

Abstract
Ion channels are multisubunit proteins responsible for the generation and propagation of action potentials in nerve, skeletal muscle, and heart as well as maintaining salt and water homeostasis in epithelium. The subunit composition and stoichiometry of these membrane protein complexes underlies their physiological function, as different cells pair ion-conducting α-subunits with specific regulatory β-subunits to produce complexes with diverse ion-conducting and gating properties. However, determining the number of α- and β-subunits in functioning ion channel complexes is challenging and often fraught with contradictory results. Here we describe the synthesis of a chemically releasable, irreversible K+ channel inhibitor and its iterative application to tally the number of β-subunits in a KCNQ1/KCNE1 K+ channel complex. Using this inhibitor in electrical recordings, we definitively show that there are two KCNE subunits in a functioning tetrameric K+ channel, breaking the apparent fourfold arrangement of the ion-conducting subunits. This digital determination rules out any measurable contribution from supra, sub, and multiple stoichiometries, providing a uniform structural picture to interpret KCNE β-subunit modulation of voltage-gated K+ channels and the inherited mutations that cause dysfunction. Moreover, the architectural asymmetry of the K+ channel complex affords a unique opportunity to therapeutically target ion channels that coassemble with KCNE β-subunits.
Keywords: charybdotoxin, KCNQ1, stoichiometry
Voltage-gated KCNQ1 K+ channels are tetrameric integral membrane proteins that open and close in response to changes in membrane potential. To regulate cellular potassium flow in both excitable and nonexcitable tissues, Q1 channels coassemble with regulatory KCNE peptides, forming membrane-embedded K+ channel complexes with various voltage-sensing and gating properties (1). The importance of forming a properly assembled Q1–KCNE complex is underscored by the mutations that give rise to long QT syndrome and congenital hearing loss (2, 3). Although the fourfold arrangement of the Q1 α-subunits along the ion conduction pathway is established and unquestioned, the number of KCNE β-subunits in the K+ channel complex has been a long-standing and heated debate (4–7). Goldstein and coworkers (6) have strongly argued that Q1 channels average two KCNE peptides per complex; however, they could not definitively rule out K+ channel complexes containing a solitary KCNE peptide. Another complicating factor is the notion that Q1 channels form complexes with KCNE β-subunits with multiple stoichiometries (5, 8). Thus, approaches that measure the average number of KCNE peptides in the Q1 channel complex at the cell surface are unable to readily discern between fixed and various stoichiometries.
To determine the stoichiometry or stoichiometries of Q1–KCNE K+ channel complexes, we devised an iterative cell surface modification scheme where the labeling reagent specifically binds to the outer vestibule of the K+ conduction pore while covalently modifying a cysteine-bearing KCNE peptide in the channel complex (Fig. 1). Once the reaction is complete, excess reagent is removed and the irreversibly bound reagent is chemically liberated, restoring K+ channel complex function, and thereby modifying one KCNE peptide. Cysteine modification renders the KCNE peptide chemically inactive, and thus the number of KCNE peptides in the K+ channel complex is simply the number of complete modification cycles (reagent in, washout, cleaved) required before the washout step in the cycle becomes reversible. If Q1 and KCNE subunits form complexes with multiple stoichiometries, then these subpopulations would manifest themselves as reversibility during the labeling steps of the modification scheme. The measured reversibility would directly correspond to the percentage of subpopulations of K+ channel complexes with sub or supra stoichiometries. However, for the outlined counting strategy to be operational, there are three absolute but experimentally testable conditions: (i) The chemical reactions must be rapid, quantitative, and cell compatible. (ii) The nonspecific bimolecular reaction between the reagent and the cysteine in the KCNE peptide must not occur. (iii) Only one KCNE peptide in the K+ channel complex may react during the exposure to the reagent.
Fig. 1.

A cartoon depiction of one modification cycle of the iterative counting strategy used to determine the number of KCNE1 subunits in a KCNQ1 K+ channel complex by using a derivatized charybdotoxin (CTX). Each complete round of chemical treatment modifies one E1 subunit, allowing for the direct counting of KCNE subunits in the K+ channel complex.
Results
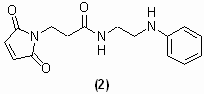
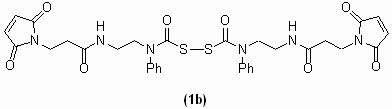
Central to our counting strategy is the chemically releasable, irreversible K+ channel inhibitor. This reagent, CTX-Clv, is composed of three functional groups: (i) a high-affinity K+ channel inhibitor tethered to (ii) a cysteine-specific modifying agent by (iii) a cleavable linker (Fig. 2a). For the K+ channel inhibitor, we chose charybdotoxin (CTX), a peptide scorpion toxin that tightly binds to the extracellular side of the potassium conduction pore with 1:1 stoichiometry (9). A wide range of K+ channels have been redesigned to bind CTX with high affinity because the outer vestibule of the pore-forming subunits is structurally conserved (6, 10, 11). A maleimide was used for the cysteine-modifying agent because it forms an irreversible thioether bond with cysteines that is stable to reducing agents. For the cleavable linker, we synthesized a homobifunctional linker, which when cleaved with reductant does not reintroduce a thiol group. We based our linker on the cyclic dithiasuccinoyl (Dts) amino-protecting group (12). The linear linker, bis(N-phenylcarbamoyl)disulfane 1a, is rapidly cleaved (<2 min) with cell-compatible reductants [dithiothreitol, 2-mercaptoethanol, and tris(2-carboxyethyl)phosphine (TCEP)] at biological pH, leaving behind secondary aryl amines with the concomitant release of two equivalents of carbonylsulfide gas (Fig. 2b). To tether the maleimide to CTX, we first synthesized the cleavable bismaleimide 1b by reacting bis(chlorocarbonyl)disulfane (13, 14) with a 3-maleimido-N-[2-(phenylamino)ethyl]propanamide [see supporting information (SI) Materials and Methods]. CTX-Clv was then synthesized by adding excess 1b to the CTX mutant R19C (Materials and Methods), which upon derivatization with cysteine-modifying reagents has been shown to maintain its high affinity for the ion conduction pore of K+ channels (15). To specifically target and modify Q1/E1 complexes with CTX-Clv, we used a variant of Q1 that binds CTX with nanomolar affinity (6) and placed a single cysteine in the N terminus of E1 (T14C) to react with the maleimide. This CTX-sensitive Q1/E1T14C complex has been previously shown to form complexes with wild-type voltage sensitivity and gating kinetics (6, 16).
Fig. 2.

Structures of the CTX reagents and the mechanism by which the cleavable linker is severed by reductant. (a) Cysteine-modifying charybdotoxins. (b) Tris(2-carboxyethyl)phosphine (TCEP) cleavage of bis(N-phenylcarbamoyl)disulfane linkers and the resultant products.
We first subjected CTX-Clv to a battery of experimental tests to assess whether it could meet the two requirements of the repetitive labeling strategy. We expressed Q1/E1T14C complexes in Xenopus oocytes and monitored the total and CTX-sensitive current because background endogenous oocyte currents are not inhibited by CTX (16, 17). Fig. 3a shows the total and normalized CTX-sensitive current elicited by a 20-mV pulse from an oocyte expressing Q1/E1T14C complexes. Treatment with 10 nM CTX-Clv followed by washout resulted in ≈85% irreversible inhibition of the total current (Fig. 3 Left). As expected, we did not observe 100% irreversible inhibition because oocytes possess CTX-insensitive endogenous Q1 channels that comigrate to the cell surface upon KCNE mRNA injection (6, 16–18). However, subtracting the endogenous CTX-insensitive current from total (Fig. 3a Right) revealed that CTX-Clv completely and irreversibly inhibits Q1/E1T14C complexes in 3 min. Subsequent treatment with 1 mM TCEP for 2 min relieves the irreversible inhibition and restores the current to pretreated levels (Fig. 3a, red trace). Irreversible inhibition requires the cysteine in E1 because CTX-Clv is an entirely reversible inhibitor with wild-type Q1/E1 complexes (Fig. 3b). These results demonstrate that each step in the modification cycle occurs quantitatively within minutes.
Fig. 3.

Characterization of CTX-Clv. (Left) Overlays of the total current elicited by a 20-mV pulse from oocytes expressing Q1/E1 and Q1/E1T14C complexes. Scale bars are 0.5 μA and 0.5 s. (Right) Normalized CTX-sensitive current (I/Imax). Oocytes were depolarized every 30 s and the data points were obtained at the end of a 5-s pulse. Gray and red data points correspond to the raw traces on the left. (a) Q1/E1T14C complexes were treated with 10 nM CTX-Clv, followed by washout and 1 mM TCEP treatment, demonstrating one complete modification cycle. (b) Irreversible inhibition of Q1/E1 complex requires an extracellular cysteine in E1 because wild-type Q1/E1 complexes are reversibly inhibited by only 10 nM CTX-Clv. (c) Irreversible inhibition of Q1/E1T14C by 10 nM CTX-Clv is completely prevented in the presence of excess (500 nM) CTX (unlabeled). (d) A tethered CTX-Clv prevents modification of the unmodified E1 peptides in Q1/E1T14C complexes by CTX-Mal.
Compared with model reactions between positively charged maleimides and thiols, the modification of E1T14C by CTX-Clv is ≈1,000-fold faster (19). This faster modification rate was expected on the basis of previous affinity-driven modifying agents, which increase the reagent's effective molarity, thereby accelerating the chemical reaction (20, 21). In addition, this accelerated reaction rate indicates that toxin binding is required for modification and suggests that the nonspecific bimolecular reaction between CTX-Clv and E1T14C does not measurably occur. To directly demonstrate that the nonspecific reaction does not occur, we performed a competition experiment in which we treated Q1/E1T14C complexes with 10 nM CTX-Clv and 500 nM unlabeled CTX for an extended duration (Fig. 3c). As predicted for a reagent that requires binding before modification, CTX-Clv did not modify the Q1/E1T14C complex in the presence of excess competitor (unlabeled CTX), as evidenced by complete washout (Fig. 3c). This result demonstrates that the bimolecular reaction between CTX-Clv and E1T14C does not contribute to the irreversible inhibition of the K+ channel complex. In addition, the vast excess of underivatized CTX inhibited the same amount of total current as 10 nM CTX-Clv (compare traces in Fig. 3 a and c Left), further demonstrating that the residual current is from endogenous CTX-insensitive channels.
To determine whether a covalently tethered CTX-Clv prevents further toxin-accelerated modifications of the other KCNE peptides in the channel complex, we performed a second competition experiment (Fig. 3d), this time using CTX-Mal (Fig. 2a), a noncleavable version of CTX-Clv. CTX-Mal irreversibly inhibits Q1/E1T14C with a modification rate similar to that of CTX-Clv (16), yet the irreversible inhibition by CTX-Mal is completely resistant to TCEP treatment (SI Fig. 5). Q1/E1T14C complexes were first irreversibly inhibited by CTX-Clv, as observed after washout (Fig. 3d). The inhibited complexes were then treated with CTX-Mal for the same duration and the reagent was washed out. A final treatment with TCEP resulted in complete restoration of the current that was measured before the toxin treatment sequence began. Because the current fully returned after TCEP treatment and CTX-Mal inhibition is not reversible by TCEP, this result confirms that K+ channel complexes with a tethered CTX-Clv are protected from subsequent toxin-accelerated modifications. Table 1 shows that CTX-Clv dissociates from the Q1/E1 channel complex with similar kinetics and percent recovery. The one exception is the competition experiment (CTX-Clv + excess CTX), where the measured recovery kinetics reflects unmodified CTX dissociation, which takes ≈50% longer than CTX-Clv.
Table 1.
Recovery parameters after CTX-Clv application
| Construct | Treatment | τrecovery, s | % recovery | n |
|---|---|---|---|---|
| Q1/E1 | CTX-Clv | 98 ± 1 | 95 ± 2 | 5 |
| Q1/E1T14C | CTX-Clv, TCEP | 97 ± 1 | 96 ± 2 | 4 |
| Q1/E1T14C | CTX-Clv + excess CTX | 155 ± 4 | 96 ± 1 | 3 |
| Q1/E1T14C | CTX-Clv, CTX-Mal, TCEP | 94 ± 3 | 94 ± 1 | 3 |
| Q1/E1T14C | Round 1, TCEP | 96 ± 2 | 96 ± 2 | 3 |
| Q1/E1T14C | Round 2, TCEP | 98 ± 3 | 98 ± 4 | 3 |
| Q1/E1T14C | Round 3 | 97 ± 1 | 94 ± 3 | 3 |
Data are from individual experiments obtained from 3–5 oocytes. Treatment describes the sequential application of chemical reagents to the indicated construct. Round 1, Round 2, and Round 3 indicates the first, second, and third rounds of subunit counting (Fig. 4a), respectively. Time constants of recovery (τrecovery) were fit to a single exponential as described in Materials and Methods. Percent recovery is a comparison between the amount of current remaining after CTX-Clv release with the amount of current before CTX-Clv application. Values are mean ± SEM.
With this now battle-tested reagent, we used CTX-Clv to count the number of KCNE peptides in the Q1 K+ channel complex. Q1/E1T14C complexes were repetitively subjected to the modification cycle: CTX-Clv treatment, washout, and TCEP reductive cleavage (Fig. 4a). The first two rounds required TCEP treatment to release CTX-Clv from the K+ channel complex. The second round of CTX-Clv treatment proceeded as smoothly as the first, demonstrating that the molecular remnants left on the KCNE peptide after TCEP cleavage do not interfere with subsequent rounds of binding and modification. In the third round, CTX-Clv once again inhibited the K+ channel complex; however, inhibition was completely reversible with a simple washout, showing that it takes two modification cycles to react with all of the KCNE peptides in a K+ channel complex. In addition, we never observed irreversible modification by CTX-Clv after the second round, nor did we observe partial reversibility during the first or second rounds, which shows that K+ channel complexes with multiple stoichiometries do not significantly contribute to the cell surface population. Moreover, the near quantitative reactions observed in each round indicate that Q1/E1 complexes at the cell surface are essentially devoid of endogenous Xenopus KCNE peptides (22), since their incorporation would result in incomplete modification cycles and reversibility with CTX-Clv. The kinetics of CTX-Clv unbinding and the percent recovery did not vary from round to round (Table 1).
Fig. 4.

KCNQ1 K+ channel complexes contain two KCNE1 peptides. (a) Iterative counting of E1T14C subunits in a Q1 K+ channel complex. (Upper) Raw data showing the total current for each round. Scale bar is 0.5 μA and 0.5 s. (Lower) Q1/E1T14C complexes were treated with two rounds (10 nM CTX-Clv, washout, 1 mM TCEP) of treatment before CTX-Clv binding became reversible. Oocytes were depolarized every 30 s and the data points were obtained at the end of a 5-s pulse. Gray and red data points correspond to the raw traces above. (b) Only one KCNE peptide reacts per CTX-Clv treatment. (Upper) CTX-sensitive current for each round. Scale bar is 0.5 μA and 0.5 s. (Lower) Chemical modification of 19% of the cell surface Q1/E1T14C complexes with 10 μM maleimide N-ethylsulfonate, sodium salt (Mal-ES), predicts that the reversibility of CTX-Clv in the second round will be 69% if one KCNE peptide reacts per round; 88% if two KCNE are modified per round (dotted lines, which correspond to two or four KCNE subunits in the K+ channel complex). Washout of CTX-Clv after the second round of treatment results in 65 ± 4% (SEM) reversibility (n = 3). I/Imax is the CTX-sensitive current that was normalized before chemical treatment. Gray and red data points correspond to the CTX-sensitive currents above.
Although we show that a CTX-Clv bound and tethered to a K+ channel complex prevents subsequent KCNE modifications by CTX-Mal (Fig. 2d), we have not directly shown that one, and only one, KCNE peptide reacts per CTX-Clv treatment. To do this, we took advantage of the difference in the probability mass function when n = 2, 4, or any even number of KCNE subunits because the results in Fig. 4a eliminate stoichiometries with an odd number of KCNE peptides (Appendix). We first pretreated Q1/E1T14C complexes with a membrane-impermeant maleimide (Mal-ES) (19) to randomly and independently react with a fraction of the E1T14C peptides. We then determined the percentage of Q1/E1T14C complexes that were fully modified by this pretreatment by measuring the reversibility of CTX-Clv after washout (Fig. 4b). Based on the 19% reversible inhibition of the CTX-sensitive current that we observed, the binomial distribution predicts that a second treatment with CTX-Clv should result in 69% reversibility if only one KCNE is modified per CTX-Clv treatment; 88%, if two KCNE peptides react per treatment. Cleavage of the previously tethered CTX-Clv, followed by another bolus of CTX-Clv, and washout resulted in 65 ± 4% (SEM) reversibility (n = 3). These results demonstrate that only one KCNE peptide is modified per exposure to CTX-Clv. In addition, this binomial determination of the stoichiometry of the complex corroborates the counting experiment in Fig. 4a, confirms the previous stoichiometric determinations by Goldstein and coworkers (4, 6), and indicates that the lack of reactivity in round 3 was not due to steric hindrance from previous CTX-Clv modifications. Moreover, these results show that CTX-Clv can accurately measure (within 5%) different subpopulations of ion channel complexes at the cell surface. Thus, the counting results in Fig. 4a demand that the vast majority (>95%) of the Q1 K+ channel complexes have only two KCNE peptides, resulting in complexes that are not fourfold symmetric.
Discussion
The 4:2 (α:β) stoichiometry of the Q1/E1 complex has several broad implications for KCNE modulation of voltage-gated K+ channels. If modulation is due to a direct KCNE–K+ channel protein–protein interaction, then KCNE peptides either interact with only two of the α-subunits in a K+ channel complex or they bifurcate adjacent α-subunits, making two unique sets of protein contacts with all four ion-conducting subunits. A recent mutagenic perturbation study of the KCNE1 transmembrane domain suggests that a stretch of residues 56–63 alternately interacts with two protein faces of the KCNQ1 channel, providing a potential bifurcation site (23). Alternatively, KCNE modulation of K+ channels could be allosteric. Likewise, mutant KCNE peptides that produce diseased K+ channel complexes will impart their deleterious effects on ion conduction and channel gating by the molecular mechanisms outlined above. Interpreting the effects of KCNE peptides on K+ channel function is made even more complex with the discovery that a single Q1 channel can assemble with two different KCNE peptides to form a functional heteromeric complex (16). However, because each K+ channel complex contains only two KCNE peptides, the number of potential heteromeric complexes that can form is greatly reduced.
How then does a K+ channel, which is destined to be tetrameric when expressed alone, incorporate only two modulatory KCNE peptides? Some insight into the Q1/E1 assembly mechanism comes from the study of K+ channel α-subunit biogenesis, which has shown that individual subunits first form dimers, then subsequently dimerize to form a tetrameric ion channel (24). In addition, kinetic modeling indicates that tetrameric proteins exclusively use a dimer-of-dimers pathway because other assembly pathways would lead to substantial amounts of partially assembled, dead-end intermediates (25). Therefore, KCNE peptides may not be breaking the symmetry of the K+ channel complex, but are instead infiltrating the dimeric assembly processes of the tetrameric ion conduction pore. For Kv-type channels, the N-terminal T1 domain tetramerizes while the nascent polypeptides are still attached to ribosomes (26). Thus, for this class of K+ channels, KCNE peptides would have to coassemble with channel subunits very early in biogenesis to use the nascent dimeric structures. For KCNQ1 channels, coassembly could occur later in biosynthesis because the subunit specificity domain for tetramerization resides in the C terminus (27). In either case, to exploit the dimer-of-dimers pathway, coassembly would have to occur in the endoplasmic reticulum, which is consistent with previous assembly studies with wild-type (28) and mutant KCNE1 peptides (29).
CTX-Clv and the iterative cell surface modification approach have enabled us to definitively determine the stoichiometry of the Q1/E1 K+ channel complex. Moreover, this counting approach has the capacity to identify and measure subpopulations of stoichiometric mixtures, which may be operational for other K+ channel complexes. The structural conservation of the outer vestibule of K+ channels allows CTX sensitivity to be transplanted into any K+ channel of interest, a strategy that we have used here, and should be applicable for most K+ channel complexes. In addition, the modular design of the reagent allows for the incorporation of different targeting ligands and chemical modifying agents. Given the vast assortment of high-affinity scorpion and spider toxins available, this iterative chemical approach can be adapted to identify and enumerate the protein subunits in a variety of multimeric ion channel complexes.
Materials and Methods
Chemical Synthesis.
Details describing the synthesis and characterization of all chemical compounds can be found in SI Materials and Methods.
Mutagenesis and in Vitro Transcription.
For optimal CTX binding to the Q1/E1 complex, CTX-sensitive Q1 and hemagglutinin-tagged E1 constructs were used (6) and subcloned into a vector containing the 5′ and 3′ UTRs from the Xenopus β-globin gene for optimal expression in Xenopus oocytes. Site-directed mutagenesis and mRNA in vitro transcription were performed as described in ref. 16.
Electrophysiology.
Standard techniques and solutions were used for the preparation of and recording from Xenopus oocytes by two-electrode voltage clamp (30–34). Oocytes were injected with a Q1:E1 mRNA ratio of 15:5 ng to ensure that E1 was in excess. Oocytes were incubated for 3–5 days with 3 mM l-glutathione and pretreated with 1 mM TCEP for 15 min before recording, and the current was measured by holding at −80 mV and pulsing to +20 mV for 5 s every 30 s.
Cell Surface Modifications.
Labeling reagents CTX-Clv and CTX-Mal (10 nM) were in ND96 (30) solution with 50 μg·ml−1 BSA and were added to the bath by a gravity-fed perfusion system with a chamber clearing time of ≈10 s. CTX-Clv cleavage was accomplished by 1 mM TCEP for ≈120 s. The recovery time constant (τrecovery) was measured by fitting the return of current after CTX-Clv washout to a single exponential. To randomly label cell surface cysteines with Mal-ES (19), TCEP-pretreated oocytes were incubated with 10 μM Mal-ES in ND96 for 500 s.
Synthesis of CTX-Clv.
Recombinant CTX R19C was purified and the methanethiosulfonate ethyltrimethylammonium (MTSET)-protected disulfide of CTX R19C was prepared as described in ref. 15. CTX-Clv was synthesized as follows: 16 nmol of CTX-MTSET in 2 ml of low-salt buffer (10 mM NaCl, 10 mM KPi, pH 7.4) was reduced with 1 mM dithiothreitol (DTT) for 45 min. Reduced CTX was purified by using a C18 HPLC column (4.6 mm × 250 mm) and eluting with solvent A [0.1% trifluoroacetic acid (TFA)} and solvent B (acetonitrile), forming a gradient of 10–40% B over 30 min. The fractions containing reduced CTX were adjusted to pH 7.0 by using 1 M KPi, pH 7.1, and a solution of 16 μmol of bismaleimide 1b in 90 μl of acetonitrile was slowly added and allowed to react for 30 min at room temperature. The reaction mixture was placed on ice for 10 min to precipitate excess unreacted bismaleimide, which was removed by filtration (GHP Acrodisc 0.45 μm, Pall Gelman Laboratory). CTX-Clv was HPLC purified as described above, using a 20–50% gradient B over 30 min. The concentration of purified CTX-Clv was determined by UV spectrometry (A280 of 1.0 = 100 μM CTX) (14) and labeling efficiency of CTX-R19C was determined to be 30 ± 8%, n = 7). The purified product was confirmed by electrospray ionization mass spectrometry (SI Fig. 6) and was aliquoted, lyophilized, and stored at −20°C. Immediately before use, individual aliquots were resuspended in ND96 containing 30% acetonitrile to ensure CTX-Clv went into solution. This solution was then diluted such that the final acetonitrile concentration was 0.3% and the final CTX-Clv concentration was 10 nM.
Supplementary Material
ACKNOWLEDGMENTS.
This work was supported by National Institutes of Health Grant GM0707650. W.R.K. was supported in part by a Burroughs–Wellcome Foundation Career Award in the Biomedical Sciences.
Appendix
The distribution of modified KCNE–K+ channel complexes generated by a partial treatment of Mal-ES can be defined by the probability mass function if each modification occurs independently:
where n is the number of KCNE peptides, k is the number of modified KCNE peptides, and F is the fraction of K+ channel complexes with all KCNE peptides modified when k = n. If one KCNE is modified per CTX-Clv treatment cycle, then Fig. 4a demands that there are two KCNE peptides in the complex. Therefore, p = 0.44 when k = 2, n = 2 and F = 0.19, which was experimentally determined after CTX-Clv washout (Fig. 4b). The complementary cumulative distribution function predicts the reversibility in the second CTX-Clv washout:
 |
where x is equal to the maximum number of Mal-ES modifications a K+ channel complex can have such that there is at least one modifiable cysteine remaining in the second cycle. For n = 2, x = 0 because all K+ channel complexes will have one CTX-Clv modification, which occurred during the measuring of F. Thus, the expected reversibility for one CTX-Clv modification per cycle with two KCNE peptides in a K+ channel complex (x = 0; n = 2, and p = 0.44) is R = 0.69. Conversely, if two KCNE modifications occur per exposure to CTX-Clv, then each complex must have four KCNE peptides (Fig. 4a) and p = 0.66 when k = 4, n = 4, and F = 0.19. In this scenario, x = 1 for the cumulative distribution function because only complexes with less than two Mal-ES modifications will have a modifiable cysteine in the second round of treatment. Thus, the expected reversibility for two CTX-Clv modifications per cycle with a four KCNE peptide–K+ channel complex (x = 1; n = 4, and p = 0.66) is R = 0.88. Stoichiometries with an odd number of KCNE peptides were not considered because they would require partial CTX-Clv irreversibility, which was not observed (Fig. 4a). Integer modifications greater than two KCNE peptides are possible; however, these would result in R > 0.95 when F = 0.19.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0710366105/DC1.
References
- 1.McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology. 2004;47:787–821. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 2.Tyson J, et al. Mutational spectrum in the cardioauditory syndrome of Jervell and Lange-Nielsen. Hum Genet. 2000;107:499–503. doi: 10.1007/s004390000402. [DOI] [PubMed] [Google Scholar]
- 3.Splawski I, et al. Spectrum of mutations in long-QT syndrome genes KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 4.Wang KW, Goldstein SA. Subunit composition of minK potassium channels. Neuron. 1995;14:1303–1309. doi: 10.1016/0896-6273(95)90277-5. [DOI] [PubMed] [Google Scholar]
- 5.Wang W, Xia J, Kass RS. MinK-KvLQT1 fusion proteins, evidence for multiple stoichiometries of the assembled IsK channel. J Biol Chem. 1998;273:34069–34074. doi: 10.1074/jbc.273.51.34069. [DOI] [PubMed] [Google Scholar]
- 6.Chen H, Kim LA, Rajan S, Xu S, Goldstein SA. Charybdotoxin binding in the IKs pore demonstrates two MinK subunits in each channel complex. Neuron. 2003;40:15–23. doi: 10.1016/s0896-6273(03)00570-1. [DOI] [PubMed] [Google Scholar]
- 7.Tzounopoulos T, Guy HR, Durell S, Adelman JP, Maylie J. min K channels form by assembly of at least 14 subunits. Proc Natl Acad Sci USA. 1995;92:9593–9597. doi: 10.1073/pnas.92.21.9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salata JJ, et al. A novel benzodiazepine that activates cardiac slow delayed rectifier K+ currents. Mol Pharmacol. 1998;54:220–230. doi: 10.1124/mol.54.1.220. [DOI] [PubMed] [Google Scholar]
- 9.Smith C, Phillips M, Miller C. Purification of charybdotoxin, a specific inhibitor of the high-conductance Ca2+-activated K+ channel. J Biol Chem. 1986;261:14607–14613. [PubMed] [Google Scholar]
- 10.MacKinnon R, Cohen SL, Kuo A, Lee A, Chait BT. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 1998;280:106–109. doi: 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein SA, Pheasant DJ, Miller C. The charybdotoxin receptor of a Shaker K+ channel: Peptide and channel residues mediating molecular recognition. Neuron. 1994;12:1377–1388. doi: 10.1016/0896-6273(94)90452-9. [DOI] [PubMed] [Google Scholar]
- 12.Barany G, Merrifield RB. A new amino protecting group removable by reduction. Chemistry of the dithiasuccinoyl (Dts) function. J Am Chem Soc. 1977;99:7363–7365. doi: 10.1021/ja00464a050. [DOI] [PubMed] [Google Scholar]
-
13.Barany G, Schroll AL, Mott AW, Halsrud DA. A general strategy for elaboration of the bithiocarbonyl functionality, –(C
 O)SS–: Application to the synthesis of bis(chlorocarbonyl)disulfane and related derivatives of thiocarbonic acids. J Org Chem. 1983;48:4750–4761. [Google Scholar]
O)SS–: Application to the synthesis of bis(chlorocarbonyl)disulfane and related derivatives of thiocarbonic acids. J Org Chem. 1983;48:4750–4761. [Google Scholar] - 14.Martinez MA, Vega JC. Synthesis of O-alkyl carbonochloridothioates. Synthesis. 1986;9:760–761. [Google Scholar]
- 15.Shimony E, Sun T, Kolmakova-Partensky L, Miller C. Engineering a uniquely reactive thiol into a cysteine-rich peptide. Protein Eng. 1994;7:503–507. doi: 10.1093/protein/7.4.503. [DOI] [PubMed] [Google Scholar]
- 16.Morin TM, Kobertz WR. A derivatized scorpion toxin reveals the functional output of heteromeric KCNQ1-KCNE K+ channel complexes. ACS Chem Biol. 2007;2:469–473. doi: 10.1021/cb700089s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobertz WR, Miller C. K+ channels lacking the ‘tetramerization’ domain: implications for pore structure. Nat Struct Biol. 1999;6:1122–1125. doi: 10.1038/70061. [DOI] [PubMed] [Google Scholar]
- 18.Sanguinetti MC, et al. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKS potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 19.Lu J, Kobertz WR, Deutsch C. Mapping the electrostatic potential within the ribosomal exit tunnel. J Mol Biol. 2007;371:1378–1391. doi: 10.1016/j.jmb.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 20.Blaustein RO. Kinetics of tethering quaternary ammonium compounds to K+ channels. J Gen Physiol. 2002;120:203–216. doi: 10.1085/jgp.20028613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levitsky K, Boersma MD, Ciolli CJ, Belshaw PJ. Exo-mechanism proximity-accelerated alkylations: investigations of linkers, electrophiles and surface mutations in engineered cyclophilin-cyclosporin systems. ChemBioChem. 2005;6:890–899. doi: 10.1002/cbic.200400383. [DOI] [PubMed] [Google Scholar]
- 22.Gordon E, Roepke TK, Abbott GW. Endogenous KCNE subunits govern Kv2.1 K+ channel activation kinetics in Xenopus oocyte studies. Biophys J. 2006;90:1223–1231. doi: 10.1529/biophysj.105.072504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Goldstein SA. Serial perturbation of MinK in IKs implies an α-helical transmembrane span traversing the channel corpus. Biophys J. 2007;93:2332–2340. doi: 10.1529/biophysj.107.109702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tu L, Deutsch C. Evidence for dimerization of dimers in K+ channel assembly. Biophys J. 1999;76:2004–2017. doi: 10.1016/S0006-3495(99)77358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Powers ET, Powers DL. A perspective on mechanisms of protein tetramer formation. Biophys J. 2003;85:3587–3599. doi: 10.1016/S0006-3495(03)74777-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J, Robinson JM, Edwards D, Deutsch C. T1–T1 interactions occur in ER membranes while nascent Kv peptides are still attached to ribosomes. Biochemistry. 2001;40:10934–10946. doi: 10.1021/bi010763e. [DOI] [PubMed] [Google Scholar]
- 27.Schwake M, Jentsch TJ, Friedrich TA. A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 2003;4:76–81. doi: 10.1038/sj.embor.embor715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandrasekhar KD, Bas T, Kobertz WR. KCNE1 subunits require co-assembly with K+ channels for efficient trafficking and cell surface expression. J Biol Chem. 2006;281:40015–40023. doi: 10.1074/jbc.M604398200. [DOI] [PubMed] [Google Scholar]
- 29.Krumerman A, et al. An LQT mutant minK alters KvLQT1 trafficking. Am J Physiol. 2004;286:C1453–C1463. doi: 10.1152/ajpcell.00275.2003. [DOI] [PubMed] [Google Scholar]
- 30.Soreq H, Seidman S. Xenopus oocyte microinjection: From gene to protein. Methods Enzymol. 1992;207:225–265. doi: 10.1016/0076-6879(92)07016-h. [DOI] [PubMed] [Google Scholar]
- 31.Goldin AL. Maintenance of Xenopus laevis and oocyte injection. Methods Enzymol. 1992;207:266–279. doi: 10.1016/0076-6879(92)07017-i. [DOI] [PubMed] [Google Scholar]
- 32.Swanson R, Folander K. In vitro synthesis of RNA for expression of ion channels in Xenopus oocytes. Methods Enzymol. 1992;207:310–319. doi: 10.1016/0076-6879(92)07020-o. [DOI] [PubMed] [Google Scholar]
- 33.Stühmer W. Electrophysiological recording from Xenopus oocytes. Methods Enzymol. 1992;207:319–339. doi: 10.1016/0076-6879(92)07021-f. [DOI] [PubMed] [Google Scholar]
- 34.Krafte DS, Lester HA. Use of stage II–III Xenopus oocytes to study voltage-dependent ion channels. Methods Enzymol. 1992;207:339–345. doi: 10.1016/0076-6879(92)07022-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}