

Abstract
ClC chloride channels, which are ubiquitously expressed in mammals, have a unique double-barreled structure, in which each monomer forms its own pore. Identification of pore-lining elements is important for understanding the conduction properties and unusual gating mechanisms of these channels. Structures of prokaryotic ClC transporters do not show an open pore, and so may not accurately represent the open state of the eukaryotic ClC channels. In this study we used cysteine-scanning mutagenesis and modification (SCAM) to screen >50 residues in the intracellular vestibule of ClC-0. We identified 14 positions sensitive to the negatively charged thiol-modifying reagents sodium (2-sulfonatoethyl)methanethiosulfonate (MTSES) or sodium 4-acetamido-4'-maleimidylstilbene-2'2-disulfonic acid (AMS) and show that 11 of these alter pore properties when modified. In addition, two MTSES-sensitive residues, on different helices and in close proximity in the prokaryotic structures, can form a disulfide bond in ClC-0. When mapped onto prokaryotic structures, MTSES/AMS-sensitive residues cluster around bound chloride ions, and the correlation is even stronger in the ClC-0 homology model developed by Corry et al. (2004). These results support the hypothesis that both secondary and tertiary structures in the intracellular vestibule are conserved among ClC family members, even in regions of very low sequence similarity.
Keywords: ClC channel, MTS modification, rectification, pore, homology model
INTRODUCTION
Members of the ClC family orchestrate the movement of chloride necessary for proper neuronal, muscular, cardiovascular, and epithelial function (Jentsch et al., 2002, 2005; Nilius and Droogmans, 2003; Uchida and Sasaki, 2005). Defects in these proteins are directly responsible for human diseases of muscle, kidney, bone, and brain (Steinmeyer et al., 1991; Lloyd et al., 1996; Simon et al., 1997; Cleiren et al., 2001; Kornak et al., 2001; Pusch, 2002; Haug et al., 2003; Naesens et al., 2004). ClC-0, found in Torpedo rays, was the first ClC to be discovered and characterized (White and Miller, 1979; Miller and Richard, 1990; Bauer et al., 1991). Since ClC-0 has a single-channel conductance higher than any other ClC yet studied (∼10 pS), it is the most thoroughly characterized ClC and serves as the family prototype (Maduke et al., 1999; Estevez and Jentsch, 2002; Pusch, 2004; Chen, 2005). The functional form of ClC-0 is a dimer in which each subunit forms its own pore (Ludewig et al., 1996, 1997a; Middleton et al., 1996). One gating mechanism, termed “slow” gating, closes both pores simultaneously. A second gating mechanism, termed “fast” gating, governs the independent opening and closing of each pore. Both gates are sensitive to transmembrane voltage, chloride, and pH (Miller, 1982; Hanke and Miller, 1983; Pusch et al., 1995, 1999; Chen and Miller, 1996; Chen and Chen, 2001), and both have unprecedented gating mechanisms in which the permeant ion plays a key role. Slow gating is energetically coupled to the transmembrane chloride gradient (Richard and Miller, 1990); fast-gating voltage dependence arises from movement of the permeant ion through the transmembrane field (Pusch et al., 1995; Chen and Miller, 1996, Ludewig et al., 1997b). Hence, gating, permeation, and chloride binding are all tightly coupled, and residues lining the ClC-0 pore are likely to be key players in these processes. To untangle this fascinating interplay between substrate and enzyme, the residues involved must first be identified.
X-ray crystal structures of Escherichia coli and Salmonella ClC homologues provide a framework for identifying residues that line the pore in ClC-0. These prokaryotic ClCs are dimers, and each subunit in the dimer has multiple chloride-binding sites (Dutzler et al., 2002, 2003), like ClC-0 (Pusch et al., 1995). In addition, highly conserved residues that affect permeation and gating in ClC-0 and ClC-1 (E166, S123, and Y512 in ClC-0) (Ludewig et al., 1996; Fahlke et al., 1997, 2001; Dutzler et al., 2003; Estevez et al., 2003; Lin and Chen, 2003; Dutzler, 2004) coordinate bound chloride ions in the prokaryotic ClC structures. Nevertheless, there are limitations to the similarity between ClC-0 and the prokaryotic ClC structures, as suggested by both the low sequence identity (∼17%) and by the differences in function. While all eukaryotic ClCs studied are chloride channels, the Escherichia coli ClC, ClC-ec1, is not a channel but a chloride–proton antiporter (Accardi and Miller, 2004). Here, the movement of chloride and protons in opposite directions is tightly coupled, and uphill movement of one ion against its electrochemical gradient can be driven by downhill movement of the other ion. Therefore, ClC-ec1 can never have an open pore, since this would prohibit such coupling. The prokaryotic ClC structures are consistent with this prediction; there is no open pore, but rather protein residues are interposed between the bound chloride ions, occluding the chloride pathway. Even a mutation at a conserved glutamate residue, which makes ClC-0 almost always open, does not reveal an open pore in the ClC-ec1 structure; the bound chlorides are still separated by protein (Dutzler et al., 2003). At best, the prokaryotic ClC structures may approximate a closed conformation of ClC-0.
ClC architecture, which is fundamentally different from that of the well-studied cation channels, makes it difficult to use the structure of the closed state to predict the structure of the open pore. The typical cation channel architecture consists of identical or homologous segments of protein that multimerize to form a symmetric or pseudosymmetric pore at the central junction of the subunits. With this design, the pore is constrained to be perpendicular to the membrane, and consequently the structure of the closed state suggests which residues line the pore in the open state. In contrast, each pore in ClC-0 is formed by only one subunit, and the lack of symmetry in this architecture confers asymmetry on the pore, which will likely snake its way through the protein. Thus, the structure of a closed state may be completely uninformative about the path of the pore in the open state.
Previous studies have identified residues as likely to line the intracellular vestibule of ClC-0: one on loop CD, one on helix D, and several on helix R (Pusch et al., 1995; Ludewig et al., 1996, 1997c; Middleton et al., 1996; Lin and Chen, 2000, 2003; Chen and Chen, 2003; Chen et al., 2003). Both mutational analysis (Ludewig et al., 1997c) and cysteine-scanning mutagenesis and modification (SCAM) (Lin and Chen, 2003) have been used to test residues on helix R, which is the only secondary structure reported to be completely scanned. To develop a more comprehensive picture of the open pore in ClC-0, we used SCAM (Akabas et al., 1992; Chen et al., 1997; Fahlke et al., 1997; Akabas, 1998; Benson et al., 1998; Becchetti et al., 1999; Lu et al., 1999; Shuck et al., 2000; Reeves et al., 2001; Smith et al., 2001; Dodier et al., 2004; Qu et al., 2004) to screen 51 candidate pore-lining residues, which were selected using the ClC-ec1 structure as a guide. We mapped our results onto prokaryotic ClC structures and a eukaryotic ClC homology model to assess how closely these resemble ClC-0. In addition, we found that two of the MTSES-sensitive residues can form a disulfide bond in ClC-0. Our results suggest that the architecture of the intracellular vestibule is conserved among ClCs, and they provide a step toward building a structural model of the open pore.
MATERIALS AND METHODS
Mutagenesis and Channel Expression
ClC-0 mutant constructs were made in a plasmid derived from the pBluescript vector (Stratagene) (Maduke et al., 1998). All constructs contained the point mutation C212S, which removes voltage-dependent slow-gate inactivation and has no other measurable effect on ClC-0 function (Lin et al., 1999). Cysteines were introduced using PCR methods, and mutations were confirmed by sequencing through the cloning cassette. Plasmids were linearized with FspI (New England Biolabs, Inc.), cleaned according to the instructions for the DNA Clean & Concentrator-5 (Zymo Research), and transcribed in vitro using the mMessage mMachine T3 RNA-polymerase transcription kit and protocol (Ambion). RNA was dissolved in RNAase free water (Invitrogen) containing 0.77 U/μL SUPERase-In (Ambion).
Excised Patch Recording
Defolliculated Xenopus oocytes were injected with 27.5 nL of ∼1 mg/ml RNA and incubated at 16°C for 2–5 d before recording. All data shown are from excised inside-out patches. Before patching, the vitelline membrane was removed manually while the oocyte bathed in shrink solution (in mM: 396 NaCl, 2 KCl, 1.8 CaCl2, 1.0 MgCl2, 5.0 HEPES, pH 7.6). The oocyte was then transferred to internal solution (in mM: 110 NMDG, 110 HCl, 5 MgCl2, 10 HEPES, 1 EGTA, brought to pH 7.3 using NaOH). Electrical contact between the recording chamber and the ground electrode was made via agar bridges. Recording electrodes were pulled from 100 μl calibrated pipettes (VWR), polished to 0.2–1.0 MΩ, and filled with external solution (in mM: 100 NMDG, 100 HCl, 5 MgCl2, 1 EGTA, 10 HEPES, brought to pH 7.3 using NaOH). Excised patches had conductances ranging from 3 to hundreds of nS; since the single-channel conductance for wild-type ClC-0 is ∼10 pS (Miller, 1982), this corresponds to hundreds to tens of thousands of channels per patch.
Data were acquired using an Axopatch 200B amplifier (Axon Instruments) interfaced with a Digidata 1322A acquisition board and a Windows-based PC running pClamp Software (Axon Instruments). The data were filtered at 1 kHz using the amplifier's low-pass Bessel filter, and sampled at 5 kHz. Data analysis was performed using IGOR Pro (WaveMetrics, Inc.).
Data Analysis
Protocol A was used to obtain the open-channel current (I) and apparent open probability (P o) as a function of voltage. In this protocol, a 50-ms prepulse to +50 mV is followed by a 170-ms test pulse, from +90 to −170 mV in 20-mV decrements, and then a 170-ms tail pulse to −100 mV. For K519C, which gates more slowly than wild type or any of the other tested mutants, the test and tail pulses were lengthened to allow the current to reach steady state (see Fig. 2, A and B).
Figure 2.
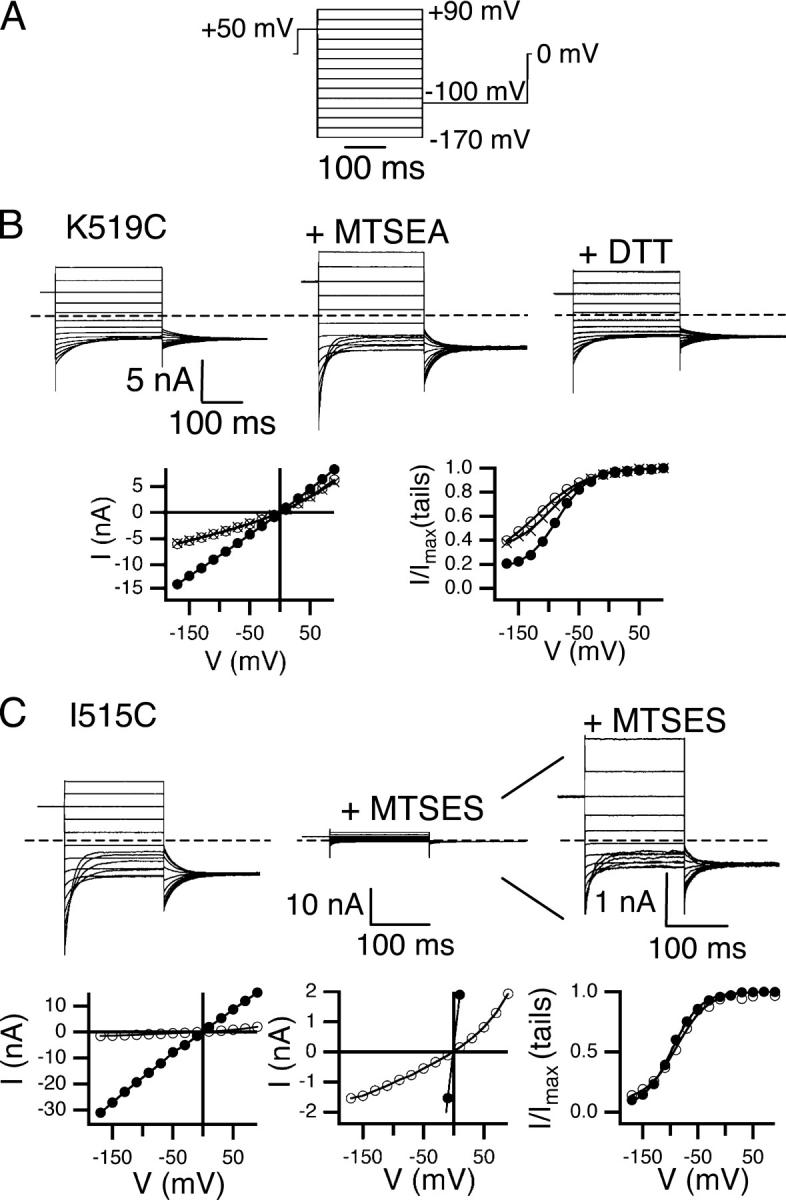
Changes in macroscopic open-channel current correspond to changes in single-channel current. Protocol A (A) and K519C current response (B) before (top left) and after (top middle) reaction with MTSEA, and after reversal with DTT (top right). The dashed line indicates zero current. The open-channel current (bottom left) and apparent open probability (bottom right) before (open circles) and after (filled circles) reaction with MTSEA, and after DTT (crosses) (derived as described in materials and methods). (C) I515C current before (top left) and after (top middle) reaction with MTSES. Top right shows current after reaction with MTSES with the y axis adjusted for ease in viewing. The bottom panels show open-channel current (bottom left and middle, same data on two scales) and voltage-dependent gating (bottom right) before (filled circles) and after (open circles) reaction with MTSES.
Tail-current analysis was used to determine the apparent open probability (P o). During each test pulse, channels were allowed to reach their steady-state open probability for that voltage; the tail pulse provides a constant driving force to compare the relative fraction of open channels in each preceding test pulse. Decay curves for each of the tails were fit to single exponentials, and these fits were then used to calculate the current at the time the voltage was stepped from the test voltage to the tail voltage. The apparent open probability is obtained from the normalized tail current, Po = I/I max, where I is the current at the test-pulse to tail-pulse transition and I max is the maximum current measured at this transition. P min (the minimum apparent open probability), z (the effective gating charge), and V o (the midpoint of the voltage–activation curve) were calculated by fitting the apparent open probability to an equation of the form
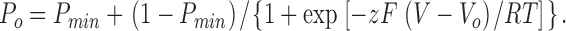 |
Gating of all mutants except S123C was well described by this equation. For S123C, the gating parameters were not evaluated because no time-dependent current decay is observed at any voltage. Also, the currents at negative voltages are much smaller than those at positive voltages. If this mutant has nonrectifying open-channel current, then these data suggest that S123C closes much faster than wild type, reaching equilibrium on a timescale faster than our recordings. Single-channel recordings also indicate that S123C closes faster than wild type and suggest that the maximum open probability does not reach unity under our conditions (Lin and Chen, 2003).
The activating +50-mV prepulse in protocol A is used to determine open-channel current. Since in wild-type ClC-0, the voltage–activation curve is saturated at +50 mV, and the maximum open probability is unity, the prepulse opens nearly all of the channels, so the current at the beginning of the ensuing test pulse is the open-channel current. In all of the mutants tested except S123C, the voltage–activation curve is near saturation at +50 mV. Currents at each of the test voltages were fit with a single exponential, and the current at the time the voltage was stepped from the prepulse voltage to the test voltage was calculated from these fits. For test pulse voltages where there is no significant decay in current, such that the trace could not be fit, the current immediately after the capacitive transient was used. The rectification metric was calculated as I−100/I+40, where I−100 and I+40 refer to the open-channel current −100 and +40 mV from the reversal potential, and normalized to the value determined for wild-type ClC-0 (−2.7), which is slightly inwardly rectifying. For the slightly asymmetric solution conditions used, the Goldman-Hodgkin-Katz current equation (Hille, 1992) predicts I−100/I+40 = −2.8.
Cysteine-modifying Reagents
2-Aminoethyl methanethiosulfonate, hydrobromide (MTSEA), MTSES (both from Toronto Research Chemicals), and AMS (Molecular Probes) were dissolved in Millipore water to make 0.1 M stock solutions, which were aliquoted and stored at −80°C until the day of use. Aliquots were thawed and kept on ice until just before use, at which point they were diluted into internal solution and flowed over the patch within 5 min from the time of dilution.
1,10-Phenanthroline monohydrate (Sigma-Aldrich) was dissolved in ethanol (Rossville gold shield, 200 proof) to make a 50 mM stock. This stock was made fresh each day and stored on ice. Anhydrous copper(II) sulfate (Cu(II)SO4; Sigma-Aldrich) was dissolved in Millipore water to make 1.5 mM stock, which was passed through a 0.2-μm filter and stored at 4°C. The 50 mM 1,10 phenanthroline and the 1.5 mM Cu(II)SO4 were diluted 104- and 1,000-fold, respectively, into internal solution lacking EGTA, and flowed over the patch within 5 min from the time of dilution.
DTT (Invitrogen) was dissolved in Millipore water to make a 1 M stock. Aliquots were frozen and stored at −20°C until the day of use, at which point they were stored on ice, diluted 100-fold into internal solution to give a final concentration of 10 mM.
Reaction with Cysteine-modifying Reagents
For each mutant, gating and rectification before and after reaction with a cysteine-modifying reagent were measured using protocol A while the patch was sitting in internal solution with no flow. This provided data with higher signal-to-noise than that obtained while solution is flowing. To monitor the effect of a reagent, protocol B was run: the patch was held at 0 mV, pulsed to +50 mV for 50 ms, and then to −150 mV for 100 ms, and this cycle was repeated every 5 s. This allowed the channels to explore both open and closed states during exposure to the reagent. The open-channel current at each 5-s interval was determined as follows: current decay during the −150-mV pulse was fit to a single exponential, and this fit was used to determine the current at the transition from +50 to −150 mV. To determine the rate of a reaction, the open-channel current was plotted versus time and fit to a single exponential (see Figs. 4 and 7). For experiments involving MTSES, AMS, or DTT, current was monitored without flow for at least 40 s, the internal solution was flowed for at least 50 s, and the solution containing the reagent was then flowed for 50–100 s, during which the chamber was exchanged with 10–15 volumes of reagent-containing solution. Flow was stopped, and the current monitored until the reaction went to at least 95% completion. Finally, the reagent was washed out with at least 10 volumes of internal solution, flow stopped, and protocol A used to measure open-channel current and voltage-dependent gating. For experiments involving pretreatment with MTSES before application of AMS, MTSES (100 μM or 1 mM) was applied until the reaction went to at least 95% completion. The chamber was then perfused with internal solution for at least 50 s, and then AMS was applied as described above. The patch was placed close to the perfusion outlet (within 3 mm) during these procedures.
Figure 4.

Time courses and rates of MTSES reactions. Protocol B was used while MTSES was flowed over the cytoplasmic side of the channels. (A and B) The open-channel current at −150 mV is plotted versus time, with arrows indicating when MTSES was introduced, and the traces fit to single exponentials. (A) Mutants that reacted faster than 0.015 s−1 (150 M−1s−1). (B) The three MTSES-sensitive mutants that reacted more slowly than 0.015 s−1, with only every 10th point plotted for clarity. For S123C, the open probability is very low at −150 mV, so the rate was based on the decrease in current at +50 mV. (C) Rates of reaction with 100 μM MTSES (mean ± SEM, with number patches in parentheses) as determined from fits such as those shown in A and B. For a second order reaction under these conditions, a rate of 0.1 s−1 corresponds to a rate constant of 103 M−1s−1. Asterisks indicate mutants that were further characterized (those that react faster than 0.015 s−1 and in which current decreased >35%), Fig. 6.
Figure 7.

Effects of AMS. (A) Chemical structures of MTSES and AMS adducts after reaction with a cysteine residue. (B–D) AMS effect on M311C. (B) The open-channel current at −150 mV is plotted versus time, with arrow indicating when 100 μM AMS was introduced, and the trace fit to a single exponential. (C) Current response to protocol A, before (left) and after (middle, right) reaction with AMS. (D) Open-channel current (left, middle) and apparent open probability (right), before (filled circles) and after (open circles) treatment with AMS. (E) Percent decrease in open-channel current −100 mV from the reversal potential, calculated as in Fig. 3. Reactions went to >95% completion except for M311C, which reached >91% completion. (F) Rates of reaction with 100 μM AMS as determined from fits such as the one shown in B. WT*, M311C*, and R312C*, shown with black bars, indicate rates of reaction with AMS for patches that had been pretreated with MTSES. (G) Rectification metric (as in Fig. 6 A) before (white bars) and after (dark bars) reaction with AMS. (H and I) Pretreatment with MTSES reduces effects of AMS. (H) Time courses as in B were used to calculate percent decrease in current at −150 mV caused by AMS with (black bars) and without (gray bars) pretreatment with MTSES. Percent decrease was calculated from time courses rather than as in E because pulse protocol A was not used between treatment with MTSES and AMS. (I) Rectification metric (as in Fig. 6 A) for untreated patches (white bars), patches treated with AMS alone (gray bars), MTSES alone (hatched bars) and MTSES followed by AMS (black bars). For R312C, the difference between AMS alone (gray bars) and MTSES followed by AMS (black bars) is significant (P = 0.027, Student's t test). All bar graphs show mean ± SEM, number patches in parentheses.
EGTA chelates copper ions and thus prevents the formation of the catalytic copper phenanthroline complex. For experiments involving copper phenanthroline, the patch was treated as above except that a perfusion with internal solution lacking EGTA was added before and after perfusion with copper phenanthroline.
When comparing voltage-dependent gating and rectification before and after reaction with MTSES, there are several sources of error that are important to consider, and which make the small effects on gating difficult to interpret. (a) For cysteine mutants where MTSES causes a significant decrease in open-channel current, unreacted channels could contribute significantly to the current measured after the reaction, thus skewing the apparent gating parameters and rectification. To test for this possibility, we subtracted the current contributed by unreacted channels, which was calculated based on the percent completion of the reaction. In all cases, subtracting this current contributed by the remaining unreacted channels (<5% of the channels in the patch) did not significantly change the derived gating parameters or the degree of outward rectification. (b) The contribution of series resistance changes as the total current in the patch changes. In all cases analyzed, correcting for the series resistance had no significant effect on the gating parameters or rectification. (c) Since ClC-0 has a minimum open probability that is significantly greater than zero, a voltage protocol cannot be used to subtract the background current (currents from endogenous channels and from leak). Background currents decrease the accuracy of P min and thus of the other gating parameters (z and V o). As seen in Fig. 6 (B–D), reaction with MTSES results in some changes in the voltage-dependent gating parameters. Because MTSES significantly reduces the current, at least some of these changes can be attributed to the increase in the ratio of background current to ClC-0 current, and MTSES-induced effects on ClC-0 gating are difficult to quantify.
Figure 6.
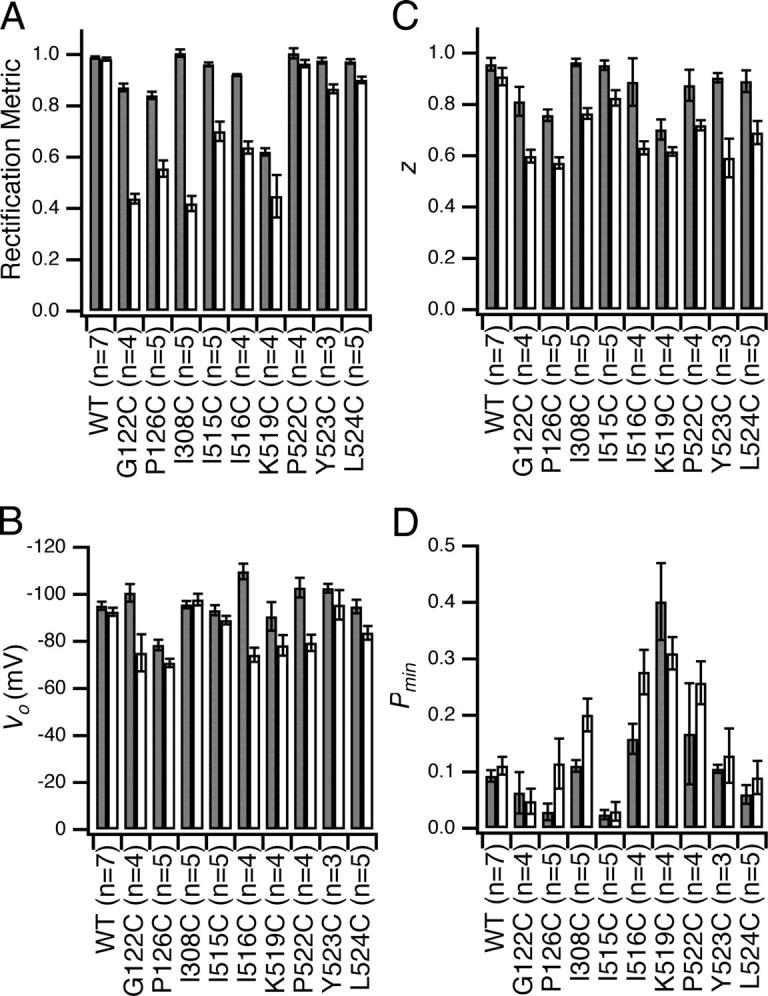
MTSES-sensitive sites: effects on voltage-dependent gating and rectification. Rectification and voltage-dependent gating parameters (derived as described in materials and methods) before (dark gray) and after (light gray) reaction with MTSES (mean ± SEM, number patches in parentheses). (A) Rectification metric; (B) V o, the midpoint of the voltage–activation curve; (C) z, the effective gating charge; (D) P min, the minimum open probability.
To use protocol A to compare open-channel current before and after reaction with a modifying reagent, the open probability at +50 mV must be the same before and after the reaction. If modification causes an extreme change in voltage-dependent gating, then our open-channel current measurements would actually reflect gating and not open-channel current. Although modification with MTSES causes subtle changes in gating (Fig. 6), the essential voltage-dependent gating mechanism, activation by depolarization, remains intact, and the open probability does plateau. Thus, it is reasonable to conclude that the measurements of open-channel current accurately reflect single-channel current both before and after treatment with a modifying reagent.
RESULTS
Candidate Pore-lining Residues in ClC-0
A cytoplasmic view of the E. coli ClC structure suggests at least six helices and two loops that might line the inner vestibule of the ClC-0 channel (Fig. 1 A, helices C, D, F, J, M, R, loop CD, DE). In addition, recent in silico studies of the prokaryotic ClC structures suggest specific residues on these sequence elements that are pore lining, and indicate that helix E may play a role in permeation (Corry et al., 2004; Faraldo-Gomez and Roux, 2004; Miloshevsky and Jordan, 2004). Previous studies have identified S123, E127, and residues along helix R as likely to be pore lining in ClC-0 (Pusch et al., 1995; Ludewig et al., 1996, 1997c; Middleton et al., 1996; Lin and Chen, 2000, 2003; Chen and Chen, 2003; Chen et al., 2003). To flesh out the picture of the intracellular vestibule, we screened three sequence elements in ClC-0 as well as nine residues on four other sequence elements hypothesized to line the intracellular vestibule (Fig. 1, A and B).
Figure 1.

Candidate pore-lining secondary structures. (A) Structure of ClC-ec1 (1OTS) as viewed from within the membrane. The subunits are colored gray and cyan, with secondary structures lining the intracellular vestibule of the gray subunit colored as follows: helix C and loop CD, orange; helix D and loop DE, magenta; helix E, light pink; helix F, periwinkle; helix J, royal blue; helix M, tan; helix R, red. The figure was created using Pymol (DeLano, 2002). (B) Alignment of secondary structures screened. ClC-ec1 and ClC-0 sequences were aligned using Clustal-W, with slight manual adjustment. Conserved residues are shaded. The regions screened are indicated by bars above the ClC-0 sequence. The beginning of helix J is omitted.
Use of Macroscopic Patch-clamp Recordings to Detect Changes in Pore Properties
To facilitate screening of a large number of residues, excised inside-out macroscopic patches were used to assess changes in pore properties, reflected in the open-channel current. Fig. 2 B shows an example of how changes in open-channel current derived from macroscopic patch data reflect the underlying single-channel current. It is known from single-channel recordings that mutating K519 to cysteine decreases single-channel current at negative voltages, where net chloride movement is outward (Middleton et al., 1996). In macroscopic recordings, K519C open-channel current is outwardly rectifying (Fig. 2 B, open circles), in contrast to wild-type open-channel current, which is essentially linear with voltage (Miller, 1982; Table I). This change in rectification reflects a decrease in single-channel current at negative voltages relative to positive voltages. Modification with MTSEA, a positively charged cysteine-modifying reagent, restores the charge at K519 and the single-channel conductance (Middleton et al., 1996), and makes the open-channel current linear with voltage, like wild type (Fig. 2 B, filled circles). When the modification with MTSEA is reversed with DTT, open-channel current is once again outwardly rectifying (Fig. 2 B, crosses). Thus, changes in the macroscopic open-channel current reflect the changes in current measured in single-channel recordings.
TABLE I.
Voltage-dependent Gating and Rectification of Cysteine Mutants
| Vo | z | Pmin | Rectification | n | |
|---|---|---|---|---|---|
| WTa | −99 ± 0.8 | 0.90 ± 0.02 | 0.076 ± 0.005 | 1.00 ± 0.004 | 68 |
| Q114C | −95 ± 2.2 | 0.87 ± 0.03 | 0.063 ± 0.013 | 0.99 ± 0.007 | 16 |
| S117C | −98 ± 3.9 | 0.93 ± 0.02 | 0.073 ± 0.013 | 0.98 ± 0.011 | 6 |
| P118C | −93 ± 2.5 | 0.91 ± 0.01 | 0.056 ± 0.012 | 0.98 ± 0.012 | 7 |
| Q119C | −106 ± 1.7 | 0.86 ± 0.02 | 0.048 ± 0.037 | 0.98 ± 0.015 | 3 |
| A120C | −86 ± 1.9 | 0.94 ± 0.02 | 0.041 ± 0.007 | 0.98 ± 0.009 | 13 |
| V121C | −91 ± 1.3 | 0.91 ± 0.02 | 0.081 ± 0.013 | 0.99 ± 0.017 | 13 |
| G122C | −94 ± 1.6 | 0.83 ± 0.02 | 0.044 ± 0.011 | 0.85 ± 0.008 | 17 |
| S123Cb | ND | ND | ND | ND | 6 |
| P126C | −80 ± 2.2 | 0.73 ± 0.02 | 0.035 ± 0.011 | 0.84 ± 0.020 | 12 |
| L128C | −94 ± 1.5 | 0.94 ± 0.11 | 0.122 ± 0.046 | 1.12 ± 0.006 | 4 |
| V136C | −97 ± 2.0 | 0.90 ± 0.02 | 0.069 ± 0.017 | 0.98 ± 0.012 | 8 |
| H138C | −90 ± 2.5 | 0.93 ± 0.04 | 0.058 ± 0.013 | 1.02 ± 0.010 | 9 |
| T142C | −90 ± 2.3 | 0.83 ± 0.04 | 0.052 ± 0.015 | 0.98 ± 0.007 | 5 |
| R144C | −91 ± 1.1 | 0.89 ± 0.02 | 0.058 ± 0.008 | 1.01 ± 0.007 | 10 |
| K149Cb | −28 ± 0.8 | 0.98 ± 0.01 | 0.042 ± 0.008 | 0.87 ± 0.007 | 19 |
| H171C | −101 ± 1.5 | 0.86 ± 0.03 | 0.069 ± 0.016 | 1.02 ± 0.009 | 12 |
| N304C | −91 ± 3.4 | 0.80 ± 0.03 | 0.051 ± 0.025 | 0.99 ± 0.010 | 3 |
| R305C | −92 ± 1.9 | 0.86 ± 0.05 | 0.055 ± 0.027 | 0.98 ± 0.024 | 3 |
| Q306C | −95 ± 1.6 | 0.94 ± 0.04 | 0.067 ± 0.022 | 0.98 ± 0.005 | 5 |
| I307C | −94 ± 1.8 | 0.88 ± 0.03 | 0.045 ± 0.017 | 0.98 ± 0.008 | 13 |
| I308C | −94 ± 1.0 | 0.95 ± 0.01 | 0.091 ± 0.006 | 1.00 ± 0.005 | 60 |
| V309C | −90 ± 1.3 | 0.93 ± 0.03 | 0.066 ± 0.011 | 1.01 ± 0.009 | 12 |
| F310C | −95 ± 1.2 | 0.89 ± 0.03 | 0.107 ± 0.030 | 0.98 ± 0.017 | 8 |
| M311C | −92 ± 0.9 | 0.94 ± 0.02 | 0.085 ± 0.017 | 0.99 ± 0.005 | 19 |
| R312C | −95 ± 1.5 | 0.89 ± 0.01 | 0.072 ± 0.006 | 0.93 ± 0.009 | 26 |
| K313C | −92 ± 1.0 | 0.95 ± 0.02 | 0.076 ± 0.011 | 1.01 ± 0.005 | 12 |
| K314C | −94 ± 0.7 | 0.91 ± 0.07 | 0.044 ± 0.023 | 1.02 ± 0.021 | 3 |
| N315C | −85 ± 2.9 | 0.87 ± 0.02 | 0.049 ± 0.006 | 0.98 ± 0.011 | 8 |
| F316C | −89 ± 1.0 | 0.94 ± 0.02 | 0.063 ± 0.008 | 0.98 ± 0.010 | 10 |
| V409C | −90 ± 0.7 | 0.93 ± 0.03 | 0.096 ± 0.011 | 0.96 ± 0.008 | 7 |
| T410C | −96 ± 0.9 | 0.75 ± 0.05 | 0.067 ± 0.019 | 0.88 ± 0.018 | 5 |
| P412C | −86 ± 1.4 | 0.83 ± 0.03 | 0.043 ± 0.012 | 0.90 ± 0.023 | 9 |
| Y512C | −98 ± 2.1 | 0.85 ± 0.02 | 0.000 ± 0.000 | 0.89 ± 0.007 | 12 |
| D513C | −92 ± 1.2 | 0.71 ± 0.01 | 0.032 ± 0.009 | 1.00 ± 0.010 | 35 |
| S514C | −98 ± 3.1 | 0.83 ± 0.02 | 0.064 ± 0.009 | 1.00 ± 0.010 | 10 |
| I515C | −87 ± 0.8 | 0.97 ± 0.01 | 0.029 ± 0.003 | 0.95 ± 0.006 | 93 |
| I516C | −101 ± 2.3 | 0.88 ± 0.05 | 0.134 ± 0.020 | 0.91 ± 0.017 | 13 |
| Q517C | −92 ± 1.1 | 0.94 ± 0.01 | 0.063 ± 0.008 | 1.02 ± 0.010 | 8 |
| I518C | −91 ± 2.0 | 0.97 ± 0.03 | 0.072 ± 0.009 | 1.02 ± 0.005 | 10 |
| K519Cb | −114 ± 1.9 | 0.65 ± 0.01 | 0.241 ± 0.017 | 0.61 ± 0.006 | 56 |
| K520C | −104 ± 1.5 | 0.84 ± 0.05 | 0.077 ± 0.039 | 1.00 ± 0.019 | 5 |
| L521Cc | |||||
| P522C | −94 ± 1.6 | 0.83 ± 0.02 | 0.115 ± 0.014 | 0.98 ± 0.005 | 26 |
| Y523C | −95 ± 1.7 | 0.92 ± 0.02 | 0.124 ± 0.012 | 0.97 ± 0.006 | 24 |
| L524C | −98 ± 1.4 | 0.88 ± 0.02 | 0.058 ± 0.009 | 0.97 ± 0.004 | 18 |
| P525C | −98 ± 1.1 | 0.90 ± 0.03 | 0.110 ± 0.010 | 0.97 ± 0.007 | 6 |
| E526C | −104 ± 1.5 | 0.84 ± 0.03 | 0.050 ± 0.021 | 0.98 ± 0.011 | 3 |
| L527C | −107 ± 2.0 | 0.87 ± 0.01 | 0.057 ± 0.017 | 1.01 ± 0.008 | 3 |
| S528C | −107 ± 2.1 | 0.88 ± 0.04 | 0.045 ± 0.021 | 1.01 ± 0.011 | 6 |
| W529C | −96 ± 1.7 | 0.85 ± 0.04 | 0.047 ± 0.019 | 0.98 ± 0.009 | 4 |
| S530C | −105 ± 2.2 | 0.85 ± 0.05 | 0.044 ± 0.028 | 1.00 ± 0.006 | 3 |
| S531C | −106 ± 1.7 | 0.92 ± 0.01 | 0.074 ± 0.006 | 1.00 ± 0.001 | 2 |
Effects of cysteine mutagenesis. Voltage-dependent gating parameters for wild type and cysteine point mutants: midpoint of the voltage–activation curve (Vo), effective gating charge (z), and minimum open probability (Pmin). Rectification is quantified as described in materials and methods and normalized to wild type, which is slightly inwardly rectifying. Values greater than one indicate inward rectification with respect to wild type; values less than one indicate outward rectification with respect to wild type. Experiments were carried out in slightly asymmetric solution conditions, such that the expected rectification is 1.04 according to the Goldman-Hodgkin-Katz current equation.
WT refers to C212S, the background for all cysteine point mutants.
Mutants with V o shifted >20 mV or rectification changed by >20% from wild type.
Mutants for which no current could be detected.
In some cases macroscopic patches reveal changes in open-channel current even when single-channel current is too small to be detected. For example, I515, predicted to be on the same face of helix R as K519, has been studied using single-channel recordings (Lin and Chen, 2003). Modification of I515C with MTSES, a negatively charged cysteine-modifying reagent, causes the single-channel current to vanish. It cannot be discerned, however, whether modification with MTSES alters gating or causes the single-channel current to decrease so that it is too small to be detected. Macroscopic patches of I515C illustrate that addition of MTSES causes a decrease in open-channel current and an increase in outward rectification, but that voltage-dependent gating remains unchanged (Fig. 2 C). Therefore, the decrease in current arises almost exclusively from effects on the open pore.
Mutations to Cysteine have Little Functional Effect
We generated 51 cysteine point mutations in the presumed intracellular vestibule of ClC-0. A previous mutational analysis of helix R showed that drastic mutations (L521K, P522G, and Y523K) affect open-channel rectification and voltage-dependent gating (Ludewig et al., 1997c). However, for most of the residues we tested, including all of helix R, mutation to cysteine had little effect on rectification or gating (Table I), which indicates that small changes can be tolerated without significantly perturbing the architecture of the pore or the gating mechanism. Aside from K519 and S123, positions at which mutations have been previously reported to affect open-channel rectification and voltage-dependent gating (Pusch et al., 1995; Middleton et al., 1996; Ludewig et al., 1996, 1997c; Accardi and Pusch, 2003; Chen and Chen, 2003; Lin and Chen, 2003), the only other major change observed was with K149, a conserved residue on helix E, where mutation to cysteine resulted in a shift in V o of ∼+70 mV.
Sensitivity to MTSES
12 mutants tested exhibited a large decrease in current (>35%) upon addition of MTSES, while the remaining 39 showed little or no change in current upon treatment with 100 μM MTSES for 1 min, similar to the wild-type control (Fig. 3). For the latter 39 mutants (with the exception of D513C and R312C; Fig. 4), the <25% decrease in current was indistinguishable from the downward drift we commonly observe in excised inside-out patches. For each of the MTSES-sensitive mutants, we determined the rate of the MTSES reaction by plotting open-channel current at −150 mV as a function of time and fitting the decay in current to a single exponential (Fig. 4). Using the rate and the MTSES exposure time, we calculated the percent completion of each reaction. Fig. 3 shows the percent inhibition for reactions that had reached at least 95% completion. Of the mutants that showed a >35% decrease in current, we further characterized the nine that reacted fastest (Fig. 4 C, asterisks) by analyzing the voltage-dependent gating and rectification after reaction with MTSES. This analysis required patches that had >100 pA current after reaction with MTSES.
Figure 3.
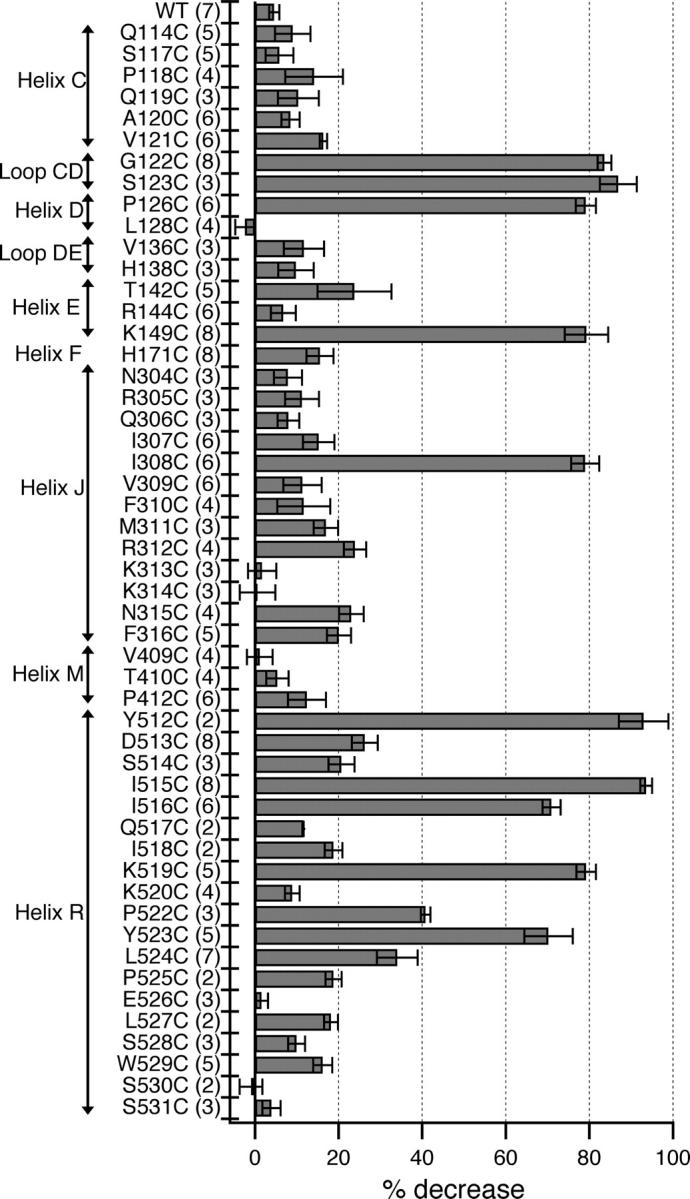
MTSES sensitivity of candidate pore-lining residues. Change in open-channel current −100 mV from the reversal potential, before (Ibefore) and after (Iafter) application of MTSES shown as percent decrease in current {100*((Ibefore − Iafter)/Ibefore)}. Bars show mean ± SEM, number of patches in parentheses. For residues for which the decrease in current was distinguishable from drift, the percent decrease is reported for reactions that had reached >95% completion. Since S123C passes very little current at negative voltages, the decrease in current at +40 mV from the reversal potential is shown.
These nine residues are on a variety of secondary structures: six on helix R, and one each on helix J, helix D, and loop CD. Fig. 5 shows primary data from the helix J mutant (I308C), and the loop CD mutant (G122C). For both, modification with MTSES caused a significant decrease in open-channel current and increase in outward rectification, as seen with I515C (helix R, Fig. 2 C). Similar effects were seen with the other seven mutants, with the change in rectification being smaller for the three MTSES-sensitive residues on the COOH-terminal end of helix R, P522C, Y523C, and L524C (Fig. 6 A). The effects of MTSES on gating are relatively minor (Fig. 6 B–D), and are similar in magnitude to those previously seen when sites on helix R are mutated to residues other than cysteine (K519E, P522G, and Y523K; Pusch et al., 1995; Ludewig et al., 1997c). Although precisely quantifying MTSES effects on gating can be problematic (see materials and methods), in every case the essential voltage-dependent gating mechanism remains intact.
Figure 5.

Effects of MTSES on open-channel current and voltage-dependent gating. Two examples of MTSES-sensitive mutants: I308C on helix J (A) and G122C on loop CD (B). Current responses to protocol A are shown before (top left) and after (top middle) exposure to MTSES. Top right shows the same data as top middle but with the y axis adjusted for ease in viewing. The open-channel current (bottom left, bottom middle with y axis adjusted) and apparent open probability (bottom right) before (filled circles) and after (open circles) reaction with MTSES.
For the three remaining mutants that showed a >35% decrease in current (S123C, K149C, and Y512C) the effects of MTSES are difficult to interpret. For S123C, we were not able to measure voltage-dependent gating (see materials and methods), so changes in open-channel current could not be distinguished from effects on gating. For K149C, the voltage dependence of gating is shifted, but the shape of the voltage–activation curve is preserved. However, because this mutant, as well as Y512C, reacts relatively slowly with MTSES, we were not able to obtain patches that were stable enough and had enough current for accurate post-reaction measurements. Furthermore, the rate of K149C reaction did not increase when the concentration of MTSES was increased 10-fold. Thus, for these three mutants, MTSES could be causing a decrease in current by altering gating or by changing the properties of the open pore, or both.
ClC-0 Secondary Structure
The 12 MTSES-sensitive sites are predicted to reside on a variety of secondary structures: 4 helices and 1 loop (Fig. 3), based on sequence alignments with the ClCs of known structure. For the two helices in which more than four residues were examined, we found patterns of sensitivity consistent with predicted secondary structures.
Helix R.
For the region predicted to be helix R, the six MTSES-sensitive residues fall onto one side of the helix (Fig. 9 A). This result supports the hypothesis that this region is helical up to residue Y523, and suggests which side of helix R faces the open pore.
Helix J.
Of the 13 residues screened in the region predicted to be helix J, I308C alone was found to be MTSES sensitive. Since helix J is steeply angled away from the axis of the bound chlorides in the ClC-ec1 structure, other residues on this helix may line a wider part of the pore. To test for this possibility, we used AMS, a negatively charged cysteine-modifying reagent that is larger than MTSES (Fig. 7 A), which has been used to probe function in a variety of membrane proteins (Lutsenko et al., 1997; Quitterer et al., 1999; Iwaki et al., 2000). As positive controls, we examined the MTSES-sensitive mutants I308C, I515C, and Y523C. These mutants showed a decrease in open-channel current upon treatment with AMS (Fig. 7 E). These changes are larger than those observed with wild type (Fig. 7 E), which exhibits a significantly slower decrease in current when treated with AMS (Fig. 7 F). We do not know the mechanism of the slow background effect on wild type, but it is unlikely to occur via modification of a native cysteine, as it is not prevented by pretreatment with MTSES (Fig. 7, F, H, and I). On helix J, we applied AMS to three MTSES-insensitive mutants (V309C, M311C, and K313C) and one mildly MTSES-sensitive mutant (R312C). For M311C, AMS causes a significant decrease in open-channel current (Fig. 7, B, C, and E) and an increase in rectification (Fig. 7 G) that occurs faster than the background effect (Fig. 7 F), but has no measurable effects on voltage-dependent gating. For R312C, although the decrease in current is similar to that observed with wild type (Fig. 7, E and H), it occurs much more rapidly (Fig. 7 F), and is accompanied by a larger change in rectification (Fig. 7 G), with no measurable effect on gating. Pretreatment of M311C or R312C with MTSES reduces the rate of current inhibition by AMS to the level of the background effect observed with wild type (Fig. 7 F), and reduces the effect on rectification (Fig. 7 I). For M311C, pretreatment with MTSES also reduces the extent of current inhibition (Fig. 7 H). Thus, reaction with MTSES protects M311C and R312C from modification by AMS, which indicates that the fast-acting effect of AMS on M311C and R312C is mediated by reaction with the introduced cysteine. When treated with AMS, V309C and K313C behaved like wild type, showing a slow decrease in current and little change in rectification (Fig. 7, E–G). This suggests that neither V309 nor K313 are as near to the pore as I308, M311, and R312. Thus, for residues on the putative helix J, the pattern of MTSES and AMS sensitivity combined is consistent with the hypothesis that this region is helical (see Fig. 9 A), at least from I308 to R312.
Figure 9.
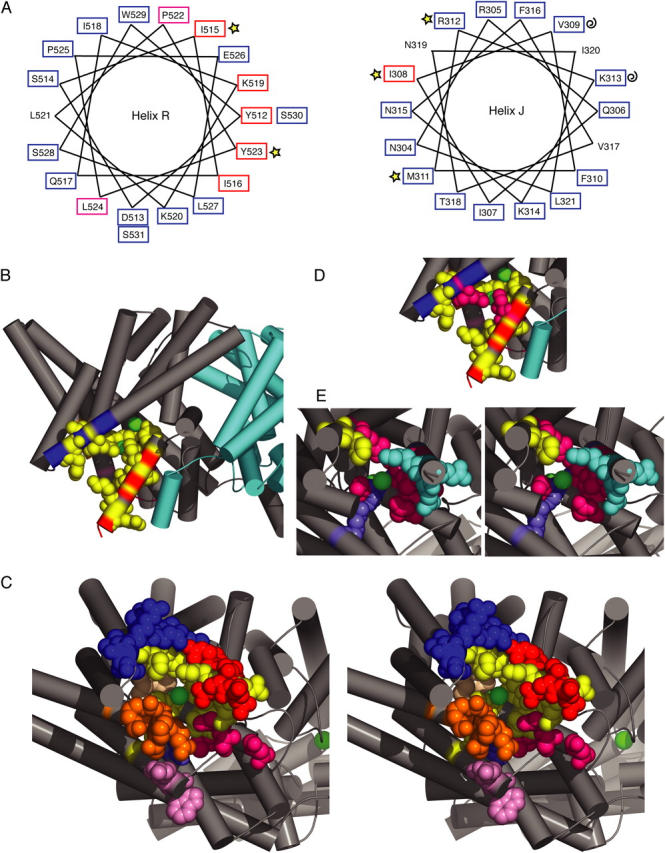
Residues sensitive to thiol modification mapped onto the ClC-ec1 structure (1OTS). (A) MTSES/AMS sensitivity correlates with predicted secondary structures. Helical wheels for regions predicted to be helix R and helix J. Tested residues are boxed according to their MTSES sensitivity reported in Fig. 3 (red, >65% decrease in current; magenta, 35–45%; blue, <35%), and marked according to their AMS sensitivity as reported in Fig. 7 (stars, >0.01 s−1 rate of AMS inhibition; curls, <0.01 s−1). (B) A view from within the membrane, with the subunits shown in gray and cyan and chloride ions spacefilled in green. In B and C, AMS/MTSES-sensitive residues are spacefilled in yellow, and all other residues tested are colored according to secondary structure (helix C and loop CD, orange; helix D and loop DE, magenta; helix E, light pink; helix F, periwinkle; helix J, royal blue; helix M, tan; helix R, red). (C) Stereoview of one subunit, viewed from inside the cell at an angle so that the bound chloride is visible. All tested residues spacefilled; AMS/MTSES-sensitive residues (yellow) cluster around bound chlorides. (D) Side view as in B, highlighting in magenta the proximity of Q277 (helix J, I308 equivalent) to I448 (helix R, I515 equivalent) in the ClC-ec1 structure. (E) Cytoplasmic view as in C showing spacefilled the AMS-sensitive residues (yellow) and the MTSES-sensitive residues reacting more slowly (periwinkle) or faster than 0.015 s−1 (150 M−1s−1) (magenta for residues with large effects on open-channel rectification, cyan for residues with smaller effects on open-channel rectification).
Cross-linking Helix R and Helix J
If helix R and helix J both line the intracellular vestibule, engineered cysteines on these helices may be close enough to form a disulfide bond, and such a cross-link would likely affect chloride permeation. The formation of disulfide bonds between pore-lining residues alters function in other ion channels (Benitah et al., 1996; Horenstein et al., 2001; Flynn and Zagotta, 2001). In the prokaryotic ClC structures, helix J and helix R are near one another, with Q277 (I308 in ClC-0) and I448 (I515) particularly close (see Fig. 9 D). The double mutant I308C-I515C, like each of the single cysteine mutants (I308C, I515C), displays current like wild type (Fig. 8 A, left). In the presence of copper phenanthroline, an oxidant commonly used to catalyze disulfide bond formation in membrane proteins (Careaga and Falke, 1992; Yu et al., 1995; Gordon et al., 1997; Wang and Kaback, 1999; Matulef and Zagotta, 2002), a significant reduction in the I308C-I515C current occurs concomitantly with an increase in outward rectification (Fig. 8, A–C) and no discernible effect on voltage-dependent gating. These effects were quickly and almost fully reversible with the subsequent addition of 10 mM DTT (Fig. 8, A–C). Treatment of the single-cysteine mutants I308C and I515C with copper phenanthroline yielded only small effects on currents that were not reversible with DTT (Fig. 8, D and E). Thus, the effect of copper phenanthroline on the double mutant I308C-I515C is likely due to a disulfide bond forming between these residues, and unlikely to have been caused by other types of oxidative damage or by formation of disulfide bonds involving native cysteines.
Figure 8.
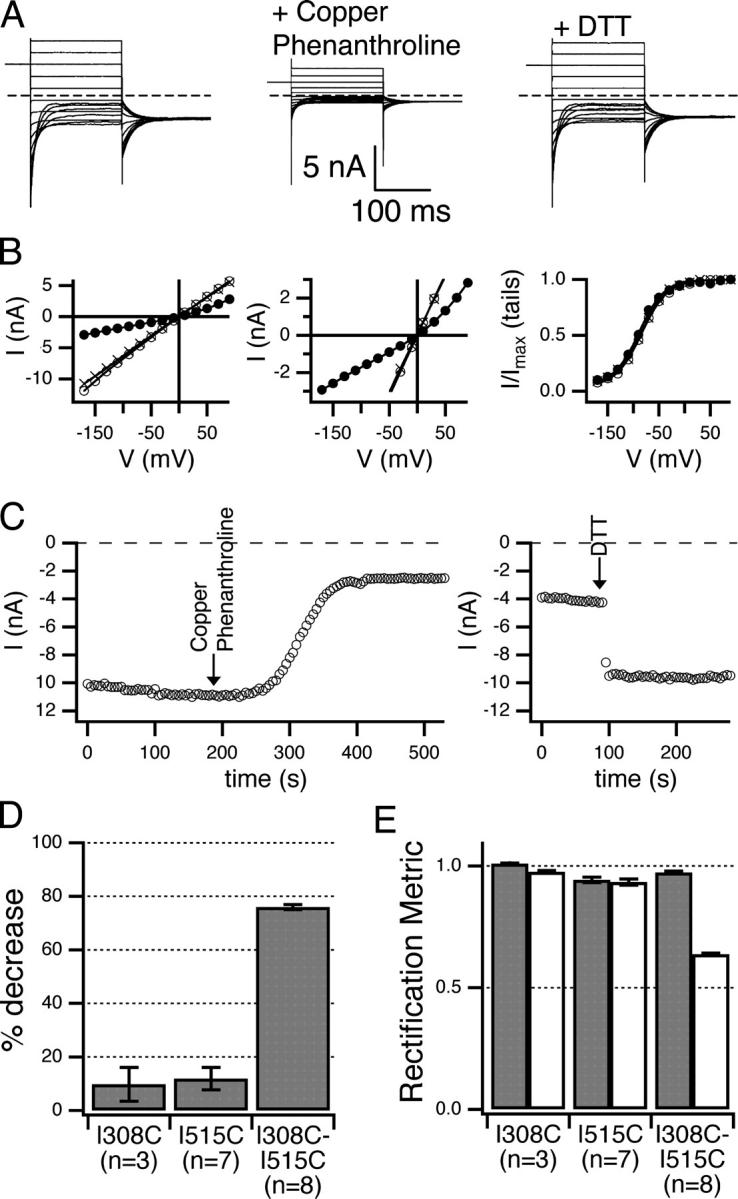
Disulfide bond formation between I308C and I515C. (A–C) Effect of copper phenanthroline and subsequent DTT on the I308C-I515C double mutant. (A) Current response to protocol A before (left) and after (middle) reaction with copper phenanthroline, and after DTT (right). (B) Open-channel current (left and middle) and open probability (right), before (open circles) and after (closed circles) copper phenanthroline, and after DTT (crosses). (C) Time courses of open-channel current at −150 mV as in Fig. 4, for the reaction catalyzed by copper phenanthroline (left) and reversed by DTT (right). Arrows indicated the time that the reagents were introduced. The time courses for the reaction induced by copper phenanthroline did not fit a first-order exponential, and the reversal by DTT was so fast as to be limited by solution exchange. (D) Percent decrease in open-channel current after treatment with copper phenanthroline, calculated as in Fig. 3. All patches included were subsequently treated with DTT, which had little effect on the I308C and I515C single mutants but restored the I308C-I515C double mutant current to near its original value. (E) The disulfide bond between I308C and I515C causes outward rectification. Rectification before (dark gray) and after (light gray) treatment with copper phenanthroline. For details on the rectification metric, see materials and methods. All bar graphs show mean ± SEM, number patches in parentheses.
To determine whether I515 is the residue on helix R that is the closest to I308, the ability of I308C to cross-link with other residues on helix R needs to be examined. However, unlike I515C, other single cysteine mutants, Q517C, I518C, K519C, and Y523C, appear to be susceptible to oxidation, as each showed a significant decrease in current upon addition of copper phenanthroline. Although there are a variety of oxidation reactions that could be causing this effect, only disulfide-bond formation is reversible with DTT. We found that for these single-cysteine mutants, the copper phenanthroline–mediated current decrease was partly reversible with DTT, which suggests that these residues may cross-link to a native cysteine. The lack of complete reversal may be due to the formation of other oxidation products or may occur because the disulfide has low accessibility or reactivity to DTT. The latter explanation is not unreasonable since reversal with DTT was at least an order of magnitude slower than for I308C-I515C (unpublished data). If this is the case, removal of one or more of the 12 native cysteine(s) may make it possible to test whether I308C can cross-link to other residues on helix R.
DISCUSSION
Using cysteine-scanning mutagenesis, we have identified 14 ClC-0 point mutants that exhibit significant MTSES- or AMS-dependent changes in current (Figs. 3, 6, and 7). For the 11 mutants in which post-reaction measurements could be examined (Figs. 6 and 7), changes in pore properties (open-channel current and rectification) suggest that these residues line the intracellular vestibule of ClC-0. Although the effects on voltage-dependent gating are relatively minor, it is not surprising that some modifications affect both permeation and gating, since these processes are tightly coupled in ClC-0.
Sites that react relatively slowly with MTSES (S123C, Y512C, K149C; periwinkle in Fig. 9 E) may have restricted accessibility to the cytoplasmic solution or may line a part of the pore where the local environment disfavors the reaction chemistry. Thus, it is possible that these residues affect conduction through some allosteric mechanism. Data from another study suggest that S123 and Y512 line a region of the pore deep in the transmembrane domain (Lin and Chen, 2003). Our results are consistent with this hypothesis. Although K149C is completely buried in the ClC-ec1 structure, two recent computational studies suggested that it plays a key role in chloride permeation (Corry et al., 2004; Faraldo-Gomez and Roux, 2004). Our results support the notion that this residue is important for permeation and gating, but further experiments are needed to determine how modification affects current and where K149 is located relative to the open pore in ClC-0.
Although 39 residues were insensitive to MTSES, some of these might line the pore. The short MTSES exposure used in our experiments identifies only relatively fast-reacting mutants, so longer exposure time might identify additional, slowly reacting sites. Many factors influence the reactivity of a cysteine residue in a protein (Karlin and Akabas, 1998; Wilson and Karlin, 2001). Lack of sensitivity to MTSES means either that the introduced cysteine side chain in that position is inaccessible or poorly reactive, or that modification with MTSES is not a significant enough perturbation to affect channel function. In the latter case, experiments using larger cysteine modifying reagents can be used to test the possibility that a residue lines a wider part of the vestibule. Using this approach, we identified two MTSES-insensitive sites on helix J for which modification with AMS affects open-channel current and rectification (Fig. 7).
For helix R, the results of this study agree with a previous cysteine-accessibility scan in which rates of modification (rather than the effects on current after modification) were used to identify residues that likely line the ClC-0 pore (Lin and Chen, 2003). The mutants D513C, I515C, I516C, K519C, P522C, and Y523C were identified based on having MTSES reaction rates faster than that of S123C, and MTSET reaction rates faster than that of Y512C (Lin and Chen, 2003). For these mutants, the ratio of MTSES to MTSET reaction rates correlates with how deep they are predicted to be in the pore based on the structure of ClC-ec1. With the exception of D513C, for which MTSES causes a small (26%) decrease in open-channel current, we find that these helix R cysteine mutants identified by Lin and Chen show a large (>35%) decrease in open-channel current upon addition of MTSES. Thus, both kinetic and post-reaction analyses of MTSES effects identify the same face of helix R as pore lining in ClC-0. We also examined residues beyond L524, and found none to be sensitive to MTSES. It may be that the region is not helical after L524 or that a kink induced by two prolines not present in the prokaryotic homologues (Fig. 1 B) causes the cytoplasmic end of the helix to be further away from the chloride path. A kink in the helix may explain why L524C is sensitive to MTSES even though it is not on the same face of helix R as the other MTSES-sensitive sites (Fig. 9 A).
This study expands the cysteine scan to include helix J, loop CD, and residues on helices C, D, E, F, and M. These results provide information needed for improving the model of the ClC-0 intracellular vestibule. Prokaryotic ClCs of known structure have only 15–20% identity to ClC-0, and since they are antiporters, not channels, are functionally in a distinct class of membrane proteins. In addition, the available structures show a closed state in which the chloride pathway is occluded. To determine how well the prokaryotic ClC structures reflect the open-pore structure of ClC-0, we mapped our results onto the ClC-ec1 structure (1OTT) (Fig. 9, B and C). All the MTSES- or AMS-sensitive residues (spacefilled in yellow) appear nearer to the bound chlorides than insensitive residues (spacefilled and colored by secondary structural element in Fig. 9 C). This suggests that the same residues that bind and transport chloride in the prokaryotic ClC transporters also help attract and move chloride across the ClC-0 pore.
To evaluate this observation more quantitatively, we calculated the distance of each residue tested to the nearest bound chloride ion in the ClC-ec1 structure. For this analysis, we used the E148A mutant structure, which is identical to the wild-type structure in the intracellular region, but has a third chloride bound extracellular to those in the wild-type structure (Dutzler et al., 2003). There is a correlation between the proximity to bound chloride and the likelihood that the equivalent residue in ClC-0 is MTSES or AMS sensitive (Table II, A). The fraction of tested residues sensitive to MTSES/AMS decreases as the distance from chloride increases, from 0.83 for residues within 7 Å of a chloride ion to 0.17 for residues >12 Å away. A similar trend is seen for residues suggested to line the intracellular vestibule in ClC-1, based on mutagenesis; of nine residues tested in ClC-1 (Fahlke et al., 1997, 2001), six correspond to residues tested in ClC-0 and are indicated in Table II (A). This supports the hypothesis that the architecture of the intracellular vestibule is conserved among the ClC family members.
TABLE II.
Proximity to Bound Chloride

Although ClC-1 is an orthologue to ClC-0 and ∼50% identical in sequence, MTSES accessibility differs between the two proteins. Three of the residues we tested in ClC-0 have also been tested with MTSES in ClC-1. Two of these (equivalent to S123C and P126C in ClC-0) are sensitive to intracellular MTS reagents in both ClC-0 and ClC-1 (Fahlke et al., 2001), while one (H171C) is only sensitive in ClC-1 (Fahlke et al., 1997). In ClC-0, H171C showed no measurable response to 1 mM MTSES (unpublished data). This suggests that even though H171 is conserved in the eukaryotic ClCs, its position or function is different in ClC-1 than in ClC-0, lending credence to the idea that the pores in these two proteins are not identical. Some differences are expected since ClC-1 differs from ClC-0 in single-channel conductance (1 pS versus 10 pS) and open-channel rectification (inwardly rectifying versus nonrectifying in symmetric solutions).
Several recent studies have used the prokaryotic ClC structures to calculate the energetics of chloride binding and permeation (Corry et al., 2004; Faraldo-Gomez and Roux, 2004; Miloshevsky and Jordan, 2004). Residues highlighted in these studies are indicated in Table II (A). The predictions regarding these residues vary in scope; some are predicted to make significant contributions to permeation energetics, while others are merely suggested to line the permeation pathway and their energetic contribution is small or not reported. While there is a core subset of residues consistently named as likely to influence pore properties, the studies vary in their predictions of what residues play the most important roles in permeation. The present study assesses the effects of mutating some of these residues. Sensitivity to modification with MTSES, manifested in changes in open-channel current and rectification, buttresses the hypothesis that certain residues highlighted in the computational studies play an important role in permeation.
Although not all the residues predicted to affect conduction in ClC-ec1 are MTSES or AMS sensitive in ClC-0, our results suggest that there is an overall structural conservation in the intracellular vestibule. First, conservation and MTSES/AMS sensitivity are correlated: 6 out of 14 MTSES/AMS sensitive residues are identical in ClC-0 and the prokaryotic ClCs; in contrast, only 3 of the 37 insensitive residues are conserved (Table II, A). Second, even in the nonconserved regions (helix R and helix J) the pattern of sensitivity suggests that secondary structure is conserved (Fig. 9 A). Therefore it is likely that the same structural elements play a role in conduction in both ClC-0 and ClC-ec1.
On the other hand, our results suggest the positions of these pore-lining elements relative to each other may be different for these two proteins. Five MTSES/AMS-sensitive residues are both nonconserved and >12 Å from a chloride in the ClC-ec1 structures. Based on the ClC-ec1 structure, these five are likely to be further out toward the cytoplasm, and therefore to line a wider part of the pore than the other MTSES-sensitive residues described here. Three of the five (P522, Y523, and L524) exhibit a milder change in open-channel rectification than the other MTSES-sensitive residues tested (Fig. 6; cyan in Fig. 9 E), while the other two (M311 and R312, yellow in Fig. 9 E) require modification by a large reagent, AMS, to significantly affect chloride permeation. Even though conduction through M311C and R312C is significantly affected only when a large modifying reagent is used, in the ClC-ec1 structures, the proximity of these two residues to bound chloride is similar to that of Y523. Assuming the size of the thiol-modifying reagent required to affect conduction is a measure of the distance between a residue and the chloride permeation pathway, our results suggest that the exact positioning of these residues in the intracellular vestibule of the ClC-0 open pore is different from what is seen in the prokaryotic ClC structures. Thus, the intracellular vestibules of ClC-0 and ClC-ec1 likely differ most at the cytoplasmic entryway.
We further assessed our data in light of a ClC-0 model that was developed by Corry et al. (2004). With this model, which like the ClC-ec1 E148A mutant structure contains three chloride ions per subunit, there was a stronger correlation between the proximity of a residue to bound chloride and its MTSES/AMS sensitivity (Table II, B). Whereas five residues shown to be AMS or MTSES sensitive in ClC-0 are >12 Å from a chloride in ClC-ec1 structures, four of these residues are within 12 Å of a chloride in the homology model. This suggests that the homology model is an improvement on prokaryotic ClC structures as a model for the open pore of ClC-0.
The results for the double mutant I308C-I515C indicate that these two residues can form a disulfide bond. In both the prokaryotic ClC structures and the homology model (Corry et al., 2004), I515 is closer to I308 than any other residue on helix R (Fig. 9 D), but not close enough to meet the geometrical requirements for disulfide bond formation (Careaga and Falke, 1992). It is possible that either of these structures represents the state where the disulfide bond is formed, since the ∼4-Å movement required could occur through breathing movements of the protein. Our results suggest that the positioning of helix R and J in ClC-0 is similar to that in the ClC-ec1 structure and in the ClC-0 homology model.
Conclusion
We have used cysteine-scanning mutagenesis to identify residues in the intracellular vestibule of ClC-0 that are sensitive to thiol-modifying reagents. Our results support the hypothesis that analogous structural elements line the intracellular vestibule in both ClC-0 and ClC-ec1, and thus plausibly in all the ClCs. When mapped onto the ClC-ec1 structure, residues that are sensitive to thiol-modifying reagents are near the bound chloride ions. This is a striking correlation given that modification affects open-pore properties and the structure of ClC-ec1 represents a closed state. The correlation is stronger in a homology model of the open state of ClC-0 that was created by making only minimal structural adjustments (Corry et al., 2004). Despite functional variation in the family, the ClC-ec1 structure is likely a useful starting model for guiding studies on other ClCs.
Acknowledgments
We thank Mila Gadzinski for technical assistance and Dr. Shin-Ho Chung for providing pdb coordinates for the ClC-0 homology model. We are grateful to members of the Aldrich lab for helpful discussions and Dr. Rob Blaustein, Dr. Kim Matulef, Dr. Joe Mindell, Dr. Richard Aldrich, Dan Kraut, and Gilbert Martinez for comments on the manuscript.
This work was supported by the Mathers Foundation. A. Engh is currently supported by an American Heart Association predoctoral fellowship and was previously supported by the National Institutes of Health Biophysics Training Grant at Stanford University.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: AMS, sodium 4-acetamido-4'-maleimidylstilbene-2'2-disulfonic acid; MTS, methanethiosulfonate; MTSEA, 2-aminoethyl methanethiosulfonate, hydrobromide; MTSES, sodium (2-sulfonatoethyl)methanethiosulfonate.
References
- Accardi, A., and C. Miller. 2004. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 427:803–807. [DOI] [PubMed] [Google Scholar]
- Accardi, A., and M. Pusch. 2003. Conformational changes in the pore of CLC-0. J. Gen. Physiol. 122:277–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akabas, M.H. 1998. Channel-lining residues in the M3 membrane-spanning segment of the cystic fibrosis transmembrane conductance regulator. Biochemistry. 37:12233–12240. [DOI] [PubMed] [Google Scholar]
- Akabas, M.H., D.A. Stauffer, M. Xu, and A. Karlin. 1992. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science. 258:307–310. [DOI] [PubMed] [Google Scholar]
- Bauer, C.K., K. Steinmeyer, J.R. Schwarz, and T.J. Jentsch. 1991. Completely functional double-barrelled chloride channel expressed from a single Torpedo cDNA. Proc. Natl. Acad. Sci. USA. 88:11052–11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becchetti, A., K. Gamel, and V. Torre. 1999. Cyclic nucleotide-gated channels. Pore topology studied through the accessibility of reporter cysteines. J. Gen. Physiol. 114:377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitah, J.-P., G.F. Tomaselli, and E. Marban. 1996. Adjacent pore-lining residues within sodium channels identified by paired cysteine mutagenesis. Proc. Natl. Acad. Sci. USA. 93:7392–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, E.L., P.D. Huynh, A. Finkelstein, and R.J. Collier. 1998. Identification of residues lining the anthrax protective antigen channel. Biochemistry. 37:3941–3948. [DOI] [PubMed] [Google Scholar]
- Careaga, C.L., and J.J. Falke. 1992. Structure and dynamics of Escherichia coli chemosensory receptors. Biophys. J. 62:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.F., and T.Y. Chen. 2001. Different fast-gate regulation by external Cl− and H+ of the muscle-type ClC chloride channels. J. Gen. Physiol. 118:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.-F., and T.Y. Chen. 2003. Side-chain charge effects and conductance determinants in the pore of ClC-0 chloride channels. J. Gen. Physiol. 122:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S., H.A. Hartmann, and G.E. Kirsch. 1997. Cysteine mapping in the ion selectivity and toxin binding region of the cardiac Na+ channel pore. J. Membr. Biol. 155:11–25. [DOI] [PubMed] [Google Scholar]
- Chen, T.-Y. 2005. Structure and function of CLC channels. Annu. Rev. Physiol. 67:809–839. [DOI] [PubMed] [Google Scholar]
- Chen, T.Y., and C. Miller. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl- channel. J. Gen. Physiol. 108:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T.-Y., M.-F. Chen, and C.-W. Lin. 2003. Electrostatic control and chloride regulation of the fast gating of ClC-0 chloride channels. J. Gen. Physiol. 122:641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleiren, E., O. Benichou, E. Van Hul, J. Gram, J. Bollerslev, F.R. Singer, K. Beaverson, A. Aledo, M.P. Whyte, T. Yoneyama, et al. 2001. Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum. Mol. Genet. 10:2861–2867. [DOI] [PubMed] [Google Scholar]
- Corry, B., M. O'Mara, and S.-H. Chung. 2004. Conduction mechanisms of chloride ions in ClC-type channels. Biophys. J. 86:846–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano, W.L. 2002. The PyMOL Molecular Graphics System. http://www.pymol.org. Accessed September 22, 2004.
- Dodier, Y., U. Banderali, H. Klein, O. Topalak, O. Dafi, M. Simoes, G. Bernatchez, R. Sauve, and L. Parent. 2004. Outer pore topology of the ECaC-TRPV5 channel by cysteine scan mutagenesis. J. Biol. Chem. 279:6853–6862. [DOI] [PubMed] [Google Scholar]
- Dutzler, R. 2004. Structural basis for ion conduction and gating in ClC chloride channels. FEBS Lett. 564:229–233. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E.B. Campbell, M. Cadene, B.T. Chalt, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E.B. Campbell, and R. Mackinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- Estevez, R., and T.J. Jentsch. 2002. CLC chloride channels: correlating structure with function. Curr. Opin. Struct. Biol. 12:531–539. [DOI] [PubMed] [Google Scholar]
- Estevez, R., B.C. Schroeder, A. Accardi, T.J. Jentsch, and M. Pusch. 2003. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 38:47–59. [DOI] [PubMed] [Google Scholar]
- Fahlke, C., H.T. Yu, C.L. Beck, T.H. Rhodes, and A.L. George Jr. 1997. Pore-forming segments in voltage-gated chloride channels. Nature. 390:529–532. [DOI] [PubMed] [Google Scholar]
- Fahlke, C., R.R. Desai, N. Gillani, and A.L. George Jr. 2001. Residues lining the inner pore vestibule of human muscle chloride channels. J. Biol. Chem. 276:1759–1765. [DOI] [PubMed] [Google Scholar]
- Faraldo-Gomez, J.D., and B. Roux. 2004. Electrostatics of ion stabilization in a ClC chloride channel homologue from Escherichia coli. J. Mol. Biol. 339:981–1000. [DOI] [PubMed] [Google Scholar]
- Flynn, G.E., and W.N. Zagotta. 2001. Conformational changes in S6 coupled to the open of cyclic nucleotide-gated channels. Neuron. 30:689–698. [DOI] [PubMed] [Google Scholar]
- Gordon, S.E., M.D. Varnum, and W.N. Zagotta. 1997. Direct interaction between amino- and carboxyl-terminal domains of cyclic nucleotide-gated channels. Neuron. 19:431–441. [DOI] [PubMed] [Google Scholar]
- Hanke, W., and C. Miller. 1983. Single chloride channels from Torpedo electroplax. Activation by protons. J. Gen. Physiol. 82:25–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug, K., M. Warnstedt, A.K. Alekov, T. Sander, A. Ramirez, B. Poser, S. Maljevic, S. Hebeisen, C. Kubisch, J. Rebstock, et al. 2003. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat. Genet. 33:527–532. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1992. Ionic Channels in Excitable Membranes. Second edition. Sinauer Associates, Inc. Sunderland, MA. 607 pp.
- Horenstein, J., D.A. Wagner, C. Czajkowski, and M.H. Akabas. 2001. Protein mobility and GABA-induced conformational changes in GABA(A) receptor pore-lining M2 segment. Nat. Neurosci. 4:477–485. [DOI] [PubMed] [Google Scholar]
- Iwaki, S., N. Tamura, T. Kimura-Someya, S. Nada, and A. Yamaguchi. 2000. Cysteine-scanning mutagenesis of transmembrane segments 4 and 5 of the Tn10-encoded metal-tetracycline/H+ antiporter reveals a permeability barrier in the middle of a transmembrane water-filled channel. J. Biol. Chem. 275:22704–22712. [DOI] [PubMed] [Google Scholar]
- Jentsch, T.J., V. Stein, F. Weinreich, and A.A. Zdebik. 2002. Molecular structure and physiological function of chloride channels. Physiol. Rev. 82:503–568. [DOI] [PubMed] [Google Scholar]
- Jentsch, T.J., M. Poet, J.C. Fuhrmann, and A.A. Zdebik. 2005. Physiological functions of CLC Cl− channels gleaned from human genetic disease and mouse models. Annu. Rev. Physiol. 67:779–807. [DOI] [PubMed] [Google Scholar]
- Karlin, A., and M.H. Akabas. 1998. Substituted-cysteine accessibility method. Methods Enzymol. 293:123–145. [DOI] [PubMed] [Google Scholar]
- Kornak, U., D. Kasper, M.R. Bosl, E. Kaiser, M. Schweizer, A. Schulz, W. Friedrich, G. Delling, and T.J. Jentsch. 2001. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 104:205–215. [DOI] [PubMed] [Google Scholar]
- Lin, C.-W., and T.-Y. Chen. 2000. Cysteine modification of a putative pore residue in ClC-0: implication for the pore stoichiometry of ClC chloride channels. J. Gen. Physiol. 116:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C.W., and T.-Y. Chen. 2003. Probing the pore of ClC-0 by substituted cysteine accessibility method using methane thiosulfonate reagents. J. Gen. Physiol. 122:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y.-W., C.-W. Lin, and T.-Y. Chen. 1999. Elimination of the slow gating of ClC-0 chloride channel by a point mutation. J. Gen. Physiol. 114:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd, S.E., S.H. Pearce, S.E. Fisher, K. Steinmeyer, B. Schwappach, S.J. Scheinman, B. Harding, A. Bolino, M. Devoto, P. Goodyer, et al. 1996. A common molecular basis for three inherited kidney stone diseases. Nature. 379:445–449. [DOI] [PubMed] [Google Scholar]
- Lu, T., T.-G. Zhu, and J. Yang. 1999. Cytoplasmic amino and carboxyl domains form a wide intracellular vestibule in an inwardly rectifying potassium channel. Proc. Natl. Acad. Sci. USA. 96:9926–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig, U., M. Pusch, and T.J. Jentsch. 1996. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature. 383:340–343. [DOI] [PubMed] [Google Scholar]
- Ludewig, U., M. Pusch, and T.J. Jentsch. 1997. a. Independent gating of single pores in CLC-0 chloride channels. Biophys. J. 73:789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig, U., T.J. Jentsch, and M. Pusch. 1997. b. Inward rectification in ClC-0 chloride channels caused by mutations in several protein regions. J. Gen. Physiol. 110:165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig, U., T.J. Jentsch, and M. Pusch. 1997. c. Analysis of a protein region involved in permeation and gating of the voltage-gated Torpedo chloride channel ClC-0. J. Physiol. 498:691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutsenko, S., S. Daoud, and J.H. Kaplan. 1997. Identification of two conformationally sensitive cysteine residues at the extracellular surface of the Na,K-ATPase α-subunit. J. Biol. Chem. 272:5249–5255. [DOI] [PubMed] [Google Scholar]
- Maduke, M., C. Williams, and C. Miller. 1998. Formation of CLC-0 chloride channels from separated transmembrane and cytoplasmic domains. Biochemistry. 37:1315–1321. [DOI] [PubMed] [Google Scholar]
- Maduke, M., D.J. Pheasant, and C. Miller. 1999. High-level expression, functional reconstitution, and quaternary structure of a prokaryotic ClC-type chloride channel. J. Gen. Physiol. 114:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulef, K., and W.N. Zagotta. 2002. Multimerizaton of the ligand binding domains of cyclic nucleotide-gated channels. Neuron. 36:93–103. [DOI] [PubMed] [Google Scholar]
- Middleton, R.E., D.J. Pheasant, and C. Miller. 1996. Homodimeric architecture of a ClC-type chloride ion channel. Nature. 383:337–383. [DOI] [PubMed] [Google Scholar]
- Miller, C. 1982. Open-state substructure of single chloride channels from Torpedo electroplax. Philos. Trans. R. Soc. Lond. B Biol. Sci. 299:401–411. [DOI] [PubMed] [Google Scholar]
- Miller, C., and E.A. Richard. 1990. The voltage-dependent chloride channel of Torpedo electroplax: intimations of molecular structure from quirks of single-channel function. In Chloride Channels and Carriers in Nerve, Muscle, and Glial Cells. F.J. Alvarez-Leefmans and J.M. Russell, editors. Plenum Publishing Corp., New York. 383–405.
- Miloshevsky, G.V., and P.C. Jordan. 2004. Anion pathway and potential energy profiles along curvilinear bacterial ClC Cl− pores: electrostatic effects of charged residues. Biophys. J. 86:825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naesens, M., P. Steels, R. Verberckmoes, Y. Vanrenterghem, and D. Kuypers. 2004. Bartter's and Gitelman's syndromes: from gene to clinic. Nephron. Physiol. 96:65–78. [DOI] [PubMed] [Google Scholar]
- Nilius, B., and G. Droogmans. 2003. Amazing chloride channels: an overview. Acta Physiol. Scand. 177:119–147. [DOI] [PubMed] [Google Scholar]
- Pusch, M. 2002. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 19:423–434. [DOI] [PubMed] [Google Scholar]
- Pusch, M. 2004. Structural insights into chloride and proton-mediated gating of CLC chloride channels. Biochemistry. 43:1135–1144. [DOI] [PubMed] [Google Scholar]
- Pusch, M., U. Ludewig, A. Rehfeldt, and T.J. Jentsch. 1995. Gating of the voltage-dependent chloride channel ClC-0 by the permeant ion. Nature. 373:527–531. [DOI] [PubMed] [Google Scholar]
- Pusch, M., S.E. Jordt, V. Stein, and T.J. Jentsch. 1999. Chloride dependence of hyperpolarization-activated chloride channel gates. J. Physiol. 515:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu, Z., R. Fischmeister, and C. Hartzell. 2004. Mouse bestrophin-2 is a bona fide Cl− channel: identification of a residue important in anion binding and conduction. J. Gen. Physiol. 123:327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quitterer, U., E. Zaki, and S. AbdAlla. 1999. Investigation of the extracellular accessibility of the connecting loop between membrane domains I and II of the bradykinin B2 receptor. J. Biol. Chem. 274:14773–14778. [DOI] [PubMed] [Google Scholar]
- Reeves, D.C., E.N. Goren, M.H. Akabas, and S.C.R. Lummis. 2001. Structural and electrostatic properties of the 5-HT3 receptor pore revealed by substituted cysteine accessibility mutagenesis. J. Biol. Chem. 276:42035–42042. [DOI] [PubMed] [Google Scholar]
- Richard, E.A., and C. Miller. 1990. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 247:1208–1210. [DOI] [PubMed] [Google Scholar]
- Shuck, K., R.A. Lamb, and L.H. Pinto. 2000. Analysis of the pore structure of the influenza A virus M(2) ion channel by the substituted-cysteine accessibility method. J. Virol. 74:7755–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, D.B., R.S. Bindra, T.A. Mansfield, C. Nelson-Williams, E. Mendonca, R. Stone, S. Schurman, A. Nayir, H. Alpay, A. Bakkaloglu, et al. 1997. Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat. Genet. 17:171–178. [DOI] [PubMed] [Google Scholar]
- Smith, S.S., X. Liu, Z.R. Zhang, F. Sun, T.E. Kriewall, N.A. McCarty, and D.C. Dawson. 2001. CFTR: covalent and noncovalent modification suggests a role for fixed charges in anion conduction. J. Gen. Physiol. 118:407–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeyer, K., R. Klocke, C. Ortland, M. Gronemeier, H. Jockusch, S. Grunder, and T.J. Jentsch. 1991. Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature. 354:304–308. [DOI] [PubMed] [Google Scholar]
- Uchida, S., and S. Sasaki. 2005. Function of chloride channels in the kidney. Annu. Rev. Physiol. 67:759–778. [DOI] [PubMed] [Google Scholar]
- Wang, Q., and H.R. Kaback. 1999. Proximity relationships between helices I and XI or XII in the lactose permease of Escherichia coli determined by site-directed thiol cross-linking. J. Mol. Biol. 291:683–692. [DOI] [PubMed] [Google Scholar]
- White, M.M., and C. Miller. 1979. A voltage-gated anion channel from the electric organ of Torpedo californica. J. Biol. Chem. 254:10161–10166. [PubMed] [Google Scholar]
- Wilson, G., and A. Karlin. 2001. Acetylcholine receptor channel structure in the resting, open, and desensitized states probed with the substituted-cysteine-accessibility method. Proc. Natl. Acad. Sci. USA. 98:1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H., M. Kono, T.D. McKee, and D.D. Oprian. 1995. A general method for mapping tertiary contacts between amino acid residues in membrane-embedded proteins. Biochemistry. 34:14963–14969. [DOI] [PubMed] [Google Scholar]