

Abstract
Heat-shock proteins (HSPs) are abundant, inducible proteins best known for their ability to maintain the conformation of proteins and to refold damaged proteins. Some HSPs, especially HSP90, can be antiapoptotic and the targets of anticancer drugs. Inositol hexakisphosphate kinase-2 (IP6K2), one of a family of enzymes generating the inositol pyrophosphate IP7 [diphosphoinositol pentakisphosphate (5-PP-IP5)], mediates apoptosis. Increased IP6K2 activity sensitizes cancer cells to stressors, whereas its depletion blocks cell death. We now show that HSP90 physiologically binds IP6K2 and inhibits its catalytic activity. Drugs and selective mutations that abolish HSP90–IP6K2 binding elicit activation of IP6K2, leading to cell death. Thus, the prosurvival actions of HSP90 reflect the inhibition of IP6K2, suggesting that selectively blocking this interaction could provide effective and safer modes of chemotherapy.
Keywords: apoptosis, cisplatin, novobiocin, inositol polyphosphate
Inositol phosphates (IPs) are major signaling molecules, with the best known, inositol 1,4,5-trisphosphate (IP3), releasing intracellular calcium (1, 2). More recently, inositol pyrophosphates (PP-IPs) have been identified, the most characterized ones being diphosphoinositol pentakisphosphate (IP7) and bis-diphosphoinositol tetrakisphosphate (IP8) (3, 4). Biosynthesis of the PP-IPs was clarified by the identification and cloning of a family of three distinct IP6 kinases (5, 6). In the IP7 formed by these enzymes, the diphosphate is at C5, affording the designation 5-PP-IP5. York and coworkers (7) described an IP7 (4/6-PP-IP5) with the diphosphate at C4 or C6, which is generated by the enzyme VIP1 (7). Hereafter, unless otherwise specified, we refer to 5-PP-IP5 as IP7(5) and to 4/6-PP-IP5 as IP7(4/6).
Insight into functions of PP-IPs derives from a variety of approaches, including functional changes after deletion of inositol hexakisphosphate kinase (IP6K) in yeast. IP6K-deficient yeast manifest major abnormalities in vesicular endocytosis (8–10) and telomere length (11, 12). Abundant evidence implicates PP-IPs in vesicular trafficking. Several proteins involved in such trafficking are binding partners of IP6 and IP7(5) (13, 14). Guanine nucleotide exchange factor for Rab3A (GRAB), a prominent binding partner of IP6K1, is involved in the regulation of vesicular exocytosis (15). In the slime mold Dictyostelium, IP7(5) regulates chemotaxis by competing with the lipid phosphatidyl inositol (PI)(3,4,5)P3 for binding to the pleckstrin homology (PH) domain of the cytosolic regulator of adenylyl cyclase (CRAC) (16). IP7(4/6) formed by VIP1 regulates phosphate homeostasis in yeast (17).
Recent advances have elucidated mechanisms whereby PP-IPs influence their targets. IP7 physiologically pyrophosphorylates a substantial number of proteins in both yeast and mammalian cells in a nonenzymatic process analogous to S-nitrosylation of proteins (18, 19).
IP6K2 has been selectively linked to mammalian cell death. Lindner and coworkers (20) screened an antisense library and uncovered IP6K2 as a proapoptotic gene. Overexpression of IP6K2 sensitizes numerous cancer cell lines to apoptotic actions of various cell stressors (20–23), whereas depletion of IP6K2 by RNAi prevents the apoptotic actions of several agents, indicating an association of IP6K2 with cell death (20, 21). IP6K2-augmented cell death is associated with an increase in DR4 [TNF-related apoptosis-inducing ligand (TRAIL) receptor] and caspase-8 expression and subsequent activation (23). IP6K2 also binds to TNF receptor-associated factor-2 (TRAF2) and interferes with the phosphorylation of transforming growth factor β-activated kinase-1 (TAK1) resulting in the inhibition of NF-κβ signaling in OVCAR-3 cells (24).
Cell death in response to anticancer drugs has been shown to involve IP6K2 (21, 23). Thus, cisplatin (CP) augments IP7(5) generation in OVCAR-3 cells, and the apoptotic influences of the drug are substantially augmented by IP6K2 overexpression (21). IP6K2-mediated cell death depends on its catalytic activity because its kinase-dead mutant prevents apoptosis. The augmentation of IP7(5) formation elicited by CP is not associated with any changes in IP6K2 protein level, suggesting that under basal conditions IP6K2 activity is inhibited by some factor.
Heat-shock proteins (HSPs) promote the refolding of denatured proteins and are increasingly appreciated as mediators of antiapoptotic signaling with substantially augmented activity during carcinogenesis (25–28). The molecular chaperone HSP90 controls the conformation, stability, activation, intracellular distribution, and turnover of numerous client proteins that are involved in cell growth, differentiation, and survival (29, 30). Several kinases, hormone receptors, mutant p53, and the catalytic subunit of telomerase hTERT are HSP90 clients (31). HSP90 is a promising target for the development of cancer chemotherapeutics because its inhibition simultaneously affects multiple oncogenic proteins (29, 30). HSP90 inhibitors broadly categorized as N-terminal- (radicicol, geldanamycin, etc.) and C-terminal-[CP, novobiocin (NB), etc.] binding compounds inhibit its chaperone activity and client–protein interaction (29–36).
We now report that HSP90 physiologically binds IP6K2 and inhibits its catalytic activity under basal conditions. IP6K2 contains a unique motif for HSP90 binding. Selective mutations within this motif abolish HSP90–IP6K2 binding, augment IP7(5) production, and cause cell death. Depletion of HSP90 by siRNA also causes increased IP6K2 catalytic activity. IP6K2 binds to the C-terminal domain of HSP90. Drugs that have been reported to interact with the C terminus of HSP90, such as CP and NB, disrupt HSP90–IP6K2 binding, leading to activation of IP6K2. Apoptotic actions of these drugs are diminished in cells depleted of IP6K2, demonstrating that increased IP6K2 activity mediates, in part, the cytotoxic actions of CP and NB. HSP90 depletion enhances the cell death elicited by IP6K2 activation. Thus, the antiapoptotic influences of HSP90 stem in major part from its binding IP6K2 and inhibiting its apoptotic effects.
Results
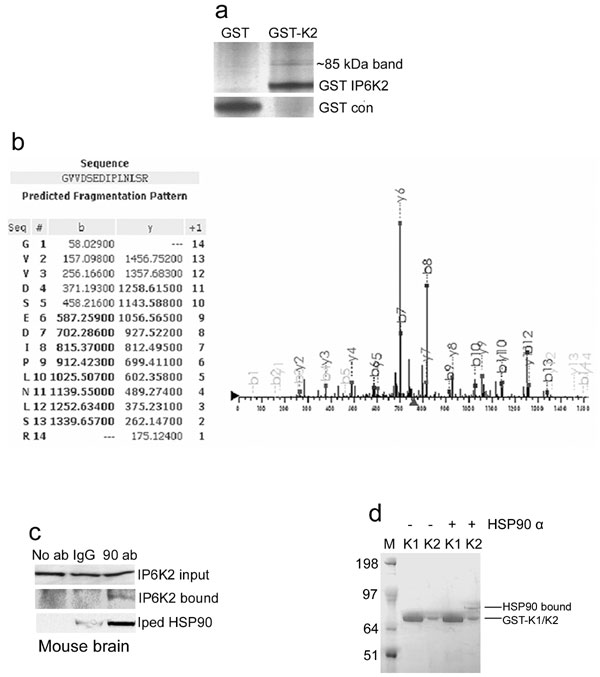
We wondered whether the rapid activation of IP6K2 in response to apoptotic stimuli reflects interaction with other proteins. We sought binding partners for IP6K2 by GST pull-down of overexpressed GST-IP6K2 from several cell lines (HEK293, HeLa, and HCT116). Tandem mass spectrometric analysis (LC/MS/MS) of the most prominent pulled-down band in HCT116 reveals HSP90 [Fig. 1a and supporting information (SI) Fig. 5 a and b]. Coprecipitation experiments in mouse brain and kidney establish that endogenous IP6K2 and HSP90 interact physiologically (Fig. 1 b and c and SI Fig. 5c). Binding between IP6K2 and native HSP90 (Fig. 1 d and e) or recombinant HSP90α (SI Fig. 5d) in vitro indicates a direct interaction.
Fig. 1.

IP6K2 binds HSP90. (a) HSP90A peptides identified by LC/MS/MS analysis. (b) Co-IP of endogenous HSP90 by endogenous IP6K2 in mouse brain. Coprecipitated HSP90 was detected by Western blotting using a monoclonal antibody. (c) HSP90 coprecipitated with IP6K2 in mouse kidney. Immunoprecipitation was done with IgG- or IP6K2-specific antibody as in brain extract. HSP90 in the immunoprecipitated sample was detected by Western blotting. (d) Direct binding of immunoprecipitated Myc-IP6K2 with exogenously added HSP90 purified from HeLa cells. Myc-vector-transfected HEK293 cells were used as negative controls. (e) Direct binding of purified recombinant His-IP6K2 with exogenously added HSP90 purified from HeLa cells.
To assess the specificity of IP6K2–HSP90 interaction, we immunoprecipitated Myc-IP6Ks and observed that IP6K2, but not IP6K1, precipitates endogenous HSP90 in HEK293 and HeLa cells (Fig. 2 a and b). Deletion mutant analysis of IP6K2 identifies a 12-aa motif critical for binding HSP90 (Fig. 2c and SI Fig. 6 a and b). The motif is not present in IP6K1, which explains the selective binding of HSP90 to IP6K2. We mutated IP6K2 in its putative HSP90-binding motif. Mutation of tryptophan-131 modestly diminishes IP6K2–HSP90 binding, whereas mutations of arginine-133 or −136 abolish binding (Fig. 2d).
Fig. 2.

HSP90 binds to a specific motif in IP6K2 by its C terminus. (a) Co-IP of endogenous HSP90 by overexpressed Myc-IP6K2 from HEK293 cells. Protein (1 mg) from each cell lysate was immunoprecipitated by anti-Myc antibody and was immunoblotted with monoclonal HSP90 antibody. Lane 1 (Con) shows the Myc-vector control. (b) Co-IP of Myc-IP6K2, not IP6K1, by endogenous HSP90 from HeLa cells. Immunoprecipitation of endogenous HSP90 by monoclonal antibody coprecipitates IP6K2, not IP6K1, as confirmed by blotting with α-Myc antibody. (c) Deletion mapping of IP6K2 to identify HSP90-binding motif. (d) Endogenous HSP90 does not coprecipitate with mutants of IP6K2 in the putative HSP90-binding region. R133A and R136A mutants do not bind, whereas W131A has little effect on binding. (e) Mapping of HSP90 to identify the IP6K2-binding region. Fragments 1–272 [N terminus (N)], 273–732 [middle and C termini (MC)], and 629–732 [C terminus (C)] were generated and cloned into pGEX vector and purified from bacteria. (f) Determination of binding region of HSP90 to IP6K2 by in vitro binding. After incubation of the proteins, the beads were washed, and bound HSP90 was analyzed by blotting with anti-HSP90 monoclonal antibody. (g) IP6K2–HSP90 interaction in cells is disrupted by CP and NB treatment. Overnight drug treatment was followed by immunoprecipitation of Myc-IP6K2 and detection of coprecipitated HSP90 by Western blotting. (h) Binding in vitro of HSP90–IP6K2 is disrupted by CP and NB, but not by AAG. HSP90 bound to Myc-IP6K2 was analyzed by blotting with anti-HSP90 monoclonal antibody.
Deletion mapping of HSP90 reveals that IP6K2 binds to HSP90's C terminus (Fig. 2 e and f). We wondered whether the apoptotic actions of anticancer drugs that inhibit HSP90 involve HSP90–IP6K2 interactions. NB and CP prevent the binding of IP6K2 and HSP90 in intact cells (Fig. 2g) and with the purified proteins (Fig. 2h), whereas the geldanamycin derivative 17-(allylamino)-17-demethoxygeldanamycin (AAG) has no effect (Fig. 2h and SI Fig. 6c). Half-maximal inhibition of binding occurs with ≈100 μM NB and 1 μM CP, comparable to their apoptotic potencies (36, 37).
We investigated whether the binding of HSP90 influences IP6K2 catalytic activity. Depletion of HSP90 by RNAi (SI Fig. 7a) in HEK293 cells increases IP6K catalytic activity ≈2.5-fold (Fig. 3a). Presumably this augmentation predominantly involves IP6K2, because IP6K1 does not bind HSP90 and because IP6K3 occurs at very low levels in these cells. To ascertain the portion of total IP6K activity attributable to IP6K2, we depleted IP6K2 by RNAi (SI Fig. 7a) and observed a 30% decrease in cellular catalytic activity. Thus, the augmentation of IP6K activity associated with IP6K2 is ≈4-fold. HSP90 RNAi in cells already depleted of IP6K2 fails to augment IP6K activity, implying that the enhancement of IP7(5) production by the depletion of HSP90 selectively reflects the catalytic activity of IP6K2 (Fig. 3a). We obtained similar results in HeLa cells (data not shown).
Fig. 3.

HSP90 inhibits IP6K2 catalytic activity. (a) IP6K2 activity in vivo is increased in the absence of endogenous HSP90. HSP90 and IP6K2 (either alone or together) were depleted by using siRNA in [3H]inositol-labeled HEK293 cells and inositol phosphates separated by HPLC. (b) IP6K activity in WT and HSP mutant S. cerevisiae in vivo. IP6 and IP7 were monitored after [3H]inositol labeling of intact cells. (c) HSP90 overexpression leads to decreased IP6K2 activity in vivo. HEK293 cells overexpressing Myc-IP6K1/Myc-IP6K2 either alone or with HA-HSP90 were labeled with [3H]inositol, and inositol phosphates were isolated by HPLC. (d) HSP90 inhibition of IP6K2 in vivo requires binding to IP6K2. IP7 labeled by [3H]inositol was assessed in HEK293 cells cotransfected with Myc-IP6K2 (WT or the mutants) and HA-HSP90. (e) IP6K activity in vitro of WT and mutant IP6K2 in the absence and presence of purified HSP90. HSP90 fails to inhibit IP6K activity of the R136A mutant that does not bind HSP90. IP6K2-W131A is catalytically inactive. (f) CP enhancement of IP7 generation in vivo is influenced by IP6K2–HSP90 binding. IP6K activity of [3H]inositol-labeled HEK293 cells was measured after 30 μM CP treatment overnight. (g) IP6K activity is increased after CP and NB treatment of HEK293 and HeLa cells. After drug treatment, extracts were prepared, and 25 μg of total protein was assayed for IP6K activity.
The yeast Saccharomyces cerevisiae possesses constitutive and inducible homologues of HSP90, designated HSC82 and HSP82, respectively. In HSC82 KO yeast, IP6K activity is ≈2.5-fold higher than WT, consistent with its physiologic binding IP6K to inhibit catalytic activity (Fig. 3b). In yeast null for HSP82, IP6K catalytic activity is increased but to a lesser extent. This observation fits with the notion that, under basal conditions, the constitutive HSC82 plays a larger role in regulating IP6K. HSP104 does not bind IP6K, and, as expected, its deletion does not alter IP6K activity.
In agreement with the augmented IP6K activity after depletion of HSP90, overexpression of HSP90 markedly reduces IP6K catalytic activity in HEK293 cells (Fig. 3c and SI Fig. 7b). This effect is selective for IP6K2 because overexpression of HSP90 does not influence catalytic activity of IP6K1. Overexpression of IP6Ks in HeLa cells provides similar results (data not shown).
We examined IP6K2 mutations that inhibit its binding to HSP90. Cells overexpressing IP6K2-R136A, which cannot bind HSP90, have higher IP6K catalytic activity than cells with IP6K2-WT (Fig. 3d). Overexpression of HSP90 (SI Fig. 7c) markedly reduces IP7(5) formation in cells with IP6K2-WT, whereas no reduction in IP7(5) formation occurs in cells overexpressing IP6K2-R136A mutant (Fig. 3d), indicating that HSP90–IP6K2 binding is required for the inhibition of IP6K activity by HSP90.
IP6K activity is markedly reduced in cells with IP6K2-W131A (Fig. 3d). To investigate the molecular mechanism underlying this loss of catalytic activity, we monitored the process in vitro with immunoprecipitated Myc-IP6K2 proteins (Fig. 3e and SI Fig. 7d). The addition of HSP90 substantially decreases the catalytic activity of IP6K2-WT. IP6K2-R136A has robust catalytic activity that is not affected by the addition of HSP90, consistent with this mutation blocking IP6K2–HSP90 binding. In contrast, the catalytic activity of IP6K2-W131A is markedly reduced in the absence or presence of HSP90. We monitored the binding of the substrate [3H]IP6 to IP6K2 (SI Fig. 7 e and f). Binding is reduced by half in IP6K2-W131A but is not affected by mutations of R133 or R136, indicating that the loss of catalytic activity of IP6K2-W131A reflects the inability of this mutant enzyme to bind IP6. Thus, HSP90 binds to a region of IP6K2 important for binding to the substrate IP6, suggesting a mechanism by which HSP90 impacts IP6K2 catalytic activity.
Drug-induced disruption of HSP90 binding impacts IP6K2 activity. Thus, in HEK293 cells with or without overexpression of IP6K2, CP augments IP6K activity (Fig. 3f). The R136A mutation increases IP6K2 catalytic activity and renders it less responsive to CP, consistent with CP's stimulatory effect reflecting, in large part, its inhibition of HSP90–IP6K2 binding, which requires R136 (Fig. 3f and SI Fig. 7g). AAG does not augment IP6K activity in intact cells, consistent with its failure to disrupt HSP90–IP6K2 binding (SI Fig. 7h). CP and NB also stimulate IP6K activity in extracts of HEK293 and HeLa cells (Fig. 3g).
We examined the influence of HSP90 on IP6K2-mediated apoptosis. Depletion of HSP90 by siRNA treatment causes a time-dependent decrease in cell survival (Fig. 4a), which is reversed by codepletion of IP6K2. IP6K2 overexpression augments cell death elicited by HSP90 depletion (Fig. 4b).
Fig. 4.

HSP90–IP6K2 interaction regulates drug-induced cell death. (a) Decreased cell survival elicited by HSP90 depletion (filled inverted triangles) is reversed by codepletion of IP6K2 (filled squares). Cotansfection of control siRNA with HSP90 siRNA has no effect (open squares). MTT assay was done after each time period of various siRNA treatments of HEK293 cells. (b) Cell death elicited by HSP90 depletion is further enhanced by IP6K2 overexpression. Cells were either transfected with HSP90 siRNA or cotransfected with Myc-IP6K2 for 48 h, and cell death was monitored by apoptotic nuclei-detection assay. (c) Cell survival (MTT) assay of WT and IP6K2-R136A-transfected HEK293 cells. Cells were transfected with various Myc-IP6K2 constructs; after each interval, the percentage survival was calculated. IP6K2-R136A mutant (filled inverted triangles) causes a significant decrease in cell survival than IP6K2-WT (open circles). (d) Increased cell death elicited by overexpression of IP6K2-R136A that does not bind to HSP90. Cells were transfected by different constructs for 72 h. Cell death caused by IP6K2-WT is reversed by HSP90 overexpression, whereas there is no effect of HSP90 on IP6K2-R136A. The catalytically impaired IP6K2-W131A does not induce cell death. (e) Caspase-3 activity is increased in cells transfected with IP6K2-R136A, which does not bind HSP90. Activity was measured after 72 h of transfection. The magnitude of increase was determined by considering OD405 of control samples as unity. (f) Death of cells overexpressing IP6K2-WT after CP treatment is reversed by HSP90 in cells overexpressing IP6K2-WT, but not IP6K2-R136A, which cannot bind HSP90. Cells overexpressing IP6K2-W131A show diminished cell death relative to WT, which is further reduced by HSP90 coexpression. (g) Cell death assay as described for panel f, after NB treatment. (h) CP- and NB-induced cell death in HEK293 cells requires IP6K2. Endogenous IP6K2 was depleted by using siRNA. CP and NB were added after 36 h of transfection for 36 and 24 h, respectively. Cell death in drug-treated cells is diminished by IP6K2 depletion.
We compared proapoptotic activities of IP6K2-R136A, which does not bind HSP90, with IP6K2-WT. Overexpression of IP6K2-R136A decreases cell survival much more than IP6K2-WT (Fig. 4c). HSP90 reverses the apoptotic effects of IP6K2-WT, but not those elicited by IP6K2-R136A (Fig. 4d). The importance of IP6K2 catalytic activity for cell death is supported by the absence of cell death augmentation after overexpression of the catalytically impaired IP6K2-W131A mutant (Fig. 4d). The apoptotic effects of IP6K2-R136A also are illustrated by a 2-fold increase in caspase-3 activity over IP6K2-WT (Fig. 4e).
If CP and NB cause cell death at least in part by disrupting an inhibitory effect of HSP90 on IP6K2, then overexpression of HSP90 should reverse drug-induced cytotoxicity. In IP6K2-WT-overexpressing cells, HSP90 overexpression markedly diminishes the cytotoxic actions of CP and NB (Fig. 4 f and g). HSP90 does not reduce drug-induced cytotoxicity in cells overexpressing IP6K2-R136A, which cannot bind HSP90. In contrast to the marked increase in drug toxicity in cells overexpressing IP6K2-WT, no augmentation is evident after overexpression of IP6K2-W131A, which has markedly diminished catalytic activity. This observation fits with the notion that CP and NB cause cell death by enhancing IP6K2 activity.
We depleted IP6K2 in HEK293 cells by RNAi. Cell death elicited by CP and NB is reduced by 50% and 40%, respectively, in IP6K2-depleted cells, confirming that IP6K2 plays a major role in CP- and NB-induced cell death (Fig. 4h). Thus, HSP90's regulation of cell survival occurs largely via its inhibition of IP6K2. As expected, the apoptotic actions of AAG are unaffected by IP6K2 depletion (data not shown).
Discussion
Our findings identify IP6K2 as an HSP90-binding partner and indicate that this interaction physiologically regulates cell survival. HSP90 binds IP6K2 and inhibits its catalytic activity, thus protecting cells from apoptosis. On the basis of the increased IP6K2 activity after disruption of HSP90 binding, we estimate that ≈30–50% of total cellular IP6K2 is bound to HSP90. Selective disruption of HSP90–IP6K2 binding markedly augments IP7 generation and cell death. The antiapoptotic effect of HSP90 is overcome by IP6K2 mutants that do not bind HSP90. We also find that the proapoptotic consequences of HSP90 knockdown require IP6K2. Thus, the antiapoptotic, cancer-promoting actions of HSP90 may reflect its inhibition of IP6K2's apoptotic influences. HSP90 might be regarded as a reservoir for IP6K2, storing it in an inactive state and releasing it as needed.
The influence of HSP90 on IP6K2 differs from typical HSP90–client interactions. IP6K2, like other inositol polyphosphate kinases, shares structural and functional similarities with protein kinases (38), a number of which are associated with HSP90, such as Akt, PDK1, the cyclin-dependent kinases Cdk4, Cdk6, and Cdk9, etc. (31, 32, 39–42). These protein kinases are activated and stabilized as a result of HSP90 association. In contrast, IP6K2 is inhibited by HSP90 binding. Activation of protein kinase clients requires the HSP90 ATPase cycle and can be disrupted by ATP-competitive HSP90 inhibitors such as geldanamycin. By contrast, the regulation of IP6K2 by HSP90 is insensitive to the N-terminal HSP90 ATPase inhibitor AAG. Protein kinase clients of HSP90 require critical residues in the middle domain of HSP90 (42), whereas IP6K2 binds to its C terminus. The 12-aa motif in IP6K2 that binds HSP90 appears to be distinct. Our database searches revealed only a few imperfect matches to this sequence, and these are not known HSP90-binding proteins.
Several HSP90 inhibitors display anticancer properties of which the best characterized are geldanamycin and its derivatives, which bind to the N-terminal ATP-binding domain of HSP90 (30, 33, 34, 43). However, CP and NB bind to the C terminus of HSP90 both in vitro and in vivo (35–37). CP binds HSP90, decreasing the transcriptional activity of androgen and glucocorticoid receptors although not affecting other HSP-regulated proteins, such as the phosphokinases Raf-1, Lck, and c-Src (37). NB binding to HSP90 can allosterically interfere with the binding to its N terminus of proteins such as mutant p53 and Raf-1 (36). Our findings indicate that CP and NB exert their cytotoxic effects, at least in part, by disrupting IP6K2–HSP90 binding, leading to augmented IP7(5) formation. CP and NB both have additional effects, most notably DNA damage via covalent adduct formation or topoisomerase II inhibition, respectively. It is difficult to ascertain the extent to which apoptotic actions of these drugs are attributable to various mechanisms. Nonetheless, the major reduction in cell death elicited by CP and NB in cells depleted of IP6K2 implies that activation of IP7(5) formation by disruption of IP6K2–HSP90 binding is an important cytotoxic mechanism for these drugs. The failure of IP6K2 depletion to influence cell death elicited by AAG fits with the failure of this drug to disrupt IP6K2–HSP90 binding.
Geldanamycin-insensitive binding of proteins to the C terminus of HSP90 is only recently becoming appreciated. FKBP38 is a peptidylprolyl cis-trans isomerase that promotes apoptosis by inhibiting interactions of Bcl-2 with calcineurin and Bad. Binding of HSP90 to FKBP38 inhibits FKBP38 catalytic activity and blocks FKBP38–Bcl-2 binding, thus preventing apoptosis (44). Like IP6K2, FKBP38 interacts with the C terminus of HSP90 in a geldanamycin-insensitive manner. In our experiments, cell death induced by C terminus-directed HSP90 inhibitors depends on IP6K2, suggesting an apoptotic mechanism independent of the HSP90–FKBP38 pathway.
Molecular mechanisms whereby IP6K2 induces cell death are unclear. IP6K2 can up-regulate DR4 and caspase 8 and inhibit NF-κβ signaling via interactions with TRAF2 (23, 24). In yeast, inositol pyrophosphates modulate cell death and telomere length by antagonizing yeast homologues of ataxia telangiectasia mutated (ATM) kinase, a regulator of the DNA damage response and cell death in higher organisms (12). IP7(5) competes with the lipid PI(3,4,5)P3 for binding to PH domains (16). Conceivably, disruption by IP7(5) of PIP3 binding to PH domains of prosurvival proteins such as Akt contributes to cell death. IP7(5) also can transfer its energetic β-phosphate to pyrophosphorylate proteins in a nonenzymatic reaction, raising the possibility that direct modification of proteins by IP7 could contribute to IP6K2-mediated apoptosis (18, 19).
Our findings may have therapeutic relevance. One could readily screen for agents that selectively block IP6K2–HSP90 binding. Such substances would be predicted to have therapeutic effects in cancer and may elicit fewer side effects than classical chemotherapeutic agents that act by mechanisms such as DNA damage. Inhibitors of HSP90–IP6K2 binding also may be more selective and less toxic than drugs that affect HSP90's ATPase activity and thereby exert more global influences.
Materials and Methods
Anti-IP6K2 polyclonal antibody was raised in rabbit by Affinity BioReagents against a synthetic peptide. Monoclonal antibodies for HSP90 and β-tubulin were from BD Biociences and Upstate Biotechnology, respectively. siRNAs against HSP90α and -β and IP6K2 were from Qiagen and Invitrogen, respectively. Purified HSP90 was from Stressgen Biotechnologies. Polyfect and Hiperfect were from Qiagen. Unless otherwise stated, chemicals were purchased from Sigma–Aldrich.
LC/MS/MS Analysis.
The 85-kDa band, pulled down by GST-IP6K2, was sent to Taplin Biological Mass Spectrometry Facility (Harvard Medical School, Boston, MA) for identification of the protein by LC/MS/MS.
Cloning of HSP90.
HSP90α and HSP90β were cloned into pCMV-HA plasmids by RT-PCR using mRNA purified from HEK293 cells.
IP7 Measurement in Vivo.
Cellular IP7 was measured as described previously (8, 21). Details are found in SI Materials and Methods.
IP6K2 Activity in Vitro.
IP6K2 activity in vitro was measured as described previously (21). Details are found in SI Materials and Methods.
Coimmunoprecipitation (Co-IP).
Cells or mouse tissues were homogenized in lysis buffer [20 mM Tris (pH 7.4), 150 mM NaCl, 0.5% Nonidet P-40, and protease inhibitor cocktails]. Equal amounts of protein were immunoprecipitated at 4°C overnight by using the protein AG agarose beads in the presence of antibody. Beads were washed (4 × 10 min) with lysis buffer. Coimmunoprecipitates were resolved by SDS/PAGE and analyzed by Western blotting.
siRNA Experiments.
HEK293 cells (50% confluent) were transfected with IP6K2, HSP90α and HSP90β together, or control siRNAs (50 nM each) for 48–72 h. After indicated time periods, cells were pelleted, and extract was prepared as described.
Binding of IP6K2 and HSP90 in Vitro.
Details of the binding assays are described in SI Materials and Methods.
Caspase-3 Activity Assay.
Caspase-3 activity was tested by using a caspase-3 colorimetric assay kit from Biovision following the manufacturer's protocol.
Cell Survival Assay.
Cell death was determined by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay protocol. HEK293 cells (4 × 105 per well) were transfected with 2 μg of Myc-IP6K2 (WT or mutants) either alone or with 2 μg of HA-HSP90 in six-well plates in triplicate. After an indicated time period, 50 μl of 2 mg/ml MTT solution was added to each well, and cells were incubated for 4 h at 37°C. Absorbance was measured at 570–630 nm on an ELISA reader.
Quantification of Apoptosis by Fluorescence Microscopy.
For cell death after overexpression of IP6K2-WT or mutants either alone or after HSP90 cooverexpression, Myc-IP6K2 (WT, R136A, and W131A) was transfected in HEK293 cells with or without HA-HSP90 (transfection efficiency was always >95%, checked by transfecting GFP-control plasmid). After 48 h of transfection, cells were treated with 30 μM CP for 24 h or 250 μM NB for 8 h. After treatment, cells were fixed rapidly with ice-cold methanol for 5 min and then stained for 5 min with Hoechst 33342 dye. After being washed with PBS, the cells were then observed under a microscope. Apoptotic cells were identified by having condensed and/or fragmented chromatin in the nuclei. At least 300 cells from randomly selected fields were counted in each experiment.
To calculate CP- or NB-induced cell death in siRNA-treated HEK293 cells, 50 nM endogenous IP6K2 was depleted by using siRNA. CP and NB were added after 36 h of siRNA treatment for 36 and 24 h, respectively, to get substantial cell death in control cells to compare with the IP6K2-depleted cells. At least 300 cells from randomly selected fields were counted in each experiment.
Statistical Analysis.
All experiments were repeated three times, and ±SEM was calculated. Significance of result was calculated by Student's t test (∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001) by using Sigmaplot software (SPSS).
Supplementary Material
ACKNOWLEDGMENTS.
We thank Adolfo Saiardi, Rashna Bhandari, Adam C. Resnick, Makoto Hara, Bindu D. Paul, and Martin S. Taylor for suggestions, discussions, and helpful comments. This work was supported by U.S. Public Health Service Grant MH18501 and Research Scientist Award DA00074 (to S.H.S.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0711168105/DC1.
References
- 1.Berridge MJ, Lipp P, Bootman MD. Signal transduction: The calcium entry pas de deux. Science. 2000;287:1604–1605. doi: 10.1126/science.287.5458.1604. [DOI] [PubMed] [Google Scholar]
- 2.Irvine RF, Schell MJ. Back in the water: The return of the inositol phosphates. Nat Rev Mol Cell Biol. 2001;2:327–338. doi: 10.1038/35073015. [DOI] [PubMed] [Google Scholar]
- 3.Menniti FS, Miller RN, Putney JW, Jr, Shears SB. Turnover of inositol polyphosphate pyrophosphates in pancreatoma cells. J Biol Chem. 1993;268:3850–3856. [PubMed] [Google Scholar]
- 4.Stephens L, et al. The detection, purification, structural characterization, and metabolism of diphosphoinositol pentakisphosphate(s) and bisdiphosphoinositol tetrakisphosphate(s). J Biol Chem. 1993;268:4009–4015. [PubMed] [Google Scholar]
- 5.Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol. 1999;9:1323–1326. doi: 10.1016/s0960-9822(00)80055-x. [DOI] [PubMed] [Google Scholar]
- 6.Saiardi A, Nagata E, Luo HR, Snowman AM, Snyder SH. Identification and characterization of a novel inositol hexakisphosphate kinase. J Biol Chem. 2001;276:39179–39185. doi: 10.1074/jbc.M106842200. [DOI] [PubMed] [Google Scholar]
- 7.Mulugu S, et al. A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science. 2007;316:106–109. doi: 10.1126/science.1139099. [DOI] [PubMed] [Google Scholar]
- 8.Dubois E, et al. In Saccharomyces cerevisiae, the inositol polyphosphate kinase activity of Kcs1p is required for resistance to salt stress, cell wall integrity, and vacuolar morphogenesis. J Biol Chem. 2002;277:23755–23763. doi: 10.1074/jbc.M202206200. [DOI] [PubMed] [Google Scholar]
- 9.Saiardi A, Sciambi C, McCaffery JM, Wendland B, Snyder SH. Inositol pyrophosphates regulate endocytic trafficking. Proc Natl Acad Sci USA. 2002;99:14206–14211. doi: 10.1073/pnas.212527899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett M, Onnebo SM, Azevedo C, Saiardi A. Inositol pyrophosphates: Metabolism and signaling. Cell Mol Life Sci. 2006;63:552–564. doi: 10.1007/s00018-005-5446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.York SJ, Armbruster BN, Greenwell P, Petes TD, York JD. Inositol diphosphate signaling regulates telomere length. J Biol Chem. 2005;280:4264–4269. doi: 10.1074/jbc.M412070200. [DOI] [PubMed] [Google Scholar]
- 12.Saiardi A, Resnick AC, Snowman AM, Wendland B, Snyder SH. Inositol pyrophosphates regulate cell death and telomere length through phosphoinositide 3-kinase-related protein kinases. Proc Natl Acad Sci USA. 2005;102:1911–1914. doi: 10.1073/pnas.0409322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleischer B, et al. Golgi coatomer binds, and forms K (+)-selective channels gated by, inositol polyphosphates. J Biol Chem. 1994;269:17826–17832. [PubMed] [Google Scholar]
- 14.Ye W, Ali N, Bembenek ME, Shears SB, Lafer EM. Inhibition of clathrin assembly by high affinity binding of specific inositol polyphosphates to the synapse-specific clathrin assembly protein AP-3. J Biol Chem. 1995;270:1564–1568. [PubMed] [Google Scholar]
- 15.Luo HR, et al. GRAB: A physiologic guanine nucleotide exchange factor for Rab3A, which interacts with inositol hexakisphosphate kinase. Neuron. 2001;31:439–451. doi: 10.1016/s0896-6273(01)00384-1. [DOI] [PubMed] [Google Scholar]
- 16.Luo HR, et al. Inositol pyrophosphates mediate chemotaxis in Dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell. 2003;114:559–572. doi: 10.1016/s0092-8674(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 17.Lee YS, Mulugu S, York JD, O'Shea EK. Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science. 2007;316:109–112. doi: 10.1126/science.1139080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhandari R, et al. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc Natl Acad Sci USA. 2007;104:15305–15310. doi: 10.1073/pnas.0707338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saiardi A, Bhandari R, Resnick AC, Snowman AM, Snyder SH. Phosphorylation of proteins by inositol pyrophosphates. Science. 2004;306:2101–2105. doi: 10.1126/science.1103344. [DOI] [PubMed] [Google Scholar]
- 20.Morrison BH, Bauer JA, Kalvakolanu DV, Lindner DJ. Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. J Biol Chem. 2001;276:24965–24970. doi: 10.1074/jbc.M101161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagata E, et al. Inositol hexakisphosphate kinase-2, a physiologic mediator of cell death. J Biol Chem. 2005;280:1634–1640. doi: 10.1074/jbc.M409416200. [DOI] [PubMed] [Google Scholar]
- 22.Morrison BH, Tang Z, Jacobs BS, Bauer JA, Lindner DJ. Apo2L/TRAIL induction and nuclear translocation of inositol hexakisphosphate kinase 2 during IFN-beta-induced apoptosis in ovarian carcinoma. Biochem J. 2005;385:595–603. doi: 10.1042/BJ20040971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrison BH, et al. Inositol hexakisphosphate kinase 2 sensitizes ovarian carcinoma cells to multiple cancer therapeutics. Oncogene. 2002;21:1882–1889. doi: 10.1038/sj/onc/1205265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrison BH, et al. Effect of inositol hexakisphosphate kinase 2 on transforming growth factor beta-activated kinase 1 and NF-kappaB activation. J Biol Chem. 2007;282:15349–15356. doi: 10.1074/jbc.M700156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruey JM, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2:645–652. doi: 10.1038/35023595. [DOI] [PubMed] [Google Scholar]
- 26.Beere HM, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 27.Dias S, Shmelkov SV, Lam G, Rafii S. VEGF (165) promotes survival of leukemic cells by Hsp90-mediated induction of Bcl-2 expression and apoptosis inhibition. Blood. 2002;99:2532–2540. doi: 10.1182/blood.v99.7.2532. [DOI] [PubMed] [Google Scholar]
- 28.Pandey P, et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000;19:4310–4322. doi: 10.1093/emboj/19.16.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;10:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 30.Sharp S, Workman P. Inhibitors of the HSP90 molecular chaperone: Current status. Adv Cancer Res. 2006;95:323–348. doi: 10.1016/S0065-230X(06)95009-X. [DOI] [PubMed] [Google Scholar]
- 31.Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: The story unfolds. Expert Opin Biol Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 32.Richter K, Buchner J. Hsp90: Chaperoning signal transduction. J Cell Physiol. 2001;188:281–290. doi: 10.1002/jcp.1131. [DOI] [PubMed] [Google Scholar]
- 33.Kamal A, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 34.Roe SM, et al. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 35.Itoh H, et al. A novel chaperone-activity-reducing mechanism of the 90-kDa molecular chaperone HSP90. Biochem J. 1999;343:697–703. [PMC free article] [PubMed] [Google Scholar]
- 36.Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 37.Rosenhagen MC, et al. The heat shock protein 90-targeting drug cisplatin selectively inhibits steroid receptor activation. Mol Endocrinol. 2003;17:1991–2001. doi: 10.1210/me.2003-0141. [DOI] [PubMed] [Google Scholar]
- 38.Gonzalez B, et al. Structure of a human inositol 1,4,5-trisphosphate 3-kinase: Substrate binding reveals why it is not a phosphoinositide 3-kinase. Mol Cell. 2004;15:689–701. doi: 10.1016/j.molcel.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 39.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci USA. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen G, Cao P, Goeddel DV. TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol Cell. 2002;9:401–410. doi: 10.1016/s1097-2765(02)00450-1. [DOI] [PubMed] [Google Scholar]
- 41.Vaughan CK, et al. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol Cell. 2006;23:697–707. doi: 10.1016/j.molcel.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schulte TW, Blagosklonny MV, Ingui C, Neckers L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem. 1995;270:24585–24588. doi: 10.1074/jbc.270.41.24585. [DOI] [PubMed] [Google Scholar]
- 43.Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–217. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 44.Edlich F, et al. The Bcl-2 regulator FKBP38-calmodulin-Ca2+ is inhibited by Hsp90. J Biol Chem. 2007;282:15341–15348. doi: 10.1074/jbc.M611594200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}