

Abstract
NF-κB activation occurs upon degradation of its inhibitor I-κB and requires prior phosphorylation of the inhibitor by I-κB kinase (IKK). Activity of IKK is governed by its noncatalytic subunit IKKγ. Signaling defects due to missense mutations in IKKγ have been correlated to its inability to either become ubiquitylated or bind ubiquitin noncovalently. Because the relative contribution of these events to signaling had remained unknown, we have studied mutations in the coil-zipper (CoZi) domain of IKKγ that either impair signaling or cause constitutive NF-κB activity. Certain signaling-deficient alleles neither bound ubiquitin nor were they ubiquitylated by TRAF6. Introducing an activating mutation into those signaling-impaired alleles restored their ubiquitylation and created mutants constitutively activating NF-κB without repairing the ubiquitin-binding defect. Constitutive activity therefore arises downstream of ubiquitin binding but upstream of ubiquitylation. Such constitutive activity reveals a signal-processing function for IKKγ beyond that of a mere ubiquitin-binding adaptor. We propose that this signal processing may involve homophilic CoZi interactions as suggested by the enhanced affinity of CoZi domains from constitutively active IKKγ.
Keywords: NF-κB, signaling, ubiquitin, Nemo
The transcription factor NF-κB plays an essential role in coordinating inflammation and immunity by controlling the expression of proinflammatory and antiapoptotic genes (1, 2). In resting cells, proteins of the I-κB family are bound to NF-κB to limit its nuclear accumulation and transactivation potential. Agonists rapidly induce NF-κB activity by triggering the ubiquitylation and the degradation of I-κB proteins (3). The ubiquitylation of I-κBs is tightly controlled and requires their prior phosphorylation by the I-κB kinase (IKK) complex (4–6). The IKK complex contains two kinases, IKKα and IKKβ (also called CHUK/IKK1 and IKK2) (7–12). IKKβ−/− mice fail to degrade I-κBα (13–15), whereas mice deficient in IKKα are born with only minor defects in I-κBα-controlled NF-κB activity (16–18). For historical reasons, the IKKβ-controlled pathway has been termed the “canonical pathway” (19). Signaling in this pathway induces catalytic activity of IKKβ via phosphorylation of its activation loop (20, 21). Recent genetic evidence identified TAK1 as the IKK acting in the canonical pathway (22, 23). TAK1 has been shown to become activated in the presence of lysine 63-linked Ub conjugates, the formation of which required the Ub ligase TRAF6 (24–26).
Besides IKKα and IKKβ, the IKK complex also contains IKKγ (also called NEMO, IKKAP, and FIP3) (27–30). IKKγ-deficient cells cannot activate IKKβ, which incapacitates the canonical pathway (31–33). Hence, IKKγ has been suggested to be the regulatory subunit of the IKK complex, but how exactly it performs this function has remained unclear. A Ub-dependent control of IKKγ seems likely given the well established role of Ub conjugates for signaling upstream of IKK, and the enhanced IKK activity in the absence of specific Ub hydrolases (34–37). Supporting this Ub-control hypothesis, exposure to NF-κB agonists causes ubiquitylation of IKKγ, and certain signaling-deficient point mutants of IKKγ fail to become ubiquitylated (38–42). A different Ub-related control mechanism has been proposed recently when it was discovered that IKKγ mutants failing to signal in response to TNFα were unable to bind ubiquitylated RIP, a protein essential for TNFα signaling (43–45). Ub binding by IKKγ was suggested to allow the recruitment of IKKα/β to specific complexes, where kinase activity might be induced. We currently do not know the relationship, if any, between these two proposed Ub-dependent control mechanisms.
Using somatic cell genetics, we have isolated a series of IKKγ alleles. We found that mutations in the coil-zipper (CoZi) domain of IKKγ can cause signaling defects or constitutive NF-κB activity. A constitutively activating IKKγ allele unable to bind Ub suggests a role for IKKγ beyond that of a Ub adaptor merely recruiting the IKK complex. Therefore, we propose that during signaling IKKγ adopts an activated state. Genetic evidence indicates that this activated state occurs downstream of Ub binding and licences IKKγ for ubiquitylation, which demonstrates a mechanistic link between IKKγ's role in sensing upstream and regulating downstream pathway activity.
Results
An Allelic Series of IKKγ Mutants.
To employ genetic analysis in mammalian somatic cells for the study of NF-κB signaling, we engineered the murine B cell line 70Z/3 into an NF-κB reporter cell line. The resulting GTPT3 cells are equipped with three NF-κB-dependent markers: GFP and rat Thy1 for fluorescence-based readouts and a pac:TK fusion gene for metabolic selection. To identify genes essential for NF-κB activation, we mutagenized GTPT3 with ICR191 and enriched for LPS-unresponsive cells in the presence of ganciclovir. Individual clones were chosen based on their inability to express GFP in response to LPS (Fig. 1A). We confirmed that the lack of GFP expression reflected a genuine defect in NF-κB activation by assessing nuclear translocation of RelA (Fig. 1B).
Fig. 1.

Defect in NF-κB signaling in F40, F29, and J77 cells due to mutations in IKKγ. (A) Cells were stimulated for 24 h with 1 μg/ml LPS before analysis for NF-κB-dependent GFP expression. (B) Nuclear extracts from cells stimulated with 10 μg/ml LPS and probed for RelA and PCNA. (C) Cells stimulated for 24 h with 1 μg/ml LPS, 10 μg/ml PGN, 250 nM CpG DNA, or 50 ng/ml PMA/1 μM Ionomycin analyzed for NF-κB-dependent GFP and Thy1 expression by flow cytometry. (D) Cell lysates were probed for IKKγ. (E) Secondary structure of IKKγ (CC1, coiled coil 1; CC2, coiled coil 2; LZ, leucine zipper; Zn, zinc finger). F40 carries a frame shift (T47fs). F29 and J77 contain point mutations (E308V and R331P, respectively). D304N occurred in a patient with EDA-ID. K270A is a designed mutation. (F) F40 cells complemented with the indicated IKKγ alleles. Cells stimulated for 24 h with 1 μg/ml LPS were analyzed for NF-κB-dependent GFP expression. Lysates were probed for AU1-tagged IKKγ.
Several mutant clones were unresponsive not only to LPS, but also to CpG DNA and phorbol 12-myristate 13-acetate (PMA)/Ionomycin (Fig. 1C). Unresponsiveness to multiple agonists suggests a defect in a downstream signaling component. We considered IKKγ a likely candidate because of its X-chromosomal localization and hence its vulnerability to mutational inactivation. Indeed, clone F40 tested negative for IKKγ by Western blot (Fig. 1D). cDNA sequencing revealed a base insertion (IKKγ137_138insG) so that the resulting protein (IKKγT47fs), even if expressed and stable, would only contain 46 amino acids (Fig. 1E). In contrast, clones F29 and J77 expressed wild-type-sized IKKγ (Fig. 1D). Sequencing of their cDNA revealed point mutations in IKKγ replacing glutamate 308 with valine (IKKγE308V) in F29 and arginine 331 with proline (IKKγR331P) in J77 (Fig. 1E). Notably, the two mutations occurred in close proximity to each other and to a further mutation exchanging aspartate with asparagine at position 311 in human IKKγ (corresponding to murine IKKγD304N), seen in a patient suffering from anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) (46). The R331P mutation is located within a leucine zipper (LZ) where the presence of a helix-breaking proline may not be tolerated (Fig. 1E) (47). This LZ has been reported to bind the adjacent CC2 region (48), suggesting that residues D304 and E308 are part of a loop connecting the two helical structures. We conclude that the region spanning CC2 and LZ forms a functionally important domain in IKKγ. We will refer to it as the CoZi domain.
To test whether the mutant IKKγ alleles caused the observed defect in NF-κB activation, we used IKKγ-deficient F40 cells. Transduction with IKKγWT restored reporter induction (Fig. 1F), which demonstrates that the absence of IKKγ protein is the only defect relevant to NF-κB signaling in F40. Transduction of F40 with IKKγD304N, IKKγE308V, or IKKγR331P did not restore NF-κB activation in response to LPS, thereby proving the alleles to be defective.
IKKγ needs to assemble with IKKβ to serve its signaling function. We wondered whether the assembly of this complex was disturbed in the mutant clones. Immunoprecipitation of IKKγ resulted in equal amounts of IKKα and IKKβ in precipitates from wild-type GTPT3 and mutant F29 or J77 cells [supporting information (SI) Fig. 7A]. In contrast, neither IKKα nor IKKβ were precipitated from F40 cells. We also tested the association of IKK subunits into higher order complexes by gel filtration (SI Fig. 7B). In lysates of wild-type GTPT3 cells, IKKβ coeluted with IKKγ in high-molecular-weight complexes, whereas in lysates of IKKγ-deficient F40 cells, IKKβ occurred in later fractions. Importantly, complexes from wild-type cells and mutant clones F29 and J77 behaved indistinguishably. These results indicate that IKKγE308V and IKKγR331P were incorporated normally into IKK complexes. Therefore, we tested their potential as dominant-negative inhibitors of NF-κB signaling (SI Fig. 7C). Similarly to dominant-negative I-κBα, the transduction of GTPT3 cells with the mutant IKKγ alleles prevented LPS-induced NF-κB reporter expression. In contrast, transduction with IKKγWT did not impair NF-κB activation. We conclude that mutations in the CoZi domain of IKKγ specifically interfere with its signaling function.
Dominant Constitutive Activity of IKKγK270A.
Because mutations in CoZi located in the LZ or the connecting loop could prevent NF-κB activation, we wondered about the role of the CC2 coil (Fig. 1E). Coils characteristically contain heptameric repeats where residues in the “a” and “d” positions form a hydrophobic binding surface (Fig. 2A). Therefore, the presence of a charged residue (K270) in a “d” position of CC2 seemed remarkable. Converting K270 into alanine (IKKγK270A) created a coil with only hydrophobic residues in the “a” and “d” positions. In contrast to IKKγWT, which barely induced NF-κB upon transduction, IKKγK270A activated NF-κB potently because integration of a single virus carrying IKKγK270A per cell caused similar levels of NF-κB activity to saturating amounts of LPS (Fig. 2B).
Fig. 2.

IKKγK270A dominantly controls NF-κB activity. (A) Helical wheel representation of CC2. Beginning (position 253) and end (position 288) of CC2 and the occurrence of K270 in a “d” position are indicated. (B) NF-κB-dependent GFP induction measured 48 h after the transduction of GTPT3 cells with a control gene (bsd) or the indicated IKKγ alleles. Cells were stimulated with or without 1 μg/ml LPS for the final 24 h as indicated. An IRES-controlled CD8 was used to distinguish transduced from nontransduced cells. (C) F40 cells containing NF-κB-dependent luciferase were transduced with IKKγ, either wild type or mutant in position 270, and carrying additional mutations as indicated. Luciferase activity was measured 24 h later. Lysates were probed for AU1-tagged IKKγ.
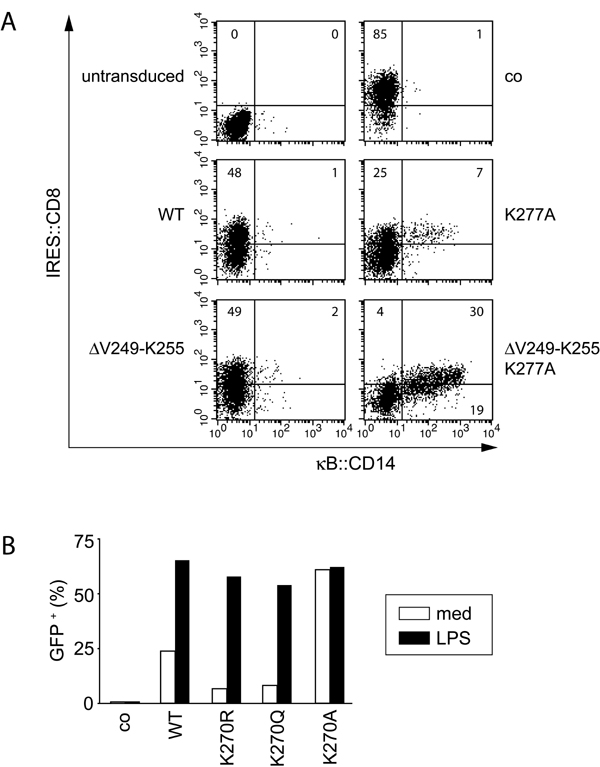
The corresponding lysine K277 in human IKKγ has been suggested to control DNA damage-induced NF-κB activation via sumoylation, ubiquitylation, and nuclear accumulation of IKKγ (38). Constitutive activity of human IKKγK277A, however, was not reported. We found that this difference between human and murine IKKγ is caused by the presence of seven additional amino acids at the beginning of CC2 (V249–K255) in the human protein because human IKKγ(ΔV249-K255)+K277A also activated NF-κB strongly (SI Fig. 8A). We further observed that the constitutive activity of IKKγK270A is caused specifically by the presence of alanine, rather than the absence of lysine, because IKKγK270R and IKKγK270Q were not constitutively activating, but supported NF-κB activation by LPS (SI Fig. 8B). The phenotype of IKKγK270A is therefore not related to the modification of K270 with Ub or related proteins.
IKKγK270A is the only known constitutively active IKKγ allele, and investigating its modus operandi may advance our understanding of physiological IKKγ activation. Deleting the binding site for IKKα and IKKβ (IKKγK270AΔN102) prevented NF-κB activation (Fig. 2C; see also Fig. 1E). In contrast, the C-terminal Zn finger and the adjacent proline-rich region (IKKγK270AΔC389 and IKKγK270AΔC338, respectively) were not required for its activity. Deleting the LZ (IKKγK270AΔC314), however, extinguished NF-κB activation. This finding indicates that the constitutive activity of IKKγK270A depends on the integrity of its CoZi domain. We therefore investigated mutations in CoZi residues essential for LPS signaling. Importantly, double mutants (IKKγK270A+D304N, IKKγK270A+E308V, and IKKγK270A+R331P) constitutively activated NF-κB (Fig. 2C). Therefore, the constitutive activity of IKKγK270A appeared dominant over mutations blocking the signaling from LPS.
Constitutive Activity of IKKγK270A Occurs Independently of Ub Binding.
Ub in the form of ubiquitylated RIP is a binding partner for IKKγ (43, 44). Lack of Ub binding by human IKKγD311N (corresponding to murine IKKγD304N) correlates with its failure to signal in response to TNFα. We therefore wondered whether other mutations in CoZi also would affect the ability of IKKγ to bind Ub. We first confirmed that IKKγ bound specifically to Ub (Fig. 3A) and that it interacted preferentially with Ub chains (Fig. 3B). We also verified that isolated CoZi domains bound Ub directly (Fig. 3C). To characterize this interaction further, we tested several Ub mutants and found that UbI44A did not bind IKKγ (Fig. 3D). Next we examined the signaling-impaired IKKγ alleles for their ability to interact with Ub (Fig. 3E). IKKγD304N and IKKγE308V failed to bind Ub, whereas IKKγR331P, despite its profound signaling defect, still bound Ub. These results are consistent with an essential role of Ub binding to CoZi for NF-κB activation, but the phenotype of IKKγR331P shows that CoZi performs another function besides Ub binding.
Fig. 3.

IKKγ binds Ub via its CoZi domain. (A and B) Purified GST fusion proteins coupled to beads were incubated with lysates of 293 cells expressing luciferase IKKγ. The ratio between luciferase activity bound to beads and present in lysates is shown. GST fusion proteins were visualized by Coomassie blue staining. (C) Purified GST fusion proteins coupled to beads were incubated with lysate from bacteria expressing His-tagged CoZi. (Upper) Eluates from beads were blotted with anti-His antibody. (Lower) GST fusion proteins were visualized by Coomassie blue staining. (D) Purified GST fusion proteins coupled to beads were incubated with lysates of 293 cells expressing a luciferase IKKγ. The ratio between luciferase activity bound to beads and present in lysates is shown. GST fusion proteins were visualized by Coomassie blue staining. (E) The 293 cells were transfected with the indicated AU1-tagged IKKγ constructs. Lysates were incubated with purified GST-tetra-Ub (GST-4xUb) bound to beads. Lysates and eluates were blotted for AU1-IKKγ.
We investigated whether changes in Ub binding might explain the constitutive activity of IKKγK270A. This allele, however, did not bind Ub any more strongly than IKKγWT (Fig. 3E). Importantly, despite their ability to induce NF-κB efficiently (Fig. 2C), IKKγK270A+D304N and IKKγK270A+E308V failed to bind Ub (Fig. 3E). Therefore, the induction of NF-κB by IKKγK270A does not require Ub binding. We conclude that IKKγ harbors dormant activation potential, which is unleashed by the K270A mutation enabling IKKγK270A to adopt an activated state. This observation suggests that an activated state downstream of Ub binding also may exist for IKKγWT and that CoZiK270A may mimic a Ub-bound CoZiWT domain.
Ubiquitylation of IKKγ.
Besides Ub binding, ubiquitylation of IKKγ also has been suggested to control IKK activity (39–42). To investigate what relationship might exist between Ub binding and ubiquitylation, we studied ubiquitylation of IKKγ upon coexpression with TRAF6 (Fig. 4A). IKKγWT and IKKγK270A became ubiquitylated to a similar extent, whereas the signaling-deficient alleles IKKγD304N, IKKγE308V, and IKKγR331P were almost completely devoid of Ub (Fig. 4B). Importantly, in all double mutants (IKKγD304N+K270A, IKKγE308V+K270A, and IKKγR331P+K270A), ubiquitylation was restored (Fig. 4C). We conclude that ubiquitylation occurs downstream of Ub binding and that K270A bypasses the need for Ub binding.
Fig. 4.

Differential ubiquitylation of IKKγ alleles. (A) The 293 cells were transfected with plasmids encoding AU1-tagged IKKγ, myc-tagged Ub, and HA-tagged TRAFs. Lysates were precipitated with an antibody against AU1. Precipitates and lysates were blotted for AU1-IKKγ, HA-TRAFs, and myc-Ub. (B) The 293 cells were transfected with plasmids encoding AU1-tagged IKKγ alleles (wild type, K270A, D304N, E308V, and R331P) and HA-tagged TRAF6 (wild type or C70A). Lysates were immunoprecipitated with an antibody against AU1. Precipitates and lysates were blotted for AU1-IKKγ and HA-TRAFs. (C) The 293 cells were transfected with plasmids encoding AU1-tagged IKKγ alleles containing either one mutation (K270A) or two mutations (K270A/D304N, K270A/E308V, and K270A/R331P) and HA-tagged TRAF6 (wild type or C70A). Lysates were immunoprecipitated with an antibody against AU1. Precipitates and lysates were blotted for AU1-IKKγ and HA-TRAFs. The asterisk indicates lanes that were removed electronically from the blot.
CoZiK270A Forms High-Affinity Dimers.
We investigated how IKKγK270A activates NF-κB and observed higher IKK activity associated with IKKγK270A than with IKKγWT (Fig. 5A). The activation of NF-κB by IKKγK270A required the presence of IKKβ but not RIP (SI Fig. 9) and was inhibited by dominant-negative alleles of IKKβ and IκBα (Fig. 5B). We conclude that cells expressing IKKγK270A harbor constitutively active IKK complexes.
Fig. 5.

K270A causes high-affinity CoZi interactions. (A) GTPT3 cells were transduced with the indicated IKKγ alleles. AU1-tagged IKKγ was precipitated, and a kinase assay was performed with GST-IκBα (amino acids 1–100) as a substrate. The expression level of AU1-tagged IKKγ in lysates was analyzed. (B) NF-κB-dependent luciferase activity in 293 cells 48 h after transfection with the indicated combinations of plasmids. IKKβDN corresponds to IKKβK44A and IκBαDN indicates to IκBαS32A+S36A. (C) (Left) CoZiWT and CoZiK270A were purified from E. coli. (Right) Mass-action-driven association was analyzed by analytical ultracentrifugation. Plots are from sedimentation equilibrium runs and indicate the formation of dimers. The Kd value for 30 ± 5 μM CoZiWT and the monomer size as determined from fitting the raw data are indicated. The latter is in excellent agreement with the theoretical value (10.9 kDa). CoZiK270A did not dissociate detectably at concentrations as low as 2 μM and showed only dimer (Mw, app, weight average apparent molecular weight). (D) The 293 cells were transfected with the indicated combinations of luciferase-tagged IKKγ (full length or CoZi) and FLAG-tagged IKKγ (only full length) either wild type or mutant in position 270. Proteins were precipitated with an antibody against Flag and eluted with Flag peptide. The ratio between luciferase activity in eluates and lysates is shown. The expression of Flag-tagged proteins was analyzed by Western blot. (E) Lysates from F40 cells transduced with IKKγWT or IKKγK270A were fractionated over Superdex 200. Fractions were tested for IKKγ.
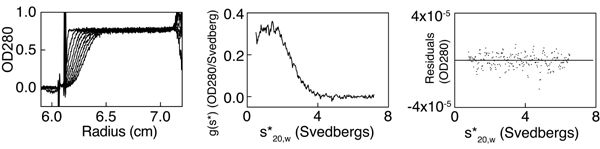
Seeking the cause of IKK activity, we wondered whether replacing the positively charged lysine at the predicted binding surface of CC2 with a hydrophobic alanine had changed the binding propensities of CoZi. To test this hypothesis, we investigated the mass-action-driven oligomerization of CoZi by analytical ultracentrifugation. Sedimentation equilibrium runs showed fully reversible dimerization of CoZiWT (Fig. 5C). The presence of dimers and the absence of higher order oligomers were confirmed by sedimentation velocity runs (SI Fig. 10). CoZiK270A also dimerized (Fig. 5C). However, in contrast to CoZiWT, which gave a Kd of 30 ± 5 μM, CoZiK270A resisted dissociation even at 2 μM, the lowest concentration measurable. The activity of IKKγK270A may therefore result from the increased homophilic interactions of its CoZiK270A domain.
We next tested whether in vivo CoZiK270A and CoZiWT differed in their ability to undergo homophilic interactions. Much stronger binding occurred between CoZiK270A and IKKγK270A than between the respective wild-type molecules (Fig. 5D). Robust binding required both partners to contain the K270A mutation. Full-length IKKγWT oligomerized potently, and, therefore, no further increase was observed for IKKγK270A. None of the above interactions required the recruitment of IKKα and IKKβ because removal of their binding site in IKKγΔN102 was inconsequential (data not shown). We conclude that the increased affinity of CoZiK270A causes homophilic binding in vivo.
Such binding may have two consequences. If it occurred between CoZi domains of separate IKK complexes, the complexes would associate into larger clusters. Alternatively, because each IKK complex already contains multiple IKKγ subunits, binding could enforce conformational changes within a preformed complex. To distinguish these scenarios, we performed gel filtration and found that IKK complexes containing IKKγWT or IKKγK270A were the same size (Fig. 5E). This result is consistent with the occurrence of a conformational change within a preformed IKK complex induced by homophilic CoZiK270A interactions.
Discussion
We have used somatic cell genetics to isolate a series of IKKγ alleles. Here, we show that the CoZi domain controls signal flow through IKKγ and that mutations in this domain can cause either loss of signaling or constitutive NF-κB activity. We demonstrate that mutant IKKγ can adopt an activated state based on a constitutively activating allele that maintains its activity even when unable to bind Ub. This finding suggests a function for IKKγ beyond binding Ub and recruiting the IKK complex to upstream signaling components. We propose that during signaling IKKγWT also adopts an activated state that occurs upstream of its ubiquitylation and could therefore link Ub binding to ubiquitylation of IKKγ and activation of NF-κB.
IKKγ is essential for NF-κB activation in the canonical pathway (31–33). It has been suggested to control IKK activity in a Ub-dependent manner. Two distinct Ub-related signaling events occur at IKKγ: noncovalent Ub binding and covalent ubiquitylation. Missense mutations that impair the signaling function of IKKγ have been demonstrated either to disturb Ub binding (43, 44) or to prevent ubiquitylation of IKKγ (38–42). Our identification of mutations in CoZi, which either prevent NF-κB signaling or constitutively activate it, indicate a crucial, possibly switch-like function for CoZi in regulating IKKγ (Fig. 6A). In the following section, we attempt to gain further insight into this function of CoZi by analyzing the ability of single and double mutants to signal, to bind Ub, and to become ubiquitylated.
Fig. 6.

Model of IKK activation. Shown are IKK complexes containing two kinase and two IKKγ subunits. Indicated are the association of IKKγ subunits, the binding of CC2 to LZ, and the binding of Ub to CoZi. Ubiquitylation also is shown, but it is not meant to indicate modification of a specific lysine residue. (A) Upon stimulation, IKKγWT binds Ub and becomes ubiquitylated, and kinase activity is induced. In contrast, IKKγD304N and IKKγE308V do not bind Ub and are not ubiquitylated by TRAF6, and kinase activity is not induced. Although IKKγR331P is able to bind Ub, it still fails to be ubiquitylated. Introducing an activating mutation, K270A, into any of those alleles restores their ubiquitylation by TRAF6 and creates mutants constitutively activating NF-κB without repairing the Ub-binding defect of IKKγD304N and IKKγE308V. (B) In unstimulated cells, kinases bound to IKKγWT remain catalytically inactive, whereas kinases bound to IKKγK270A are active. The heightened affinity of CoZiK270A domains for homophilic interactions suggests a mechanism for inducing kinase activity in wild-type cells, where binding of Ub chains to CoZiWT may be required to stabilize weaker homophilic CoZiWT interactions.
Binding of IKKγ to Ub (in the form of ubiquitylated RIP) occurs in TNFα signaling (43, 44). We extended this result by demonstrating that CoZi, like most Ub-binding domains (49, 50), requires I44 in Ub for binding. We also confirmed that IKKγD304N cannot bind Ub, and we demonstrated a similar defect for IKKγE308V. These data are consistent with LPS and CpG DNA requiring Ub binding by IKKγ to activate NF-κB. The constitutive activity of IKKγK270A, however, is not due to increased Ub binding. Notably, the introduction of K270A into alleles unable to bind Ub (IKKγK270A+D304N and IKKγK270A+E308V) constitutively activated NF-κB, but did not restore Ub binding. K270A therefore bypasses the need for Ub binding, suggesting that it affects CoZi downstream of Ub binding. K270A also acts downstream of R331P because the transduction of IKKγK270A+R331P caused NF-κB activity. Because IKKγR331P, in contrast to IKKγD304N and IKKγE308V, still bound tetra-Ub, CoZi must participate in two early signaling events. Consistent with this conclusion would be a bipartite interaction of IKKγ with a ubiquitylated ligand, in which D304N and E308V interfere with Ub binding, while R331P prevents recognition of the non-Ub part of the ligand. The identity of the ubiquitylated ligand in LPS signaling is unknown because this pathway does not require RIP (51, 52).
Ubiquitylation of IKKγ accompanies activation of the IKK complex, whereas impaired signaling due to missense mutations in IKKγ correlates with a lack of ubiquitylation (38–42). Concordant with these observations, TRAF6 failed to ubiquitylate IKKγD304N, IKKγE308V, and IKKγR331P. Introducing K270A into these alleles restored TRAF6-induced ubiquitylation. Restored ubiquitylation, but sustained lack of Ub binding, in IKKγK270A+D304N and IKKγK270A+E308V identifies the ubiquitylation defect as an indirect consequence of the D304N and E308V mutations. We conclude that Ub binding occurs upstream of ubiquitylation and that K270A bypasses the need for Ub binding. This order of events is entirely consistent with functions attributed previously to Ub binding and ubiquitylation, i.e., sensing and regulating pathway activity, respectively.
How the sensor and regulatory functions of IKKγ are linked remains largely unknown. Binding of IKKγ to Ub was suggested to recruit the IKK complex into the proximity of activated upstream signaling components, leading to IKK activation (43, 44). However, IKKγ alleles that constitutively activate NF-κB even when unable to bind Ub (IKKγK270A+D304N and IKKγK270A+E308V) challenge the notion of IKKγ as a mere Ub adaptor, rather suggesting that the mutant protein has adopted an activated state and IKKγWT harbors dormant activation potential. Constitutive activity of IKKγK270A without Ub binding is consistent with thr activation of IKKγWT occurring downstream of Ub binding. Therefore, Ub binding may serve a dual function: It may recruit the IKK complex into the proximity of upstream signaling components (43, 44) and it also may induce an activated state in IKKγ.
How could Ub binding activate IKKγ (Fig. 6B)? The constitutive activity of IKKγK270A correlates with the high affinity of its CoZiK270A domain for homophilic binding. If this interaction does cause activation, then Ub binding may serve to stabilize the weaker interaction of CoZiWT domains. IKKγ binds preferentially to Ub chains, which, due to their multivalency, may drive contacts between CoZiWT domains similar to the homophilic binding of CoZiK270A domains in IKKγK270A.
Homophilic CoZiK270A interactions could occur between IKKγ subunits of separate IKK complexes, thereby causing their clustering. Alternatively, because each complex contains at least two IKKγ subunits, CoZiK270A interactions could cause a conformational change within a preformed complex. IKK complexes containing IKKγK270A were no larger than those containing IKKγWT. Therefore, CoZiK270A interactions seem to occur preferentially between IKKγ subunits within one IKK complex. This result supports the notion of a conformational change in IKK complexes containing IKKγK270A as the cause of constitutive NF-κB activity. It is tempting to speculate that binding of a ubiquitylated ligand causes a similar conformational change in wild-type IKK complexes during signaling. Ultimately, structural work may be required to test this hypothesis.
Materials and Methods
Reagents.
Antibodies were from BD PharMingen (IKKγ, CD8, and Thy1.1), Imgenex (IKKα and IKKβ), Abcam (rabbit HA11, AU1, and myc), Covance (murine AU1), Santa Cruz Biotechnology (Ub), and Dabco (HRP-conjugated reagents). LPS (Escherichia coli O127:B8), PMA, and Ionomycin were from Sigma–Aldrich, peptidoglycan was from Fluka, and CpG DNA (ODN1668 TCCATGACGTTCCTGATGCT) was from Operon.
Plasmids.
TRAFs were expressed from pEAK8. All other genes were in M5P (53) or M6P8, an M5P derivative containing IRES-controlled CD8. Numbering in IKKγ constructs refers to NM_178590. IKKγ in GTPT3, from which the cDNA for this study was derived, contains asparagine at position 285.
Cell Culture and Mutagenesis.
An NF-κB-dependent promoter (45) was used to control the expression of reporter genes (GFP, rat Thy1, nd puromycin acetyltransferase fused to thymidine kinase). GTPT3, a clone derived from 70Z/3 cells stably transfected with all three reporter genes, was chosen for its low constitutive- and high LPS-stimulated reporter expression. Mutagenesis was performed as described in ref. 54. After recovery, cells were stimulated with 1 μg/ml LPS and selected with 0.5 μM ganciclovir (Sigma–Aldrich). Mutant clones were identified by the absence of GFP and Thy1 expression upon LPS stimulation.
M35 cells lack IKKβ and were isolated from GTPT3 based on their unresponsiveness to CpG DNA after random mutagenesis as above. SVT35 Jurkat cells carry an NF-κB-controlled CD14 reporter. These cells and an RIP-deficient subclone were provided by Adrian Ting (45).
Reporter Assays.
NF-κB-dependent GFP activity was analyzed on a FACSCalibur (BD Biosciences). Luciferase activity was measured with Bright-Glo (Promega).
Chromatography.
After swelling in hypotonic buffer [10 mM Tris·HCl (pH 7.4), 1 mM KCl, and 10 mM MgCl2], cells were disrupted with a tight Dounce homogenizer. NaCl was added to a final concentration of 150 mM, and insoluble cell remnants were pelleted at 100,000 × g. Supernatants were fractionated on a Superdex 200 column (Amersham Pharmacia).
Immunoprecipitation.
Postnuclear supernatants from cells lysed in 0.5% Triton X-100, 20 mM Tris·HCl (pH 7.4), 150 mM NaCl, and 1 mM EDTA were incubated for 2 h with 1–2 μg/ml primary antibody, followed by incubation for 2 h with protein G Sepharose. After washing, samples were eluted with SDS buffer.
Kinase Assay.
After immunoprecipitation in the presence of protease and phosphatase inhibitors, beads were washed in 20 mM Mops (pH 7.4), 1% Triton X-100, 0.1 mM EDTA, 1 mM EGTA, and 1 mM DTT. Immune complexes were incubated for 20 min at 30°C in a 20-μl reaction mixture containing 25 μM cold ATP, 3 μCi [32]ATP, 12 mM MgCl2, and 3 μg of GST-IκBα 1–100. The reaction was stopped with SDS buffer.
Analytical Ultracentrifugation.
The CoZi domain of IKKγ (amino acids 250–339) was expressed from pETM11. After purification on Ni-NTA agarose (Qiagen), the His tag was cleaved off with TEV protease, and the resulting material was repurified over Ni-NTA and Superdex 75 columns. Sedimentation equilibrium/velocity experiments were carried out as described in ref. 55.
Ub Binding.
GST fusion proteins expressed in E. coli were coupled onto GSH beads. For LUMIER assays (56), Renilla luciferase fused to IKKγ was expressed in 293ET cells. Binding was performed for 2 h in 20 mM Tris·HCl (pH 7.4), 150 mM NaCl, and 0.1% Triton. Proteins were eluted with glutathione. The ratio between luciferase activity in eluates and lysates is presented as fold binding over a control reaction.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Olga Perisic and Allan Warren (Medical Research Council Laboratory of Molecular Biology) for TEV protease and help with chromatography, Aarie Geerlof (European Molecular Biology Laboratory, Heidelberg) for pETM plasmids, Hiroyasu Nakano (Juntendo University Medical School, Tokyo) for pGEX-IκBα, Adrian Ting (Mount Sinai Medical Center, New York) for RIP-deficient cells, and Alexander Betz and Mariann Bienz for reading the manuscript.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0706552105/DC1.
References
- 1.Ghosh S, Karin M. Cell. 2002;109(Suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 2.Hayden MS, Ghosh S. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 3.Karin M, Ben-Neriah Y. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 4.Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 6.DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connelly MA, Marcu KB. Cell Mol Biol Res. 1995;41:537–549. [PubMed] [Google Scholar]
- 8.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 9.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 10.Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 11.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 12.Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- 14.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Science. 1999;284:321–325. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 16.Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 17.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 18.Li Q, Lu Q, Hwang JY, Buscher D, Lee KF, Izpisua-Belmonte JC, Verma IM. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 20.Ling L, Cao Z, Goeddel DV. Proc Natl Acad Sci USA. 1998;95:3792–3797. doi: 10.1073/pnas.95.7.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delhase M, Hayakawa M, Chen Y, Karin M. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 22.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 23.Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, et al. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 25.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 26.Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. Mol Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 28.Rothwarf DM, Zandi E, Natoli G, Karin M. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Kang J, Friedman J, Tarassishin L, Ye J, Kovalenko A, Wallach D, Horwitz MS. Proc Natl Acad Sci USA. 1999;96:1042–1047. doi: 10.1073/pnas.96.3.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mercurio F, Murray BW, Shevchenko A, Bennett BL, Young DB, Li JW, Pascual G, Motiwala A, Zhu H, Mann M, Manning AM. Mol Cell Biol. 1999;19:1526–1538. doi: 10.1128/mcb.19.2.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, Rajewsky K, Pasparakis M. Mol Cell. 2000;5:981–992. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- 32.Makris C, Godfrey VL, Krahn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M. Mol Cell. 2000;5:969–979. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 33.Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW. Genes Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 34.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 36.Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 37.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 38.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Cell. 2003;115:565–576. doi: 10.1016/s0092-8674(03)00895-x. [DOI] [PubMed] [Google Scholar]
- 39.Tang ED, Wang CY, Xiong Y, Guan KL. J Biol Chem. 2003;278:37297–37305. doi: 10.1074/jbc.M303389200. [DOI] [PubMed] [Google Scholar]
- 40.Zhou H, Wertz I, O'Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. Nature. 2004;427:167–171. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]
- 41.Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 42.Abbott DW, Wilkins A, Asara JM, Cantley LC. Curr Biol. 2004;14:2217–2227. doi: 10.1016/j.cub.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 43.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 44.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 45.Ting AT, Pimentel-Muinos FX, Seed B. EMBO J. 1996;15:6189–6196. [PMC free article] [PubMed] [Google Scholar]
- 46.Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S, et al. Nat Genet. 2001;27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 47.Makris C, Roberts JL, Karin M. Mol Cell Biol. 2002;22:6573–6581. doi: 10.1128/MCB.22.18.6573-6581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agou F, Traincard F, Vinolo E, Courtois G, Yamaoka S, Israel A, Veron M. J Biol Chem. 2004;279:27861–27869. doi: 10.1074/jbc.M314278200. [DOI] [PubMed] [Google Scholar]
- 49.Hicke L, Schubert HL, Hill CP. Nat Rev Mol Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 50.Haglund K, Dikic I. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J. Nat Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 52.Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA. J Biol Chem. 2005;280:36560–36566. doi: 10.1074/jbc.M506831200. [DOI] [PubMed] [Google Scholar]
- 53.Randow F, Sale JE. Subcell Biochem. 2006;40:383–386. doi: 10.1007/978-1-4020-4896-8_30. [DOI] [PubMed] [Google Scholar]
- 54.Randow F, Seed B. Nat Cell Biol. 2001;3:891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 55.Chaillan-Huntington C, Butler PJ, Huntington JA, Akin D, Feldherr C, Stewart M. J Mol Biol. 2001;314:465–477. doi: 10.1006/jmbi.2001.5136. [DOI] [PubMed] [Google Scholar]
- 56.Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW, et al. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}