 1993; Lommel, 2001; Adriaensen et al., 2003) or by mast cells during de-granulation in an allergic response may serve as a paracrine stimulant of SMCs. The possibility that 5-HT serves as a putative regulator of SMCs is supported by the ability of SMCs to transport and metabolize 5-HT; actions that would inactive 5-HT (Dodson et al., 2004).
Another interesting aspect of lung slices that requires further investigation is the influence, if any, of the airway epithelial cells on the contractile responses of the airway SMCs. While epithelial cells can release mediators such as prostaglandin E2 and nitric oxide that may relax SMCs (Folkerts and Nijkamp, 1998), we did not observe any major changes in [Ca2+]i in the epithelial cells in response to ACH, 5-HT, or KCl that might stimulate this release. In addition, in our previous studies with lung slices, we found little or no effect of ATP on the release of relaxing factors from epithelial cells (Bergner and Sanderson, 2002b).
In conclusion, intrapulmonary airways respond to 5-HT and ACH with a contraction that is maintained by high frequency Ca2+ oscillations within the SMCs that arise from repetitive cycles of Ca2+ release and uptake by the SR and require extracellular Ca2+ for store refilling. By contrast, KCl-induced twitching of SMCs results from low frequency Ca2+ oscillations produced by an overfilling and uncontrolled release of internal Ca2+. Most importantly, the magnitude of the contraction of airway SMCs is regulated by the frequency of the Ca2+ oscillations.
Supplemental Material
Acknowledgments
This work was supported by the National Institutes of Health grant HL71930 to M.J. Sanderson.
Lawrence G. Palmer served as editor.
Abbreviations used in this paper: ACH, acetylcholine; CPA, cyclopiazonic acid; 5-HT, 5-hydroxytrypamine; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; RyR, ryanodine receptor; SMC, smooth muscle cell.
References
- Abdullah, N.A., M. Hirata, K. Matsumoto, H. Aizawa, R. Inoue, S. Hamano, S. Ikeda, Z. Xie, N. Hara, and Y. Ito. 1994. Contraction and depolarization induced by fetal bovine serum in airway smooth muscle. Am. J. Physiol. 266:L528–L535. [DOI] [PubMed] [Google Scholar]
- Adler, A., E.A. Cowley, J.H. Bates, and D.H. Eidelman. 1998. Airway-parenchymal interdependence after airway contraction in rat lung explants. J. Appl. Physiol. 85:231–237. [DOI] [PubMed] [Google Scholar]
- Adriaensen, D., I. Brouns, J. Van Genechten, and J.P. Timmermans. 2003. Functional morphology of pulmonary neuroepithelial bodies: extremely complex airway receptors. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 270:25–40. [DOI] [PubMed] [Google Scholar]
- Ay, B., Y.S. Prakash, C.M. Pabelick, and G.C. Sieck. 2004. Store-operated Ca2+ entry in porcine airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 286:L909–L917. [DOI] [PubMed] [Google Scholar]
- Ben-Harari, R.R., A. Parent Ermini, and J. Kleinerman. 1990. Metabolism of 5-hydroxytryptophan in the isolated perfused rat lung. Pharmacology. 41:272–279. [DOI] [PubMed] [Google Scholar]
- Bergner, A., and M.J. Sanderson. 2002. a. Acetylcholine-induced calcium signaling and contraction of airway smooth muscle cells in lung slices. J. Gen. Physiol. 119:187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergner, A., and M.J. Sanderson. 2002. b. ATP stimulates Ca2+ oscillations and contraction in airway smooth muscle cells of mouse lung slices. Am. J. Physiol. Lung Cell. Mol. Physiol. 283:L1271–L1279. [DOI] [PubMed] [Google Scholar]
- Bergner, A., and M.J. Sanderson. 2003. Airway contractility and smooth muscle Ca2+ signaling in lung slices from different mouse strains. J. Appl. Physiol. 95:1325–1332. [DOI] [PubMed] [Google Scholar]
- Berridge, M.J., M.D. Bootman, and H.L. Roderick. 2003. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4:517–529. [DOI] [PubMed] [Google Scholar]
- Bolton, T.B., S.A. Prestwich, A.V. Zholos, and D.V. Gordienko. 1999. a. Excitation-contraction coupling in gastrointestinal and other smooth muscles. Annu. Rev. Physiol. 61:85–115. [DOI] [PubMed] [Google Scholar]
- Bolton, T.B., S.A. Prestwich, A.V. Zholos, and D.V. Gordienko. 1999. b. Excitation-contraction coupling in gastrointestinal and other smooth muscles. Annu. Rev. Physiol. 61:85–115. [DOI] [PubMed] [Google Scholar]
- Cheng, H., M.R. Lederer, W.J. Lederer, and M.B. Cannell. 1996. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am. J. Physiol. 270:C148–C159. [DOI] [PubMed] [Google Scholar]
- Cheng, H., W.J. Lederer, and M.B. Cannell. 1993. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 262:740–744. [DOI] [PubMed] [Google Scholar]
- Cushley, M.J., L.H. Wee, and S.T. Holgate. 1986. The effect of inhaled 5-hydroxytryptamine (5-HT, serotonin) on airway calibre in man. Br. J. Clin. Pharmacol. 22:487–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutz, E., V. Speirs, H. Yeger, C. Newman, D. Wang, and D.G. Perrin. 1993. Cell biology of pulmonary neuroepithelial bodies–validation of an in vitro model. I. Effects of hypoxia and Ca2+ ionophore on serotonin content and exocytosis of dense core vesicles. Anat. Rec. 236:41–52. [DOI] [PubMed] [Google Scholar]
- Dandurand, R.J., C.G. Wang, N.C. Phillips, and D.H. Eidelman. 1993. Responsiveness of individual airways to methacholine in adult rat lung explants. J. Appl. Physiol. 75:364–372. [DOI] [PubMed] [Google Scholar]
- Ding, D.J., J.G. Martin, and P.T. Macklem. 1987. Effects of lung volume on maximal methacholine-induced bronchoconstriction in normal humans. J. Appl. Physiol. 62:1324–1330. [DOI] [PubMed] [Google Scholar]
- Dodson, A.M., G.M. Anderson, and K.J. Rhoden. 2004. Serotonin uptake and metabolism by cultured guinea pig airway smooth muscle cells. Pulm. Pharmacol. Ther. 17:19–25. [DOI] [PubMed] [Google Scholar]
- Duguet, A., C.G. Wang, R. Gomes, H. Ghezzo, D.H. Eidelman, and R.S. Tepper. 2001. Greater velocity and magnitude of airway narrowing in immature than in mature rabbit lung explants. Am. J. Respir. Crit. Care Med. 164:1728–1733. [DOI] [PubMed] [Google Scholar]
- Dupont, L.J., J.L. Pype, M.G. Demedts, P. De Leyn, G. Deneffe, and G.M. Verleden. 1999. The effects of 5-HT on cholinergic contraction in human airways in vitro. Eur. Respir. J. 14:642–649. [DOI] [PubMed] [Google Scholar]
- Eum, S.Y., X. Norel, J. Lefort, C. Labat, B.B. Vargaftig, and C. Brink. 1999. Anaphylactic bronchoconstriction in BP2 mice: interactions between serotonin and acetylcholine. Br. J. Pharmacol. 126:312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez, V.E., V. McCaskill, N.D. Atkins, and A. Wanner. 1999. Variability of airway responses in mice. Lung. 177:355–366. [DOI] [PubMed] [Google Scholar]
- Folkerts, G., and F.P. Nijkamp. 1998. Airway epithelium: more than just a barrier! Trends Pharmacol. Sci. 19:334–341. [DOI] [PubMed] [Google Scholar]
- Gonmori, K., K.S. Rao, and H.M. Mehendale. 1986. Pulmonary synthesis of 5-hydroxytryptamine in isolated perfused rabbit and rat lung preparations. Exp. Lung Res. 11:295–305. [DOI] [PubMed] [Google Scholar]
- Hall, I.P. 2004. The β-agonist controversy revisited. Lancet. 363:183–184. [DOI] [PubMed] [Google Scholar]
- Held, H.D., C. Martin, and S. Uhlig. 1999. Characterization of airway and vascular responses in murine lungs. Br. J. Pharmacol. 126:1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer, D., J.P. Hannon, and G.R. Martin. 2002. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav. 71:533–554. [DOI] [PubMed] [Google Scholar]
- Janssen, L.J. 1997. T-type and L-type Ca2+ currents in canine bronchial smooth muscle: characterization and physiological roles. Am. J. Physiol. 272:C1757–C1765. [DOI] [PubMed] [Google Scholar]
- Janssen, L.J. 2002. Ionic mechanisms and Ca2+ regulation in airway smooth muscle contraction: do the data contradict dogma? Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L1161–L1178. [DOI] [PubMed] [Google Scholar]
- Junod, A.F. 1975. Metabolism, production, and release of hormones and mediators in the lung. Am. Rev. Respir. Dis. 112:93–108. [DOI] [PubMed] [Google Scholar]
- Kotlikoff, M.I., M.S. Kannan, J. Solway, K.Y. Deng, D.A. Deshpande, M. Dowell, M. Feldman, K.S. Green, G. Ji, R. Johnston, et al. 2004. Methodologic advancements in the study of airway smooth muscle. J. Allergy Clin. Immunol. 114:S18–S31. [DOI] [PubMed] [Google Scholar]
- Kuo, K.H., J. Dai, C.Y. Seow, C.H. Lee, and C. van Breemen. 2003. Relationship between asynchronous Ca2+ waves and force development in intact smooth muscle bundles of the porcine trachea. Am. J. Physiol. Lung Cell. Mol. Physiol. 285:L1345–L1353. [DOI] [PubMed] [Google Scholar]
- Lauweryns, J.M., J. Cokelaere, and P. Theunynck. 1973. Serotonin producing neuroepithelial bodies in rabbit respiratory mucosa. Science. 180:410–413. [DOI] [PubMed] [Google Scholar]
- Lechin, F., B. van der Dijs, B. Orozco, M. Lechin, and A.E. Lechin. 1996. Increased levels of free serotonin in plasma of symptomatic asthmatic patients. Ann. Allergy Asthma Immunol. 77:245–253. [DOI] [PubMed] [Google Scholar]
- Levitt, R.C., and W. Mitzner. 1989. Autosomal recessive inheritance of airway hyperreactivity to 5-hydroxytryptamine. J. Appl. Physiol. 67:1125–1132. [DOI] [PubMed] [Google Scholar]
- Li, S., J. Westwick, and C. Poll. 2003. Transient receptor potential (TRP) channels as potential drug targets in respiratory disease. Cell Calcium. 33:551–558. [DOI] [PubMed] [Google Scholar]
- Lommel, A.V. 2001. Pulmonary neuroendocrine cells (PNEC) and neuroepithelial bodies (NEB): chemoreceptors and regulators of lung development. Paediatr. Respir. Rev. 2:171–176. [DOI] [PubMed] [Google Scholar]
- Martin, C., S. Uhlig, and V. Ullrich. 1996. Videomicroscopy of methacholine-induced contraction of individual airways in precision-cut lung slices. Eur. Respir. J. 9:2479–2487. [DOI] [PubMed] [Google Scholar]
- Martin, C., S. Uhlig, and V. Ullrich. 2001. Cytokine-induced bronchoconstriction in precision-cut lung slices is dependent upon cyclooxygenase-2 and thromboxane receptor activation. Am. J. Respir. Cell Mol. Biol. 24:139–145. [DOI] [PubMed] [Google Scholar]
- Minshall, E., C.G. Wang, R. Dandurand, and D. Eidelman. 1997. Heterogeneity of responsiveness of individual airways in cultured lung explants. Can. J. Physiol. Pharmacol. 75:911–916. [PubMed] [Google Scholar]
- Moffatt, J.D., T.M. Cocks, and C.P. Page. 2004. Role of the epithelium and acetylcholine in mediating the contraction to 5-hydroxytryptamine in the mouse isolated trachea. Br. J. Pharmacol. 141:1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabelick, C.M., G.C. Sieck, and Y.S. Prakash. 2001. Invited review: significance of spatial and temporal heterogeneity of calcium transients in smooth muscle. J. Appl. Physiol. 91:488–496. [DOI] [PubMed] [Google Scholar]
- Perez, J.F., and M.J. Sanderson. 2005. The contraction of smooth muscle cells of intrapulmonary arterioles is determined by the frequency of Ca2+ oscillations induced by 5-HT and KCl. J. Gen. Physiol. 125:555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash, Y.S., M.S. Kannan, and G.C. Sieck. 1997. Regulation of intracellular calcium oscillations in porcine tracheal smooth muscle cells. Am. J. Physiol. 272:C966–C975. [DOI] [PubMed] [Google Scholar]
- Prakash, Y.S., C.M. Pabelick, M.S. Kannan, and G.C. Sieck. 2000. Spatial and temporal aspects of ACh-induced [Ca2+]i oscillations in porcine tracheal smooth muscle. Cell Calcium. 27:153–162. [DOI] [PubMed] [Google Scholar]
- Prasada Rao, K.S., and H.M. Mehendale. 1987. Precursor utilization of 5-hydroxytryptophan for 5-hydroxytryptamine biosynthesis in isolated and perfused rabbit and rat lungs. Can. J. Physiol. Pharmacol. 65:2117–2123. [DOI] [PubMed] [Google Scholar]
- Raffestin, B., J. Cerrina, C. Boullet, C. Labat, J. Benveniste, and C. Brink. 1985. Response and sensitivity of isolated human pulmonary muscle preparations to pharmacological agents. J. Pharmacol. Exp. Ther. 233:186–194. [PubMed] [Google Scholar]
- Reeves, D.C., and S.C. Lummis. 2002. The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel (review). Mol. Membr. Biol. 19:11–26. [DOI] [PubMed] [Google Scholar]
- Roux, E., C. Guibert, J.P. Savineau, and R. Marthan. 1997. [Ca2+]i oscillations induced by muscarinic stimulation in airway smooth muscle cells: receptor subtypes and correlation with the mechanical activity. Br. J. Pharmacol. 120:1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson, M.J. 2004. Acquisition of multiple real-time images for laser scanning microscopy. Microscopy and Analysis. 18:17–23. [Google Scholar]
- Sanderson, M.J., and I. Parker. 2003. Video-rate confocal microscopy. Methods Enzymol. 360:447–481. [DOI] [PubMed] [Google Scholar]
- Tolloczko, B., Y.L. Jia, and J.G. Martin. 1995. Serotonin-evoked calcium transients in airway smooth muscle cells. Am. J. Physiol. 269:L234–L240. [DOI] [PubMed] [Google Scholar]
- Tolloczko, B., Y.L. Jia, and J.G. Martin. 1997. Effects of cAMP on serotonin evoked calcium transients in cultured rat airway smooth muscle cells. Am. J. Physiol. 272:L865–L871. [DOI] [PubMed] [Google Scholar]
- Wang, S.Q., C. Wei, G. Zhao, D.X. Brochet, J. Shen, L.S. Song, W. Wang, D. Yang, and H. Cheng. 2004. Imaging microdomain Ca2+ in muscle cells. Circ. Res. 94:1011–1022. [DOI] [PubMed] [Google Scholar]
- Wasserman, S.I. 1994. Mast cells and airway inflammation in asthma. Am. J. Respir. Crit. Care Med. 150:S39–S41. [DOI] [PubMed] [Google Scholar]
- Wellman, G.C., and M.T. Nelson. 2003. Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium. 34:211–229. [DOI] [PubMed] [Google Scholar]
- Wohlsen, A., S. Uhlig, and C. Martin. 2001. Immediate allergic response in small airways. Am. J. Respir. Crit. Care Med. 163:1462–1469. [DOI] [PubMed] [Google Scholar]
- Yamakage, M., X. Chen, N. Tsujiguchi, Y. Kamada, and A. Namiki. 2001. Different inhibitory effects of volatile anesthetics on T- and L-type voltage-dependent Ca2+ channels in porcine tracheal and bronchial smooth muscles. Anesthesiology. 94:683–693. [DOI] [PubMed] [Google Scholar]
- Yang, C.M. 1998. Dissociation of intracellular Ca2+ release and Ca2+ entry response to 5-hydroxytryptamine in cultured canine tracheal smooth muscle cells. Cell. Signal. 10:735–742. [DOI] [PubMed] [Google Scholar]
- Yang, C.M., L.W. Fen, H.L. Tsao, and C.T. Chiu. 1997. Inhibition of 5-hydroxytryptamine-induced phosphoinositide hydrolysis and Ca2+ mobilization in canine cultured tracheal smooth muscle cells by phorbol ester. Br. J. Pharmacol. 121:853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuge, R., K.E. Fogarty, S.P. Baker, J.G. McCarron, R.A. Tuft, L.M. Lifshitz, and J.V. Walsh Jr. 2004. Ca2+ spark sites in smooth muscle cells are numerous and differ in number of ryanodine receptors, BK channels and coupling ratio between them. Am. J. Physiol. Cell Physiol. 287:C1577–C1588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.