

Abstract
Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most common cause of Parkinson's disease (PD). LRRK2 contains a Ras of complex proteins (ROC) domain that may act as a GTPase to regulate its protein kinase activity. The structure of ROC and the mechanism(s) by which it regulates kinase activity are not known. Here, we report the crystal structure of the LRRK2 ROC domain in complex with GDP-Mg2+ at 2.0-Å resolution. The structure displays a dimeric fold generated by extensive domain-swapping, resulting in a pair of active sites constructed with essential functional groups contributed from both monomers. Two PD-associated pathogenic residues, R1441 and I1371, are located at the interface of two monomers and provide exquisite interactions to stabilize the ROC dimer. The structure demonstrates that loss of stabilizing forces in the ROC dimer is likely related to decreased GTPase activity resulting from mutations at these sites. Our data suggest that the ROC domain may regulate LRRK2 kinase activity as a dimer, possibly via the C-terminal of ROC (COR) domain as a molecular hinge. The structure of the LRRK2 ROC domain also represents a signature from a previously undescribed class of GTPases from complex proteins and results may provide a unique molecular target for therapeutics in PD.
Parkinson's disease (PD) is a common, age-related neurodegenerative disorder for which only symptomatic treatment is available. The etiology of PD is poorly understood, but over the past decade it has become clear that there are rare families with Mendelian inheritance (1). Of the genes that cause PD, dominantly inherited mutations in leucine-rich repeat kinase 2 (LRRK2) are numerically the most common and account for an appreciable fraction of apparently sporadic PD (reviewed in ref. 2). LRRK2 encodes a large (2,527-aa) multidomain protein originally identified as a unique kinase with leucine-rich repeats. LRRK2 is a member of a superfamily of proteins, named ROCO, that includes at least three other human proteins: leucine-rich repeat kinase 1 (LRRK1), death-associated protein kinase (DAPK1), and malignant fibrous histiocytoma-amplified sequences with leucine-rich tandem repeat-1 (MASL1) (3). A unique feature of all ROCO proteins is a 200- to 250-aa Ras-related GTPase or Ras of complex proteins (ROC) domain, followed by a C-terminal of ROC (COR) domain immediately before the kinase domain.
Both LRRK2 and LRRK1 have been shown to be active protein kinases in vitro (4–7), and some mutations are found in the kinase domain. These mutations generally increase kinase activity, although there are some discrepancies in different studies as to whether all mutations increase kinase function (4, 6, 8–11). However, the kinase activity of LRRK2 is required for the ability of the mutant protein to cause neuronal damage, at least in cell culture models (5, 10), suggesting that kinase inhibitors may represent a therapeutic avenue for PD.
Although the kinase domain therefore is important in understanding pathogenesis, mutations also are found in other regions of the protein, and understanding why these mutations cause disease is difficult. Mutations at position R1441 in LRRK2 (R1441C, R1441G, and R1441H) are all pathogenic for PD (12, 13), and another variant also is reported at the adjacent residue [A1442P (14)] in the ROC domain. Several studies have shown that GTP binding at the ROC domain regulates kinase activity (4, 7, 10, 15, 16). In other recent data, ROC domain mutants have been shown to have lower GTPase activity than wild-type LRRK2 (15, 17, 18), although it is likely that the GTPase activity is quite low as other groups did not find measurable GTPase activity in full-length LRRK2 (11) unless the protein was mutated to residues that are similar to Ras (16). This finding suggests that mutations outside of the kinase domain may indirectly impact enzyme function but leave open the question of how these mutations affect the structure of the protein and, in turn, how this impacts communication between the GTP-binding region and the kinase domain.
In the current study, we determined the structure of the ROC domain of LRRK2 in complex with GDP-Mg2+ at 2.0-Å resolution. We have tested several key predictions from this structural information that impact protein function. The structure suggests a molecular basis for pathogenic mutations in the ROC domain of human LRRK2, which lead to PD. Our data also suggest that the COR domain of LRRK2 may serve as a molecular hinge to convey signal from the ROC domain to the kinase domain through a GTP/GDP-bound cycle.
Results
The LRRK2 ROC Domain Has a Unique Dimeric Structure.
The structure of the LRRK2 ROC domain displays a unique homodimer with extensive domain-swapping (Fig. 1A). Each monomer contains five α-helices and six β-strands with loops in between, displaying three subdomains: head, neck, and body. The head domain consists of β1, α1, β2, and β3. The loop between α2 and β2, as well as that between β2 and β3, are disordered. The neck domain is composed mainly of a bent helix, α2. The body domain is structured with β4, α3, β5, α4, β6, and α5 with defined loops in between (Fig. 1 A and B).
Fig. 1.

The unique dimeric ROC GTPase. (A) Stereoview of the domain-swapped dimer. The two individual monomers are shown in yellow and green. The GDP-Mg2+ ligands are shown in ball-and-stick format. (B) Ribbon representation of a single monomer. The three head, neck, and body subdomains are indicated, along with the labeled secondary structures. The P-loop, G3/Switch II, and G4 and G5 loops are indicated in orange, pink, red, and cyan, respectively. The disordered G2 loop is shown as a black dotted curve. (C) Surface representation highlighting the GDP-Mg2+ binding pocket on the surface of the dimer that is contributed from both monomers. The pair of functional units are shown as ROCs1 and ROCs2, respectively.
The head domain and the first half of the neck domain from one monomer pair with the body domain from the other, forming two compact units (ROCs1 and ROCs2; Fig. 1C), each with a six-stranded β-sheet (parallel except the β2 strand) that is sandwiched by five α-helices. The two monomers are related to each other via a pseudo-twofold axis. Through water-mediated hydrogen bonding, the two six-stranded β-sheets are linked together, forming a 12-stranded β-saddle. The two monomers adopt near-identical conformation with a 0.33-Å Cα root-mean-square deviation (rmsd) on 153 residues superimposed. The dimer interactions are stabilized through extensive hydrogen bondings and hydrophobic interactions, burying ≈5,888 Å2 of solvent-accessible surface area (Fig. 1C). A pair of ligand-binding sites were identified on the surface of the ROC dimer, with well defined GDP molecule together with a Mg2+ metal ion in a groove at each site. Therefore, the current domain-swapped structure serves as one functional unit, displaying unique nucleotide and Mg2+ binding sites at the dimer interface with contributions from both monomers.
A fold homology search using the DALI server (19) with one of the noncontiguous catalytic domains (ROCs1 via domain-swapping; Fig. 1) of ROC revealed that the overall fold of ROC has the highest similarity to the G domain of elongation factor EF-Tu (20) (PDB ID 1efc), a Rho-related GTP-binding protein (PDB ID 2cls), and p21Ras (21) (PDB ID 1ctq), even though the shared amino acid sequence identity is <24% in each pairwise comparison. The rmsd of 146 equivalent Cα atoms between ROCs1 and the top Z-scoring Escherichia coli elongation factor EF-Tu (PDB ID 1efc, Z score = 16.5) is ≈2.3 Å. All of these proteins share a canonical GTPase fold (21–23) [supporting information (SI) Fig. 5]. However, the catalytic core of the LRRK2-ROC domain adopts an unusual noncontiguous topology because of domain-swapping. The β1, P-loop, α1, β2, and β3 from the head domain, G3/switch II loop, and the following first half of the α2 from the neck domain are contributed from one monomer peptide. Additional key components that represent the canonical GTPase fold come from the body domain of the second monomer peptide (β4, α3, β5, α4, β6, α5, G4, and G5 loops). As the first member of the ROCO superfamily (3) to be reported, this structure could serve as a fingerprint for the whole class of ROC GTPases.
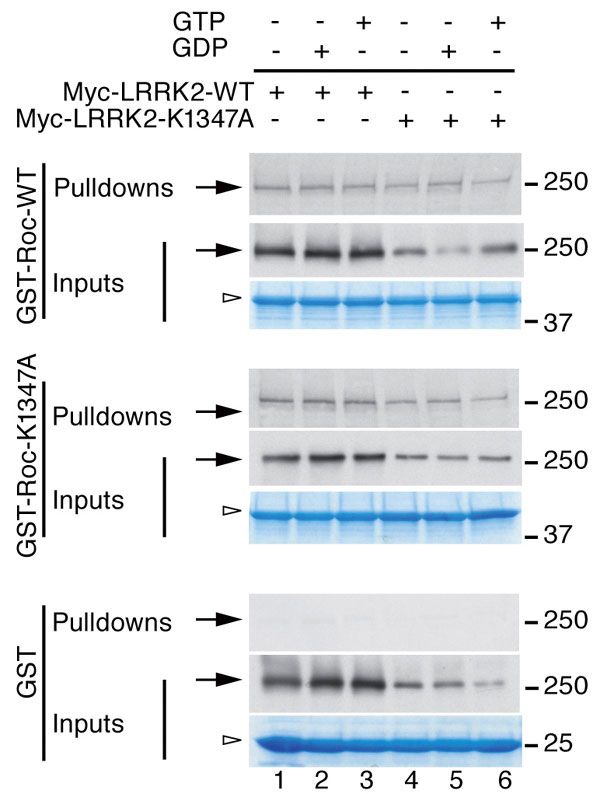
To verify that LRRK2 forms a dimer with contributions from residues within the ROC domain, we used fusion proteins of GST with the ROC domain of LRRK2 to pull down full-length LRRK2 expressed in mammalian cells. We found that the ROC domain alone was capable of interacting with full-length LRRK2 (SI Fig. 6). Nucleotide binding was not necessary for LRRK2 self-interaction because similar results were seen in the presence or absence of GDP or GTP. In addition, mutating a key residue in the nucleotide-binding pocket to prevent GTP binding (K1347A; refs. 4, 10, 15, and 16; see also SI Fig. 5) did not disrupt the interaction. These data support the structural model that the dimer is formed largely by interactions outside of the nucleotide pocket, although GTP/GDP binding could further stabilize the protein as demonstrated in other GTPases (22, 23).
Furthermore, we engineered a double cysteine mutant (K1336C/A1413C) in the ROC domain, based on the geometry and environment around these two residues at the dimer interface using the current model. The modeled mutant has the potential to form an intermolecular disulfide bond, should the domain-crossed dimer form exist in solution. The purified double cysteine mutant displayed a dimer band on nonreducing SDS gel that was independent of nucleotide or Mg2+ binding (SI Fig. 7). In addition, we also have crystallized ROC domain in complex with GDP-Mg2+ in a second crystal form (space group C2), and we have solved the structure to 2.6-Å resolution (not refined) by the molecular replacement method using one of the peptide chains from the current structure as a searching model. This second structure also displayed a domain-swapped dimer and adopted nearly the same conformation as the current model, except the two monomers are identical to each other and are related by twofold crystallographic symmetry (data not shown). Therefore, multiple lines of evidence support the hypothesis that the intrinsic form of LRRK2 is a dimer.
Pathogenic Mutations in the ROC Domain Weaken the Dimer Structure.
The pathogenic residue R1441 is located at the end of helix α3 and interacts with helix α2 from the other paired monomer peptide chain at the dimer interface (Fig. 2A). The guanidium group of R1441 is hydrogen-bonded with the backbone carbonyl oxygen of F1401 and the hydroxyl group of T1404 on helix α2 from the other peptide chain, respectively. The same guanidinium group, the six-membered ring of W1434, together with the side chain rings of F1401 and P1406 from the other chain, stack on each other alternately, forming a hydrophobic “greasy” zipper across the dimer interface. Therefore, R1441 stabilizes dimer formation, and any mutation at this position with a shorter side chain will disrupt both the exquisite hydrogen bonding and stacking interactions provided by the arginine side chain. Loss of these interactions from R1441 would destabilize G3 and α2, leading to the displacement of R1398 and D1394, two residues that could be important for interaction with the γ-phosphate of GTP and Mg2+ ion (see below). It is likely that substitution of A1442 for a proline (14) also would disrupt this region, suggesting that this is a pathogenic mutation with a similar mechanism to R1441 mutations.
Fig. 2.

Structural basis of PD-associated mutations in ROC. (A) R1441 and W1434 from one monomer together with F1401 and P1406 from the other stack on each other alternately, forming a hydrophobic zipper at the dimer interface. The guanidinium group of R1441 also is hydrogen-bonded with the backbone carbonyl oxygen of F1401 and the hydroxyl group of T1404 on helix α2 from the other peptide chain. 2mFo − DFc electron density map is shown in blue. (B) I1371 is inserted in a hydrophobic cavity, which is constructed by residues from both monomers at the dimer interface. I1371 is shown in stick format and colored in orange. The surrounding residues are shown in stick format within the semitransparent surface representation. The color scheme is the same as that in Fig. 1. Note the side-chain methyl group of T1404 is pointing directly to the tip of I1371, forming a favorable van der Waals' interaction. (C) R1441C (lane 3), as a prototypical mutation at the dimer interface, decreases interaction with the full-length wild-type LRRK2 protein compared with wild-type GST fusions (lane 2); no interaction was seen with GST alone (lane 1). (D) Pull-down assays were quantified and corrected for the amount of LRRK2 protein in the inputs (middle blots). *, P < 0.0001; **, P < 0.01 compared with GST alone (one-way ANOVA with Student–Newman–Kuell's post hoc test; n = 3).
Another PD associated mutation, I1371V, is located on the β2 strand that pairs with the other ROC chain, resulting in a 12-stranded β-saddle in the dimer. The backbones of both β2 strands from each chain are connected via water-mediated hydrogen bonding. The two I1371 residues are related by a pseudo-twofold axis at the dimer interface and are surrounded by a cluster of hydrophobic side chains from β2 strands (V1373), β3 strands (W1393, F1395), and α2 helices (methylene groups of E1400 side chain, methyl group of T1404 side chain, and M1409). I1371 is buried in a hydrophobic pocket constructed by these residues (Fig. 2B). The side wall of the cavity is composed of V1373 side chains (from both monomers) and the side chain methylene groups of E1400. The floor of the cavity is constructed with the side chains from residues W1393 (from both monomers), F1395, and M1409. The methyl group on the side chain of T1404 is pointing directly to the Cδ of I1371, providing optimal van der Waals' interactions (T1404 Cγ-I1371 Cδ distances are 3.56 Å and 3.67 Å for each monomer, respectively). Thus, T1404 Cγ seals the hydrophobic pocket as a plug. These clustered hydrophobic contacts at the dimer interface provide a remarkable “gluing” force to stabilize the structure. A valine residue in the PD associated mutant (I1371V) would not fit this cavity as well as an isoleucine, because of the loss of the interactions provided by the Cδ methyl group on the isoleucine residue.
Our data suggest that mutations within the ROC domain disrupt dimer formation. We therefore compared the interaction of wild-type or R1441C ROC domain against full-length (wild-type) LRRK2 and observed that the mutation decreased the interaction, consistent with these predictions (Fig. 2 C and D).
The LRRK2 ROC Domain is an Active GTPase.
Although GTP binding by LRRK2 has been reported by several groups, whether significant amounts of GTP hydrolysis occurs has been unclear, with both negative (11, 16) and positive (15, 17, 18) results reported depending on experimental conditions. Our structural experiments strongly suggested that the ROC domain of LRRK2 does have intrinsic GTPase activity because GTP-Mg2+ was incubated together with ROC protein during crystallization but only GDP-Mg2+ was observed in the structure (Fig. 3). Furthermore, the overall binding mode of the GDP-Mg2+ in the current structure resembles those observed in other small GTPase structures, although with some unusual features that may stabilize GDP within the LRRK2 ROC domain after hydrolysis. The Mg2+ ion adopts near-perfect octahedral geometry involving six direct ligands: Oβ3 atom on the β-phosphate group, hydroxyl oxygen (Oγ1) of T1348 from the P-loop, and four water molecules. Through this water-mediated hydrogen bonding, the Mg2+ ion is further linked to T1368 Oγ1 from the G2/switch I and a carboxyl oxygen atom (Oδ1) of D1394 from the G3/switch II. K1347 Nζ forms bifurcated hydrogen bonding with Oβ1 on the β-phosphate and a direct water ligand from the Mg2+. The β-phosphate group (Oβ1 and Oβ2) and the nonbridging oxygen atom from α-phosphate are embraced by four contiguous backbone amides from the P-loop (G1344, S1345, G1346, and K1347) through extensive hydrogen bonding. The α-phosphate is more solvent-exposed. Oα1 on the α-phosphate is hydrogen-bonded with both backbone amide and the side chain hydroxyl groups of T1349 on α1. The sugar ring adopts a C2′-endo conformation and is solvent-exposed. A preference for guanine is obtained through hydrogen bonding interactions from the Watson–Crick base-pairing face on the guanine base ring. In particular, the exocyclic keto oxygen at position 6 of the guanine ring forms hydrogen bonds with two main-chain amides: H1453 in loop G4 and N1489 in the opposite loop G5, respectively. In Ras or EF-Tu GTPase structures, only one backbone amide is involved in hydrogen bonding with O6 atom on the guanine ring (23, 24). The N7 atom on the guanine ring in ROC has no interactions, compared with those in Ras and EF-Tu structures, which are stabilized by a nearby Asn residue (20, 24). In addition, D1455 from loop G4 is hydrogen-bonded with N1 and amino group at position 2 on the guanine ring, providing exquisite guanine recognition. Interestingly, the guanine ring in ROC is stabilized by unique stacking interactions, being sandwiched between a ROC family conserved His residue (3) (H1453 from loop G4) and T1491 from loop G5. In all other GTPase structures, a highly conserved lysine residue is occupying the position of H1453 (23). H1453 thus represents a sequence structural signature for the ROC GTPase superfamily.
Fig. 3.

The nucleotide-binding site and a stereoview of the GDP-Mg2+ binding site. The GDP-Mg2+ ligand is shown in ball-and-stick format. The Mg2+ is colored in purple, and its direct water ligands are colored in cyan. The essential interacting residues from the dimer ROC are shown in stick format and are colored with the same scheme as in Fig. 1.
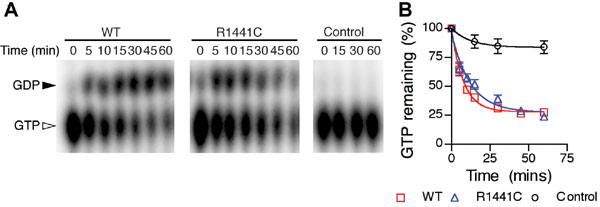
These observations suggest that the LRRK2 ROC domain should have all of the catalytic residues required for GTP hydrolysis, although some differences in the switch regions may affect overall efficiency of the enzyme. To test this finding, we used the recombinant LRRK2 ROC domain in in vitro GTPase assays and confirmed it as an authentic GTPase (SI Fig. 8). The R1441C mutation had a slightly decreased rate of GTP hydrolysis, although at a less dramatic scale in the context of the recombinant protein compared with other recent data by using full-length protein expressed in cells (15, 17, 18).
The LRRK2 ROC Domain Regulates Its Kinase Activity.
LRRK2 is an unusual kinase in that the two enzymatic domains within a single polypeptide communicate to control activity (12, 13). Specifically, the kinase activity of LRRK2 is stimulated upon GTP binding to ROC (7, 16). Several laboratories have shown that K1347A or other mutations that cannot bind GTP decrease kinase activity (4, 10, 15, 16). Although the mechanism of GTP regulation of kinase activity is unclear, it is reasonable to postulate that ROC regulates the kinase activity by alternating its conformations through a GTP-bound (active) and GDP-bound (inactive) cycle, which suggests that loss of binding of GTP or increasing turnover of GTP to GDP should result in lowered kinase activity. A direct comparison of the current structure with Ras (bound to a nonhydrolyzable GTP analogue) shows that two critical residues in this region, T1343 and R1398, occupy equivalent positions to Ras G13 and Q61 (although with a different orientation), suggesting their potential roles in interactions with the γ-phosphate of substrate GTP (Fig. 4A). Therefore, ROC might have a different mechanism for GTP hydrolysis compared with Ras. We introduced mutations T1343G and R1398Q either singly or together into full-length LRRK2 and measured kinase activity by using autophosphorylation (Fig. 4B). Neither mutation alone affected kinase activity, suggesting that both were tolerated in the structure. However, the double TG/RQ mutant lowered the kinase activity, presumably because of more significant structural perturbation in GTP binding.
Fig. 4.

LRRK2 kinase regulation by ROC. (A) Stereo model of the LRRK2 functional GTPase unit (yellow and green for each monomer) superimposed on Ras (PDB ID 1ctq, red). Two key residues important in the regulation of Ras activity after GTP binding are Q61 and G12 (shown in red, labeled in red); the equivalent residues in LRRK2 are R1398 and T1343, respectively (shown in yellow, labeled in black). (B) Ras-like mutations R1398Q and T1343G were made individually and together (TQ/RG, lane 5) in the full-length LRRK2 construct, and their effects on autophosphorylation of the kinase were tested. (Upper) Autoradiogram with 32P. (Lower) A blot for LRRK2. Quantification (n = 3, graph) shows that although each Ras-like mutation alone has no effect on kinase activity, the double TG/RQ caused a decrease in net kinase activity. **, P < 0.01; ***, P < 0.001 by ANOVA with Bonferroni's multiple comparison test. (C) ROC domain has intrinsic association with COR domain. Truncated domain proteins used for interaction assay are as follows: ROC–COR–kinase protein (lane 1), COR domain only (lane 2), and kinase domain only (lane 3), respectively, blotted with a myc tag. Their corresponding pull-down results with ROC domain as a GST fusion protein are shown in lanes 4–6, respectively. GST was used as negative control in lanes 7–9. (D) Model for ROC regulation of kinase activity. Dimeric ROC GTPase (yellow and green ovals) act as binary switches in regulation of kinase activation. Upon binding of GTP (stars), the activated ROC induces dimerization of the COR domain (barrels). The dimerization of COR domain further induces self-association of kinase domain, resulting in its autophosphorylation and activation (red ovals). Through hydrolysis of GTP to GDP (black oval), conformational changes in ROC disrupt the dimeric association of COR and kinase domain, inactivating the kinase (blue ovals). GTPase activating protein (GAP) and guanine nucleotide exchange factor (GEF) are not known.
The basis by which the two domains communicate is unclear. We considered whether the ROC and kinase domains might interact physically but saw little evidence of this. However, in these experiments, we did see that the COR and ROC domains interacted strongly (Fig. 4C), suggesting that the COR domain might act as a molecular link between the ROC and kinase domains (Fig. 4D).
Discussion
In this study, we have shown that the ROC domain of LRRK2, as a prototype for the ROCO class of proteins, forms a strong dimer that also acts as a GTPase to regulate kinase activity. An intrinsic self-association of full-length LRRK2 was suggested by Gloeckner et al. (8), and we also have found that full-length LRRK2 purified from mammalian cells forms a dimer using size-exclusion chromatography (E.G., I. Zambrano, and A. Kaganovich, unpublished data). Therefore, it is likely that LRRK2 and perhaps other ROCO proteins are functional dimers.
Sequence analyses indicate that the ROC domain usually is followed by a COR domain in ROCO superfamily (3). We found the ROC domain has intrinsic interactions with the COR domain. It should be noted that the interaction between the ROC and COR domains in this context is stronger than the interaction between the ROC domain and full-length protein (data not shown), suggesting that the ROC–COR interaction might be more direct. There are several other protein–protein interaction domains in LRRK2 (ankyrin repeat, leucine-rich repeat, and WD40 domains), which potentially play additional roles in the overall topology of the protein. These results suggest that within the intact full-length LRRK2 dimer, the dynamic regulation of GTP conversion impacts kinase activity. Upon activation of the dimeric ROC domains by GTP binding, the switch I and switch II in the ROC domain likely adopt specific conformations. The switch I (G2) loop is largely disordered in our structure, which indicates its inherent flexibility. In fact, it has been shown in other GTPase structures that this region adopts significantly different conformations between GTP- and GDP-bound forms and is the interaction region with their effectors (22, 23). During the GTP/GDP binding cycle, the conformational changes in the ROC domain may be conveyed to the kinase domain through the COR domain either directly or indirectly as a molecular hinge, leading to the dimerization of the kinase domain and subsequently its autophosphorylation and activation.
The data presented here show that LRRK2, as a prototype for other ROCO proteins, has an active GTPase domain that has a unique dimeric fold by domain-swapping. Our results suggest that mutations in the ROC domain of LRRK2 that are pathogenic for PD, such as R1441C and I1371V mutations, have the effect of partially disrupting the tertiary structure of the protein at the dimer interface, which will result in the decrease of the GTP hydrolysis and therefore prolong GTP-mediated activation of the kinase.
The structural model is consistent with several known aspects of the biochemistry of LRRK2 and may have further implications both for understanding how complex multidomain proteins function. Because the topology is different from any other known small GTPases, and because the ROCO family of proteins is relatively small, our structural data may also allow rational design of compounds that prevent the hyperactivation of LRRK2.
Materials and Methods
Recombinant Protein Production, Crystallization, and Structure Determination.
The human LRRK2 ROC domain (amino acids 1,333–1,516) was cloned into a modified pET28 b vector (pSKB3) with an N-terminal 6×His tag and a tobacco etch virus protease cleavage site. The recombinant ROC domain and the selenomethionine-substituted protein were expressed in E. coli and purified as previously described (25). The protein was concentrated to 13 mg·ml−1 in the presence of 5 mM GTP and 20 mM Mg2+. Multiple thin-plated crystals were obtained under conditions containing 30% PEG 4000, 100 mM Mg2+, and 0.1 M Tris (pH 8.0) in a week. Single crystals were obtained by microseeding (26). Twenty percent glycerol was added to the crystallization condition for cryoprotection. A set of data was collected to 2.0-Å resolution at low temperature (100 K) by using the synchrotron radiation source at wavelength of selenium K-absorption edge (Advanced Photon Source, beamline 19-ID, Argonne National Laboratory, Argonne, IL). The crystal belongs to space group P1 with two molecules in the asymmetric unit. The data were processed and the initial phasing was done at the beamline with program HKL3000 (27). After iterative noncrystallographic symmetry averaging and density modification by programs DM (28) and RESOLVE (29), an initial model was obtained by using program Arp/Warp (30) with 25% coverage. Subsequent manual modeling was done with the program Coot (31). The structure was refined with REFMAC5 (32), and PROCHECK (33) was used for the final model analysis. The current models are of excellent geometry and refinement statistics (SI Table 1). All molecular graphic figures were generated with PyMOL (34).
The double cysteine mutagenesis (K1336C/A1413C) was generated by standard fragment-overlapping PCR amplification. The mutant was cloned in the pSKB3 vector, and the protein expression and purification were carried out as outlined above.
Enzyme Assays.
For in vitro GTPase assays, recombinant ROC domains were diluted to 3 μM in 20 μl of reaction buffer (20 mM Hepes, pH 7.2/2 mM MgCl2/10 mM DTT/0.005% BSA). After addition of 5 μCi of [α32P]-GTP (GE Healthcare), aliquots were incubated at 30°C, and 1-μl samples were removed and spotted onto thin-layer chromatography plates (Cellulose PEI; Sigma). After times up to 60 min, nucleotides were separated by rising chromatography with 1 M formic acid and 1.2 M LiCl for 90 min. TLC plates were exposed to phospho-screens, and the results were analyzed on a Storm 840 scanner (GE Healthcare). Kinase assays were performed as previously described (5, 15) with the minor modification that Ponceau staining was used to correct for LRRK2 protein loading.
Protein Pull-Down Assays.
HEK293FT were transfected with Myc-tagged LRRK2 and lysed in 20 mM Tris·HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, protease inhibitor mixture (Roche), and phosphatase inhibitors (Pierce). Glutathione Sepharose beads with purified proteins were incubated overnight at 4°C with total cell lysates. After four washes (PBS, 150 mM NaCl, 1% Triton, and protease inhibitors), copurified proteins were analyzed by Western blotting with anti-Myc antibody.
Supplementary Material
ACKNOWLEDGMENTS.
We thank the staff of beamline 19ID at the Advanced Photon Source for their support. This work was supported by the Division of Agricultural Sciences and Natural Resources at Oklahoma State University (J.D.). This work was funded in part by the Intramural Research Program of the National Institutes of Health, National Institute on Aging (M.R.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2ZEJ).
This article contains supporting information online at www.pnas.org/cgi/content/full/0709098105/DC1.
References
- 1.Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson's disease and parkinsonism. Ann Neurol. 2006;60:389–398. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- 2.Cookson MR, Xiromerisiou G, Singleton A. How genetics research in Parkinson's disease is enhancing understanding of the common idiopathic forms of the disease. Curr Opin Neurol. 2005;18:706–711. doi: 10.1097/01.wco.0000186841.43505.e6. [DOI] [PubMed] [Google Scholar]
- 3.Bosgraaf L, Van Haastert PJ. ROC, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 4.West AB, et al. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greggio E, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Jaleel M, et al. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korr D, et al. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal. 2006;18:910–920. doi: 10.1016/j.cellsig.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 8.Gloeckner CJ, et al. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- 9.Luzón-Toro B, de la Torre ER, Delgado A, Pérez-Tur J, Hilfiker S. Mechanistic insight into the dominant mode of the Parkinson's disease-associated G2019S LRRK2 mutation. Hum Mol Genet. 2007;16:2031–2039. doi: 10.1093/hmg/ddm151. [DOI] [PubMed] [Google Scholar]
- 10.Smith WW, et al. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 11.West AB, et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 12.Marin I. The Parkinson disease gene LRRK2: Evolutionary and structural insights. Mol Biol Evol. 2006;23:2423–2433. doi: 10.1093/molbev/msl114. [DOI] [PubMed] [Google Scholar]
- 13.Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y, et al. Prevalence and clinical features of common LRRK2 mutations in Australians with Parkinson's disease. Mov Disord. 2007;22:982–989. doi: 10.1002/mds.21477. [DOI] [PubMed] [Google Scholar]
- 15.Lewis PA, Greggio E, Beilina A, Jain S, Baker A, Cookson MR. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. 2007;357:668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito G, et al. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- 17.Guo L, et al. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase thatstimulates kinase activity. Exp Cell Res. 2007;313:3658–3670. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, et al. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 20.Song H, Parsons MR, Rowsell S, Leonard G, Phillips SE. Crystal structure of intact elongation factor EF-Tu from Escherichia coli in GDP conformation at 2.05 Å resolution. J Mol Biol. 1999;285:1245–1256. doi: 10.1006/jmbi.1998.2387. [DOI] [PubMed] [Google Scholar]
- 21.Scheidig AJ, Burmester C, Goody RS. The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure (London) 1999;7:1311–1324. doi: 10.1016/s0969-2126(00)80021-0. [DOI] [PubMed] [Google Scholar]
- 22.Paduch M, Jelen F, Otlewski J. Structure of small G proteins and their regulators. Acta Biochim Pol. 2001;48:829–850. [PubMed] [Google Scholar]
- 23.Sprang SR. G protein mechanisms: insights from structural analysis. Annu Rev Biochem. 1997;66:639–678. doi: 10.1146/annurev.biochem.66.1.639. [DOI] [PubMed] [Google Scholar]
- 24.Milburn MV, et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 25.Deng J, Ernst NL, Turley S, Stuart KD, Hol WG. Structural basis for UTP specificity of RNA editing TUTases from Trypanosoma brucei. EMBO J. 2005;24:4007–4017. doi: 10.1038/sj.emboj.7600861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng J, Schnaufer A, Salavati R, Stuart KD, Hol WG. High resolution crystal structure of a key editosome enzyme from Trypanosoma brucei: RNA editing ligase 1. J Mol Biol. 2004;343:601–613. doi: 10.1016/j.jmb.2004.08.041. [DOI] [PubMed] [Google Scholar]
- 27.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: The integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 28.Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 29.Terwilliger TC. SOLVE and RESOLVE: Automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 30.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the sterochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 34.DeLano WL. Palo Alto, CA: DeLano Scientific; 2002. The PyMOL Molecular Graphics System. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}