

Abstract
Axon formation is the initial step in establishing neuronal polarity. We examine here the role of microtubule dynamics in neuronal polarization using hippocampal neurons in culture. We see increased microtubule stability along the shaft in a single neurite before axon formation and in the axon of morphologically polarized cells. Loss of polarity or formation of multiple axons after manipulation of neuronal polarity regulators, synapses of amphids defective (SAD) kinases, and glycogen synthase kinase-3β correlates with characteristic changes in microtubule turnover. Consistently, changing the microtubule dynamics is sufficient to alter neuronal polarization. Application of low doses of the microtubule-destabilizing drug nocodazole selectively reduces the formation of future dendrites. Conversely, low doses of the microtubule-stabilizing drug taxol shift polymerizing microtubules from neurite shafts to process tips and lead to the formation of multiple axons. Finally, local stabilization of microtubules using a photoactivatable analogue of taxol induces axon formation from the activated area. Thus, local microtubule stabilization in one neurite is a physiological signal specifying neuronal polarization.
Introduction
Neurons are highly polarized cells that typically have one thin, long process to transmit information (the axon) and several tapered, shorter processes to receive information (dendrites; Craig and Banker, 1994). One of the key questions of neurobiology is how a neuron acquires these polar structures, which provide the basis for unidirectional signal transmission. Previous studies focused on the actin cytoskeleton and its modulators, including Rho, Cdc42, profilin, cofilin, and T-lymphoma and metastasis 1 protein, which perform an important regulatory function in the process of neuronal polarization (Bradke and Dotti, 1999; Bito et al., 2000; Kunda et al., 2001; Garvalov et al., 2007).
Nevertheless, a growing number of studies identified polarity regulators that appear to act through processes independent of actin dynamics, including synapses of amphids defective (SAD) kinases (Kishi et al., 2005), collapsin response mediator protein 2 (CRMP-2; Inagaki et al., 2001), and glycogen synthase kinase-3β (GSK-3β; Jiang et al., 2005; Yoshimura et al., 2005). This underscores the notion that additional intracellular mechanisms underlie neuronal polarization. Although these nonactin regulating proteins are involved in multiple processes, one common denominator appears to be their direct or indirect involvement in the control of microtubule (MT) dynamics. GSK-3β, for example, a multitarget protein kinase regulating many metabolic, signaling, and structural proteins (for review see Doble and Woodgett, 2003) is involved in the establishment and maintenance of neuronal polarity (Jiang et al., 2005; Yoshimura et al., 2005). Among its many functions, GSK-3β also modulates MT dynamics, e.g., by phosphorylating MT-associated proteins (MAPs; Goold et al., 1999; for review see Doble and Woodgett, 2003), whose binding to MTs is essential for neurite formation (Caceres and Kosik, 1990). It is noteworthy that some MAPs, including adenomatous polyposis coli protein, are inhibited by GSK-3β phosphorylation, whereas others, including MAP1B, are activated (Goold et al., 1999; for review see Doble and Woodgett, 2003). Consistent with the complex effects on MAPs, GSK-3β can either support or inhibit axonal growth depending on the extent of its inhibition (Kim et al., 2006). Overexpression of CRMP-2, another target of GSK-3β implicated in the regulation of MT dynamics and endocytosis, induces multiple axons in later developmental stages, which suggests a role for CRMP-2 in axon formation and maintenance (Inagaki et al., 2001). Another example is the SAD kinases, homologues of the conserved partitioning defective-1 (PAR-1) serine/threonine kinase, which acts in a variety of polarity events in species ranging from nematodes and flies to mammals (Wodarz, 2002). SAD kinases are required for neuronal polarization (Kishi et al., 2005) and modulate presynaptic vesicle clustering (Crump et al., 2001) but also phosphorylate MAPs (Kishi et al., 2005). Recent work has also shown that activated c-Jun N-terminal kinase, so far known to be implicated in the regulation of gene transcription, cell death, and survival (for review see Bogoyevitch and Kobe, 2006), might play a role in axon formation (Oliva et al., 2006). c-Jun N-terminal kinase targets a wide variety of nuclear and cytoplasmic proteins, including transcription factors and actin-regulating proteins but also MAPs (for review see Bogoyevitch and Kobe, 2006).
In summary, despite the wealth of polarity regulators identified in the past years, our knowledge regarding the intracellular mechanisms that establish neuronal polarization has remained fragmentary. Given that some identified regulators of neuronal polarity appear to act indirectly or directly on MTs, the possibility arises that, in addition to the well-established function of the actin cytoskeleton, MTs may play a pivotal role in axon formation. The actual task of MTs in the establishment of neuronal polarity, however, has remained unclear and is still poorly understood. We therefore aimed to characterize whether MTs play an active role in neuronal polarization. Here, we find that the future axon has more stable MTs in its shaft and that stabilization of MTs is sufficient to induce axon formation. Our data show that MTs and the regulation of their stability play an instructive role in the initial polarization of neuronal cells.
Results
MT stability is increased in axons and one process of a subpopulation of morphologically unpolarized neurons
Earlier studies have yielded divergent results concerning the distribution of stabilized MTs in developing neuronal cells (Arregui et al., 1991; Dotti and Banker, 1991). We therefore first aimed to assess MT stability in developing axons and minor neurites using cultured rodent hippocampal neurons (Dotti et al., 1988). Upon plating, these cells form lamellipodia around the cell body (stage 1) that, after 12–24 h, condense to four to five processes, the minor neurites (stage 2). From this pool of morphologically indistinguishable processes, one starts to grow out quickly to become the axon (stage 3; Dotti et al., 1988). This first morphological sign of axon formation is a crucial hallmark of neuronal polarization, as it marks the initial break in symmetry during neuronal development (Craig and Banker, 1994).
As a first means to study MT stability, we extracted cells to remove unpolymerized tubulin subunits to assess MTs only and stained the cells for acetylated and tyrosinated α-tubulin, which are markers for stable and dynamic MTs, respectively (for review see Westermann and Weber, 2003). Acetylation of α-tubulin or enzymatic removal of its C-terminal tyrosine residue (detyrosination) gradually occurs in the MT polymer and is therefore found in long-lived, stable MTs, i.e., MTs with a low turnover that undergo few catastrophic events. In contrast, the presence of tyrosinated α-tubulin in MTs that has not yet undergone detyrosination denotes a recent assembly, i.e., tyrosinated α-tubulin is found in dynamic MTs with a high turnover. Expectedly, we found that tyrosinated MTs were predominant in the growth cones of all processes, which reflects their dynamic state required for steering and extension (Fig. 1, C and D; Tanaka et al., 1995). When we analyzed FRAP using neurons transfected with α-tubulin fused to GFP, we found that the GFP signal recovered faster in the growth cone compared with the axonal shaft. This further points to a high turnover of MTs in the growth cones (Video S1, available at http://www.jcb.org/cgi/content/full/jcb.200707042/DC1).
Figure 1.

Differential distribution of acetylated and tyrosinated MTs in hippocampal neurons. (A–H) Polarized stage 3 (A–D) and morphologically unpolarized stage 2 (E–H) rat hippocampal neurons stained for acetylated (B and F) and tyrosinated (C and G) α-tubulin (arrows, axons; arrowheads, minor neurites). Cells were permeabilized during fixation to remove unpolymerized tubulin subunits, therefore only tubulin incorporated in MTs was assessed. In stage 3 neurons, a high ratio of acetylated to tyrosinated α-tubulin is found in MTs in the axonal shaft (D, arrow) in comparison to MTs of minor neurites (D, arrowheads, and I). In 35.0 ± 6.1% of morphologically unpolarized stage 2 neurons, the ratio of acetylated/tyrosinated α-tubulin is significantly increased in one of the minor neurites (H, white arrowhead with asterisk; P < 0.05 by Hampel outlier test). The areas boxed in H are shown in higher magnification in K and M. (I and J) Ratio quantification of fluorescence intensities of acetylated and tyrosinated α-tubulin in MTs of stage 2 (J) and 3 (I) neurons (mean ± SEM; n > 105 neurons from three independent experiments for each stage 2 and 3). Values are normalized to the mean of nonaxonal or nonmaximal processes for stage 3 and 2, respectively. ***, P < 0.001 by t test. (K–N) Higher magnification views (K and M) and profiles of immunofluorescence intensity (in arbitrary units; L and N) of acetylated and tyrosinated α-tubulin of the neurites marked in H. Bars: (A–H) 20 μm; (K and M) 10 μm.
Morphologically polarized neurons (stage 3; Fig. 1 A) showed an enrichment of stable acetylated MTs in the axonal shaft compared with the shafts of minor neurites in 83.5 ± 1.0% of the cases (mean ± SEM, n = 709 neurons from three independent experiments; Fig. 1, B and D; and Fig. S1, A–D, available at http://www.jcb.org/cgi/content/full/jcb.200707042/DC1). On average from all stage 3 neurons, the axonal shaft showed a 3.2 ± 0.5-fold ratio increase of the fluorescence intensities of acetylated versus tyrosinated MTs compared with the shafts of minor neurites (P < 0.001 by t test; Fig. 1 I). Similar results were obtained by comparing acetylated to total α-tubulin integrated in MTs (Fig. S1, A–D and I).
Such differences in MT turnover already occur in morphologically unpolarized neurons (stage 2; Fig. 1 E). In 35.0 ± 6.1% of all stage 2 neurons, one neurite singled out and exhibited a significantly higher ratio of acetylated to tyrosinated MTs compared with the mean of the remaining neurites (P < 0.05 by Hampel outlier test; Fig. 1, F–H [arrowhead with asterisk], K [arrow], and M), which indicates that MT stabilization in one neurite precedes axon formation in morphologically still-unpolarized cells. Similar results were obtained by comparing acetylated to total MTs (Fig. S1, E–H and J). On average from all stage 2 neurons, the ratio of acetylated versus tyrosinated MTs was increased 1.9 ± 0.3-fold in the minor neurite with the highest ratio compared with the mean of the other neurites (P < 0.001 by t test; Fig. 1 J) and also significantly different from the neurite with the second-highest ratio alone (P < 0.001 by t test). The increased ratio was caused by a relative increase of acetylated over tyrosinated or total MTs within the process (Fig. 1, L and N; and Fig. S1, J–L). Thus, the axon of stage 3 neurons and one minor neurite of a subpopulation of stage 2 neurons show markers of lower MT turnover.
This differential distribution of posttranslational modifications also reflects an actual stability difference of MTs in axons and minor neurites. In 81.8 ± 5.9% of nocodazole short term–treated polarized neurons (stage 3), MTs of minor neurites retracted more than axonal MTs (7.6 ± 0.9 and 11.9 ± 0.8 μm in axons and minor neurites, respectively; P < 0.001 by t test; Fig. 2, C, D, and I; and compare A and B). Similarly, morphologically unpolarized neurons (stage 2) showed a distinct difference between MT retraction in one neurite compared with the remaining neurites (3.1 ± 0.7 and 9.9 ± 1.0 μm, respectively; P < 0.001 by t test; Fig. 2, G, H, and J; and compare E and F), which suggests an early polarization of stabilized MTs during neuronal development. The MTs that resisted depolymerization were acetylated (Fig. 2, K, L, and N), whereas tyrosinated MTs had vanished after nocodazole treatment (Fig. 2, K, M, and N). Collectively, these data show that MT stability is increased in the axon of stage 3 neurons and one minor neurite of a subpopulation of stage 2 neurons.
Figure 2.

Stable MTs are enriched in axons. (A–H) To consider actual differences in MT stability only, MTs (green) were partially depolymerized by treatment with nocodazole and their retraction from the distal end of the processes toward the cell body was examined (see I and J). F-actin (red) outlining the neuron was used as a reference point for measuring MT retraction. In DMSO-treated control cells, MTs reach close to the distal end of both axon (B, arrow) and minor neurites (B and F, arrowheads). In nocodazole-treated cells, MTs of minor neurites retract toward the cell body (D and H, arrowheads). MTs in the axon of stage 3 cells (D, arrow) and in one of the minor neurites of stage 2 cells (H, arrowhead with asterisk) are more resistant to depolymerization. Arrows, axons; arrowheads, minor neurites. (I and J) MT retraction after nocodazole treatment in polarized stage 3 (I) and morphologically unpolarized stage 2 (J) neurons (mean ± SEM; n = 119 and 50 neurons from five and three independent experiments, respectively; ***, P < 0.001 by t test). (K–N) Polarized rat hippocampal neurons with one axon (arrow) and several minor neurites (arrowheads) after 2 DIV before (K) and after (L–N) treatment with nocodazole (5 μM for 5 min). Tyrosinated (M and N, red) and acetylated (L and N, green) MTs were assessed. Bars, 20 μm.
MT stability is changed in neurons whose polarity is affected by manipulation of GSK-3β or SAD kinases
Next, we assessed whether molecules that regulate neuronal polarity affect MT stability in polarizing neurons. Neurons treated with the GSK-3β inhibitor SB415286 (10–20 μM), which has been reported to induce multiple axons per cell (Jiang et al., 2005; Yoshimura et al., 2005), showed an enrichment of stable, acetylated MTs in the supernumerary axons similar to wild-type axons (Fig. 3 A; compare with Figs. 1 D and 3 D). Notably, MT acetylation already increased upon GSK-3β inhibition in stage 2 neurons, which indicates an induction of MT stabilization by GSK-3β inhibition before axon formation (Fig. 3, B and C). For comparison to the multiaxonal phenotype induced by GSK-3β inhibition, we analyzed neurons from mice deficient in the PAR-1 homologues SAD A and B. These neurons show a polarity defect and lack a mature axon but generate multiple processes of similar lengths containing both the dendritic marker MAP2 as well as the axonal marker Tau-1 (Kishi et al., 2005). We found that SAD A/B kinase double knockout neurons lacked the specific enrichment of stable, acetylated MTs in a single process found in wild-type neurons as well as littermate control cultures (Fig. 3, E–G). Some of these neurons had neurites with MTs acetylated to a similar extent as in axons, whereas a majority had an acetylation level that rather resembled minor neurites. Despite this cell-to-cell variation (Fig. 3, E and F), however, acetylation levels were rather uniform within a given cell. The acetylation/tyrosination ratio of the longest versus the second longest neurite per cell was not significantly different in SAD A−/− B−/− neurons (1.09 ± 0.03, P > 0.25 by t test), whereas control neurons showed a clear distinction (2.06 ± 0.05, P < 0.01 by t test; Fig. 3 H). Thus, specific alterations of neuronal polarity, including the formation of supernumerary axons or a loss of polarity, correlate with characteristic changes in MT stability.
Figure 3.

Altered neuronal polarity correlates with changed MT stability. (A and B) Rat hippocampal neurons grown in the presence of the GSK-3β inhibitor SB 415286 for 72 (A) and 24 h (B). Stage 3 neurons have formed multiple axons that feature an increased MT stability (A, arrows) compared with minor neurites (A, arrowhead). A rise in MT acetylation in several minor neurites (B, arrowheads with asterisk) precedes the formation of multiple axons in morphologically still unpolarized stage 2 neurons treated with GSK-3β inhibitor. Arrows, axons; arrowhead, minor neurites. Bars, 20 μm. (C) Ratio quantification of fluorescence intensities of acetylated and tyrosinated α-tubulin in MTs of unpolarized stage 2 neurons. The normalized average ratio of the five longest neurites is shown. Approximately 5 h after treatment with SB 415286, stage 2 neurons show a trend toward increased MT acetylation (23.0 ± 10.6%), which indicates a rise in MT stability. The increase is significant after ∼8 h (47.4 ± 8.7%; mean ± SEM; n > 50 neurons per condition and time point from three independent experiments; *, P < 0.05 by t test). (D) Rat hippocampal neurons (3 DIV) treated with the GSK-3β inhibitor SB 415286 (10–20 μM treatment 6–8 h after plating) formed 2.1 ± 0.1 axons on average. These supernumerary axons show an increased ratio of acetylated to tyrosinated α-tubulin equal (P > 0.35 by t test) to that of the single axon of stage 3 control neurons (treatment with 0.04% DMSO; mean ± SEM; n > 35 neurons from three independent experiments). (E and F) Hippocampal neurons (3 DIV) derived from mice deficient for SAD A and B kinase show disturbed polarity and lack a defined axon. Instead, SAD A/B knockout neurons form multiple processes of similar length and uniform tubulin acetylation levels (see G), yet a high cell-to-cell variability. Bars, 20 μm. (G) Ratio quantification of fluorescence intensities of acetylated and tyrosinated α-tubulin in MTs. Processes of SAD A−/−B−/− neurons are short of the specific enrichment of acetylated MTs in one process found in wild-type as well as littermate control neurons (SAD A+/+B+/−; mean ± SEM; n = 66 and 27 neurons from three independent experiments for SAD A−/− SAD B−/− and control, respectively). Note that the acetylation/tyrosination ratio varies slightly in control cells between species (rat vs. mouse; D and G). (H) Ratio of the acetylation/tyrosination ratios of the longest versus second longest process per cell for SAD A−/−B−/− and control neurons (mean ± SEM; ***, P < 0.001 by t test).
Moderate MT destabilization selectively reduces the formation of minor neurites
SAD kinases and GSK-3β can act upstream of MT dynamics by controlling the affinity of MAPs (Jiang et al., 2005; Kishi et al., 2005; Yoshimura et al., 2005), which are known modulators of MT stability, yet they have various other cellular functions including the regulation of metabolism, signaling, gene transcription, and endocytosis (Crump et al., 2001; Grimes and Jope, 2001). We therefore wondered whether the observed differences in MT stability between axons and minor neurites play a direct role in polarization. We hypothesized that if MT stability is critical in distinguishing future axonal and dendritic fate, manipulations of MT dynamics should affect polarization. To test this assumption, we cultured neurons in the presence of low concentrations of the MT-destabilizing drug nocodazole, which moderately increase the catastrophe rate of MTs (15–75 nM; Vasquez et al., 1997). In control cells, the mean number of processes almost doubled from 1 (2.2 ± 0.2 processes per cell) to 3 d in vitro (DIV; 4.0 ± 0.1 processes per cell; P < 0.001 by analysis of variance [ANOVA]; Fig. 4 E), which reflects the formation of an axon and several minor neurites (Fig. 4, A and B). Cells grown in the presence of low concentrations of nocodazole were able to form and extend an axon likewise (Fig. 4, C and D, arrow). The number of minor neurites, however, was significantly reduced in nocodazole-treated neurons (Fig. 4, C and D [arrowhead], and E). This reduction was not caused by a general growth inhibition by nocodazole (Fig. 4 F) but by the development of many neurons with just one or two processes (Fig. 4, C–E). Interestingly, axon extension was not impaired in contrast to minor neurite extension (132.1 ± 6.1 and 134.0 ± 14.7 μm for 0.02% DMSO and 45 nM nocodazole, respectively; P > 0.5 by t test; Fig. 4 F). Expectedly, higher concentrations of nocodazole started to inhibit axonal outgrowth as well (Fig. 4 F). Collectively, our data suggest that the future axon is able to overcome moderate MT-destabilizing conditions, whereas the outgrowth of minor neurites is impaired.
Figure 4.

Moderate MT destabilization selectively blocks the formation of minor neurites. (A–D) Rat hippocampal neurons (3 DIV) cultured in the presence of various low concentrations of nocodazole or 0.02% DMSO (treatment after 1 DIV) stained for Tau-1 (B and D). (A and B) Control neurons have formed one axon (A, arrow) with the typical Tau-1 gradient toward its distal part (B, arrow) and several Tau-1–negative minor neurites (arrowheads). (C and D) The number of minor neurites (arrowhead) is reduced under growth conditions that slightly destabilize MTs, however, neurons are still able to form an axon (arrow). Bars, 20 μm. (E) Nocodazole reduces the number of minor neurites formed in a concentration-dependent manner (neurites per cell: 4.0 ± 0.1, 3.6 ± 0.1, 2.6 ± 0.1, and 2.2 ± 0.2 for 0.02% DMSO and 15, 45, and 75 nM nocodazole, respectively; P < 0.001 by ANOVA; n > 750 neurons from three independent experiments per condition). (F) Neurite extension is not blocked by low concentrations of nocodazole. Total neurite length increased from day 1 to 3 under all conditions (P < 0.001 by t test), though to a lesser extent when treated with 45 or 75 nM nocodazole (increase from 57.5 ± 7.8 to 255.8 ± 9.7, 209.3 ± 8.6, or 130.3 ± 3.6 μm for 0.02% DMSO and 45 or 75 nM nocodazole, respectively; n > 275 neurons per condition from three independent experiments). Data is presented as mean ± SEM.
MT stabilization is sufficient to induce axon formation
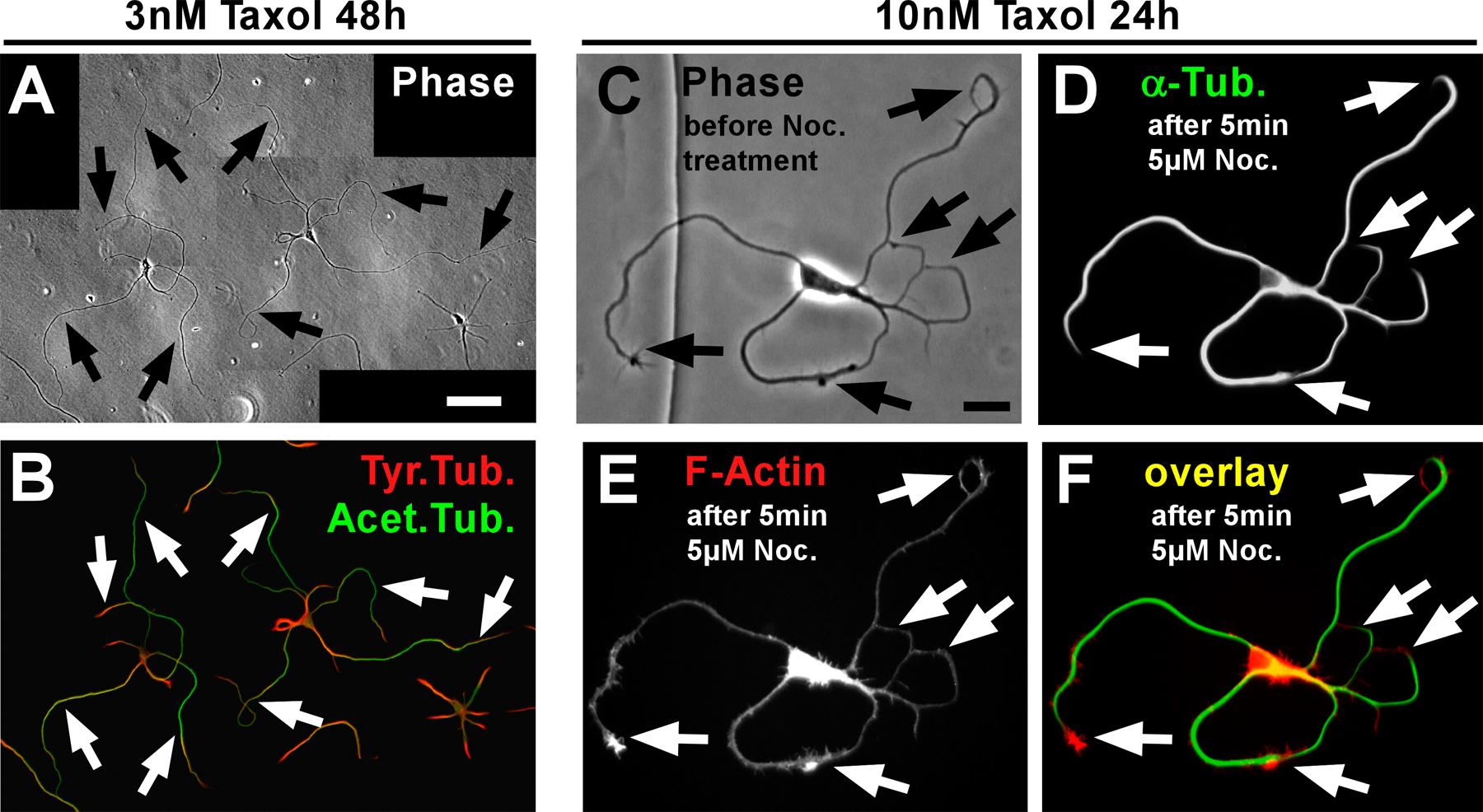
To assess whether MT stabilization itself is sufficient to induce axon formation, we treated hippocampal neurons after 1 DIV with the MT-stabilizing drug taxol for varying times. Taxol-treated neurons showed a drastic increase in neurite outgrowth (Fig. 5, A and E; and Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200707042/DC1). The number of processes per cell exceeding 60 μm, a morphological characteristic of early axons, had increased more than twofold in the presence of low concentrations of taxol that favor MT polymerization (3–10 nM; Derry et al., 1995) for 2 d in comparison to control neurons (Fig. 5, A, C, and E). Taxol-induced processes showed a high ratio of acetylated to tyrosinated MTs resembling axonal characteristics (Fig. S2 B, compare with Fig. 1 D) and were indistinguishable from axons in terms of their MT stability (Fig. S2, C–F; compare with Fig. 2). Still, with the low taxol concentrations used, tyrosinated MTs prevailed in the growth cones of these processes (Fig. S2 B), which indicates that MT dynamics were not completely abolished. In contrast, higher concentrations of taxol blocked MT dynamics and axonal growth completely (unpublished data; Dehmelt et al., 2003).
Figure 5.

Taxol-induced MT stabilization triggers the formation of multiple axons. (A–D) Rat hippocampal neurons (3 DIV) grown in the presence of low concentrations of taxol (3 nM) or DMSO (treatment after 1 DIV) stained for the axonal marker Tau-1. Taxol induces the formation of multiple elongated processes (A, arrows) positive for Tau-1 (B, arrows). Control neurons have formed one axon (arrow) and several minor neurites (arrowheads; C and D). (E) The number of neurites longer than 60 μm is increased after 2 d of taxol treatment (mean ± SEM; P < 0.001 by t test; n > 170 neurons per condition from three independent experiments). DMSO (0.02%), vehicle; Cyto.D (1 μM Cytochalasin D), positive control (Bradke and Dotti, 1999). (F) The number of cells with two or more Tau-1–positive processes increases in a concentration-dependent manner in taxol-treated neurons (mean ± SEM; P < 0.001 by ANOVA; n > 800 neurons per condition from at least three independent experiments). (G–J) Rat hippocampal neurons (5 DIV) grown in the presence of taxol (3 nM; G and H) or DMSO (treatment after 1 DIV; I and J) and stained for Tau-1 (H and J, red) and the dendritic marker MAP2 (H and J, green). DMSO-treated neurons have formed one axon (arrows) and several dendrites (arrowheads; I) Taxol-induced processes (G, arrows) show a proximal-distal gradient of Tau-1 (H, arrows) like control axons (J, arrow). The MAP2 signal is restricted to dendrites (J, white arrowheads) and the proximal part of axons and taxol-induced processes (H and J, open arrowheads). Bars: (A–D) 20 μm; (G–J) 25 μm.
Taxol-induced processes also showed other typical axonal features. After 3 DIV, the number of cells with two or more processes positive for the axonal marker Tau-1 was increased fourfold in taxol-treated neurons compared with control neurons (Fig. 5, B, D, and F). At later stages of development, when axonal and dendritic proteins become segregated (Craig and Banker, 1994), the taxol-induced Tau-1–positive processes also showed the restriction of the dendritic marker MAP2 to the proximal part of the process as in control axons (Fig. 5, G–J, open arrowheads). In addition to the supernumerary axons, most of the cells had at least one or two dendrites. The taxol-induced processes clustered the presynaptic marker synapsin 1 later in development (10–12 DIV; Fig. 6), which was not observed in dendrites, further confirming their axonal character. Our results therefore indicate that MT stabilization causes the formation of multiple axons.
Figure 6.

Multiaxonal neurons mature and form neuronal networks. (A–E) Maturation of taxol-treated neurons. To unequivocally identify the origin of axons in the dense neuronal network neurons establish, we used cultures of mouse hippocampal neurons in which a subset of cells expressed EGFP under the control of a ubiquitously active promoter (Okabe et al., 1997). Mixed wild-type/GFP cultures containing 1–3% of neurons expressing GFP allow individual neurons to be followed in dense networks (A and B). Taxol (final concentration 3 μm) was added to the medium of the mixed culture after 1 DIV, neurons were further grown in the presence of the drug, fixed after 11 DIV, and immunostained for GFP (green) and the presynaptic marker synapsin 1 (blue). (A and B) Taxol-treated neurons have formed multiple axons (arrows) in 81.3 ± 2.2% of the cases (n > 120 neurons). (C–E) Higher magnifications of the regions marked in A and B. The multiaxonal GFP-positive neurons cluster the presynaptic marker synapsin 1 in their axons. Magnifications of the regions boxed in C–E are shown as insets to better visualize the localization of synapsin 1 on GFP-positive axons (arrows). Bars: (A and B) 100 μm; (C–E) 20 μm.
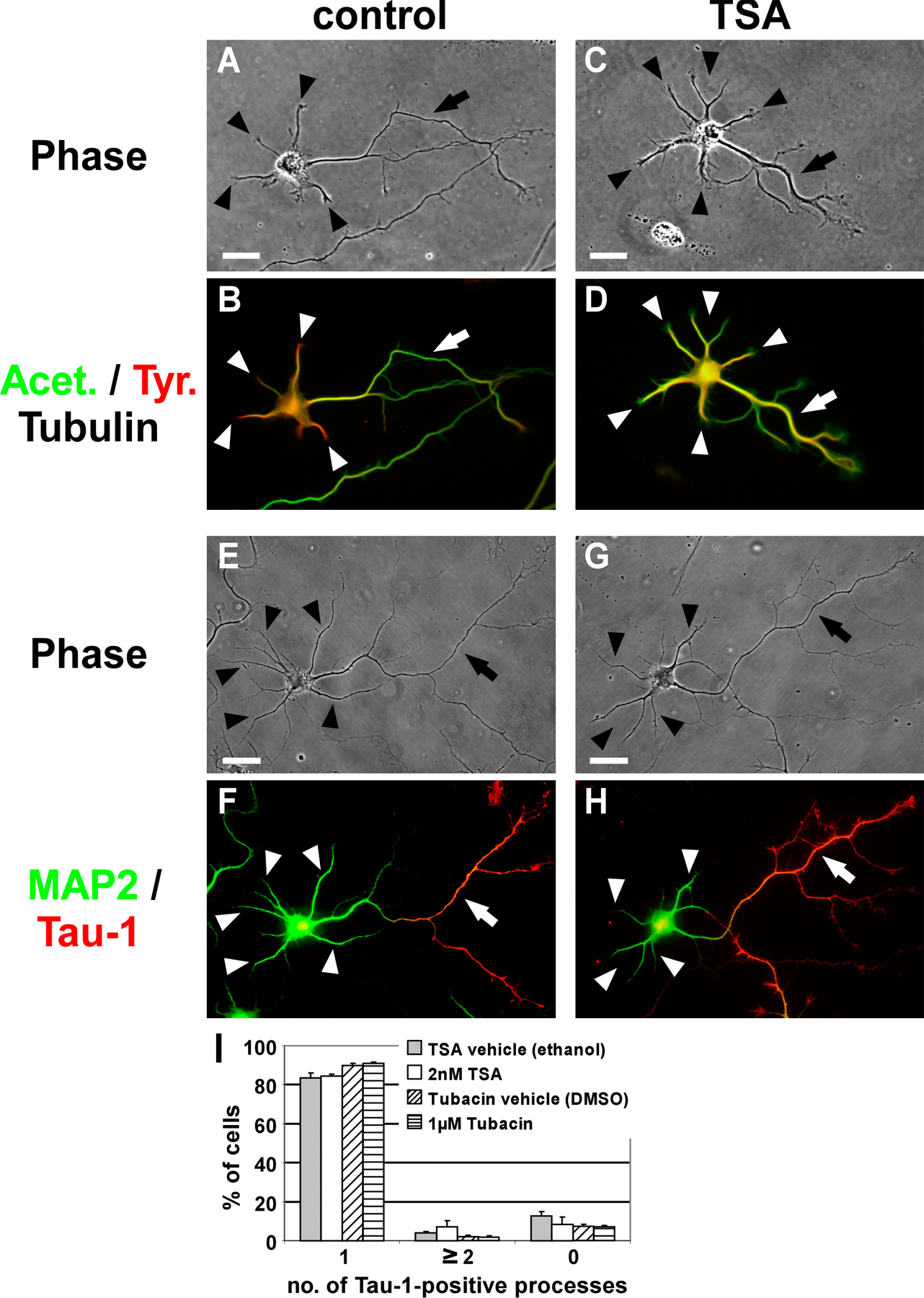
Because MT acetylation was recently reported to promote kinesin-1–mediated cargo transport to specific neurites (Reed et al., 2006), we wondered whether the axon-inducing effect of taxol is related to its MT stabilizing function or the consecutive acetylation of MTs (Fig. S2 B). Neurons cultured in the presence of the deacetylase inhibitors tubacin (1 μM) and trichostatin A (TSA; 2 nM) featured a clear increase of MT acetylation in minor neurites similar to that of axons (Fig. S3, A–D, available at http://www.jcb.org/cgi/content/full/jcb.200707042/DC1). Polarity, however, was not altered in these neurons; they showed no increase in supernumerary axons compared with control neurons (Fig. S3 I) and the distribution of dephosphorylated Tau and MAP2 was indistinguishable from control cells (Fig. S3, E–H). Thus, the axon-inducing effect of taxol appears to be linked to MT stabilization itself. Increasing acetylation of MTs does not per se affect neuronal polarity.
Taxol application rapidly stabilizes MTs and shifts dynamic MTs to the tips of processes
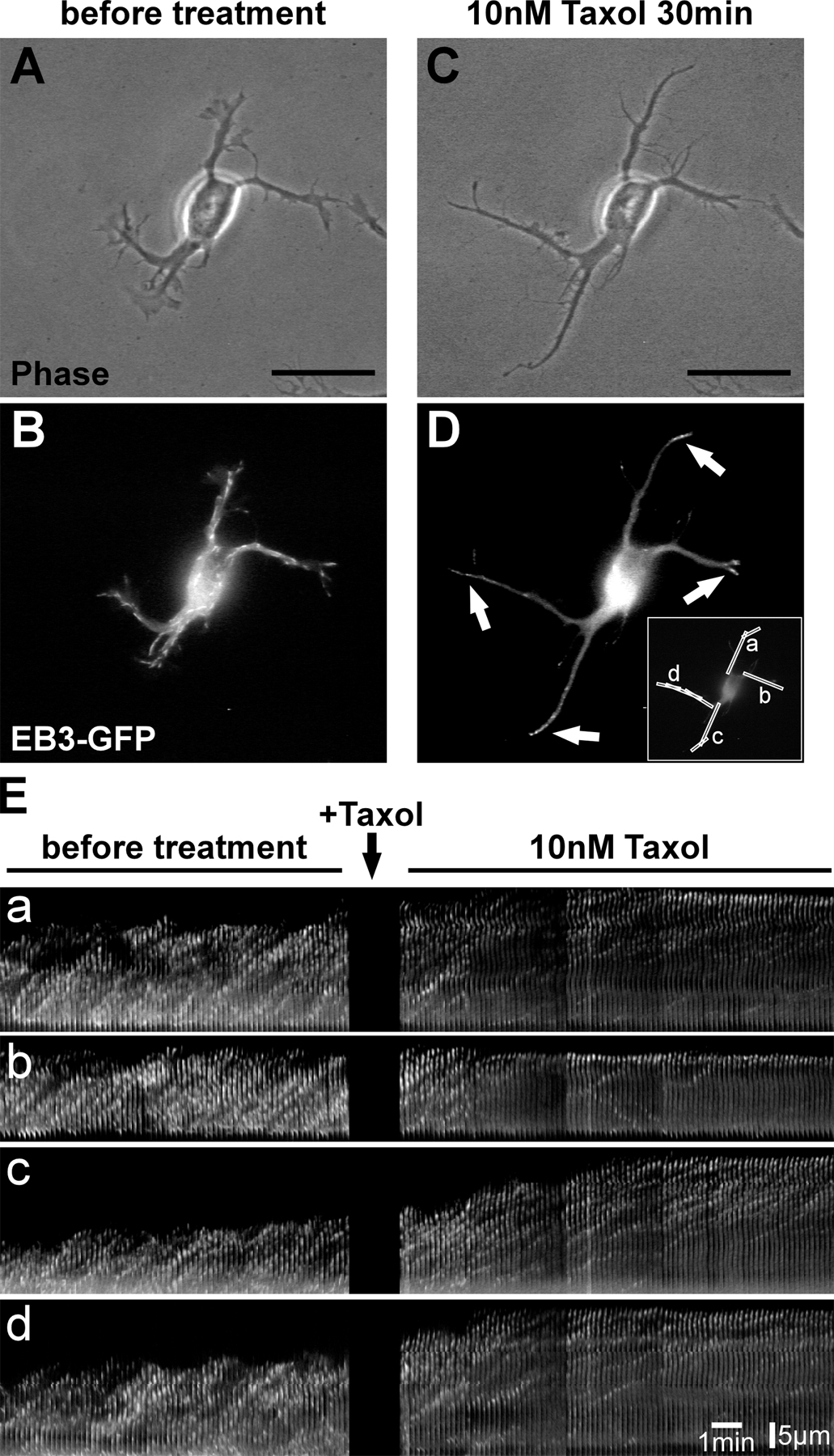
To assess MT dynamics during axon formation, polymerizing MT plus ends were visualized by transfecting neurons with the GFP-tagged MT plus end–binding protein 3 (EB3; Stepanova et al., 2003). Overall, the application of low concentrations of taxol (10 nM) reduced the dynamicity of MTs (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200707042/DC1). We observed a reduction of moving EB3-GFP particles in neurites to 51.0% of controls (≥500 moving EB3 particles of at least five neurons per condition quantified). Low concentrations of taxol, however, did not completely abolish MT dynamics like higher taxol concentrations used in earlier studies (unpublished data; Stepanova et al., 2003). Instead, low doses of taxol caused an accumulation of EB3-GFP at the tips of all minor neurites after taxol application in 84.6 ± 12.0% of the cases (Fig. 7 A–D; and Fig. S4). This directional shift toward the tip indicates the protrusion of polymerizing dynamic MTs to the growth cone periphery and was accompanied by neurite outgrowth (Fig. S4 and Video 1). Interestingly, in polarizing control neurons (Fig. 7, E and F), we observed in 64 ± 12% of the cases an enrichment of EB3-GFP in the presumptive future axonal growth cone, which was identified by its size and dynamics (Fig. 7, F and G, top, asterisk; Bradke and Dotti, 1997, 1999). Such enrichment was not seen in the growth cones of other neurites (Fig. 7 G).
Figure 7.

Taxol directs growing MT plus ends toward the tips of processes. (A–D) After 1 DIV, unpolarized neurons transfected with EB3-GFP (A) were subjected to low doses of taxol. The effect on MT dynamics was examined 30 min after treatment by monitoring the distribution of EB3-GFP (B–D). n = 52 neurons from eight independent experiments. (C) Higher magnification of the growth cones marked in A and B. EB3-GFP is mainly localized at the tips of neurites after a 30-min treatment with 10 nM taxol (a'–d') in comparison to a more even distribution before treatment (a–d). (D) Profiles of EB3-GFP immunofluorescence intensity (arbitrary units) of a representative neurite (neurite “c”) before (red) and after (green) taxol treatment. (E–G) Dynamic MT plus ends in polarizing neurons (1 DIV) visualized by transfection with EB3-GFP. (G) Higher magnification of the growth cones marked in F. The growth cone of the future axon (F and G, asterisk) harbors a high amount of dynamic MTs in comparison to the growth cones of the remaining minor neurites in 64 ± 12% of the cases (n = 14 neurons from more than five independent experiments). Bars: (A and B) 20 μm; (C) 5 μm; (E and F) 20 μm; (G) 5 μm.
We conclude that manipulations of MT stability cause drastic changes in neuronal polarity. Stabilizing MTs using taxol promotes polymerization at their plus ends, leading to an accumulation of dynamic MT plus ends at the neurite tips, which results in outgrowth of multiple axons.
Local MT stabilization is sufficient to bias the site of axon formation
During initial neuronal polarization, one out of several neurites is singled out to become the axon. It has been postulated that this selection is triggered by a positive feedback loop that allows sustained growth of the future axon (for reviews see Bradke and Dotti, 2000; Arimura and Kaibuchi, 2007). Hence, we analyzed whether MT stabilization could be part of such a proposed feedback loop by testing whether transient MT stabilization in one neurite is sufficient to trigger axon formation. One minor neurite of individual, unpolarized neurons (stage 2, 1 DIV) was randomly chosen and a membrane-permeable, photoactivatable (caged) form of taxol (1–10 nM paclitaxel,2′-[4,5-dimethoxy-2-nitrobenzyl]carbonate; Buck and Zheng, 2002) was locally activated by applying UV pulses to the selected growth cone or the region directly adjacent to it (Fig. 8 A, neurite VI, circle) for 10–15 min (Fig. 8 B). Neurons were further cultured, reimaged 2 d after uncaging (Fig. 8 E), fixed, and stained for Tau-1 and MAP2 to unambiguously identify the site of axon formation (Fig. 8 F).
Figure 8.

Local MT stabilization promotes axon formation. (A and B) Rat hippocampal neuron (1 DIV) before (A) and after (B) UV-mediated photoactivation (circle) of caged taxol at the tip of one randomly chosen minor neurite. (C and D) Photoactivation did not interfere with the overall growth cone dynamics; most growth cones, including the pulsed one (arrow), are active. (E and F) 2 d after uncaging, the pulsed process had become the axon (arrow), which is Tau-1–positive (red) and MAP2-negative (green; F). (G) Probability of axon formation in the targeted area doubles after local activation of caged taxol compared with that expected by random chance (mean ± SEM; **, P < 0.01 by χ2 test). Control treatment (DMSO and UV) does not influence randomized axon formation (P > 0.8 by χ2 test). (H–L) Caged taxol was locally activated at the tip of a nongrowing neurite (circle) of an EB3-GFP–transfected neuron at 1 DIV. Before uncaging, the chosen neurite does not grow and shows little MT dynamics (H and I [arrow], and L, 1), whereas another neurite is rapidly growing (H and I, arrowhead; also see J and K). During uncaging (L, 2 and 3), the process becomes activated, visualized by enrichment of EB3-GFP at its tip (L, 3, arrow). After uncaging, the pulsed neurite shows increased thickness (J and K). Dynamic MTs keep protruding to the peripheral part of the process, promoting its outgrowth (K and L, 4 and 5, arrow). The asterisk in H and J indicates the initial position of the neurite tip in H. Bars: (A, B, and H–K) 20 μm; (C and D) 5 μm; (E and F) 50 μm; (L) 10 μm.
Local uncaging did not interfere with growth cone dynamics (Fig. 8, C and D). To quantify the extent of site-directed axon formation caused by local activation of caged taxol, we measured the deviation of the site of axon formation from the pulsed area two days after uncaging. From 56 cells that underwent focal taxol activation, six died and nine did not develop further within the next 48 h; therefore they were excluded from the analysis. From the remaining 41 cells, 38 formed a single axon and only the other 3 neurons formed multiple axons. 68.3 ± 7.5% of the developing neurons formed an axon from the pulsed neurite or close by (within a sector deviating <30° from the pulsed area) compared with 32.9 ± 1.9% expected by random chance (P < 0.01 by χ2 test, 12 independent experiments; Fig. 8 G). In contrast, in vehicle (DMSO)-treated cells, UV pulses did not influence randomized axon formation (32.1 ± 7.8% observed vs. 34.9 ± 1.7% expected; P > 0.8 by χ2 test; n = 28 neurons from nine independent experiments). The outcome of local uncaging in terms of site-directed axon formation was independent of the size of the growth cone and the length of the neurite receiving the UV pulses. Pulsed neurites could become the axon even if the neuron had other longer neurites with bigger growth cones that had a higher probability to form the axon (Fig. 8 A; Goslin and Banker, 1989; Bradke and Dotti, 1997, 1999), a phenomenon hardly observed in vehicle control experiments. Thus, a short trigger (10–15 min) of MT stabilization is already enough to bias the site of axon formation.
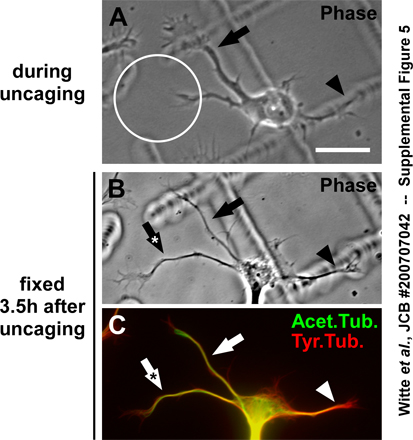
The immediate effects of local uncaging were visualized by performing the uncaging experiment with EB3-GFP–transfected neurons. Caged taxol was activated in a restricted area at the tip of a nongrowing process (Fig. 8, H and I, circle) with limited MT dynamics (visualized by the absence of EB3-GFP; Fig. 8 L, 1). Photoactivation of caged taxol promoted the protrusion of polymerizing MTs to the distal part of the process (Fig. 8, K and L, 2 and 3, arrow). This effect persisted after withdrawal of the trigger, i.e., after uncaging, and resulted in process outgrowth (Fig. 8, H, J, and L, 4 and 5). Local MT stabilization could also initiate outgrowth of a process when the cell already had another rapidly growing process (Fig. 8, H and J, arrowhead). Further assessment showed that MT acetylation increased in the uncaged neurite before axon formation took place, which indicates an increase in MT stability (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200707042/DC1). We conclude that transient local stabilization of MTs is sufficient to induce axon formation from this site.
Discussion
The recent years have seen the identification of a rapidly expanding number of molecules involved in neuronal polarization (for review see Arimura and Kaibuchi, 2007). Although these studies have led to a better understanding of the specific signaling pathways mediating neuronal polarity, the number of intracellular events that these molecules could potentially regulate has increased as well. In this respect, a substantial amount of research has so far focused on the role of the actin cytoskeleton in axon formation. Our goal was to investigate which fundamental process, besides actin dynamics, could regulate neuronal polarization.
Our data show that MT stabilization plays an active role during the specification of axonal fate in early neuronal development. Axon formation correlates with increased MT stability, and stabilizing MTs is sufficient to induce axon formation. We show how molecules and pathways that act independent of actin dynamics can govern neuronal polarization by selective alteration of MT stability.
MT stability correlates with axon formation
Previous studies analyzing the MT network in developing neuronal cells had yielded differing results. One study found stable MTs just in the proximal part of the axon of cerebellar macroneurons (Arregui et al., 1991) but another study could not confirm the confinement of stable acetylated MTs to the axon in hippocampal neurons (Dotti and Banker, 1991). By assessing the ratio of stable versus dynamic MTs in neuronal processes instead of individual posttranslational modifications of tubulin alone, we were able to reevaluate MT stability in developing neurons. We found that stable MTs are indeed not restricted to the axon but nevertheless predominate in the axonal shaft in comparison to minor neurites. The higher resistance of axonal MTs to depolymerization, which we observe, confirms increased MT stability in axons.
When we analyzed neurons with altered polarity, we further substantiated the correlation that we had found between polarized MT stability and axonal identity. Neurons deficient in the PAR-1 homologues SAD A and B (Kishi et al., 2005) showed a loss of polarity that was accompanied by a loss of polarized MT stability. In contrast, when we induced multiple axons by inhibition of GSK-3β (Jiang et al., 2005; Yoshimura et al., 2005), we found that they exhibit a prevalence of stable MTs like normal axons. Among many other targets, SAD kinases and GSK-3β regulate the affinity of MAPs to MTs (Goold et al., 1999; Kishi et al., 2005), which, in turn, control MT dynamics. Moderate inhibition of GSK-3β, for example, reduces phosphorylation-dependent inactivation of specific MAPs (for review see Doble and Woodgett, 2003; Kim et al., 2006), leading to a stabilization of MTs by increased MAP binding. Our data show that polarization of MT stability and neuronal polarization are parallel events. Moreover, interfering with the regulation of MT stability disrupts proper establishment of neuronal polarity.
MT stabilization causes axon formation
When we treated neurons with very low concentrations of taxol that favor MT polymerization yet do not block MT dynamics completely (Derry et al., 1995), we found that MT stabilization itself is sufficient to induce axon formation. Taxol-treated neurons formed multiple axons with increased axon-like MT stability. High concentrations of taxol that hyperstabilize MTs and render them completely static, however, impede axon formation as previously described (unpublished data; Dehmelt et al., 2003), which indicates that a slight, balanced shift of MT dynamics toward more stable MTs is necessary to induce axon formation. Using EB3-GFP as a marker for polymerizing MTs in living neurons, we showed that stabilization enables MTs to polymerize more distally, resulting in net elongation of the axon.
Various pathways may act on MTs to promote MT stabilization in polarizing neurons. On the one hand, signaling pathways may stabilize MTs by regulating the affinity of MAPs to MTs and thereby changing their catastrophe rates, e.g., SAD kinases or GSK-3β. On the other hand, MT stabilization can also be achieved by a reduction of active depolymerization. For instance, overexpression of the Rac activator dedicator of cytokinesis 7 (DOCK7) induces multiple axons, which may be caused by DOCK7-mediated down-regulation of the MT-depolymerizing activity of stathmin (Watabe-Uchida et al., 2006). Unlike MAPs, stathmin acts on the plus ends of MTs yet its inactivation has the same outcome, a reduced catastrophe rate. CRMP-2, in turn, promotes MT assembly in the growing axon and induces multiple axons upon overexpression (Fukata et al., 2002). We propose that the fundamental process these pathways converge on is increased MT stability in the axon that allows MTs to protrude with their dynamic ends more distally.
Currently, it is still unclear how morphological and molecular polarization are linked. MT stabilization could on the one hand enhance neurite extension, thereby triggering maturation of the axon, including molecular and functional polarization. However, MT stabilization could act more directly on the polarization process itself.
MT stabilization precedes axon formation
During initial neuronal development, one out of several seemingly equal neurites is singled out to become the axon. Our data suggest that MT stabilization in this neurite precedes morphological polarization based on the following observations: (a) MT stability is increased in one neurite of a subpopulation of morphologically unpolarized neurons; (b) neurons grown under mild MT-destabilizing conditions are able to form an axon, whereas formation of minor neurites is impaired, which suggests that only one minor neurite is able to overcome the destabilizing environment; and (c) local MT stabilization in one minor neurite by focal activation of a caged form of taxol strongly biases the site of axon formation. Consistent with our findings, the overexpressed kinesin-1 motor domain, which preferably binds to MTs containing markers of lower turnover (Reed et al., 2006), accumulates in the future axon before morphological polarization occurs (Jacobson et al., 2006). Our data argue that changes in MT dynamics are the primary cause of axon formation and that acetylation of MTs alone is not sufficient to induce initial axon formation.
How could local MT stabilization be achieved during axon formation? Presumably, either an environmental cue (Adler et al., 2006) or an internal signal like centrosome localization (Zmuda and Rivas, 1998; de Anda et al., 2005) could initiate a local imbalance inside the MT network. In the nematode Caenorhabditis elegans, for example, directional netrin cues define the site of axon initiation of a subtype of motor neurons by promoting a phosphoinositide 3-kinase (PI3K) signaling–dependent asymmetrical distribution of the actin regulator MIG-10/lamellipodin (Adler et al., 2006). Interestingly, asymmetrical PI3K signaling itself plays an important role in neuronal polarization (Menager et al., 2004) and promotes MT stabilization in migrating fibroblasts (Onishi et al., 2007). Thus, netrin-induced axon formation may function, at least in part, via regulation of MT dynamics, which is in line with the netrin turning response depending on MTs (Buck and Zheng, 2002) and MAP1B (Del Rio et al., 2004). Similarly, local inactivation of GSK-3β in one neurite of an unpolarized neuron may induce axon outgrowth by MAP-mediated MT stabilization (Kim et al., 2006). Interestingly, the small GTPase R-Ras, which can inhibit GSK-3β via PI3K and Akt-kinase, is selectively localized to a single neurite of unpolarized neurons (Oinuma et al., 2007) and might therefore account for local GSK-3β inactivation.
During early developmental stages, the centrosome and the Golgi apparatus cluster close to the area where neurons form their first neurite, which will later become the axon (Zmuda and Rivas, 1998; de Anda et al., 2005). The localization of the centrosome may therefore be responsible for an early bias of MT stability between different neurites. As the centrosome position is controlled by PI3K and Cdc42 (Etienne-Manneville and Hall, 2003), however, it may not be the cause for local MT stabilization but rather a result of axon-inducing signaling (for review see Arimura and Kaibuchi, 2007). Alternatively, a bias could come from CLIP-associated protein (CLASP)–mediated nucleation of noncentrosomal MTs at the Golgi network (Efimov et al., 2007) or other localized MT-stabilizing factors including EB1 or EB3, adenomatous polyposis coli protein, and mDia (Nakagawa et al., 2000; Wen et al., 2004; Votin et al., 2005).
Interestingly, the local MT stabilization in morphologically unpolarized neurons, which we observe, also offers an explanation for the increased membrane traffic that precedes axon formation (Bradke and Dotti, 1997). MT-dependent motor proteins show a higher affinity toward stabilized MTs (Liao and Gundersen, 1998; Reed et al., 2006), which is in line with enhanced vesicle transport on stable MTs (Lin et al., 2002; Nakata and Hirokawa, 2003). Increased MT stability in the future axon may therefore lead to polarized membrane flow and contribute to determining the site of axon formation.
In summary, we present here for the first time direct evidence that MT stability is an active determinant of neuronal polarization. Recent work has shown that actin and MTs mutually influence each other (Basu and Chang, 2007). Taking into account our findings about the instructive role of MTs in neuronal polarization, we hypothesize that the initial trigger for axon formation could derive from both actin and MTs. Their reciprocal regulation in turn may drive a positive feedback loop that sustains axonal growth. The next challenge will be to characterize this molecular interplay of MTs and the actin cytoskeleton during neuronal polarization. Moreover, molecules involved in the regulation of MT dynamics in processes like cell migration or growth cone steering should be reassessed for a potential role in neuronal polarization. Interestingly, we recently showed that MT organization is a crucial factor for the formation of the two distinct structures after axonal lesioning: actively protruding growth cones and nongrowing retraction bulbs (Ertürk et al., 2007). We therefore believe that the ability of taxol to convert a nongrowing minor neurite into a growing axon should be explored in situations where process growth is restrained, e.g., after central nervous system lesioning.
Materials and methods
Cell culture
Primary hippocampal neurons derived from rat embryos were cultured as described previously (de Hoop et al., 1997). In brief, the hippocampi of embryonic day (E) 18 rats or mice were dissected, trypsinized (0.05% Trypsin-EDTA; Invitrogen) and washed in HBSS. Cells were then dissociated with glass Pasteur pipettes and 1–1.3 × 105 cells were plated onto poly-lysine–coated glass coverslips in 6-cm Petri dishes containing MEM and 10% heat-inactivated horse serum. The cells were kept in 5% CO2 at 36.5°C. After 6–12 h, the coverslips were transferred to a 6-cm dish containing astrocytes in MEM and N2 supplements. Hippocampal neurons from E18 SAD A SAD B knockout mice (provided by J. Sanes, Harvard Medical School, Boston, MA) were cultured as described previously using wild-type astrocytes. For mixed wild-type/GFP neuronal cultures, we combined wild-type mouse hippocampal neurons with 1–3% of hippocampal neurons isolated from mice expressing EGFP (provided by M. Götz, German Research Center for Environmental Health, Neuherberg, Germany; Okabe et al., 1997).
Video microscopy and image acquisition
Living neurons were kept at 36°C on the stage of a microscope (Axiovert 135TV; Carl Zeiss, Inc.). Glass-bottom dishes (MatTek Corporation) filled with Hepes-buffered HBSS were used for observation with LD A-Plan 32× NA 0.4 or Plan Apochromat 40× NA 1.0 objectives (both from Carl Zeiss, Inc.). Alternatively, custom-made cell chambers served for live observation of neurons with a Plan Apochromat 63× NA 1.4 objective (Carl Zeiss, Inc.). When necessary, drugs were diluted to 4× concentration in HBSS and added on stage during the observation. Cells were illuminated with 100 W, 12 V halogen light or fluorescence light from a 103W/2 mercury short arc bulb. Halogen light was set to minimal intensity to avoid phototoxicity and fluorescence radiation was reduced to 5 or 25% with transmission filters to avoid phototoxicity and reduce bleaching.
Images were captured using a camera (4912 series; Cohu) at room temperature (fixed samples) or 36°C (living neurons). The camera was connected to a charge-coiled device camera control panel (C 2741; Hamamatsu). Pictures were recorded using an image grabber (LG3) and Scion Image Beta 4.0.2 (both from Scion Corp.).
FRAP
FRAP experiments were performed with a life cell imaging setup (DeltaVision RT; Applied Precision) based on an inverted fluorescence microscope (IX71; Olympus) with a UPlanApo 100× NA 1.35 objective (Olympus). The setup included a quantifiable laser module (488-nm diode laser) for bleaching GFP and an incubation chamber (Solent Scientific). Living neurons were imaged in Hepes-buffered HBSS at 36°C. Images were acquired using a camera (Photometrics CoolSnap HQ; Roper Scientific). SoftWoRx 3.5.0 (Applied Precision) was used for image recording, deconvolution (based on measured point spread function and the additive iteration method), and analysis of the FRAP experiments.
Transfection and protein expression
Neurons were transfected before plating with the Amaxa Nucleofector system using highly purified DNA (EndoFree Maxi Prep; QIAGEN). Directly after isolation of hippocampal neurons, 3 μg of pEGFP-N1-EB3 or 7.5 μg of pEGFP-T7/C1–α-tubulin plasmid DNA was used for electroporation of 500,000 cells according to the manufacturer's instructions. Subsequently, neurons were plated in 6-cm dishes containing MEM and 10% heat-inactivated horse serum and further cultured as described in Cell culture. pEGFP N1 EB3 was provided by A. Akhmanova (Erasmus Medical Center, Rotterdam, Netherlands) and V. Small (Institute of Molecular Biotechnology of the Austrian Academy of Sciences, Vienna, Austria). pEGFP-T7/C1–α-tubulin was a gift from E. Nigg and S. Nagel (Max Planck Institute of Biochemistry, Martinsried, Germany).
Immunocytochemistry
To stain for Tau-1, MAP2, GFP, or synapsin 1, cells were fixed in 4% paraformaldehyde, quenched in 50 mM ammonium chloride, and permeabilized with 0.1% Triton X-100 for 3–5 min
To assess acetylated, tyrosinated, and total tubulin integrated in MTs without unpolymerized tubulin subunits, cells were simultaneously fixed and permeabilized in PHEM buffer (60 mM Pipes, 25 mM Hepes, 5 mM EGTA, and 1 mM MgCl) containing 0.25% glutaraldehyde, 3.7% paraformaldehyde, 3.7% sucrose, and 0.1% Triton X-100 (adapted from Smith, 1994) and quenched as above.
The neurons were then blocked at room temperature for 1 h in a solution containing 2% fetal bovine serum (Invitrogen), 2% bovine serum albumin (Sigma-Aldrich), and 0.2% fish gelatin (Sigma-Aldrich) dissolved in phosphate-buffered saline. Subsequently, cells were incubated with primary antibodies diluted in 10% blocking solution. The primary antibodies used were: Tau-1 (1:5,000; Millipore), anti-MAP2 (1:6,000; Millipore), anti–α-tubulin (clone B-5-1-2, 1:20,000; Sigma-Aldrich), anti-tubulin (rabbit polyclonal, 1:200; Sigma-Aldrich), anti-acetylated tubulin (clone 6-11B-1, 1:50,000; Sigma-Aldrich), anti-tyrosinated tubulin YL1/2 (1:40,000; Abcam), anti-GFP (1:5,000; United States Biological), or anti-synapsin 1 (1:200; Millipore).
For visualization of F-actin, 4 U/ml rhodamine-coupled phalloidin (stored as a 200 U/ml methanol stock solution at −20°C) was used (Invitrogen).
As secondary antibodies, Alexa Fluor 350, 488, 555, or 568–conjugated goat anti–mouse, anti–rabbit, or anti–rat IgG antibodies or Alexa Fluor 488 or 555–conjugated donkey anti–mouse or anti–goat (all 1:500; Invitrogen) were used.
Drug treatment
Depending on the experiment, 0.3–100 nM taxol (Sigma-Aldrich), 1 μM cytochalasin D (Sigma-Aldrich), 15–225 nM nocodazole (Sigma-Aldrich), 10–20 μM SB415286 (Tocris Bioscience), 2 nM TSA (Cell Signaling Technology), and 1 μM tubacin were added to culture medium 6–24 h after plating and cells were further incubated at 36.5°C in the presence of the drug. Drugs were kept as stock solutions in DMSO (5 mM taxol, 10 mM cytochalasin D, 6.67 mM nocodazole, 25 mM SB 415286, or 20 mM tubacin) or ethanol (4 mM TSA) at −20°C. Tubacin was provided by R. Mazitschek and S. Schreiber (Harvard University and Massachusetts Institute of Technology, Cambridge, MA).
To assess MT stability, neurons grown on a coverslip with a relocation grid (Laser Zentrum Hannover) were transferred to Hepes-buffered HBSS, located, and imaged. The cells were subsequently treated with 0.075% DMSO or nocodazole (3.3–5 μM) for 5 min and then simultaneously extracted and fixed to remove unpolymerized tubulin subunits and allow clear visualization of MTs. F-actin outlining the neuron was used as a reference point to measure the retraction of MTs from the distal end of the processes toward the cell body after partial depolymerization of the MT cytoskeleton.
Image analysis and quantification
Length and intensity measurements were performed using Scion Image Beta 4.0.2 for Microsoft Windows or ImageJ analysis software. Plot profiles of fluorescence intensity were created with ImageJ. Average neurite length in experiments with long term nocodazole treatment was determined taking into account neurons without processes. To analyze movement of fluorescent particles in EB3-GFP–transfected neurons, both phase and fluorescent images were acquired in 5–10-s intervals during the experiment. Subsequently, kymographs from the regions of interest were made from the individual images using Scion Image and a purpose-written macro. EB3-particles were only considered for analysis if they could be followed clearly for three or more frames in the kymographs. The ratio of acetylated versus tyrosinated α-tubulin was determined from the mean fluorescence intensity of both channels in a square of 3 × 3 to 5 × 5 pixels in the medial part of each process after background subtraction with Photoshop (Adobe).
Focal photoactivation of caged taxol
Near-UV light (λ = 365 nm) was focused on a spot of 20–30 μm in diameter using the fluorescence iris of a microscope (Axiovert 135 TV) equipped with a Plan Apochromat 40× oil immersion objective (both from Carl Zeiss, Inc.). For the uncaging experiment with EB3-GFP–transfected neurons, a Plan Apochromat 63× oil immersion objective (Carl Zeiss, Inc.) was used instead and the cells were handled as described in Video microscopy and image aquisition. Under phase light, one dynamic growth cone of an unpolarized stage 2 neuron grown on a relocation coverslip was randomly chosen. Subsequently, caged taxol or DMSO was added to the neurons on the microscope stage to a final concentration of 1–10 nM or 0.02%, respectively. After equilibration, the tip of the selected growth cone or the region directly adjacent to it was pulsed for 10–15 min with near-UV light (pulse duration 50–100 ms; frequency 0.2–0.33 Hz). After uncaging, the neurons were incubated for another 2 d in the incubator.
Neurons were then fixed, stained for Tau-1 and MAP2, relocated, and the site of axon formation was determined. Only neurons that had formed an axon during the 48 h after uncaging were taken into account. For analysis, we measured the angle between the edge of the uncaged area, the center of the cell body, and the site of axon formation.
For calculation of the probability of random axon formation, the number of growth cones within the sector in question was divided by the total number of the cells' growth cones.
Online supplemental material
Fig. S1 shows the distribution of acetylated versus total tubulin in hippocampal neurons. Fig. S2 illustrates the induction of axon-like processes with increased MT stability by taxol. Fig. S3 shows that deacetylase inhibitor–induced acetylation of MTs alone is not sufficient to induce axon formation. Fig. S4 shows that taxol directs growing MT plus ends toward the tips of processes. Fig. S5 illustrates an increase of MT acetylation in the pulsed neurite after local uncaging of photoactivatable taxol. Video 1 demonstrates a high MT turnover in neuronal growth cones by assessing FRAP in α-tubulin–GFP–transfected neurons. Video 2 visualizes MT dynamics in EB3-GFP–transfected neurons upon taxol addition. Online supplemental materials is available at http://www.jcb.org/cgi/content/full/jcb.200707042/DC1.
Supplemental Material
Acknowledgments
We are deeply indebted to Ms. Liane Meyn for preparing hippocampal neurons. We would like to thank Drs. Axel Borst, Cord Brakebusch, Reinhard Fässler, Boyan Garvalov, Farida Hellal, Rüdiger Klein, Thomas Mayer, Gaia Tavosanis, Hans Thoenen, and Bhavna Ylera for discussing the manuscript. We are also deeply indebted to Dr. Magdalena Götz for providing transgenic GFP mice, Dr. Joshua Sanes for providing SAD A/B knockout mice, and Drs. Ralph Mazitschek and Stuart Schreiber, who provided us with tubacin through funding of the Initiative for Chemical Genetics at the National Cancer Institute. We would also like to thank Dr. Susana Gomis-Rüth for establishing mixed wild-type/GFP neuronal cultures as well as Drs. Anna Akhmanova and Vic Small for the EB3-GFP expression plasmid and Dr. Erich Nigg and Ms. Susanna Nagel for the tubulin-GFP construct. We are very grateful to Drs. Michael Schleicher and Roger Davis for support and to Dr. Thomas Waldbach for synthesis of caged taxol and further backing during the experiment. We would also like to thank Ralf Zenke for help with the FRAP experiments and Thomas Morton from Scion Corporation and Annette Dell'Aere for continued support.
Frank Bradke is a recipient of a Career Development Award from the Human Frontier Science Program (CDA0030/2003-C). This work was supported by the Max Planck Society and additional grants from the Deutsche Forschungsgemeinschaft (SFB 391).
Abbreviations used in this paper: ANOVA, analysis of variance; CRMP-2, collapsin response mediator protein 2; DIV, days in vitro; EB3, end-binding protein 3; GSK-3β, glycogen synthase kinase-3β; MAP, microtubule-associated protein; MT, microtubule; PAR-1, partitioning defective-1; PI3K, phosphoinositide 3-kinase; SAD, synapses of amphids defective; TSA, trichostatin A.
References
- Adler, C.E., R.D. Fetter, and C.I. Bargmann. 2006. UNC-6/Netrin induces neuronal asymmetry and defines the site of axon formation. Nat. Neurosci. 9:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura, N., and K. Kaibuchi. 2007. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 8:194–205. [DOI] [PubMed] [Google Scholar]
- Arregui, C., J. Busciglio, A. Caceres, and H.S. Barra. 1991. Tyrosinated and detyrosinated microtubules in axonal processes of cerebellar macroneurons grown in culture. J. Neurosci. Res. 28:171–181. [DOI] [PubMed] [Google Scholar]
- Basu, R., and F. Chang. 2007. Shaping the actin cytoskeleton using microtubule tips. Curr. Opin. Cell Biol. 19:88–94. [DOI] [PubMed] [Google Scholar]
- Bito, H., T. Furuyashiki, H. Ishihara, Y. Shibasaki, K. Ohashi, K. Mizuno, M. Maekawa, T. Ishizaki, and S. Narumiya. 2000. A critical role for a Rho-associated kinase, p160ROCK, in determining axon outgrowth in mammalian CNS neurons. Neuron. 26:431–441. [DOI] [PubMed] [Google Scholar]
- Bogoyevitch, M.A., and B. Kobe. 2006. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 70:1061–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradke, F., and C.G. Dotti. 1997. Neuronal polarity: vectorial cytoplasmic flow precedes axon formation. Neuron. 19:1175–1186. [DOI] [PubMed] [Google Scholar]
- Bradke, F., and C.G. Dotti. 1999. The role of local actin instability in axon formation. Science. 283:1931–1934. [DOI] [PubMed] [Google Scholar]
- Bradke, F., and C.G. Dotti. 2000. Establishment of neuronal polarity: lessons from cultured hippocampal neurons. Curr. Opin. Neurobiol. 10:574–581. [DOI] [PubMed] [Google Scholar]
- Buck, K.B., and J.Q. Zheng. 2002. Growth cone turning induced by direct local modification of microtubule dynamics. J. Neurosci. 22:9358–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres, A., and K.S. Kosik. 1990. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 343:461–463. [DOI] [PubMed] [Google Scholar]
- Craig, A.M., and G. Banker. 1994. Neuronal polarity. Annu. Rev. Neurosci. 17:267–310. [DOI] [PubMed] [Google Scholar]
- Crump, J.G., M. Zhen, Y. Jin, and C.I. Bargmann. 2001. The SAD-1 kinase regulates presynaptic vesicle clustering and axon termination. Neuron. 29:115–129. [DOI] [PubMed] [Google Scholar]
- de Anda, F.C., G. Pollarolo, J.S. Da Silva, P.G. Camoletto, F. Feiguin, and C.G. Dotti. 2005. Centrosome localization determines neuronal polarity. Nature. 436:704–708. [DOI] [PubMed] [Google Scholar]
- de Hoop, M.J., L. Meyn, and C.G. Dotti. 1997. Culturing hippocampal neurons and astrocytes from fetal rodent brain. In Cell Biology: A Laboratory Handbook. J.E. Celis, editor. Academic Press, San Diego, CA. 154–163.
- Dehmelt, L., F.M. Smart, R.S. Ozer, and S. Halpain. 2003. The role of microtubule-associated protein 2c in the reorganization of microtubules and lamellipodia during neurite initiation. J. Neurosci. 23:9479–9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio, J.A., C. Gonzalez-Billault, J.M. Urena, E.M. Jimenez, M.J. Barallobre, M. Pascual, L. Pujadas, S. Simo, A. La Torre, F. Wandosell, et al. 2004. MAP1B is required for Netrin 1 signaling in neuronal migration and axonal guidance. Curr. Biol. 14:840–850. [DOI] [PubMed] [Google Scholar]
- Derry, W.B., L. Wilson, and M.A. Jordan. 1995. Substoichiometric binding of taxol suppresses microtubule dynamics. Biochemistry. 34:2203–2211. [DOI] [PubMed] [Google Scholar]
- Doble, B.W., and J.R. Woodgett. 2003. GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti, C.G., and G. Banker. 1991. Intracellular organization of hippocampal neurons during the development of neuronal polarity. J. Cell Sci. Suppl. 15:75–84. [DOI] [PubMed] [Google Scholar]
- Dotti, C.G., C.A. Sullivan, and G.A. Banker. 1988. The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 8:1454–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov, A., A. Kharitonov, N. Efimova, J. Loncarek, P.M. Miller, N. Andreyeva, P. Gleeson, N. Galjart, A.R. Maia, I.X. McLeod, et al. 2007. Asymmetric CLASP-dependent nucleation of noncentrosomal microtubules at the trans-Golgi network. Dev. Cell. 12:917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk, A., F. Hellal, J. Enes, and F. Bradke. 2007. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J. Neurosci. 27:9169–9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2003. Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature. 421:753–756. [DOI] [PubMed] [Google Scholar]
- Fukata, Y., T.J. Itoh, T. Kimura, C. Menager, T. Nishimura, T. Shiromizu, H. Watanabe, N. Inagaki, A. Iwamatsu, H. Hotani, and K. Kaibuchi. 2002. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4:583–591. [DOI] [PubMed] [Google Scholar]
- Garvalov, B.K., K.C. Flynn, D. Neukirchen, L. Meyn, N. Teusch, X. Wu, C. Brakebusch, J.R. Bamburg, and F. Bradke. 2007. Cdc42 regulates cofilin during the establishment of neuronal polarity. J. Neurosci. 27:13117–13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold, R.G., R. Owen, and P.R. Gordon-Weeks. 1999. Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J. Cell Sci. 112(Pt 19):3373–3384. [DOI] [PubMed] [Google Scholar]
- Goslin, K., and G. Banker. 1989. Experimental observations on the development of polarity by hippocampal neurons in culture. J. Cell Biol. 108:1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes, C.A., and R.S. Jope. 2001. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 65:391–426. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., K. Chihara, N. Arimura, C. Menager, Y. Kawano, N. Matsuo, T. Nishimura, M. Amano, and K. Kaibuchi. 2001. CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 4:781–782. [DOI] [PubMed] [Google Scholar]
- Jacobson, C., B. Schnapp, and G.A. Banker. 2006. A change in the selective translocation of the Kinesin-1 motor domain marks the initial specification of the axon. Neuron. 49:797–804. [DOI] [PubMed] [Google Scholar]
- Jiang, H., W. Guo, X. Liang, and Y. Rao. 2005. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 120:123–135. [DOI] [PubMed] [Google Scholar]
- Kim, W.Y., F.Q. Zhou, J. Zhou, Y. Yokota, Y.M. Wang, T. Yoshimura, K. Kaibuchi, J.R. Woodgett, E.S. Anton, and W.D. Snider. 2006. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 52:981–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi, M., Y.A. Pan, J.G. Crump, and J.R. Sanes. 2005. Mammalian SAD kinases are required for neuronal polarization. Science. 307:929–932. [DOI] [PubMed] [Google Scholar]
- Kunda, P., G. Paglini, S. Quiroga, K. Kosik, and A. Caceres. 2001. Evidence for the involvement of Tiam1 in axon formation. J. Neurosci. 21:2361–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, G., and G.G. Gundersen. 1998. Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J. Biol. Chem. 273:9797–9803. [DOI] [PubMed] [Google Scholar]
- Lin, S.X., G.G. Gundersen, and F.R. Maxfield. 2002. Export from pericentriolar endocytic recycling compartment to cell surface depends on stable, detyrosinated (glu) microtubules and kinesin. Mol. Biol. Cell. 13:96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menager, C., N. Arimura, Y. Fukata, and K. Kaibuchi. 2004. PIP3 is involved in neuronal polarization and axon formation. J. Neurochem. 89:109–118. [DOI] [PubMed] [Google Scholar]
- Nakagawa, H., K. Koyama, Y. Murata, M. Morito, T. Akiyama, and Y. Nakamura. 2000. EB3, a novel member of the EB1 family preferentially expressed in the central nervous system, binds to a CNS-specific APC homologue. Oncogene. 19:210–216. [DOI] [PubMed] [Google Scholar]
- Nakata, T., and N. Hirokawa. 2003. Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J. Cell Biol. 162:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oinuma, I., H. Katoh, and M. Negishi. 2007. R-Ras controls axon specification upstream of glycogen synthase kinase-3beta through integrin-linked kinase. J. Biol. Chem. 282:303–318. [DOI] [PubMed] [Google Scholar]
- Okabe, M., M. Ikawa, K. Kominami, T. Nakanishi, and Y. Nishimune. 1997. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 407:313–319. [DOI] [PubMed] [Google Scholar]
- Oliva, A.A. Jr., C.M. Atkins, L. Copenagle, and G.A. Banker. 2006. Activated c-Jun N-terminal kinase is required for axon formation. J. Neurosci. 26:9462–9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi, K., M. Higuchi, T. Asakura, N. Masuyama, and Y. Gotoh. 2007. The PI3K-Akt pathway promotes microtubule stabilization in migrating fibroblasts. Genes Cells. 12:535–546. [DOI] [PubMed] [Google Scholar]
- Reed, N.A., D. Cai, T.L. Blasius, G.T. Jih, E. Meyhofer, J. Gaertig, and K.J. Verhey. 2006. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 16:2166–2172. [DOI] [PubMed] [Google Scholar]
- Smith, C.L. 1994. Cytoskeletal movements and substrate interactions during initiation of neurite outgrowth by sympathetic neurons in vitro. J. Neurosci. 14:384–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova, T., J. Slemmer, C.C. Hoogenraad, G. Lansbergen, B. Dortland, C.I. De Zeeuw, F. Grosveld, G. van Cappellen, A. Akhmanova, and N. Galjart. 2003. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J. Neurosci. 23:2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, E., T. Ho, and M.W. Kirschner. 1995. The role of microtubule dynamics in growth cone motility and axonal growth. J. Cell Biol. 128:139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez, R.J., B. Howell, A.M. Yvon, P. Wadsworth, and L. Cassimeris. 1997. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol. Biol. Cell. 8:973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votin, V., W.J. Nelson, and A.I. Barth. 2005. Neurite outgrowth involves adenomatous polyposis coli protein and beta-catenin. J. Cell Sci. 118:5699–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida, M., K.A. John, J.A. Janas, S.E. Newey, and L. Van Aelst. 2006. The Rac activator DOCK7 regulates neuronal polarity through local phosphorylation of stathmin/Op18. Neuron. 51:727–739. [DOI] [PubMed] [Google Scholar]
- Wen, Y., C.H. Eng, J. Schmoranzer, N. Cabrera-Poch, E.J. Morris, M. Chen, B.J. Wallar, A.S. Alberts, and G.G. Gundersen. 2004. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 6:820–830. [DOI] [PubMed] [Google Scholar]
- Westermann, S., and K. Weber. 2003. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 4:938–947. [DOI] [PubMed] [Google Scholar]
- Wodarz, A. 2002. Establishing cell polarity in development. Nat. Cell Biol. 4:E39–E44. [DOI] [PubMed] [Google Scholar]
- Yoshimura, T., Y. Kawano, N. Arimura, S. Kawabata, A. Kikuchi, and K. Kaibuchi. 2005. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 120:137–149. [DOI] [PubMed] [Google Scholar]
- Zmuda, J.F., and R.J. Rivas. 1998. The Golgi apparatus and the centrosome are localized to the sites of newly emerging axons in cerebellar granule neurons in vitro. Cell Motil. Cytoskeleton. 41:18–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}