

Abstract
Melanocortin receptors are considered promising candidates for the treatment of behavioral and metabolic disorders ranging from obesity to anorexia and cachexia. These experiments examined the response of mice to peripheral injections of two compounds. PG932 is a derivative of SHU9119 which is non-selective antagonist of melanocortin-3 and melanocortin-4 receptors (Mc3r, Mc4r). PG946 is a derivative of a hybrid of alpha- and beta-MSH, and is a moderately selective Mc3r antagonist. SHU9119 increases food intake when administered intracerebroventricularly but is without effect when injected into the periphery. In contrast, PG932 was found to be highly effective at stimulating food intake when administered peripherally by intraperitoneal injection. The orexigenic effect of PG932 required functional Mc4r, suggesting that inhibition of this receptor is involved in the stimulation of food intake. PG946 did not significantly affect on feeding behavior. PG932 is thus a useful new compound for studies examining the regulation of appetite and energy balance, and may also prove useful for the treatment of cachectic conditions.
Keywords: Melanocortin receptor, antagonist, anorexia, appetite, endotoxin, proopiomelanocortin
1. Introduction
Melanocortin neurons are a critical component of the gut-brain and adipose-brain circuits involved in energy homeostasis, the long term balancing of energy intake and expenditure to maintain body weight at a constant level [2, 9]. Neurons secreting the endogenous ligands for melanocortin receptors expressed in the brain are located in the arcuate nucleus of the hypothalamus and the nucleus tractus solitarius in the brainstem [9, 14, 37]. The melanocortins α- and β-melanocyte stimulating hormone (α-MSH), products of the proteolytic cleavage of proopiomelanocortin (POMC), have been implicated in the control of energy balance [9]. Central administration of the α-MSH derivative melanotan-II (MT-II) reduces food intake and increases oxygen consumption through activation of Mc4r [8, 28]. Conversely, central administration of non-selective antagonists of the MC3R and Mc4r, such as the endogenous antagonist agouti-related peptide (AgRP) or the synthetic melanocortin analog SHU9119, increase food intake and body weight when administered intracerebroventricularly [10, 32]. The regulation of feeding behavior and energy expenditure by MSH is through to primarily involve Mc4r [8, 28]. Analysis of Mc3r and Mc4r knockout mice indicate a minimal role for the Mc3r in regulating food intake [5]. However, recent data suggests that stimulation of MC3R by peripheral administration of a moderately selective agonist D-trp8-γMSH can increase food intake, perhaps through stimulation of neurons co-expressing NPY and AgRP [26].
Pharmacological manipulation of the balance of caloric intake and daily energy expenditure is considered an important goal in the treatment of diseases ranging from obesity and insulin resistance to cachexia and anorexia associated with infection, renal failure, or cancer [24, 36]. Several circulating and neural peptides have been identified that act on sites in the hypothalamus and brainstem to regulate energy homeostasis [13, 37]. The hypothalamic melanocortin system is a critical target for many of the peripheral and central factors that regulate ingestive behavior and metabolism [5, 9]. Non-selective agonists of the Mc3r and Mc4r suppress food intake and increase energy expenditure, primarily through Mc4r [8, 28]. Cytokine stimulation of proopiomelanocortin neurons in the hypothalamus is thought to have an important role in weight loss associated with chronic illness [33]. Administration of antagonists of the Mc3r and Mc4r, or genetic blockade of Mc4r, are effective at ameliorating cachexia induced by bacterial endotoxin (lipopolysaccharide, or LPS) or cancer in several animal models [25, 27, 35, 36].
In this report, we present data from experiments testing the biological activity of two MSH derivates on food intake. PG-932 is a derivative of SHU9119 (SHU9119: Ac-Nle-c[Asp-His-D-Nal(2′)-Arg-Trp-Lys]-NH2, PG-932: Ac-Nle-c[Asp-Pro-D-Nal(2′)-Arg-Trp-Lys]-Pro-Val-NH2). PG932 has the same affinity as SHU9119 at the Mc3r, with evidence for a modest (7-fold) selectivity for the Mc4r [15]. The second peptide, PG946, is a cyclic α-MSH/β-MSH hybrid that is a selective Mc3r antagonist [3]. Since the peripheral administration of D-trp8-γMSH can increase food intake [26], we were interested in determining whether PG-932 administered peripherally would have an anorectic effect. We demonstrate herein that unlike SHU9119, PG932 dose-dependently increases food intake when administered peripherally, most likely through antagonism at Mc4r. Moreover, PG932 administered peripherally also attenuated the anorexic and illness-induced behavioral effects of LPS. PG932 thus appears to be novel potent orexigenic peptide that stimulates food intake when administered peripherally, and which could be useful in the treatment of anorectic and cachectic disorders.
2. Materials and Methods
2.1 Animal husbandry
The Pennington Biomedical Research Center Institutional Animal Care and Use Committee reviewed and approved these experiments. All mice utilized in this study, unless indicated otherwise, were approximately 8 wks of age. C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mc4r knockout (Mc4rKO) mice backcrossed 12+ generations onto the B6 background were obtained from a colony maintained at the Pennington Biomedical Research Center. Offspring of heterozygote by heterozygote matings were genotyped as described previously [1]. Mice were housed on a 12 h light: dark period (lights on: 0700–1900 h) and given ad libitum access to standard Laboratory Rodent Diet (Purina Mills, St Louis, MO). Naïve mice were used for all of these studies.
2.2 Drug Treatments
SHU9119 was purchased from Phoenix Pharmaceuticals (Belmont, CA); LPS was purchased from Sigma (Escherichia coli 055:B5, Sigma, St. Louis, MO.). PG932 was synthesized, and purified, as previously described, except for the peptide cyclization, which was induced with DIC/HOBt in DMF [15]. For food intake studies, animals were housed for 2 weeks in wire mesh cages to allow for adaptation. Mice were given ad libitum access to a low fat diet (Research Diets low fat diet 12450B, 10% kJ/fat) to minimize hyperphagia. A known amount of food was added to a hopper at the front of the cage and a tray was anchored below the cage to catch any spillage. Food intake was calculated by subtracting the amount of food remaining in the hopper after a defined period, and adjusted for the amount of spillage.
For the dose response experiment, mice were food deprived overnight and administered an i.p. dose of either PG932 (0.0, 0.4, 1.2, 2.0 or 4.0 mg/kg, n=6/group) or saline soon after lights on (0700). A measured amount of food was given to the mice at the time of injection, and intake measured 1, 2, 4 and 6 hr post injection.
For chronic studies, a single i.p. injection of a selective Mc4r antagonists, PG932 (0.4 mg/kg), or a selective Mc3r antagonist, PG946 (0.4 and 1.5 mg/kg), or saline, was administered at lights out (1800) and food intake was measured soon after lights on (0800–1000) for 9 consecutive days (n=8/group).
For PG932/SHU9119 comparison experiments, both SHU9119 and PG932 were solubilized in 0.9% saline. Mice were food deprived overnight and a dose of 4 mg/kg PG932, or 4 mg/kg SHU9119, or saline (n=6/group) was administered soon after lights on (0700) the following morning.
The mechanism of PG932-induced hyperphagia was examined using Mc4rKO mice [21]. Knockout and age and weight matched wild-type littermates were individually housed in wire mesh cages. Mice were food deprived overnight and either PG932 (4 mg/kg) or saline was administered i.p. after lights on and food intake was monitored at 1 and 2 hours post injection (n=6/group).
Effects of PG932 on endotoxin-induced anorexia and malaise
For LPS administration, animals were housed individually in cages and provided free access to running wheels (Mini-Mitter Co., Bend, OR) for 14 days prior to experimental testing. LPS was dissolved in 0.09% saline and administered i.p. In the first experiment, a single dose of LPS (100 μg/kg) or saline was administered 2 hours prior to lights out. One hour before lights out, a single dose of PG932 (4 mg/kg) or saline (n=6/group) was administered and food intake was monitored for 1, 2, and 4 hours. Wheel running activity was monitored by magnetic switch closures and analyzed using Vital View (Mini-Mitter, Bend, OR) software. In a second experiment, 32 C57BL/6J mice were acclimated to housing in wheel cages. LPS or saline was administered to 16 mice 2 hr prior to lights out. Half of each group receiving LPS or saline where then injected with two injections of PG932 (4 mg/kg) or saline, one injection given 1 hour prior to lights out and another given 30 minutes before lights out. Food intake and activity were monitored as described above.
2.3 Statistics
All data are presented as mean ± SE. Sigmastat software (SPSS Inc., Chicago, IL) was used for statistical analysis. Analysis of two or multiple groups of data used either Student’s t-test, or analysis of variance (ANOVA) respectively for normally distributed data. Food intake data was analyzed using 2-way ANOVA (genotype, treatment) with repeated measures (treated, untreated). Student-Newman-Keuls post hoc tests were performed after ANOVA analysis. Statistical significance was assumed for p values < 0.05.
3. Results
3.1 Effect of acute and chronic administration of PG932 on food intake
The first experiment was a dose-response study performed to determine if peripheral injection of PG932, a SHU9119 derivative with modest selectivity for Mc4r over Mc3r [15], would acutely effect on food intake. We also wanted to address whether PG932 could regulate food intake in the presence of elevated hypothalamic NPY/Agrp expression as a consequence of an overnight fast (8). Food intake was monitored 1, 2, 4, and 6 h after a single intraperitoneal injection of 0.4, 1.2, 2.0, or 4.0 mg/kg PG932 to overnight food deprived mice. The dose range was based on previous studies where melanocortin ligands were administered peripherally ([7] and our unpublished data). PG932 had a significant and dose-dependent effect on food intake at the 1 h time point compared to saline (Fig. 1A). The 4 mg/kg dose also increased food intake, compared to all other treatment groups, at the 2 h time point (Fig 1A). However, by 4 h the effect of PG932 was no longer significant. Thus, a single peripheral injection of PG932 results in a short-term increase of food intake, with the magnitude and duration of the increase correlating with dose.
Figure 1. Effect of chronic and acute administration of PG932 on food intake.

(A) Dose-dependent increase of food intake by PG932. The data show food intake 1, 2 and 4 h after a single intraperitoneal injection of PG932 at 0.4, 1.2, 2.0 or 4.0 mg/kg (n=6/group) to male B6 mice, compared to saline treated controls, after an overnight fast. Significantly different from saline controls, * P<0.05, † P<0.01. (B). Daily administration of a single i.p. injection of 0.4 mg/kg PG932 for 8 d significantly increased cumulative food intake compared to saline treated controls (* P<0.05). Cumulative food intake for the pre-treatment period was not significantly different between groups, the treatment period is indicated by arrows.
The second experiment involved four groups of C57/B6 mice (n=8), matched for body weight. The initial objective was to examine the physiologic response to long term peripheral treatment with a sub-threshold dose of PG932. A second objective was to examine the effect of PG946, a cyclic α-MSH/β-MSH hybrid that is a selective Mc3r antagonist [3], on food intake. Food intake and body weight during the lead-in period, the 7 days prior to treatment, were not significantly different between groups (Fig. 1B, and data not shown). Over time, mice treated with PG932 consumed approximately 20% more food than saline-treated controls (Fig. 1B) (cumulative food intake over 9d in kJ: saline, 344 ± 21 kJ; 0.4 mg/kg PG932, 425 ± 19 kJ; P<0.05 vs. saline). However, the 20% increase in cumulative food intake in mice treated with PG932 was not associated with significantly increased weight gain (data not shown). PG946 at 0.4 and 1.5 mg/kg did not significantly affect food intake compared to saline controls (387 ± 15 and 387 ± 16 kJ, respectively), or body weight (data not shown).
3.2 Comparison of PG932 and its derivative, SHU9119, on food intake after peripheral injection
The results from the first experiment indicate that PG932 is a more biologically potent compared to the other frequently used melanocortin receptor antagonists, SHU9119 and AgRP, which require intracerebroventricular administration to increase food intake. In a third experiment, we directly compared the effects of a single peripheral injection of PG932 with SHU9119, which is a potent orexigen when administered centrally but is without effect when administered peripherally [10]. We compared our highest dose of PG932 with a slightly elevated dose of SHU9119 when molecular weights of the compounds are factored in to the dosing regimen (PG932 1230 M.W.; SHU9119, 1074 M.W.). While 4 mg/kg of PG932 significantly, and transiently, increased food intake as observed in the dose-response study, SHU9119 given in an equivalent dose had no effect on food intake (Fig. 2).
Figure 2. PG932 but not SHU9119 increases food intake when administered peripherally.

Food intake after a single i.p. injection of saline, PG932 (4 mg/kg) or SHU9119 (4 mg/kg) in 16 hr food deprived mice (n=6/group). Food intake was monitored at 1, 2, and 4 hr after injection. PG932 treatment significantly increased food intake at the 2 h time point. * P<0.01 compared to saline, P<0.05 compared to SHU9119.
3.3 Simulation of food intake by PG932 requires a functional Mc4r
The majority of published data from studies indicate that the acute effects of melanocortin agonists on feeding behavior require Mc4r [7, 8, 12, 28]. To test specificity of the effect of PG932 on feeding behavior, we examined its effects on food intake in wild type and Mc4rKO mice. Weight and age matched male Mc4rKO, along with wild type littermates were food deprived overnight, and given either 4 mg/kg PG932 (determined from dose response studies carried out above) or saline on test day (n=6/group). Figure 3 shows that PG932 treated wild type mice ate significantly more than their saline injected controls at the 1 hr time point (7.2 ± 1.0 vs 12.2 ± 0.91, P>0.01). In contrast, peripheral injection of PG932 in Mc4rKO did not increase food intake in these mice (6.07 ± 0.76 vs 6.35 ± 1.17). These results demonstrate that PG932 requires functional Mc4r to affect food intake.
Figure 3. Increased food intake in mice receiving PG932 injections (solid bar) compared to saline (open bar) requires functional MC4R.
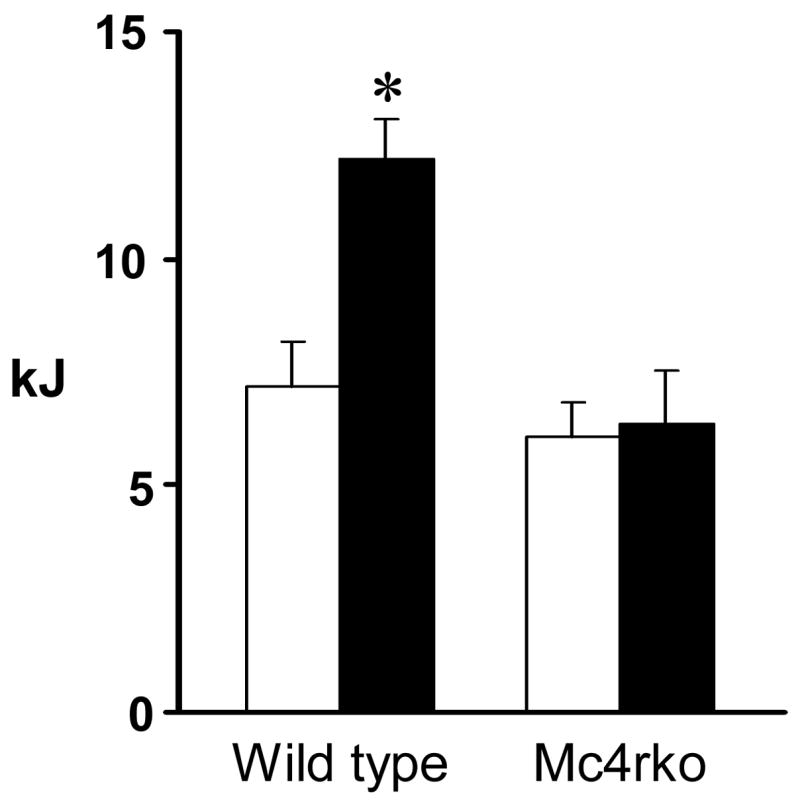
Food intake was monitored after an overnight fast in either C57BL/6J wild type mice, or mice null for the MC4R given a single i.p. dose of PG932 (4 mg/kg). PG 932 had a significant effect on food intake at the 1 h time point with no effect observed in MC4R knock out mice (n=6/group). * P<0.05 compared to saline treated controls.
3.4 Peripheral injections of PG932 transiently reduce anorexia and illness behavior associated with lipopolysaccharide treatment
Mc4r are involved in the anorexia associated with lipopolysaccaride and tumor-induced cachexia [20, 25, 27, 35, 36]. To determine if antagonism of Mc4r by PG932 can attenuate illness behavior, we assessed food intake and spontaneous physical activity of mice treated with LPS (Fig. 4, Fig. 5). Mice were given free access to running wheels and, after 14 d to establish baseline activity mice were separated into four treatment groups (n=6 for each group; saline, LPS, PG932, or a combination of both PG932 and LPS).
Figure 4. Peripheral injections of PG932 transiently inhibit lipopolysaccharide induced anorexia.

C57BL/6J mice were singly housed in shoebox cages and allowed free access to a running wheel. Two hours before lights out, LPS (100 μg/kg) was injected followed by an injection of PG932 administered either singly, 1 h before lights out (exp.1), or with an additional injection just before lights out (exp. 2). (A). PG932 had a significant effect on LPS-induced hypophagia as well as increasing basal food intake when given alone at the 1 hr time point. An additional injection of PG932 did not have an additive effect on food intake either in the presence of LPS or when given alone. (B). No significant increase in food intake was observed at the 2 hr time point either alone or in the presence of LPS. Columns not sharing letters are significantly different, P<0.01. N=8/group.
Figure 5. Peripheral injections of PG932 transiently inhibit the reduction in physical activity associated with lipopolysaccharide injections.
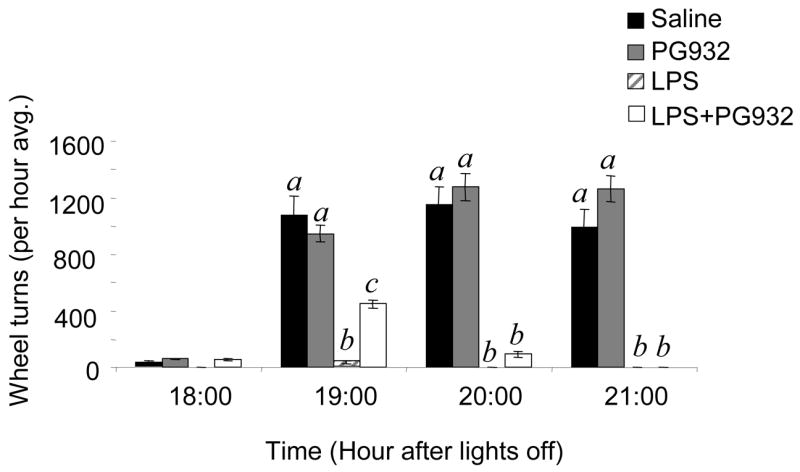
C57BL/6J mice were singly housed in shoebox cages and allowed free access to a running wheel. LPS injection was associated with a marked reduction in wheel running activity. Two injections of PG932, 1hr and 30 min prior to lights out had a transient but significant effect to improve wheel running behavior during the first 3 h of the dark period (1900–2100 h) in mice treated with LPS. Columns not sharing letters are significantly different, P<0.01. N=6/group.
Along with wheel running behavior, we also measured PG932 effects on food intake behavior using our high dose of PG932. A single injection of PG932 (4 mg/kg, given 1 hour before lights out) increased food intake to a similar degree as observed previously (Fig 4A, exp1). LPS treatment (100 μg/kg given 2 hours prior to lights out) significantly reduced food intake compared to saline treated mice (6.9 ± 0.8 vs 2.2± 0.6 kJ, P>0.01). A single injection of PG932 (4 mg/kg), given 1 h before lights out in the presence of LPS significantly increased food intake at the 1 hr time point after lights out compared to LPS treatment alone (5.3 ± 1.0 vs 2.2 ± 0.6 kJ, P<0.01). As expected, this dosing regimen increased food intake in the PG932 group comparable to saline-treated controls (6.9 ± 1.0 vs 9.9 ± 1.1 kJ, P<0.05). No effect of PG932 was observed at the 2 hr time point (i.e., 3 hrs post PG932 injection), at which time PG932/LPS injected mice appeared to have a similar degree of anorexia as LPS treatment alone (Figure 4B, exp. 1).
A single injection of PG932 did not significantly affect wheel running behavior, either alone or in terms of preventing suppression of spontaneous physical activity associated with LPS injection (data not shown). To determine if extending the duration of treatment results in a significant improvement of both feeding behavior and activity, a second experiment was performed where mice were administered two injections of PG932 (4 mg/kg), the first injection administered 1 hour after LPS administration with a second injection being administered 30 minutes before lights out. Two injections of PG932 did not appear to be more effective than one injection in preventing LPS-induced anorexia (Fig 4A, exp.2, 3.4 ± 1.1 vs 7.6 ± 1.5 kJ, P<0.01). However, a significant (P<0.01) but transient increase in wheel running behavior was observed for 3 h in mice receiving LPS and two injections of PG932, compared to mice treated with LPS alone (Fig. 5). No difference in wheel running behavior was observed between untreated and PG932 treated mice.
4. Discussion
The main finding presented in this report is that PG932, a derivative of SHU9119, a non-selective antagonist of the Mc3r and Mc4r [19], significantly increases food intake when administered by peripheral injections. In fasted mice, a single peripheral injection of PG932 increased food intake, and inhibited the anorexia and malaise associated with LPS injection. The magnitude and duration of the response was dose-dependent. As would be predicted, the stimulation of food intake by PG932 was found to depend on a functional Mc4r, suggesting antagonism of Mc4r as the mechanism involved in the stimulation of food intake.
SHU9119 was discovered in a screen of analogs of MTII, a potent cyclic lactam non-selective melanocortin receptor agonist (Ac-Nle-c[Asp-His-D-Phe-Arg-Trp-Lys]-NH2) [19]. Hruby et al. reported that the substitution of the D-Phenylalanine in MTII with D-Naphthylalanine results in a compound which is a functional antagonist at the MC3R and Mc4r [19]. SHU9119 increases food intake when administered intracerebroventricularly, but not peripherally [10]. PG932 differs from SHU9119 in that the histidine has been replaced by a proline, with an extra two amino acids (proline, valine) added to the C-terminus [15]. Using cell-based assays, these substitutions were found not to affect the affinity for human MC3R, MC4R, or MC5R, and the antagonism at the human MC3R and MC4R is retained. However, the results from the current experiments suggest a dramatic, and significant, increase in biological potency such that behavioral effects are observed with peripheral administration.
The current study did not address the site of action of PG932 to increase food intake, although the effect was shown to be dependent on functional Mc4r. Peripheral administration of MTII, a non-selective agonist of the Mc3r and Mc4r, suppresses food intake at high doses. The effect of MTII on food intake when administered either centrally or peripherally is dependent on functional Mc4r [7, 8, 34], however the sites of action when administered peripherally are not clear. Peripherally administered MTII does not induce the same pattern of c-Fos expression, an indicator of neuronal activity, observed following intracerebroventricular MTII injections [34]. In the same study, analysis of the uptake of peripherally administered iodinated MTII also indicated limited penetrance of this peptide into the brain. Overall, the analysis by Trivedi et al. suggested that peripherally administered MTII likely regulates food intake at sites outside the blood brain barrier, or in circumventricular organs which (subfornical organ, median eminence, area postrema and choroid plexus), or through other peripheral systems [34]. Mc4r mRNA has been reported in the subfornical organ, median eminence, and area postrema [22, 23, 30]. However, the results from the analysis of mutant mice where expression of Mc4r can be conditionally rescued indicate that receptors in paraventricular nucleus of the hypothalamus and amygdale are critical for the anorectic effects associated with treatment with melanocortin receptor agonists [4].
These studies do not address why PG932 is more physiologically potent than SHU9119 when administered peripherally. The affinities of PG932 and SHU9119 for melanocortin receptors are similar; therefore, one possible hypothesis to explain the increased potency of PG932 would be an increase in stability and bioavailability. One could speculate that increased stability of PG932 in the circulation allows for passage to circumventricular organs, or across the blood brain barrier, into the central nervous system. Indeed, the regulation of energy homeostasis by Mc4r involves the central nervous system, with peripheral Mc4r having little effect on energy homeostasis [4]. Peptides are allowed access to receptors located in neuronal circuits in two main areas of the brain, the median eminence located at the level of the hypothalamus, which receives projections from the arcuate nucleus of the hypothalamus, and at the level of the area postrema in the brainstem. Both of these regions contain neurons that express Mc4r and that could influence feeding behavior [22, 23, 30]. Future studies involving radioligand binding to determine the specific hypothalamic and hindbrain regions as well as specific neuronal phenotypes in these regions would be useful in elucidating the central mechanism involved in the biological activity of PG932.
Another potential mechanism for the effects of PG932 on energy balance could be through the regulation of insulin. Mc4r have been located in many central brain regions that control glucose homeostasis such as the lateral hypothalamic area (LH), ventromedial hypothalamus (VMH), and paraventricular nucleus (PVN) [30]. Central and peripheral injections of Mc4r agonists have been shown to reduce serum insulin levels through melanocortin regulation of sympathetic outflow to the pancreas [11]. SHU9119 has been shown to effect insulin levels when given peripherally, but has no reported effect on food intake [10]. Future studies are warranted to determine if different derivatives of melanocortin antagonists have differential effects on serum insulin through blockade of central Mc4r.
The increase of food intake associated with PG932 injections in normal and during conditions of LPS-induced anorexia was transient and, at the doses employed for these studies, did not last much more then 2 hours. The duration of hyperphagia did not appear to be prolonged when two doses of PG932 were given within a 90 min time span, although the addition of the second dose did improve wheel running behavior.
It is also interesting to note that the lowest dose of PG932, 0.4 mg/kg, while not effective at increasing food intake acutely did result in a modest increase in food intake when administered over 9 days. It is possible that the transient effects of PG932 on food intake allow compensatory mechanisms to prevent weight gain associated with the 20% increase in food intake. However, there may also be significant differences in the mechanisms by which PG932 and other antagonists affect homeostasis. For example, it is known that central administration of a single picomole dose of AgRP, and SHU9119, increase food intake for up to seven days, possibly due to long term alterations in the activity of neurons involved in reward and satiety [16, 17]. AgRP is also an inverse agonist at the Mc4r, and it not known whether PG932 has inverse agonist activity [18, 31]. At this time it is not clear whether PG932 administered peripherally can access the same neurons as SHU9119 or AgRP administered centrally. However, continuous infusion of PG932 may be required for weight gain to occur.
The second compound tested, PG946, did not significantly affect feeding behavior. PG946 is a moderately selective Mc3r antagonist [3]. Peripheral injections of an Mc3r agonist were recently reported to increase food intake, perhaps through regulation of neurons expressing the potent orexigens Neuropeptide Y and AgRP in the arcuate nucleus [26]. The Mc3r may also be involved in the stimulation of food intake by AgRP, which is an inverse agonist and antagonist at the Mc3r and Mc4r [29]. However, the analysis of feeding behavior in Mc3r knockout mice has produced inconsistent results ranging from hypophagia [7] and normal food intake [6] on chow diets to modest hyperphagia [5]. The current data may suggest that injection of an antagonist to the Mc3r is not an effective approach to reducing food intake. However, it is also possible that the bioavailability of PG946 is, like that of SHU9119, low when compared to PG932, and that this compound does not efficiently access sites in central nervous system areas involved in appetite regulation.
In conclusion, these studies demonstrate that a derivative of the non-selective melanocortin receptor antagonist SHU9119 has potent efficacy in attenuating LPS induced malaise and increases basal food intake behavior in male C57BL/6J mice. This effect was not observed in Mc4r null mice, supporting the conclusion that this peptide mainly functions to stimulate food intake by inhibiting Mc4r within the CNS. No effect of food intake was observed with equivalent doses of either the selective Mc3/4r antagonist, SHU9119, or the selective peptide antagonists for the Mc3r, PG946. This is the first report characterizing a functional peptide derivative of an antagonist at Mc4r that may have important value in clinical or basic science research where i.c.v. administration is unwanted or not possible.
Acknowledgments
We thank the staff of the Department of Comparative Biology at the Pennington Biomedical Research Center for assistance with the preparation of this manuscript. Dr Dennis Huszar (Millennium Pharmaceuticals) and Dr Roger Cone (Vollum Institute) kindly provided the MC4R knockout mice used in these studies.
Grants: AB has been or is currently supported by grants from the Health Excellence Fund (HEF) of the Lousiana State University Board of Regents, the Metabolife Settlement Fund, and the American Diabetes Association (1-04-JF09). AB is partially supported by a CNRU Center Grant # 1P30 DK072476 entitled “Nutritional Programming: Environmental and Molecular Interactions” sponsored by NIDDK. This work was also supported by the National Institutes of Diabetes, Digestive and Kidney Diseases (DK-068330 to AB; DK-17420 to VJH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Gregory M. Sutton, Neuropeptides Laboratory, Pennington Biomedical Research Center, Lousiana State University System, LA
M. Josephine Babin, Neuropeptides Laboratory, Pennington Biomedical Research Center, Lousiana State University System, LA.
Xuyuan Gu, Department of Chemistry, University of Arizona, AZ.
Victor J. Hruby, Department of Chemistry, University of Arizona, AZ
Andrew A. Butler, Neuropeptides Laboratory, Pennington Biomedical Research Center, Lousiana State University System, LA
References
- 1.Albarado DC, McClaine J, Stephens JM, Mynatt RL, Ye J, Bannon AW, Richards WG, Butler AA. Impaired coordination of nutrient intake and substrate oxidation in melanocortin-4 receptor knockout mice. Endocrinology. 2004;145:243–52. doi: 10.1210/en.2003-0452. [DOI] [PubMed] [Google Scholar]
- 2.Badman MK, Flier JS. The gut and energy balance: visceral allies in the obesity wars. Science. 2005;307:1909–14. doi: 10.1126/science.1109951. [DOI] [PubMed] [Google Scholar]
- 3.Balse-Srinivasan P, Grieco P, Cai M, Trivedi D, Hruby VJ. Structure-activity relationships of novel cyclic alpha-MSH/beta-MSH hybrid analogues that lead to potent and selective ligands for the human MC3R and human MC5R. J Med Chem. 2003;46:3728–33. doi: 10.1021/jm030111j. [DOI] [PubMed] [Google Scholar]
- 4.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 5.Butler AA. The melanocortin system and energy balance. Peptides. 2006;27:301–309. doi: 10.1016/j.peptides.2005.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–21. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 7.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 8.Chen AS, Metzger JM, Trumbauer ME, Guan XM, Yu H, Frazier EG, Marsh DJ, Forrest MJ, Gopal-Truter S, Fisher J, Camacho RE, Strack AM, Mellin TN, MacIntyre DE, Chen HY, Van der Ploeg LH. Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res. 2000;9:145–54. doi: 10.1023/a:1008983615045. [DOI] [PubMed] [Google Scholar]
- 9.Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 10.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–8. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 11.Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. 2000;141:3072–9. doi: 10.1210/endo.141.9.7665. [DOI] [PubMed] [Google Scholar]
- 12.Fan W, Ellacott KL, Halatchev IG, Takahashi K, Yu P, Cone RD. Cholecystokinin-mediated suppression of feeding involves the brainstem melanocortin system. Nat Neurosci. 2004 doi: 10.1038/nn1214. [DOI] [PubMed] [Google Scholar]
- 13.Flier JS. Obesity wars. Molecular progress confronts an expanding epidemic. Cell. 2004;116:337–50. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- 14.Friedman JM. The function of leptin in nutrition, weight, and physiology. Nutr Rev. 2002;60:S1–14. doi: 10.1301/002966402320634878. discussion S68–84, 85–7. [DOI] [PubMed] [Google Scholar]
- 15.Grieco P, Balse-Srinivasan P, Han G, Weinberg D, MacNeil T, Van der Ploeg LH, Hruby VJ. Extensive structure-activity studies of lactam derivatives of MT-II and SHU-9119: their activity and selectivity at human melanocortin receptors 3, 4, and 5. J Pept Res. 2003;62:199–206. doi: 10.1034/j.1399-3011.2003.00087.x. [DOI] [PubMed] [Google Scholar]
- 16.Hagan MM, Benoit SC, Rushing PA, Pritchard LM, Woods SC, Seeley RJ. Immediate and prolonged patterns of Agouti-related peptide-(83--132)- induced c-Fos activation in hypothalamic and extrahypothalamic sites. Endocrinology. 2001;142:1050–6. doi: 10.1210/endo.142.3.8018. [DOI] [PubMed] [Google Scholar]
- 17.Hagan MM, Rushing PA, Pritchard LM, Schwartz MW, Strack AM, Van Der Ploeg LH, Woods SC, Seeley RJ. Long-term orexigenic effects of AgRP-(83---132) involve mechanisms other than melanocortin receptor blockade. Am J Physiol Regul Integr Comp Physiol. 2000;279:R47–52. doi: 10.1152/ajpregu.2000.279.1.R47. [DOI] [PubMed] [Google Scholar]
- 18.Haskell-Luevano C, Monck EK. Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regul Pept. 2001;99:1–7. doi: 10.1016/s0167-0115(01)00234-8. [DOI] [PubMed] [Google Scholar]
- 19.Hruby VJ, Lu D, Sharma SD, Castrucci AL, Kesterson RA, al-Obeidi FA, Hadley ME, Cone RD. Cyclic lactam alpha-melanotropin analogues of Ac-Nle4-cyclo[Asp5, D-Phe7, Lys10] alpha-melanocyte-stimulating hormone-(4-10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–61. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 20.Huang QH, Hruby VJ, Tatro JB. Role of central melanocortins in endotoxin-induced anorexia. Am J Physiol. 1999;276:R864–71. doi: 10.1152/ajpregu.1999.276.3.R864. [DOI] [PubMed] [Google Scholar]
- 21.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–41. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 22.Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457:213–35. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- 23.Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J Neurosci. 2003;23:7143–54. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marks DL, Butler AA, Cone RD. Melanocortin pathway: animal models of obesity and disease. Ann Endocrinol (Paris) 2002;63:121–4. [PubMed] [Google Scholar]
- 25.Marks DL, Butler AA, Turner R, Brookhart G, Cone RD. Differential role of melanocortin receptor subtypes in cachexia. Endocrinology. 2003;144:1513–23. doi: 10.1210/en.2002-221099. [DOI] [PubMed] [Google Scholar]
- 26.Marks DL, Hruby V, Brookhart G, Cone RD. The regulation of food intake by selective stimulation of the type 3 melanocortin receptor (MC3R) Peptides. 2005 doi: 10.1016/j.peptides.2005.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marks DL, Ling N, Cone RD. Role of the central melanocortin system in cachexia. Cancer Res. 2001;61:1432–8. [PubMed] [Google Scholar]
- 28.Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21:119–22. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 29.Marsh DJ, Miura GI, Yagaloff KA, Schwartz MW, Barsh GS, Palmiter RD. Effects of neuropeptide Y deficiency on hypothalamic agouti-related protein expression and responsiveness to melanocortin analogues. Brain Res. 1999;848:66–77. doi: 10.1016/s0006-8993(99)01962-9. [DOI] [PubMed] [Google Scholar]
- 30.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 31.Nijenhuis WA, Oosterom J, Adan RA. AgRP(83-132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol Endocrinol. 2001;15:164–71. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 32.Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D, Abusnana S, Goldstone AP, Russell SH, Stanley SA, Smith DM, Yagaloff K, Ghatei MA, Bloom SR. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology. 1998;139:4428–31. doi: 10.1210/endo.139.10.6332. [DOI] [PubMed] [Google Scholar]
- 33.Scarlett JM, Jobst EE, Enriori PJ, Bowe DD, Batra AK, Grant WF, Cowley MA, Marks DL. Regulation of central melanocortin signaling by interleukin-1 beta. Endocrinology. 2007;148:4217–25. doi: 10.1210/en.2007-0017. [DOI] [PubMed] [Google Scholar]
- 34.Trivedi P, Jiang M, Tamvakopoulos CC, Shen X, Yu H, Mock S, Fenyk-Melody J, Van der Ploeg LH, Guan XM. Exploring the site of anorectic action of peripherally administered synthetic melanocortin peptide MT-II in rats. Brain Res. 2003;977:221–30. doi: 10.1016/s0006-8993(03)02683-0. [DOI] [PubMed] [Google Scholar]
- 35.Wisse BE, Frayo RS, Schwartz MW, Cummings DE. Reversal of cancer anorexia by blockade of central melanocortin receptors in rats. Endocrinology. 2001;142:3292–301. doi: 10.1210/endo.142.8.8324. [DOI] [PubMed] [Google Scholar]
- 36.Wisse BE, Schwartz MW, Cummings DE. Melanocortin signaling and anorexia in chronic disease states. Ann N Y Acad Sci. 2003;994:275–81. doi: 10.1111/j.1749-6632.2003.tb03190.x. [DOI] [PubMed] [Google Scholar]
- 37.Zigman JM, Elmquist JK. Minireview: From anorexia to obesity--the yin and yang of body weight control. Endocrinology. 2003;144:3749–56. doi: 10.1210/en.2003-0241. [DOI] [PubMed] [Google Scholar]