

Abstract
Many actin binding proteins have a modular architecture, and calponin-homology (CH) domains are one such structurally conserved module found in numerous proteins that interact with F-actin. The manner in which CH-domains bind F-actin has been controversial. Using cryo-EM and a single-particle approach to helical reconstruction, we have generated 12-Å-resolution maps of F-actin alone and F-actin decorated with a fragment of human fimbrin (L-plastin) containing tandem CH-domains. The high resolution allows an unambiguous fit of the crystal structure of fimbrin into the map. The interaction between fimbrin ABD2 (actin binding domain 2) and F-actin is different from any interaction previously observed or proposed for tandem CH-domain proteins, showing that the structural conservation of the CH-domains does not lead to a conserved mode of interaction with F-actin. Both the stapling of adjacent actin protomers and the additional closure of the nucleotide binding cleft in F-actin when the fimbrin fragment binds may explain how fimbrin can stabilize actin filaments. A mechanism is proposed where ABD1 of fimbrin becomes activated for binding a second actin filament after ABD2 is bound to a first filament, and this can explain how mutations of residues buried in the interface between ABD2 and ABD1 can rescue temperature-sensitive defects in actin.
Keywords: cytoskeleton, electron microscopy, helical polymers
Almost a dozen proteins involved in cell signaling and cytoskeletal structure have been shown to contain structurally conserved calponin-homology (CH) domains (1). Based on this structural conservation, it has been suggested that many actin binding proteins containing CH-domains interact with F-actin in a similar way (2–4). Fimbrin (called plastin in humans) is involved in actin bundle formation in microvilli and stereocilia, and is composed of two actin binding domains (ABD1 and ABD2), each containing tandem CH-domains. A model of actin-fimbrin bundles (5) assumed that ABD2 interacts with actin filaments in the same manner observed for ABD1 (2). Alternatively, we have previously suggested that CH-containing proteins bind to F-actin polymorphically (6). Biochemical studies suggested that ABD1 and ABD2 of plastin interact with actin in different manners (7). We have also shown that the CH-domain within the eponymous protein of this family, calponin, does not actually contact actin when calponin is bound to F-actin (8).
It has been observed that interactions of many actin binding proteins with actin filaments are highly cooperative and involve structural transitions that propagate along the actin filament (9, 10). The uniform arrays seen in fimbrin–F-actin sheets (5) suggest that there are cooperative mechanisms that dictate how cross-bridging occurs simultaneously with polymerization. Structural changes in actin upon binding of ABD1 to F-actin (11) have been observed and were suggested to explain this cooperativity (5). To evaluate the structural changes in F-actin induced by the binding of fimbrin ABD2, we need to have a high-resolution model for the structure of F-actin alone.
Our understanding of F-actin structure has been hampered by the fact that F-actin cannot be crystallized, given that the symmetry of the filament is incompatible with any crystal symmetry. Studies using EM have been plagued by the fact that the helical symmetry of F-actin is not fixed but allows for a large variability in the angle between adjacent subunits (12). A high-resolution reconstruction of a tightly cross-linked bundle of actin filaments (13) has confirmed the variability in twist as well as tilt (14) of subunits seen for free actin filaments, but every actin subunit in this bundle is bound by an additional protein. X-ray fiber diffraction from oriented gels of F-actin has been used with success (15), but the variable twist of F-actin and the model-building approach needed result in solutions that are not necessarily unique at higher resolution (16).
The single-particle approach, iterative helical real space reconstruction (IHRSR), to electron microscopic three-dimensional reconstruction of helical structures having intrinsic disorder (such as F-actin and RecA) (17) allows us to sort the short segments extracted from such filaments into relatively homogeneous classes. As classes become more homogeneous, they can be reconstructed at higher resolution. In contrast, the heterogeneity in such filaments means that techniques that average over long lengths will suffer from poor resolution. In addition to the twist disorder (12), F-actin possesses structural polymorphism. One form of polymorphism involves tilted subunits that also undergo a substantial opening of the nucleotide binding cleft (14). One of the most variable parts of actin among different crystal structures is subdomain-2 (SD2), and within F-actin, SD2 can be largely disordered (18). We have therefore used a single-particle approach to reconstruct both pure F-actin filaments as well as F-actin filaments decorated with ABD2 of fimbrin. We have achieved an unprecedented resolution for reconstruction of both filaments by using cryo-EM of unstained samples and by image processing to overcome the structural heterogeneity.
Results
Pure F-Actin Reconstruction.
As a control for understanding the complex of ABD2 with F-actin, we have polymerized and imaged pure F-actin under similar conditions to those used for the complex. We found a substantial heterogeneity within such pure actin filaments. In addition to the variable twist (12, 13), F-actin shows other structural heterogeneity. Starting with 28,134 filament segments (each ≈480 Å long) obtained from cryo-EM images (Fig. 1a), we found similar conformational states of F-actin in the frozen-hydrated filaments as had previously been found by using negative stain: ≈31% of segments had subdomain-2 (SD2) disordered (19) (and thus density for this region was missing in the reconstruction), and ≈28% were found in the “tilted” state (14). The largest class (41%) was the “canonical” state, and this class was reconstructed separately (Fig. 1c).
Fig. 1.

Cryo-EM of the pure F-actin control. A typical segment used for image processing (480 Å long) is shown within the red box in the cryo-EM micrograph of pure F-actin (a). Actin filaments decorated with ABD2 of fimbrin (b) are noticeably thicker than pure actin filaments. (Scale bar: 500 Å.) Three-dimensional reconstruction of F-actin in the “canonical” state (c) was generated with the IHRSR method (17), using a subset (n = 10,986) of the total segments collected after sorting to exclude structural variability, such as tilted actin and segments where subdomain 2 was disordered. Actin subdomains are labeled with red numbers. To improve the fitting, the actin monomer was divided into three parts: SD1 (residues 2–33, 70–145, and 336–375), SD2 (34–69), and SD34 (146–335). These parts were fit as rigid bodies, and the corresponding symmetry was imposed to build a model filament (c, blue ribbons). To evaluate the similarity between the model and the actual reconstruction, the atomic model was filtered to 12 Å (d, transparent surface) so that it may be compared with the original volume (c, transparent surface). Our model is in agreement with the contacts between the protomers in the Holmes model (15). Residues that maintain longitudinal contacts (e) are 243–245 (blue) and 322–325 (cyan); 202–204 (yellow) and 286–289 (green); and 41–45 (red) and 166–169,375 (magenta). Lateral contacts are shown in f: 110–112 (magenta) bridges with (195–197 (cyan), while the tip of the hydrophobic loop (266–269, green) would contribute to the bridge of density with a hydrophobic pocket (166, 169, 171, 173, 285, and 289 shown in yellow, and 40–45 and 63–64 shown in red) formed by residues on the opposite strand. Because we have not rotated the loop from the body of the actin subunit, some of the red residues appear far from the loop.
The resolution was determined to be 12.5 Å by using a conservative measure [see supporting information (SI) Fig. 4 and Methods]. The best measure of resolution, however, is the quality of the map itself, and an atomic model of the actin filament filtered to 12 Å is almost indistinguishable from the reconstruction (Fig. 1d). The resolution in our reconstruction is not homogeneous, and this reflects different degrees of disorder in different parts of the actin protomer within the filament. Some parts of the map have better than 12 Å resolution, as can be seen by the helix followed by a loop in actin's SD4 (residues 221–237), which is resolved at 10 Å resolution (SI Fig. 4c).
Actin protomers possess a relatively closed ATP binding cleft in the reconstruction; thus, a crystal structure of G-actin with a closed cleft was used for atomic modeling (20). When the actin subdomains were fit separately (Fig. 1), only small perturbations from the original crystal structure (≈2 Å rmsd) were required to generate a better fit. The conformation of the protomers in our atomic model of the “canonical” state of F-actin is closer to the original model proposed by Holmes and colleagues (15) (rmsd of ≈2.7 Å) than to a refined model (21) (rmsd of ≈2.9 Å). However, because we observe a multiplicity of structural states in F-actin, an atomic model of each structural state will need to be generated, and we are not suggesting that there is one atomic model of the actin filament.
Actin–ABD2 Reconstruction.
Using similar methods, we have been able to generate a three-dimensional reconstruction of F-actin decorated with ABD2 of human fimbrin (L-plastin) at 12-Å resolution (Fig. 2a). Analysis of the twist distribution within these decorated filaments shows that the binding of ABD2 reduces the variability in twist found in naked F-actin (data not shown). The ABD2 fragment that we have used contains the tandem CH3 and CH4 domains (7). The decoration of F-actin is very clear in the original images (Fig. 1b), because ABD2 produces “chevrons” on F-actin that are somewhat reminiscent of myosin S1 decoration of actin. The reconstruction clearly displays these chevrons projecting out from F-actin at an angle of ≈45°. At this resolution, the asymmetry between the two CH-domains within ABD2 is great enough that they can be assigned unambiguously by using a crystal structure for the fimbrin core. Because no crystal structure exists for the plastin ABD2, we have used the crystal structure (22) of the Arabidopsis thaliana fimbrin core (which contains both ABD1 and ABD2). The justification for this is that there is a 38% sequence identity (66% similarity) within ABD2 between human L-plastin and A. thaliana fimbrin, and there are no insertions or deletions of more than three amino acids. The CH3-domain is located at the smaller radius and produces an extensive contact with SD2 of one actin protomer and the lower portion of SD1 of an adjacent protomer within the same long-pitch helical strand (Fig. 2 a and c). The CH4-domain, which is positioned at a higher radius (>55 Å), has only a weak contact with SD1 (residues 22–25) of actin (Fig. 2 a and c). Strikingly, the binding of ABD2 that we observe is different from any binding previously observed or proposed for other tandem CH-domain proteins, such as utrophin (6), α-actinin (23), and even ABD1 of T-fimbrin (2). The differences in binding cannot be due to potential artifacts associated with negatively stained samples (6), because at 20-Å resolution, a reconstruction of negatively stained ABD2-decorated F-actin is indistinguishable from that of the cryo-EM reconstruction (SI Fig. 5).
Fig. 2.

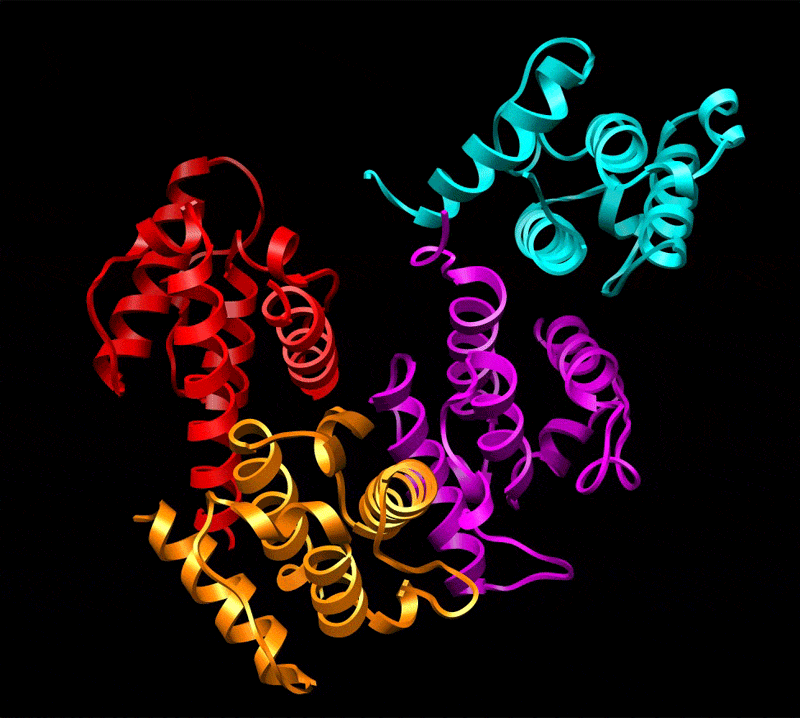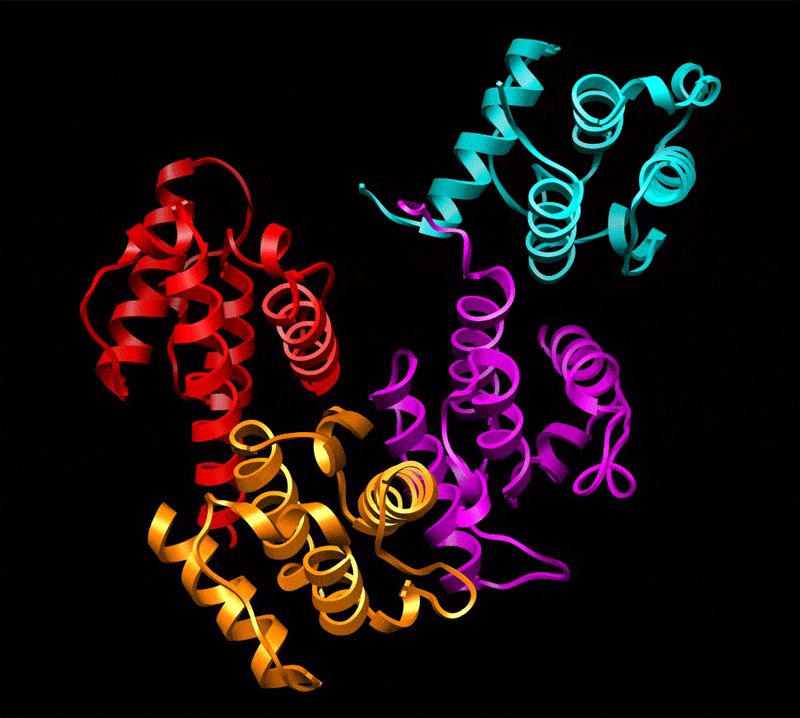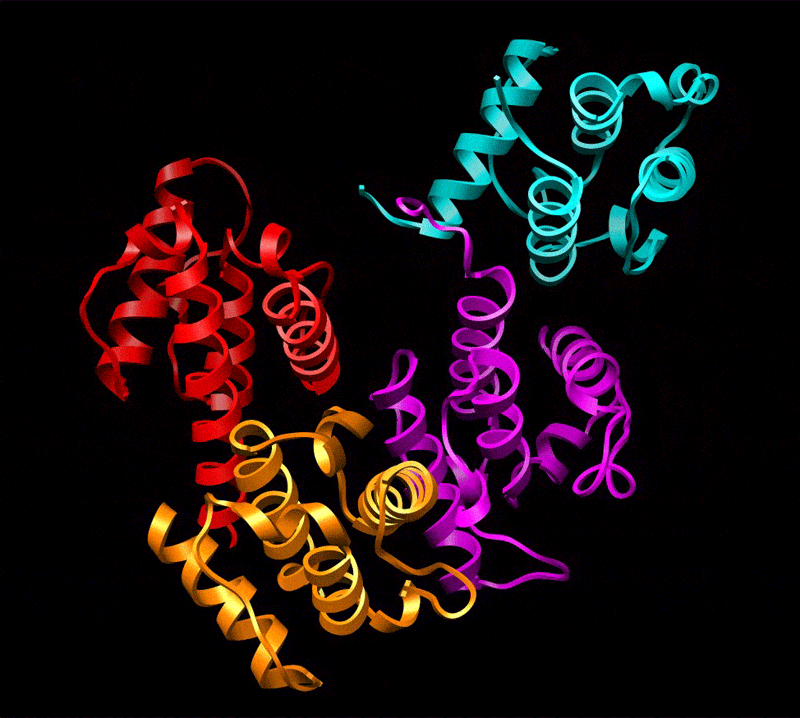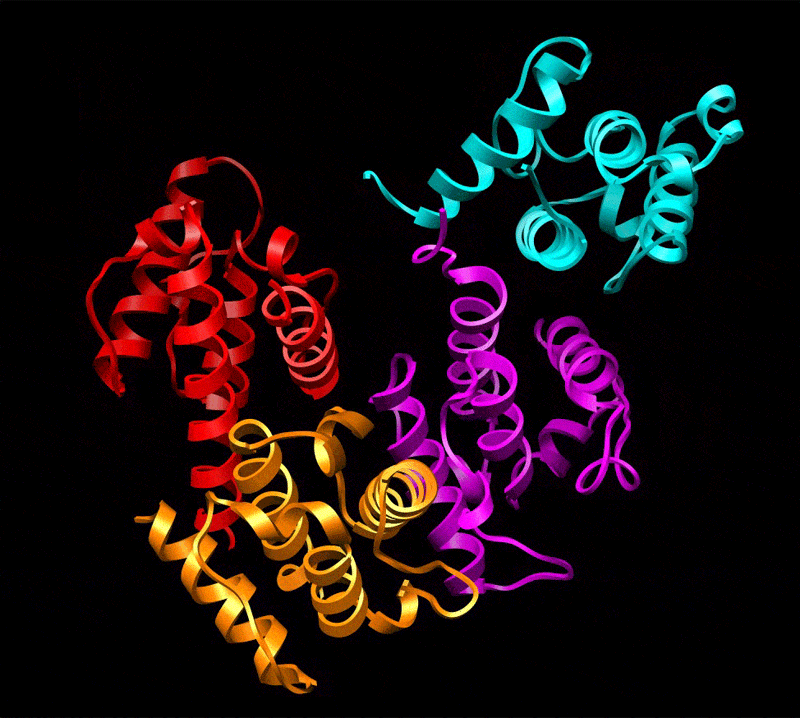
Three-dimensional reconstruction of ABD2–F-actin complex generated from a subset of segments (n = 8,357) chosen to eliminate heterogeneity. The volume has been filtered to 12-Å resolution and is shown with docked atomic models of actin and ABD2 (a). Actin subdomains are labeled with red numbers, and CH domains of ABD2 are labeled in black. Docking of actin was performed as described in Fig. 1, and CH3 and CH4 of ABD2 were docked independently. The atomic model of ABD2–F-actin complex is filtered to 12-Å resolution (b, blue mesh) and is superimposed on the actual map (b, transparent gray surface). Residues of actin shown to interact with fimbrin are shown as green spheres (c and d) and numbered in red (d), and residues of ABD2 important for interacting with F-actin are shown as yellow spheres (c and d) and numbered in black (d). The actin binding core of fimbrin in the conformation found in the crystal (22) is superimposed on the atomic model of actin filament derived from the ABD2–F-actin complex (d). CH-domains are marked as follows: CH1, magenta; CH2, cyan; CH3, red; CH4, orange.
Comparison of the reconstruction and the atomic model (containing actin, CH3, and CH4) filtered to 12 Å shows that the actin part of the map and the CH3 region have an excellent agreement with the atomic model (Fig. 2b). The docking of CH4 is unambiguous at 16-Å resolution and provides an excellent match to the reconstruction at that resolution (data not shown), but the CH4 region of the reconstruction does not extend to 12-Å resolution. Such a steep drop in resolution at higher radius suggests that CH4 is partially disordered. Resolution measurements show that the overall map has a Fourier shell correlation (FSC) of 0.5 at 16.7 Å, whereas radial truncation (removing CH4) improves the resolution to 13.3 Å (SI Fig. 4d). Care was taken in such radial truncation to use Gaussian masks of different sizes for the volumes being compared to prevent the mask function from introducing a spurious correlation. Again, this estimation is overly pessimistic because we have a nearly perfect match between the atomic model and the actual reconstruction at 12-Å resolution when CH4 is excluded. To improve the fit of CH4, we had to move the most distal region of CH4 by 14 Å away from its position in the crystal structure (Fig. 3b). Such a movement is consistent with the fact that there is a 3-Å difference in the position of CH4 between two copies of the fimbrin core (when measured at the same distal loop) within the crystal unit cell (22) (SI Movie 1), suggesting flexibility in the interface between the CH3- and CH4-domains.
Fig. 3.

Perturbations of atomic models were needed to match the reconstruction. The ATP binding cleft of actin in the ABD2–F-actin complex is more closed (a, blue ribbon) than that in the pure F-actin reconstruction (a, green ribbon). This is due to an apparent rotation of subdomain 1 by ≈12° and a shift of subdomain 2 by ≈3.5 Å. To better fit CH4 into the reconstruction (b, green ribbon), it was rotated with respect to CH3 so that the most distal loop was shifted by 14 Å from its position in the crystal (b, red ribbon).
The position of ABD2 on F-actin in our atomic model is supported by mutagenesis data. All actin residues (K50, D51, K61, E99, and E100) shown to interact with fimbrin (24) are at the interface with ABD2 of fimbrin (Fig. 2 c and d). Interestingly, it was shown that these residues are involved in interaction with both ABDs of fimbrin. Residues R395, K461, F539, and K589 within ABD2 of Arabidopsis fimbrin [using the alignment of Klein et al. (22)] are expected to affect the binding of fimbrin to F-actin, based on mutations in the corresponding residues within yeast fimbrin (A414, D484, F560, and K610, respectively) (25). In our reconstruction, K461 interacts with the upper portion of SD2 of actin (Fig. 2c, red arrows), whereas F539 forms a bridge of density with actin residues 22–25 (Fig. 2c, green arrows). Mutation of R395 may introduce structural alterations in the actin binding site of ABD2 because of its proximity to K461 (Fig. 2d). K589, on the other hand, is located at the most distal tip of CH4 and cannot be involved in any direct interaction with actin.
We can take the entire fimbrin core crystal structure (containing CH1, CH2, CH3, and CH4) and dock it to F-actin based on the binding of CH3 and CH4 that we have established. It can be seen when this is done that K589 (Fig. 2d, black asterisk) is buried at the interface between CH1 (Fig. 2d, magenta ribbons) and CH4 (Fig. 2d, orange ribbons) of fimbrin. Because our atomic model of the ABD2–F-actin complex shows that K589 is not involved in the interaction with F-actin, the most likely explanation for the yeast K610 (A. thaliana K589) mutagenesis result is the proposal that rearrangements of the two ABDs of fimbrin are important for bundling (22). Thus, A. thaliana K589 may govern a conformational change within ABD1 (CH1 and CH2) to make it active in the binding to a second actin filament. When a second actin filament is placed 120 Å away from the filament to which ABD2 is bound (5), CH1 of ABD1 is in close proximity to the binding site on actin previously described (2, 11) for ABD1.
Another important conclusion derived from our reconstruction is that the ATP-binding cleft of actin is more closed than in the control undecorated actin filament (Fig. 3a). In fact, the cleft in actin is more closed than in all existing crystals because we have needed to shift subdomain 2 inwards by 3.5 Å and rotate subdomain 1 by ≈12° with respect to their positions in G-actin (20). The opening of the cleft has been shown to destabilize F-actin (26), and the potential stabilization of F-actin by a closure of the cleft is in excellent agreement with biochemical data that either full-sized fimbrin or ABD2 alone can stabilize actin filaments (7, 27). An additional means of stabilization of F-actin may arise from the fact that ABD2 is bound to two adjacent protomers along the same long-pitch helical strand in F-actin, and this stapling of adjacent protomers may have an important effect on filament stability. Because it is expected that bundling of actin filaments by fimbrin proceeds simultaneously with polymerization of these filaments (5), the stabilization of nascent F-actin filaments by fimbrin may be an important element in the coordination of polymerization with bundling.
Actin–ABD1 Complexes.
The docking of the fimbrin core crystal structure to an ABD2-decorated F-actin filament (Fig. 2d) provides a starting point for understanding cross-linking of adjacent actin filaments by fimbrin. The obvious question is whether ABD1 (containing CH1 and CH2) can bind to a second filament that is 120 Å away from the filament to which ABD2 is bound while in the same conformation seen in the crystal structure of the fimbrin core (containing both ABD1 and ABD2). We have therefore attempted to decorate isolated F-actin filaments with ABD1 alone to perform a similar analysis to that used for the ABD2-decorated filaments. Using the estimated dissociation constant of 0.34 μM for the binding of ABD1 to F-actin (7), we incubated 1 μM F-actin with 2–4 μM ABD1 expecting to see saturated binding. Image analysis and IHRSR reconstruction from negatively stained specimens showed essentially naked F-actin filaments. Using 1 μM F-actin with 10–15 μM ABD1, we were able to obtain substantial decoration of F-actin filaments by ABD1 (SI Fig. 6a), but the overall reconstruction generated (SI Fig. 6c) was uninterpretable in terms of what was actin and what was ABD1. This suggested to us that this overall reconstruction was largely artifactual because of the misalignment of both actin and ABD1 in the process of averaging together segments that are structurally diverse and inhomogeneous. Because we have previously shown that polymorphic binding to F-actin can be detected and different modes can be separated (6, 8, 28–31), considerable effort was invested in attempting to separate these segments into homogeneous classes. All such efforts failed.
The simplest explanation is that unlike ABD2, where the tandem CH-domains bind to F-actin in a compact conformation similar to that seen in the fimbrin core crystal structure, the tandem CH-domains in ABD1 can separate, resulting in a multiplicity of different conformations when bound to F-actin. This is consistent with what has been seen in the fimbrin core crystal structures, where two copies of the core (unrelated by crystal symmetry) are present in each asymmetric unit. Although there is a relatively fixed interaction between CH3 and CH4 when comparing these two molecules within the asymmetric unit (SI Movie 1), there is no conservation of the CH1–CH2 interface. The inability to separate out distinct modes of binding for these CH-domains, when such modes could be separated for the tandem CH-domains present in utrophin (6, 30), suggests that there is little cooperativity in the modes of fimbrin ABD1 binding to F-actin within any given segment, whereas substantial cooperativity must exist in the case of utrophin.
Discussion
There has been an extensive debate in the literature about whether tandem CH-domains bind to F-actin in an “open” or “closed” conformation, and whether the interaction of all CH-domains with F-actin is conserved (2, 3, 5, 6, 8, 30, 32–34). The structural conservation that has been seen among CH-domains led to the reasonable suggestion that there would be a conserved functional interaction between CH-domains and actin. However, we have shown that the CH-domain within calponin is never actually in contact with actin when calponin is bound to F-actin (8), suggesting that there may have been a divergence between structure and function in the evolution of proteins containing CH-domains. We have also examined the tandem CH-domains in utrophin and shown that the binding to F-actin is polymorphic because the two CH-domains do not maintain a single fixed interaction with each other when attached to F-actin (6, 30). We now show that in contrast to the polymorphism in the utrophin tandem CH-domains, the tandem CH-domains in fimbrin ABD2 bind F-actin in a rather fixed manner.
The stoichiometric decoration of F-actin by ABD2 is reminiscent of the decoration of F-actin by myosin S1, but we have been able to achieve a higher resolution in reconstructing this complex than has previously been achieved for any acto-S1 reconstruction (35–37). An interesting question that we cannot answer at this point is whether the higher resolution is due to the fact that the actin–ABD2 complex is more ordered than acto-S1, with less variability in twist and polymorphism within F-actin, or whether the improvement comes from the image processing scheme that we use (17). The reduction in variability of twist when ABD2 is bound, and the closure of actin's nucleotide binding pocket, may be coupled. Bacterial ParM is a structural homolog of actin (38) that has a very similar fold with two major domains surrounding a nucleotide binding pocket. We have suggested that the much larger variability in twist present in ParM filaments than exists in F-actin is due to the much greater opening of the nucleotide binding pocket in ParM than occurs in F-actin (39).
In contrast to the fixed and stoichiometric binding of ABD2 to F-actin, the binding of ABD1 alone is polymorphic and partial. In fact, we have found that the binding is much more polymorphic than the binding of the utrophin tandem CH-domains to F-actin (6), and we have been unable to reconstruct any ABD1–actin complexes. The partial decoration of F-actin by ABD1 that we have seen is very similar to the partial occupancy that has been reported previously (2, 11). However, in those previous reports it was assumed that ABD1 bound in a single mode, albeit at low occupancy, and so a pseudo-atomic model of ABD1 bound to F-actin was constructed from those data (2). Previous evidence for the polymorphic nature of the ABD1 binding comes from the fact that when two different methods were used to construct a difference map between the ABD1–actin complex and pure F-actin, two different results were obtained (2, 40). The partial and disordered binding of ABD1 to F-actin, in contrast to the stoichiometric and ordered binding of ABD2, is consistent with the fact that ABD2 significantly protects actin filaments from depolymerization whereas ABD1 provides very little protection (7).
What do our present results tell us about fimbrin's role in bundling actin filaments? The inability to define the interaction between ABD1 and actin means that some speculation is required, but the observations presented here are suggestive. We propose that ABD2 is bound first to one F-actin filament, which induces the closure of the actin cleft that we observe. As with many structural perturbations within F-actin, we expect that there is a cooperativity that results in the stabilization of a region in the nascent actin filament by the transmission of this conformational change to adjacent subunits within the same filament (41, 42). In addition, the binding of ABD2 to actin “activates” ABD1 so that it may bind in a more ordered manner than that observed for free ABD1 to another actin filament with high affinity. The fact that mutagenesis of the residue (K610 in yeast) equivalent to K589 in Arabidopsis fimbrin ABD2 (Fig. 2d) can suppress a temperature-sensitive mutation in yeast actin is quite consistent with our picture that the binding of ABD2 to one F-actin filament may be needed to activate ABD1 to bind to an adjacent filament. The shift of CH4 that we observe from the fimbrin core crystal structure when ABD2 is bound to F-actin might be part of the mechanism for the activation of ABD1. Alternatively, mutation of K610 in yeast fimbrin (K589 in A. thaliana fimbrin) may change the tightly packed interface seen between CH4 and CH1 in the fimbrin core structure and thus activate ABD1 (CH1 and CH2) in the absence of ABD2 binding to F-actin. Thus, the yeast K610R mutation may increase the overall affinity between actin and fimbrin (25), which will rescue a defect in actin.
As databases of gene sequences grow exponentially, more tools are needed for annotation to provide biological meaning. Many, if not most, proteins have been found to have a modular structure, and the similarity of modules is being used to assign similar functions to common modules in different proteins. We show here that the structural similarity among CH1, CH2, CH3, and CH4 in the fimbrin core does not necessarily dictate similar properties or functions. In fact, the presence of CH-domains in proteins only directly tells us about the evolutionary origins of this module and not about the function of this module in any protein of interest. Further detailed studies of fimbrin, including a high-resolution structure of the ABD1–F-actin complex, are still required to build an atomic model of actin–fimbrin bundles.
Methods
Sample Preparation and Microscopy.
Actin (2.5 μM) was polymerized in 20 mM Mops buffer (pH 7.2), 50 mM KCl, 1 mM MgCl2, 0.5 mM EGTA, 0.5 mM DTT, and 0.2 mM ATP for 2.5 h before grids were prepared. Human L-plastin fragments ABD1 and ABD2 were prepared as described in ref. 7. ABD2 was used at 2–4 μM concentrations when incubated with F-actin at 1 μM concentration. All cryo-EM was done on a Tecnai F20 FEG microscope operated at 200 keV at a magnification of ×50,000, and negatively stained samples were imaged on a Tecnai T12 microscope operated at 120 keV at a magnification of ×30,000.
Image Processing.
The SPIDER software package (43) was used for most image processing, but the EMAN package (44) was used to determine the defocus values in the micrographs and to extract filament images from micrographs.
Pure F-actin.
A Nikon COOLPIX 8000 scanner was used to digitize 74 cryo-EM micrographs having a defocus range from −1.5 to −3.8 μm at a raster of 2.38 Å per pixel. Initial correction for the contrast transfer function (CTF) was made by multiplying each image by its theoretical CTF. From these CTF-corrected images, 28,134 short (200 pixels long) overlapping segments were extracted. Three model volumes were created by using crystal structures of G-actin (20) having actin protomers in the “canonical” state, with missing SD2, and finally in the “tilted” state (14). These volumes were scaled to 4.76 Å per pixel and projected into 100 × 100-pixel images with an azimuthal rotational increment of 4°, generating 270 reference projections (3 × 90). The F-actin segments were down-sampled to 4.76 Å per pixel and cross-correlated with the 270 reference projections. To check the quality of the sorting, the model volumes were azimuthally rotated by 2° and a second set of reference images was created and used for a new cross-correlation sorting of the image segments. Only 25,567 F-actin segments were assigned to the same class in both cross-correlations, and these were used for the final reconstruction. A set of 10,986 segments (sampled at 2.38 Å per pixel), selected as similar to the “canonical” state of F-actin, was reconstructed with the IHRSR method (17) and after 30 iterations yielded a stable solution of 166.6°/27.6 Å. The volume was corrected for the CTF (because images had been effectively multiplied by the CTF twice, once by the microscope and once by us) by using a Wiener filter assuming that the signal-to-noise ratio in the volume was very large, and a range of negative B-factors was used to amplify high frequencies in the reconstruction that were damped by the envelope function of the microscope. The most reasonable volume was used to generate an atomic model. The spherically averaged power spectra of the actual reconstruction and the atomic model were then compared and used to adjust structure factor amplitudes in the reconstruction in a resolution-dependent manner, and this new volume was used for the final modeling. UCSF Chimera software (45) was used to fit crystal structures into the experimental maps. Atomic coordinates from crystal structures were converted to density maps, and these were filtered to the resolution of the experimental map and docked both manually and by using the Chimera automated procedure. Both approaches gave the same result.
The conservative FSC = 0.5 criterion was used for resolution determination. A widely used approach has been to split an aligned data set into two halves yielding two volumes for FSC comparison, but this method can yield an overly optimistic resolution value due to alignment of noise (46, 47). We have modified the conventional approach by dividing the images into two sets and then using the IHRSR procedure on these two sets starting each from a different helical symmetry. The two structures converge to a common symmetry (SI Fig. 4a), and the resultant volumes do not have noise aligned to a common reference. However, the smaller number of images present in each half data set suggests that the resolution of each half set will be less than the resolution of the combined reconstruction under conditions where the resolution is limited by the number of particles. Thus, the 12.5-Å resolution that we measure by this method in our undecorated F-actin map (SI Fig. 4b) is the most pessimistic resolution estimation.
ABD2–F-actin complex.
A set of 65 micrographs (defocus range of −1.3 to −4.5 μm) was digitized as described for pure F-actin, corrected for CTF, and used to collect 22,210 short segments (200 pixels long). The overall reconstruction suggested that all filaments were fully occupied because the full density of ABD2 was visible without any prior sorting. However, the resolution of the overall reconstruction was ≈18 Å. To improve the resolution, three model volumes having actin protomers in different structural states were created, but ABD2 was attached to each as observed in the overall reconstruction. In the first model, the ATP-binding cleft was closed; in the second model, it was wide open; and in the third model, actin protomers were in the “tilted” state. The model volumes were used to create a reference set of images as described earlier. The majority of segments yielded the best correlation with the model having the ATP binding cleft closed (n = 9,051), and images that had reasonably small in-plane rotation angles (<10°) were used for the reconstruction (n = 8,357). These segments were then reconstructed by using a featureless solid cylinder as an initial starting model. After 60 iterations of the IHRSR procedure, the set yielded a stable solution of 166.5°/27.8 Å. The same solid cylinder was used as an initial reference for reconstructing the other sets. The reconstruction of segments that possessed the best correlation with the model having the open ATP binding cleft showed that CH4 of fimbrin ABD2 was less ordered, and the occupancy was somewhat lower than that in the first set. The third set yielded a reconstruction suggesting that mostly noisy and disordered filaments were assigned to that class. Correction for the CTF in the final volume and model building was done similarly to the naked F-actin map. The coordinates for the ABD2–actin model have been deposited in the Protein Data Bank (PDB ID code 3BYH).
Negatively stained ABD2–F-actin complex.
We collected 8,960 segments of F-actin decorated with ABD2 from negatively stained samples, each 100 pixels long and sampled at 4.16 Å per pixel. This set was reconstructed by using the IHRSR approach and after 60 iterations yielded a symmetry of 166°/27.7 Å.
Negatively stained ABD1–F-actin complex.
A set of 4,210 segments (100 pixels long, 4.16 Å per pixel) was used in the IHRSR procedure and converged to a symmetry of 165.3°/29.5 Å. Not only was this symmetry unexpected (given the fixed axial rise in F-actin of ≈27.3 Å), but the reconstruction was uninterpretable in terms of what was actin and what was ABD1. Attempts to use reconstructions of F-actin decorated with utrophin (6), α-actinin (unpublished data), and ABD2 of fimbrin to sort this set into more homogeneous classes did not succeed.
Supplementary Material
ACKNOWLEDGMENTS.
This work was supported by National Institutes of Health Grant GM081303 (to E.H.E.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3BYH).
This article contains supporting information online at www.pnas.org/cgi/content/full/0708667105/DC1.
References
- 1.Gimona M, Djinovic-Carugo K, Kranewitter WJ, Winder SJ. FEBS Lett. 2002;513:98–106. doi: 10.1016/s0014-5793(01)03240-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanein D, Volkmann N, Goldsmith S, Michon AM, Lehman W, Craig R, DeRosier D, Almo S, Matsudaira P. Nat Struct Biol. 1998;5:787–792. doi: 10.1038/1828. [DOI] [PubMed] [Google Scholar]
- 3.Lehman W, Craig R, Kendrick-Jones J, Sutherland-Smith AJ. J Muscle Res Cell Motil. 2004;25:351–358. doi: 10.1007/s10974-004-0690-7. [DOI] [PubMed] [Google Scholar]
- 4.Bramham J, Hodgkinson JL, Smith BO, Uhrin D, Barlow PN, Winder SJ. Structure. 2002;10:249–258. doi: 10.1016/s0969-2126(02)00703-7. [DOI] [PubMed] [Google Scholar]
- 5.Volkmann N, DeRosier D, Matsudaira P, Hanein D. J Cell Biol. 2001;153:947–956. doi: 10.1083/jcb.153.5.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galkin VE, Orlova A, VanLoock MS, Rybakova IN, Ervasti JM, Egelman EH. J Cell Biol. 2002;157:243–251. doi: 10.1083/jcb.200111097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lebart MC, Hubert F, Boiteau C, Venteo S, Roustan C, Benyamin Y. Biochemistry. 2004;43:2428–2437. doi: 10.1021/bi030151p. [DOI] [PubMed] [Google Scholar]
- 8.Galkin VE, Orlova A, Fattoum A, Walsh MP, Egelman EH. J Mol Biol. 2006;359:478–485. doi: 10.1016/j.jmb.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 9.Orlova A, Egelman EH. J Mol Biol. 1997;265:469–474. doi: 10.1006/jmbi.1996.0761. [DOI] [PubMed] [Google Scholar]
- 10.Orlova A, Rybakova IN, Prochniewicz E, Thomas DD, Ervasti JM, Egelman EH. Biophys J. 2001;80:1926–1931. doi: 10.1016/S0006-3495(01)76162-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanein D, Matsudaira P, DeRosier DJ. J Cell Biol. 1997;139:387–396. doi: 10.1083/jcb.139.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egelman EH, Francis N, DeRosier DJ. Nature. 1982;298:131–135. doi: 10.1038/298131a0. [DOI] [PubMed] [Google Scholar]
- 13.Schmid MF, Sherman MB, Matsudaira P, Chiu W. Nature. 2004;431:104–107. doi: 10.1038/nature02881. [DOI] [PubMed] [Google Scholar]
- 14.Galkin VE, VanLoock MS, Orlova A, Egelman EH. Curr Biol. 2002;12:570–575. doi: 10.1016/s0960-9822(02)00742-x. [DOI] [PubMed] [Google Scholar]
- 15.Holmes KC, Popp D, Gebhard W, Kabsch W. Nature. 1990;347:44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- 16.Lorenz M, Popp D, Holmes KC. J Mol Biol. 1993;234:826–836. doi: 10.1006/jmbi.1993.1628. [DOI] [PubMed] [Google Scholar]
- 17.Egelman EH. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- 18.Orlova A, Egelman EH. J Mol Biol. 1992;227:1043–1053. doi: 10.1016/0022-2836(92)90520-t. [DOI] [PubMed] [Google Scholar]
- 19.Orlova A, Egelman EH. J Mol Biol. 1993;232:334–341. doi: 10.1006/jmbi.1993.1393. [DOI] [PubMed] [Google Scholar]
- 20.Schutt CE, Myslik JC, Rozycki MD, Goonesekere NCW, Lindberg U. Nature. 1993;365:810–816. doi: 10.1038/365810a0. [DOI] [PubMed] [Google Scholar]
- 21.Tirion MM, ben Avraham D. J Mol Biol. 1993;230:186–195. doi: 10.1006/jmbi.1993.1135. [DOI] [PubMed] [Google Scholar]
- 22.Klein MG, Shi W, Ramagopal U, Tseng Y, Wirtz D, Kovar DR, Staiger CJ, Almo SC. Structure. 2004;12:999–1013. doi: 10.1016/j.str.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 23.McGough A, Way M, DeRosier D. J Cell Biol. 1994;126:433–443. doi: 10.1083/jcb.126.2.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Honts JE, Sandrock TS, Brower SM, O'Dell JL, Adams AE. J Cell Biol. 1994;126:413–422. doi: 10.1083/jcb.126.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brower SM, Honts JE, Adams AE. Genetics. 1995;140:91–101. doi: 10.1093/genetics/140.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belmont LD, Orlova A, Drubin DG, Egelman EH. Proc Natl Acad Sci USA. 1999;96:29–34. doi: 10.1073/pnas.96.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng D, Marner J, Rubenstein PA. J Biol Chem. 1999;274:35873–35880. doi: 10.1074/jbc.274.50.35873. [DOI] [PubMed] [Google Scholar]
- 28.Galkin VE, Orlova A, Lukoyanova N, Wriggers W, Egelman EH. J Cell Biol. 2001;153:75–86. doi: 10.1083/jcb.153.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukoyanova N, VanLoock MS, Orlova A, Galkin VE, Wang K, Egelman EH. Cur Biol. 2002;12:383–388. doi: 10.1016/s0960-9822(02)00678-4. [DOI] [PubMed] [Google Scholar]
- 30.Galkin VE, Orlova A, VanLoock MS, Egelman EH. J Mol Biol. 2003;331:967–972. doi: 10.1016/s0022-2836(03)00842-8. [DOI] [PubMed] [Google Scholar]
- 31.Galkin VE, Orlova A, Koleske AJ, Egelman EH. J Mol Biol. 2005;346:565–575. doi: 10.1016/j.jmb.2004.11.078. [DOI] [PubMed] [Google Scholar]
- 32.Moores CA, Keep NH, Kendrick-Jones J. J Mol Biol. 2000;297:465–480. doi: 10.1006/jmbi.2000.3583. [DOI] [PubMed] [Google Scholar]
- 33.Sutherland-Smith AJ, Moores CA, Norwood FL, Hatch V, Craig R, Kendrick-Jones J, Lehman W. J Mol Biol. 2003;329:15–33. doi: 10.1016/s0022-2836(03)00422-4. [DOI] [PubMed] [Google Scholar]
- 34.Bramham J, Hodgkinson JL, Smith BO, Uhrin D, Barlow PN, Winder SJ. Structure. 2002;10:249–258. doi: 10.1016/s0969-2126(02)00703-7. [DOI] [PubMed] [Google Scholar]
- 35.Holmes KC, Angert I, Kull FJ, Jahn W, Schroder RR. Nature. 2003;425:423–427. doi: 10.1038/nature02005. [DOI] [PubMed] [Google Scholar]
- 36.Jontes JD, Ostap EM, Pollard TD, Milligan RA. J Cell Biol. 1998;141:155–162. doi: 10.1083/jcb.141.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volkmann N, Liu H, Hazelwood L, Krementsova EB, Lowey S, Trybus KM, Hanein D. Mol Cell. 2005;19:595–605. doi: 10.1016/j.molcel.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 38.van den Ent F, Moller-Jensen J, Amos LA, Gerdes K, Lowe J. EMBO J. 2002;21:6935–6943. doi: 10.1093/emboj/cdf672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orlova A, Garner EC, Galkin VE, Heuser J, Mullins RD, Egelman EH. Nat Struct Mol Biol. 2007;14:921–926. doi: 10.1038/nsmb1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rost LE, Hanein D, DeRosier DJ. Ultramicroscopy. 1998;72:187–197. doi: 10.1016/s0304-3991(98)00017-5. [DOI] [PubMed] [Google Scholar]
- 41.Orlova A, Prochniewicz E, Egelman EH. J Mol Biol. 1995;245:598–607. doi: 10.1006/jmbi.1994.0049. [DOI] [PubMed] [Google Scholar]
- 42.Bobkov AA, Muhlrad A, Pavlov DA, Kokabi K, Yilmaz A, Reisler E. J Mol Biol. 2006;356:325–334. doi: 10.1016/j.jmb.2005.11.072. [DOI] [PubMed] [Google Scholar]
- 43.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 44.Ludtke SJ, Baldwin PR, Chiu W. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 45.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Grigorieff N. Acta Crystallogr D. 2000;56:1270–1277. doi: 10.1107/s0907444900009549. [DOI] [PubMed] [Google Scholar]
- 47.Yang S, Yu X, Galkin VE, Egelman EH. J Struct Biol. 2003;144:162–171. doi: 10.1016/j.jsb.2003.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}