

Summary
Age is the single most important prognostic factor in the development of many cancers. The major reason for this age-dependence is thought to be the progressive accumulation of oncogenic mutations and epigenetic changes. Similarly, mutagens are thought to be carcinogenic primarily by engendering oncogenic mutations. Yet while the accumulation of heritable somatic changes is expected to augment the incidence of oncogenic mutations, a major effect of increased mutation load is reduced fitness. We propose that the fitness of progenitor cell compartments substantially impacts on the selective advantage conferred by particular mutations. We hypothesize that reduced cellular fitness within aged stem cell pools can select for adaptive oncogenic events and thereby promote the initiation of cancer. Thus, certain oncogenic mutations may be adaptive within aged but not young stem cell pools. We further argue that accumulating genetic alterations with age or mutagen exposure might promote cancer not only by causing oncogenic hits within cells but also by leading to eventual reduction in stem cell fitness, which then selects for oncogenic events. Therefore, initial stages of cancer development may not be limited by the incidence of initiating oncogenic changes, but instead by contexts of reduced cellular fitness that select for these changes.
Keywords: mutation, evolution, cancer, fitness, Bcr-Abl, aging
Introduction
Initiation and progression of cancers can be reconciled in terms of Darwinian evolution [1–4]. If an oncogenic mutation confers a selective advantage to a mutated cell, this advantage should allow for clonal expansion, initiating the first required step for tumorigenesis. Clonal expansion substantially increases the chances of secondary oncogenic mutations occurring in the cell clone harboring the initiating oncogenic event. If a secondary mutation provides an additional selective advantage, a second round of clonal selection ensues; repeating this cycle can eventually lead to cancer. Thus, cancer progression can be reconstructed from an evolutionary perspective as a number of consecutive mutational changes that progressively increase the selective advantage of the mutated cells.
Age is the largest risk factor for cancer development in mammals. The incidence of many human cancers rises exponentially with age, dramatically increasing after midlife. As a result, more than 80% of human malignancies are diagnosed after 50 years of age [5–7]. Overwhelming evidence supports the concept of the multistage evolution of cancer involving a series of oncogenic epigenetic changes and mutations leading to oncogene activation or tumor suppressor gene inhibition [8, 9]. Therefore, the prevailing logic linking aging and cancer rates is straightforward: increasing age leads to the accumulation of genomic changes, and since some of these mutations and epigenetic events are expected to be oncogenic, the chance of developing a malignancy should increase with age dependent on the buildup of oncogenic events [10, 11] (see Box 1 for definitions).
Box 1: Definitions for the purposes of our model.
Fitness
The capacity of a cell to survive and proliferate relative to other cells competing in the same stem cell niche (i.e. whether representation of the cell clone within the progenitor cell pool increases or decreases). For our model, we are concerned with the fitness of cells with self-renewal capacity.
Mutation
We will often refer to heritable epigenetic and genetic mutational changes generally as “mutations”.
Oncogenic mutations or events
Heritable epigenetic and genetic changes, including the activation of oncogenes or inactivation of tumor suppressor genes, which have the potential to contribute to cancer initiation or progression.
Adaptive mutation
A mutation that provides a cell with improved fitness relative to the population average. Selection for adaptive mutations should become stronger as the fitness of a population declines.
Progenitor cells
Stem and progenitor cell populations with at least some self-renewal capacity, even if not for the life of the organism. To serve as targets for oncogenesis, progenitor cells should possess sufficient self-renewal to allow for the accumulation of additional oncogenic events (unless an early event conferred self-renewal).
Because current views of aging-associated cancer postulate that the progressive accrual of mutations in oncogenes and tumor suppressors account for increased cancer rates, most human cancers are thought to develop over decades [9]. The exponentially increased incidence of most human cancers late in life has led to mathematical models predicting the requirement for 5–7 rare events (mutations) over many years. Prevailing views consider the incidence of oncogenic mutations to be the rate-limiting step in tumorigenesis [12].
Since achieving a reasonable probability of acquiring secondary oncogenic mutations requires substantial expansion of the mutant clone, and since single oncogenic mutations do not appear sufficient for cellular transformation, initiating oncogenic events capable of generating a tumor may need to occur in stem and progenitor cells that possess substantial self-renewal capacity. From an evolutionary perspective, initiating oncogenic mutations have to provide a positive fitness gain in order to be fixed and trigger clonal expansion. Importantly, this fitness gain is relative to other cells in a population, and for the initial stages of tumorigenesis these other cells are the normal cells occupying and competing for the same niches as the initiated cells. That cells within a progenitor cell population are in competition has perhaps been best studied in Drosophila, where a dominant clone can even actively eliminate competing cells with reduced fitness [13, 14]. Progenitor cells in mammals should also compete with each other for niches, nutrients and growth factors, and at least in an adult, progenitor pools are homeostatically maintained at relatively constant numbers. We would therefore argue that understanding the initial stages of tumor evolution from an evolutionary perspective requires careful consideration of the change in fitness conferred by an oncogenic mutation relative to the fitness of other cells in the same progenitor cell population.
It is often presumed that particular initiating oncogenic mutations invariably lead to clonal expansion once they occur in the appropriate cell type. However, selection of oncogenic mutations appears to be channeled by multiple constraints imposed by the tissue environment [15]. An additional important consideration is that cancer is a rare disease in young, healthy individuals, and thus mammals have clearly evolved robust mechanisms to greatly limit cancer development until old age. We will argue that the high fitness of a stem cell population in a young, healthy individual represents a powerful barrier to tumor development, by creating an environment that is not conducive for selection of oncogenic mutations.
We propose an Adaptive Oncogenesis model whereby declining cellular fitness promotes cancer initiation by selecting for adaptive oncogenic mutations (Figure 1). The fitness of progenitor cell populations is reduced with age as the result of deleterious cell-intrinsic alterations (both genetic mutations and heritable epigenetic changes) and as the result of age-related changes in the stem and progenitor cell environment. The buffering capacity of cells (eg. via genetic redundancy) and tissues (eg. excess stem cell reserves) should result in a lag in the fitness decline despite accumulating mutations. In our model, declining stem cell pool fitness creates selective pressure for adaptive mutations, which can be oncogenic, increasing the probability of an initial round of clonal expansion which in turn translates into increased cancer incidence late in life. Therefore, the same oncogenic mutation that may be non-adaptive and thus not fixed within a youthful stem pool can become adaptive in an aged stem pool, promoting cancer initiation.
Figure 1. Adaptive Oncogenesis.

In this model, conditions that reduce cellular fitness (such as aging or mutagen exposure) within progenitor cell pools select for adaptive mutations which can be oncogenic. When mutations and epigenetic changes accumulate to the point where the buffering capacity of the progenitor cell pool is exhausted (denoted by dotted line), the fitness of the population will begin to decline. Declining fitness will then increase selective pressure for adaptive oncogenic mutations that in turn promote cancer initiation. Note that the shape of the fitness curve is hypothetical and not based on data or mathematical modeling.
Thus, we argue that the increased incidence of cancer with aging might be limited not only by the incidence of oncogenic mutations, but also by the creation of an environment that is permissive for the clonal expansion of mutant cells. We will build support for this model, which could radically change our understanding of links between aging, mutation accumulation and cancer (see Box 2 for the major points of our argument). Notably, much of the support for our model will be from the reinterpretation of some of the same observations used to support the current model of cancer as the result of the slow progressive accumulation of oncogenic events, including 1) the exponential increased cancer incidence with advancing age, 2) the accumulation of mutations and epigenetic changes with age, and 3) increased cancer incidence resulting from exposure to mutagens or in individuals with DNA repair deficiencies.
Box 2: Major points of our argument.
1) A major effect of accumulating mutations and epigenetic changes with age is the reduction of fitness in populations of progenitor cells.
2) Mutations or epigenetic changes in a cell causing phenotypic alterations should rarely be advantageous within a young, healthy stem cell population. Reductions of cellular fitness should increase the selective impact of certain oncogenic mutations, leading to expansion of cell clones with adaptive oncogenic mutations.
3) Cancer may not be limited by the accumulation of oncogenic mutations or epigenetic changes, which occur in susceptible cells throughout our lifetime. Rather, differential selection for these events under contexts of poor fitness (such as in old age) may promote the fixation of oncogenic mutations or epigenetic events in a population.
4) Cancer development may be more rapid than previously thought. Earlier exposure to agents causing genomic damage may predispose to cancer decades later not by directly inducing oncogenic mutations, but by reducing the pool of fit stem and progenitor cells (leading to premature loss of fitness within these pools).
Mutations and epigenetic changes accumulate with age
Over a lifetime, our cells are exposed to DNA damaging products of cellular metabolism and environmental mutagens. Since DNA repair mechanisms possess less than 100% fidelity, this exposure, together with rare replication errors, leads to the accumulation of mutations with age. Indeed, while different experimental approaches produce different estimates of the magnitude of the mutational load, overwhelming evidence supports the idea that mutations do accumulate with age on both chromosomal and gene levels, while the rate of accumulation does not appear to change [16]. Substantial accumulation of mutations has also been reported for mitochondrial DNA [17, 18].
Moreover, the extent of accumulation of heritable epigenetic changes may far exceed the accumulation of genomic mutations. While DNA methylation patterns are heritable, the replication fidelity of these patterns (ca. 10−5 error rate for CpG sites) is several orders of magnitude less than that for the replication of the DNA base sequence [19]. Consequently, DNA methylation patterns deteriorate with age [20]. While most CpG sites (which start out mostly methylated) become progressively hypomethylated with aging [21], hypermethylation of CpG islands in promoter regions (which start out mostly unmethylated) occurs with increased numbers of cell divisions [19].
Mutations and epigenetic changes reduce average cellular fitness
The most widely accepted idea of the nature of aging is that aging is caused by the accumulation of somatic mutations that decrease the fitness of cells, therefore reducing the fitness of tissues, organs and the organism as a whole [22–27]. According to the hypothesis known as Muller’s Ratchet proposed by Hermann Muller over 40 years ago, an asexual population should inevitably accumulate deleterious mutations, leading to unavoidable, ratchet-like reduction of fitness of the most fit clone, progressively reducing the overall fitness of the population [28]. This hypothesis has been experimentally validated in unicellular organisms: in the absence of recombination populations of microbes exhaust and become extinct or enter a state of senescence [29, 30].
Muller’s ratchet should also apply to somatic cells of multicellular organisms. Accumulating genomic damage with aging should pave a one-way road to decreased fitness. Importantly, decreased cellular fitness is a much more likely outcome for random mutations and epigenetic changes than activation of oncogenes or inactivation of tumor-suppressor genes, as random changes are far more likely to decrease fitness than to produce a trait that can be favored by selection (especially in a highly adapted population) [31]. The applicability of Muller’s ratchet is clearly not limited to heritable changes associated with aging. Increased exposure to environmental mutagens or increased mutation rates due to cell-intrinsic factors, such as augmented production of reactive oxygen species or inherited defects in DNA repair efficiency, should lead to similar declines in fitness. Indeed, experimental increases in random mutation load have been reported to cause faster loss of fitness for E. coli, with a strong correlation between the number of mutations and loss of fitness [32]. While direct testing of the applicability of Muller’s ratchet to somatic cells of multicellular organisms is difficult, it has been demonstrated that increased mutation rates result in a competitive disadvantage for hematopoietic stem cells (HSC) [33–35], arguing that increased mutational load results in fitness reductions in mammalian progenitor cells.
Decisions that determine cellular fate, such as those controlling proliferation, differentiation, and stem cell maintenance, are governed by sophisticated intracellular circuitry including the orchestrated expression of hundreds of genes. Therefore, accumulation of random mutations and epigenetic changes in coding and regulatory sequences are expected to alter the fine-tuning of this regulation. One consequence of random genomic alterations might be the reported increase in cell-to-cell variation in gene expression [36]; while the average level of expression within the population for any given gene does not change significantly, variability increases. As increased cell-to-cell variability in gene expression can be recapitulated by treatment of cells with hydrogen peroxide, the authors postulate that accumulating genomic damage with age disrupts nuclear architecture and patterns of gene expression [36]. Others have also suggested that “noise” in gene expression patterns could reduce cellular fitness [37]. DNA damage and the resulting repair, both during aging and following genotoxic exposures, can also modify epigenetic programming and nuclear architecture, leading to altered gene expression [38]. While some have speculated that age related methylation changes may lead to altered expression of tumor suppressor genes and oncogenes, leading to the selection of hyperproliferative clones and cancer [21], we instead argue that stochastic epigenetic changes will be more likely to decrease cellular fitness, as random unselected change tends to be disadvantageous.
Still, mutations in many genes important for cellular function can be surprisingly tolerated, due in part to a diploid genome. Moreover, compensatory pathways can sometimes assume the functions of a missing component. Therefore, while random mutations and epigenetic changes occur from the beginning of life, this plasticity may allow for maintenance of high organismal fitness through a sufficient age to allow for reproductive success. However, when too many components are altered or disabled, the buffering capacity of this plasticity becomes exhausted, and as a result, cellular fitness drops (Figure 1).
The fitness of stem cell populations might also be reduced as a result of alterations in their niche. Accumulating genomic damage should impair the normal function of cells and tissues that constitute the stem cell microenvironment. Indeed, aging is associated with substantial changes in the architecture of most tissues [39]. Extensive telomere shortening, normally associated with aging, has been shown to diminish lymphopoiesis and HSC function non-cell autonomously by altering the bone marrow environment [40]. Thus, even if stem cells themselves manage to preserve genetic/epigenetic integrity, they will remain adapted to an environment that no longer exists, while their adaptation to the aged environment is suboptimal. Conversely, rejuvenation of the environment might result in restoration of stem cell fitness. Indeed, serum from young animals or transplantation into a young host can rejuvenate muscle stem cells from old rodents [41, 42], highlighting the importance of non-cell autonomous influences of aging on stem cells.
Mutations or epigenetic changes in a cell causing phenotypic alteration should not be advantageous within a fit cell population
Initiating oncogenic mutations are often presumed to provide a selective advantage should they occur in the relevant progenitor cell type. We would argue otherwise. As multicellular organisms with long life-span have been evolving under selective pressure to prevent tumorigenesis, the majority of oncogenic mutations that have the potential to promote rogue expansion should be coupled to a fitness disadvantage. Indeed, most oncogenic mutations that stimulate cellular proliferation engage powerful intrinsic tumor suppressor mechanisms, such as by activating programs of apoptosis or senescence [43].
Stabilizing selection as a tumor suppressive mechanism
We argue that oncogenic mutations may also engage an additional powerful inherent tumor suppressor mechanism that causes a mutant cell to lose its stem-cell properties and therefore to be eliminated from the stem cell pool. The inherent organization of stem cell niches with complex regulatory interactions (including contacts with other cells, the extracellular matrix and diffusible factors) creates a very tight set of requirements for stem cell maintenance. From analyzing intra- and inter-species variation in gene expression patterns, multiple studies indicate that stabilizing selection greatly constrains gene expression divergence (i.e. changes in gene expression frequently reduce the fitness of the organism) [44]. Stabilizing selection is the evolutionary principle whereby large changes in an organismal trait are selected against, limiting genetic diversity [45]. As described above, gene expression becomes more variable with age. We propose that the complex organization of stem cell niches drives strong stabilizing selection that renders the wild-type phenotype to be the most adapted, thereby ensuring that most initiating oncogenic mutations bring about fitness decline (Figure 2, top panel). Even if an oncogenic mutation provides both strong proliferative and survival advantages, but fails to meet all of the strict requirements for remaining in the niche, such a mutation will most likely be lost from the stem cell niche. For example, while deletion of the PTEN tumor suppressor gene in adult HSC stimulates their proliferation, PTEN loss also leads to HSC depletion via cell-autonomous mechanisms [46, 47]. Similarly, overexpression of the c-Myc oncogene in HSC induces proliferation, but leads to loss of self-renewal activity presumably due to disrupted adhesion within the niche [48]. Some level of stem cell quiescence appears required for maintenance in the stem cell pool [49]. Thus, the complicated niche requirements of stem cells should represent a potent tumor suppressor mechanism, in that stem cells are highly adapted to their niches in a young healthy individual, minimizing selective pressure for heritable adaptive changes.
Figure 2. Stabilizing selection prevents oncogenesis in a fit stem cell pool, but can select for adaptive oncogenic mutations in a poorly fit pool.
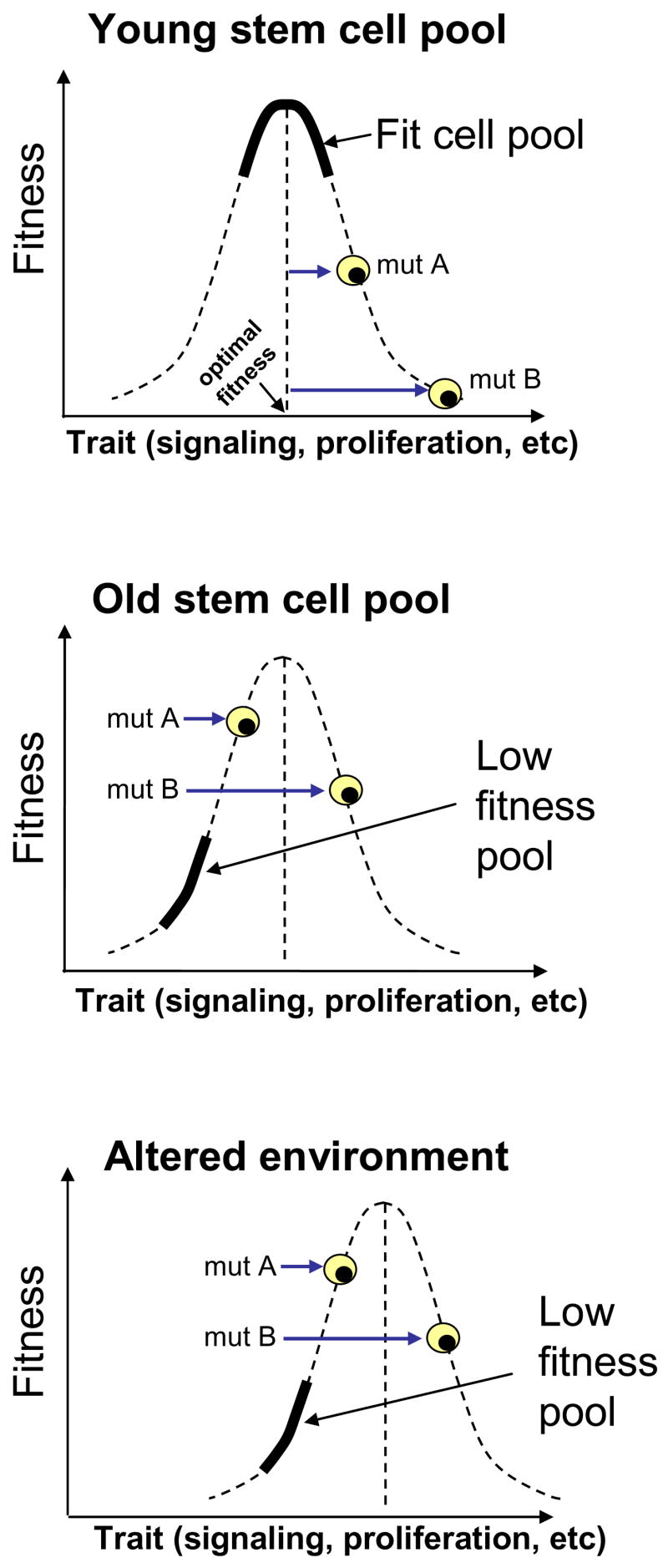
Top: A young, fit progenitor cell pool (solid curve) should possess traits that confer close to the optimal fitness for cellular maintenance in that pool. The dashed curve describes the relationship between trait and fitness. Oncogenic and non-oncogenic mutations that change the trait (mutations A and B) will tend to move the cell away from the optimum towards lower fitness. Middle: In contrast, the same oncogenic mutations in a low fitness pool may become adaptive, such as by promotion of signaling in a signaling deficient background. The low fitness pool is represented by a solid curve. In this example, the average trait value is reduced, and fitness is now limited by the trait level. In this low fitness pool, oncogenic mutations that increase the intensity of the trait become adaptive, leading to net improvement in fitness relative to the population average. Bottom: Likewise, following degradation of the stem cell environment (without necessarily directly damaging the stem cells), the relationship between trait and fitness is altered (dashed curve shifted right), and now the wild-type stem cell phenotype no longer confers optimal fitness, causing certain oncogenic mutations to become adaptive.
Reduction in cellular fitness should select for oncogenic mutations
We have thus far argued that accumulating heritable changes will lead to reduced stem cell fitness. But why should reduction in cellular fitness lead to selection of clones with oncogenic mutations? One consequence of alterations of the fine-tuned cellular circuitry might be loss of normal responsiveness to proliferation signals. For example, aged B cell progenitors display dramatically reduced proliferation in response to interleukin-7 stimulation [50]. One can easily envision how in such a scenario oncogenic events that confer partial independence from growth factors might result in a net increase in fitness compared to a scenario of normal responsiveness to growth stimuli. In addition, decreased stem cell fitness and consequent inadequate production of mature progeny are also expected to result in increased homeostatic demand, leading to higher levels of pro-survival growth factor signaling and thereby increasing thresholds for triggering apoptosis or senescence by oncogenic mutations.
Likewise, multiple reports have demonstrated increases of apoptosis with aging [51], which could lead to selection for oncogenic mutations that confer resistance to apoptosis, such as p53 mutation. The idea that impaired survival associated with aging might select for particular oncogenic mutations is analogous to the idea that carcinogen and chemotherapeutic exposures select for particular oncogenic mutations. Seventy years ago Haddow proposed that carcinogens may promote cancer by inhibiting cell proliferation: “the production of cancer by such physical agents is a reaction by which the cell emancipates itself from their inhibitory effects” [52]. More recently, others have proposed that carcinogenic treatments may create strong selective pressure for oncogenic mutations, thereby initiating tumorigenesis [53–55]. Finally, another group has suggested that decreased cellular proliferation with aging could favor the expansion of cancer cells [56].
We would therefore argue that reductions in cellular fitness that result from aging or mutagen exposure might tilt the balance between negative and positive fitness effects of oncogenic mutations, leading to increases in the net positive selective advantage for particular mutations (Figure 2, middle panel), thereby allowing for an increased probability for the clonal expansion of mutated cells that would typically not occur in young fit populations. Stabilizing selection would not be limited to stem cell pools with intrinsic damage accumulation, as degradation of the stem cell niche should also select for potentially oncogenic genetic changes conferring adaptation to the altered niche (Figure 2, bottom panel). In other words, when cellular fitness is reduced as a result of cell-intrinsic changes or changes in the environment, the stem cell population no longer possesses maximal fitness and certain oncogenic mutations have an increased chance of being adaptive and hence advantageous.
But what about cancers in children, which though rare, still do occur? We would speculate that any given oncogenic event will have some probability of being clonally expanded within a progenitor cell population, possibly being fixed. We propose that as the fitness of the progenitor population declines, that this probability will increase but only when the oncogenic event is adaptive to that context of reduced fitness. Thus, cancer can be initiated in a young individual with generally fit progenitor populations, but we argue that this initiation is more common in old age due to the greatly increased chance that an initiated cell will be adaptive and competitively expand. Furthermore, animal models involving large numbers of initiating events could still successfully generate cancer, particularly for transgenic and knockout models where all cells in a tissue possess the oncogenic event, as the fixation of the event has already occurred (even if the oncogenic event would normally exhibit a small probability of fixation in a more competitive context).
Childhood leukemias may nonetheless represent a unique scenario, particularly for hematologic malignancies bearing particular oncogenic translocations that contribute to childhood leukemias but rarely adult malignancies [57]. Many translocations that cause early childhood leukemias are already detectable in peripheral blood at birth [57]. If an oncogenic mutation occurs in a hematopoietic stem or progenitor cell during fetal development, the mutant clone could undergo substantial clonal expansion even if the fitness gain is neutral or negative (as long a the mutation is not highly deleterious), owing to the dramatic expansion of the progenitor cell population. Thus, even when the expansion of a mutant clone with negative fitness gain is lower than the expansion of competing “wild-type” clones, this expansion might still be sufficient to substantially increase the probability of secondary malignant events. An analogy can again be drawn from evolutionary biology: there are contexts when natural selection appears to be temporarily suspended, such as when a colonizing population finds abundant resources and minimal competition [58]. Nonetheless, we recognize that there may be other explanations for cancer initiation in childhood that do not relate to relative progenitor fitness.
While Natural Selection has been proposed to be the major driving force in both early and late stages of tumorigenesis [59, 60], current paradigms consider the incidence of oncogenic mutations (including initiating mutations) to be rate-limiting in tumorigenesis. In contrast, we argue that intrinsic tumor suppressor mechanisms as well as stabilizing selection create powerful barriers to cancer initiation, in most cases preventing clonal expansion of oncogenically initiated cells. In order for cancer to arise, these barriers must be breached. Breaching may occur when fitness gain resulting from an oncogenic mutation outweighs the disadvantages coupled to the mutation. Therefore, we speculate that conditions that increase the selective advantage of initiating oncogenic mutations, including reduced fitness of stem cell populations, might be the rate-limiting step for many cancers. It should be noted though that while the occurrence of initiating oncogenic mutations might typically not be a rate-limiting step in the initial stages of cancer development, the situation might change in later stages of tumor development. Following sufficient clonal expansion of oncogenically initiated cells under permissive conditions, selective pressures for additional oncogenic events that counteract intrinsic tumor suppressor mechanisms (such as apoptotic sensitivity) or that provide an advantage in the face of hypoxia (or other features of a growing tumor) may become inevitable. Thus, with progressive tumor development, the occurrence of oncogenic mutations that might relieve these pressures may become rate-limiting in cancer evolution.
Evidence used in support of the occurrence of oncogenic mutations as limiting cancer initiation is also compatible with reduced cellular fitness being the primary driver of tumor initiation
Does carcinogenic exposure lead to cancer by directly causing oncogenic mutations, or by reducing cellular fitness which subsequently selects for oncogenic changes?
Links between aging, exposure to mutagens, smoking, ionizing radiation and increased cancer incidence have been used as support for the theory that tumorigenesis is driven primarily by enhancing frequencies of oncogenic mutations [9]. For example, the 20–30 year lag between increases in smoking in the United States and the onset of lung cancers has been used to argue that the evolution of cancer from initiating oncogenic mutations requires several decades for the progressive accumulation of additional growth deregulating mutations [9, 61]. Similarly, ionizing radiation is an established breast cancer risk factor, but with a more than a 10 year lag period between exposure and increased cancer risk [62, 63]; again ionizing radiation is presumed to be the instigator of initiating mutations. Exposure to ionizing radiation can also increase the risk of chronic myelogenous leukemia (CML) but the disease usually occurs after a prolonged latency of 4–11 years [64]. Increased cancer risk is thought to be mediated by the introduction of initiating mutations that drive initial clonal expansion, with eventual accumulation of secondary events. However, as mutagenic exposures are expected to reduce stem cell fitness, we would argue that the occurrence of oncogenic mutations might be insufficient to initiate the cancers, and that both the presence of initiating oncogenic events and reductions of fitness that enables selection of these mutations might be required for oncogenesis. Importantly, fitness decline need not be immediate, but irradiation or other mutagenic insults may sap part of the stem cell reservoir, such that fitness decline will be accelerated or premature (Figure 3). Declining fitness in stem cell pools would then provide selective pressure for adaptive mutations, driving clonal expansion of preexisting mutants or mutants introduced with mutagenic exposure.
Figure 3. Changing mutation rates can alter cancer incidence by influencing fitness.

Inherited or environmental conditions that increase mutation rates should accelerate fitness decline in progenitor cell populations, leading to more rapid and penetrant development of cancer (top right). On the other hand, reducing mutation rates should help maintain progenitor cell fitness, decreasing the selective pressure for adaptive mutations and cancer development (bottom right). Increasing mutation rates should also increase the chance of a cell acquiring an initiating oncogenic mutation, but we argue that clonal expansion of an initiated cell is usually limited by selection rather than oncogenic mutation.
Impaired stem cell fitness in individuals with DNA repair deficiency could contribute to increased cancer rates
Accelerated aging and dramatically increased cancer rates are often observed in mice and humans with inherited mutations that disable DNA repair pathways [16, 65]. Conversely, conditions that extend life-span (such as caloric restriction) are associated with decreased mutation load and reduced incidence of cancers [66–68]. These observations have been used to both support the role of mutation accumulation in aging and the importance of accumulating mutations in critical proliferation control genes as the key determinants for cancer [69, 70]. But a higher mutation load should also exhaust stem cell fitness more rapidly, leading to faster and greater mounting selection for adaptive mutations, some of which will be oncogenic (Figure 3). Indeed, decreased cellular fitness is evident in individuals with DNA repair or damage recognition deficiencies [69]. For example, ataxia-telangiectasia mutated (ATM) gene deficient mice exhibit highly defective hematopoiesis in general and T lymphopoiesis in particular [70, 71], coinciding with a high risk for the development of T cell lymphomas. Thus, in our model, accumulating genomic damage contributes to both premature aging and cancer through the same mechanism: reduction of cellular fitness. Of course, our explanation need not be incompatible with increased chances for oncogenic mutations resulting from elevated mutation rates also contributing to higher cancer rates. But while oncogenic mutations are certainly required for initiation of cancer, without reductions of fitness or other mechanism to select for the expansion of oncogene bearing clones, these oncogenic events should not be sufficient to drive clonal expansion with consequent increased cancer risk.
Cancer: gradual evolution or delayed-accelerated origin?
Another implication of our model is that cancer development may not be as gradual as previously thought. If selection for initiating adaptive oncogenic mutations is more important for cancer evolution than the rate of oncogenic mutations themselves, then cancers may develop relatively rapidly late in life as the result of a lifetime of accumulating genomic damage leading to reduced stem cell fitness (Figure 4). Environmental or genetic factors that increase genomic damage would accelerate fitness decline, and thus promote the adaptive evolution of cancers. This delayed-accelerated model does not imply that oncogenic mutations can never persist for long periods of time or that cancer evolution is always rapid, but that as stem cell fitness declines in old age, the selective pressure for adaptive oncogenic mutations will be much greater than in youth, typically leading to much more rapid evolution of cancer (in years instead of decades). Thus our hypothesis would predict substantial increases in oncogenically-initiated clonal expansions in aged individuals.
Figure 4. Contrasting the gradual and delayed-accelerated models of cancer evolution.

Left: A simplistic interpretation of the prevailing view of cancer evolution as the gradual accumulation of rare oncogenic events over decades. Earlier exposure to carcinogens would increase the incidence of later cancers by providing initiating oncogenic mutations. Right: The delayed-accelerated model. A lifetime of accumulating genomic damage leads to the eventual reduction of stem cell fitness, providing selective pressure for adaptive oncogenic mutations. As selection does not occur until stem cell fitness has declined, cancer evolution occurs late in life and over a relatively short time period. Cancer may also rapidly evolve long after carcinogen exposure, facilitated by reduced fitness in stem cell pools set in motion by these genotoxic insults decades earlier. The delayed-accelerated model is analogous to the theory of Punctuated Equilibrium for species evolution championed by Eldredge and Gould [102].
Evidence that clonal expansion of oncogenically mutated cells result from permissive conditions
While the evidence presented above is consistent both with the classical view of tumorigenesis as being limited by oncogenic mutations as well as our model of adaptive oncogenesis, distinguishing between the two possibilities requires the introduction of oncogenic events into a small fraction of a progenitor cell population, with subsequent analysis of the competitive expansion of this population under different fitness contexts. Using this approach, we have shown that genetic or pharmacological inhibition of the proliferation of hematopoietic progenitor cells creates selective pressure for oncogenic mutations which restore cell cycle progression [72]. In particular, impairing DNA replication either by hydroxyurea treatment or E2F mutation dramatically enhances the competitive advantage provided by the expression of Bcr-Abl, a chromosomal translocation necessary for the initiation of CML and some acute lymphoblastic leukemias. The preferential expansion of Bcr-Abl expressing cells when DNA replication is inhibited leads to substantially increased leukemia rates in mouse models. In contrast, Bcr-Abl expression is rarely selected for in a healthy progenitor pool, preventing leukemia in most cases. Thus, reductions in DNA replication result in reduced progenitor cell fitness, and Bcr-Abl is adaptive in this context by restoring S phase progression. Our more recent studies indicate that aged hematopoiesis similarly promotes leukemias initiated by Bcr-Abl by impairing progenitor cell fitness (Marusyk and DeGregori, unpublished data). Importantly, introduction of young progenitors can reverse the increased leukemia incidence caused by Bcr-Abl expression in old progenitors, suggesting that reduced competitiveness of aged progenitors can contribute to aging-associated increases in leukemias.
Furthermore, multiple reports support the idea that the occurrence of oncogenic mutations within tissues far outpaces the incidence of cancers thought to be initiated by these oncogenic events. For example, expression of Bcr-Abl is detected at very low levels in >30% of asymptomatic humans for protracted periods of several months to years [73, 74], while the incidence of spontaneous disease is about 1:100,000. Although it could be rationalized that the detection of Bcr-Abl comes from cells devoid of long-term proliferative capacity, kinetic data considerations favor the explanation that Bcr-Abl translocation in human HSC is not sufficient to cause CML [75]. Our model would suggest that induction of CML may be limited not by the Bcr-Abl translocation (albeit certainly dependent on it), but instead stem cells with Bcr-Abl translocations are usually only clonally expanded within an aged or otherwise poorly fit pool. While bone marrow progenitors retrovirally transduced with Bcr-Abl can cause rapid leukemia development in recipient mice, leukemia induction requires high efficiency transduction [53, 76, 77]. Thus, even if any given Bcr-Abl expressing progenitor has a low probability of initiating leukemia, transfer of a large number of these Bcr-Abl+ progenitors can lead to leukemia. Moreover, hematopoietic reconstitution after bone marrow transplant may itself promote leukemia, given the massive expansion of progenitors required and the at least temporary paucity of competition.
Cell clones with mutational or epigenetic inactivation of the PTEN or INK4A tumor suppressor genes are commonly found in histologically normal endometria and breast (respectively) of cancer free women [78, 79], far outpacing the incidence of cancers in these tissues, suggesting that at least these initial oncogenic events are not rate limiting. As INK4A gene expression increases in aged tissues, and disruption of INK4A in mice results in some restoration of the regenerative capacity for particular tissues [80–82], inhibition of INK4A may be selected for in aged tissues. Reduced fitness independent of age may also be important. For instance, Maley and Reid [60] have suggested that premalignant esophageal clones with INK4A loss may be selected by the abnormal environment due to gastric reflux. Importantly, while frequent observations of mutant cell clones might be viewed as evidence for selective advantage conferred by the oncogenic alteration, given that these clones largely consist of differentiated cells, they could also represent differentiated progeny of oncogenically altered stem cells for which the heritable alteration did not provide a sustained advantage within the progenitor pool.
Zarbl and colleagues [83] showed that the carcinogen N-nitroso-N-methylurea induced mammary tumors arise from cell clones with pre-existing oncogenic H-Ras mutations. Thus, this carcinogen appears to promote breast tumors not by inducing oncogenic mutations in H-Ras, but perhaps by promoting the expansion of cells with pre-existing H-Ras mutations. Similarly, studies of ultraviolet (UV) light exposed and non-exposed human and mouse skin have argued that UV light exposure increases the numbers and size of p53 mutant clones [84, 85]. Although disputed by others [86], Rodin and Rodin [87] have argued that smoking induced physiological stress selects for cells with p53 mutations, rather than p53 mutations being directly induced by the mutagenic qualities of cigarette smoke. Moreover, similar frequencies of allelic loss (including of tumor suppressor genes) were detected in histologically normal and abnormal bronchial epithelial biopsies from both current smokers and former smokers, despite the significant decrease in the incidence of lung cancer in the former smokers [88, 89]. Following the logic of our model, continued smoking may further reduce the fitness of lung epithelial progenitors, promoting continued expansion of preneoplastic clones with adaptive mutations. Of course, smoking may also increase mutation rates, leading to increased chances for additional oncogenic mutations in addition to impacts on cellular fitness.
These studies and the lack of solid evidence demonstrating carcinogen induced mutations (with notable exceptions such as following UV light exposure), have led to proposals that carcinogens may select for preexisting oncogenic mutations, such as by stimulating the growth of preneoplastic lesions (i.e. by “promotion”) [55, 90, 91].
The complex relationship between aging, carcinogen exposure and cancer
While we argue that an aged environment primarily promotes tumor initiation by decreasing the fitness of stem cell populations, there is evidence that an aged environment can positively promote malignant growth. For example, liver carcinoma cells transplanted into livers of young rats differentiate into hepatocytes; in contrast, when the same cells are transplanted into livers of old animals, these cells produce tumors consisting of undifferentiated cells [92]. In fact, age-related changes in tissue environment substantially affect carcinogen induced tumorigenesis in many rodent model systems [93]. Moreover, one of the consequences of aging is the accumulation of senescent cells in many tissues [94–96], and senescent cells secrete a number of active molecules that can promote tumor growth [97, 98]. Finally, given that chronic inflammation is a well-established tumor promoter [9, 99], persistence of chronic inflammation in aged individual might be an additional factor driving tumorigenesis.
We have further argued that carcinogenic insults promote cancer at least in part by creating conditions that facilitate clonal expansion of cells with preexisting oncogenic mutations, by increasing the selective advantage of these mutations. Our hypothesis does not challenge the idea of mutagens directly causing oncogenic mutations. Indeed, carcinogenic exposure has been demonstrated to directly result in oncogenic mutations, at least in some contexts, as evidenced by the distinct mutational spectra apparent in the p16Ink4a and p53 tumor suppressor genes in melanomas and skin cancers, respectively, consistent with UV light induced mutagenesis [100, 101]. Thus, while UV light could potentially promote the clonal selection of cells with p16Ink4a and p53 mutations, the mutagenic effects of UV light directly on these tumor suppressor genes also contributes to skin carcinogenesis. Still, given that mutagenic exposure inevitably results in reduced cellular fitness, and that the bulk of the evidence used to support the rate-limiting role of initiating oncogenic events is equally consistent with the reduction in cellular fitness playing a crucial role, we would argue that the latter alternative should be given equally thorough consideration as the former one.
Conclusions
We have proposed that accumulation of genomic damage and epigenetic changes occurring naturally with age or following exposure to environmental mutagens leads to reduced cellular fitness in stem cell pools, creating selective pressure for the adaptive oncogenic mutations required for cancer evolution. We do not claim that reduced cellular fitness is the only explanation for increased cancer incidence observed in aged individuals. But we do argue that our Adaptive Oncogenesis model is at least as consistent with the available data to explain rising cancers with age as the current paradigm that the progressive accumulation of oncogenic hits limits cancer initiation. Clearly, experiments are required in order to distinguish between the two possibilities, although conclusions will be complicated by the fact that genotoxic agents lead to both increased oncogenic mutations and reduced cellular fitness. Given that cancer is not one disease, it would not be surprising if different mechanisms predominate for different cancers, with impaired progenitor fitness important for particular classes of cancers, declining immune surveillance influencing another subset, the occurrence of initiating oncogenic mutations limiting the development of yet another class, and increased inflammation with age contributing to others. Then again, all or some of the mechanisms could act in concert to contribute to the evolution of any given cancer, which may make our jobs as researchers trying to understand cancer all the more challenging.
Acknowledgments
JD is supported by the National Institutes of Health (RO1 CA109657). We thank Drs. Robert Sclafani, Jeffrey Kieft, Avinash Bhandoola, Heide Ford and Mel Greaves for their critical review of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 2.Greaves M. Darwinian medicine: a case for cancer. Nat Rev Cancer. 2007;7:213–21. doi: 10.1038/nrc2071. [DOI] [PubMed] [Google Scholar]
- 3.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–35. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 4.Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 5.Balducci L, Beghe C. Cancer and age in the USA. Crit Rev Oncol Hematol. 2001;37:137–45. doi: 10.1016/s1040-8428(00)00109-8. [DOI] [PubMed] [Google Scholar]
- 6.Balducci L, Lyman GH. Cancer in the elderly. Epidemiologic and clinical implications. Clin Geriatr Med. 1997;13:1–14. [PubMed] [Google Scholar]
- 7.DePinho RA. The age of cancer. Nature. 2000;408:248–54. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- 8.Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–41. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 9.Weinberg RA. The Biology of Cancer. Garland Science; New York: 2007. [Google Scholar]
- 10.Sharpless NE, Depinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–13. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 11.Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol. 2007;8:715–22. doi: 10.1038/nrm2242. [DOI] [PubMed] [Google Scholar]
- 12.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci U S A. 2003;100:776–81. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz B, Moreno E. The competitive nature of cells. Exp Cell Res. 2005;306:317–22. doi: 10.1016/j.yexcr.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 14.Abrams JM. Competition and compensation: coupled to death in development and cancer. Cell. 2002;110:403–6. doi: 10.1016/s0092-8674(02)00904-2. [DOI] [PubMed] [Google Scholar]
- 15.Sieber OM, Tomlinson SR, Tomlinson IP. Tissue, cell and stage specificity of (epi)mutations in cancers. Nat Rev Cancer. 2005;5:649–55. doi: 10.1038/nrc1674. [DOI] [PubMed] [Google Scholar]
- 16.Vijg J. Somatic mutations and aging: a re-evaluation. Mutat Res. 2000;447:117–35. doi: 10.1016/s0027-5107(99)00202-x. [DOI] [PubMed] [Google Scholar]
- 17.Khrapko K, Bodyak N, Thilly WG, van Orsouw NJ, Zhang X, Coller HA, Perls TT, Upton M, Vijg J, Wei JY. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27:2434–41. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopsidas G, Kovalenko SA, Kelso JM, Linnane AW. An age-associated correlation between cellular bioenergy decline and mtDNA rearrangements in human skeletal muscle. Mutat Res. 1998;421:27–36. doi: 10.1016/s0027-5107(98)00150-x. [DOI] [PubMed] [Google Scholar]
- 19.Shibata D, Tavare S. Counting divisions in a human somatic cell tree: how. what and why? Cell Cycle. 2006;5:610–4. doi: 10.4161/cc.5.6.2570. [DOI] [PubMed] [Google Scholar]
- 20.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu L, Wylie RC, Andrews LG, Tollefsbol TO. Aging, cancer and nutrition: the DNA methylation connection. Mech Ageing Dev. 2003;124:989–98. doi: 10.1016/j.mad.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Szilard L. On the Nature of the Aging Process. Proc Natl Acad Sci U S A. 1959;45:30–45. doi: 10.1073/pnas.45.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Failla G. The aging process and cancerogenesis. Ann N Y Acad Sci. 1958;71:1124–40. doi: 10.1111/j.1749-6632.1958.tb46828.x. [DOI] [PubMed] [Google Scholar]
- 24.Medawar P. An unsolved problem of biology. HK Lewis; London: 1952. [Google Scholar]
- 25.Strehler BL. Deletional mutations are the basic cause of aging: historical perspectives. Mutat Res. 1995;338:3–17. doi: 10.1016/0921-8734(95)00006-r. [DOI] [PubMed] [Google Scholar]
- 26.Vijg J, Dolle ME. Large genome rearrangements as a primary cause of aging. Mech Ageing Dev. 2002;123:907–15. doi: 10.1016/s0047-6374(02)00028-3. [DOI] [PubMed] [Google Scholar]
- 27.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–47. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 28.Muller HJ. The Relation of Recombination to Mutational Advance. Mutat Res. 1964;106:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- 29.Andersson DI, Hughes D. Muller’s ratchet decreases fitness of a DNA-based microbe. Proc Natl Acad Sci U S A. 1996;93:906–7. doi: 10.1073/pnas.93.2.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kibota TT, Lynch M. Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature. 1996;381:694–6. doi: 10.1038/381694a0. [DOI] [PubMed] [Google Scholar]
- 31.Eyre-Walker A, Keightley PD. The distribution of fitness effects of new mutations. Nat Rev Genet. 2007;8:610–8. doi: 10.1038/nrg2146. [DOI] [PubMed] [Google Scholar]
- 32.Elena SF, Lenski RE. Test of synergistic interactions among deleterious mutations in bacteria. Nature. 1997;390:395–8. doi: 10.1038/37108. [DOI] [PubMed] [Google Scholar]
- 33.Reese JS, Liu L, Gerson SL. Repopulating defect of mismatch repair-deficient hematopoietic stem cells. Blood. 2003;102:1626–1633. doi: 10.1182/blood-2002-10-3035. [DOI] [PubMed] [Google Scholar]
- 34.Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, Rodrigues NP, Crockford TL, Cabuy E, Vindigni A, Enver T, Bell JI, Slijepcevic P, Goodnow CC, Jeggo PA, Cornall RJ. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447:686–90. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- 35.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–9. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 36.Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441:1011–4. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- 37.Fraser HB, Hirsh AE, Giaever G, Kumm J, Eisen MB. Noise minimization in eukaryotic gene expression. PLoS Biol. 2004;2:e137. doi: 10.1371/journal.pbio.0020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberdoerffer P, Sinclair DA. The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol. 2007;8:692–702. doi: 10.1038/nrm2238. [DOI] [PubMed] [Google Scholar]
- 39.Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441:1080–6. doi: 10.1038/nature04958. [DOI] [PubMed] [Google Scholar]
- 40.Ju Z, Jiang H, Jaworski M, Rathinam C, Gompf A, Klein C, Trumpp A, Lenhard Rudolph K. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat Med. 2007;13:742–7. doi: 10.1038/nm1578. [DOI] [PubMed] [Google Scholar]
- 41.Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–4. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 42.Carlson BM, Faulkner JA. Muscle transplantation between young and old rats: age of host determines recovery. Am J Physiol. 1989;256:C1262–6. doi: 10.1152/ajpcell.1989.256.6.C1262. [DOI] [PubMed] [Google Scholar]
- 43.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–15. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 44.Gilad Y, Oshlack A, Rifkin SA. Natural selection on gene expression. Trends Genet. 2006;22:456–61. doi: 10.1016/j.tig.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 45.Fisher RA. The Genetical Theory of Natural Selection. Oxford University Press; Oxford: 1930. [Google Scholar]
- 46.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM, Wu H, Li L. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–22. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 48.Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, Macdonald HR, Trumpp A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18:2747–63. doi: 10.1101/gad.313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–8. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 50.Stephan RP, Lill-Elghanian DA, Witte PL. Development of B cells in aged mice: decline in the ability of pro-B cells to respond to IL-7 but not to other growth factors. J Immunol. 1997;158:1598–609. [PubMed] [Google Scholar]
- 51.Zhang Y, Herman B. Ageing and apoptosis. Mech Ageing Dev. 2002;123:245–60. doi: 10.1016/s0047-6374(01)00349-9. [DOI] [PubMed] [Google Scholar]
- 52.Haddow A. Cellular inhibition and the origin of cancer. Acta Unio Int Contra Cancrum. 1938;3:342–352. [Google Scholar]
- 53.Bilousova G, Marusyk A, Porter CC, Cardiff RD, DeGregori J. Impaired DNA replication within progenitor cell pools promotes leukemogenesis. PLoS Biol. 2005;3:e401. doi: 10.1371/journal.pbio.0030401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farber E. Cell proliferation as a major risk factor for cancer: a concept of doubtful validity. Cancer Res. 1995;55:3759–62. [PubMed] [Google Scholar]
- 55.Blagosklonny MV. Carcinogenesis, cancer therapy and chemoprevention. Cell Death Differ. 2005;12:592–602. doi: 10.1038/sj.cdd.4401610. [DOI] [PubMed] [Google Scholar]
- 56.Ukraintseva SV, Yashin AI. Treating cancer with embryonic stem cells: rationale comes from aging studies. Front Biosci. 2005;10:588–95. doi: 10.2741/1555. [DOI] [PubMed] [Google Scholar]
- 57.Greaves MF, Wiemels J. Origins of Chromosome Translocations in Childhood Leukemia. Nat Rev Cancer. 2003;3:1–10. doi: 10.1038/nrc1164. [DOI] [PubMed] [Google Scholar]
- 58.Templeton AR. Experimental evidence for the genetic-transilience model of speciation. Evolution. 1996;50:909–915. doi: 10.1111/j.1558-5646.1996.tb03899.x. [DOI] [PubMed] [Google Scholar]
- 59.Sieber OM, Heinimann K, Tomlinson IP. Genomic instability--the engine of tumorigenesis? Nat Rev Cancer. 2003;3:701–8. doi: 10.1038/nrc1170. [DOI] [PubMed] [Google Scholar]
- 60.Maley CC, Reid BJ. Natural selection in neoplastic progression of Barrett’s esophagus. Semin Cancer Biol. 2005;15:474–83. doi: 10.1016/j.semcancer.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 61.Proctor RN. Tobacco and the global lung cancer epidemic. Nat Rev Cancer. 2001;1:82–6. doi: 10.1038/35094091. [DOI] [PubMed] [Google Scholar]
- 62.Ronckers CM, Erdmann CA, Land CE. Radiation and breast cancer: a review of current evidence. Breast Cancer Res. 2005;7:21–32. doi: 10.1186/bcr970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carmichael A, Sami AS, Dixon JM. Breast cancer risk among the survivors of atomic bomb and patients exposed to therapeutic ionising radiation. Eur J Surg Oncol. 2003;29:475–9. doi: 10.1016/s0748-7983(03)00010-6. [DOI] [PubMed] [Google Scholar]
- 64.Lichtman JLLMA. In: Williams Hematology. Beutler Ernest, M.D., M.A.L.M.D., Coller Barry S, M.D., Kipps Thomas J, M.D. Ph.D., Seligsohn Uri., M.D., editors. McGraw-Hill Professional; 2001. pp. 1085–1125. [Google Scholar]
- 65.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 66.Bronson RT, Lipman RD. Reduction in rate of occurrence of age related lesions in dietary restricted laboratory mice. Growth Dev Aging. 1991;55:169–84. [PubMed] [Google Scholar]
- 67.Roe FJ, Lee PN, Conybeare G, Kelly D, Matter B, Prentice D, Tobin G. The Biosure Study: influence of composition of diet and food consumption on longevity, degenerative diseases and neoplasia in Wistar rats studied for up to 30 months post weaning. Food Chem Toxicol. 1995;33(Suppl 1):1S–100S. doi: 10.1016/0278-6915(95)80200-2. [DOI] [PubMed] [Google Scholar]
- 68.Weindruch R, Walford RL, Fligiel S, Guthrie D. The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. J Nutr. 1986;116:641–54. doi: 10.1093/jn/116.4.641. [DOI] [PubMed] [Google Scholar]
- 69.Thompson LH, Schild D. Recombinational DNA repair and human disease. Mutat Res. 2002;509:49–78. doi: 10.1016/s0027-5107(02)00224-5. [DOI] [PubMed] [Google Scholar]
- 70.Matei IR, Guidos CJ, Danska JS. ATM-dependent DNA damage surveillance in T-cell development and leukemogenesis: the DSB connection. Immunol Rev. 2006;209:142–58. doi: 10.1111/j.0105-2896.2006.00361.x. [DOI] [PubMed] [Google Scholar]
- 71.Bagley J, Cortes ML, Breakefield XO, Iacomini J. Bone marrow transplantation restores immune system function and prevents lymphoma in Atm-deficient mice. Blood. 2004;104:572–8. doi: 10.1182/blood-2003-12-4226. [DOI] [PubMed] [Google Scholar]
- 72.Bilousova G, Marusyk A, Porter CC, Cardiff RD, DeGregori J. Impaired DNA Replication within Progenitor Cell Pools Promotes Leukemogenesis. PLoS Biology. 2005;3 doi: 10.1371/journal.pbio.0030401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bose S, Deininger M, Gora-Tybor J, Goldman JM, Melo JV. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood. 1998;92:3362–7. [PubMed] [Google Scholar]
- 74.Biernaux C, Loos M, Sels A, Huez G, Stryckmans P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood. 1995;86:3118–22. [PubMed] [Google Scholar]
- 75.Matioli GT. BCR-ABL insufficiency for the transformation of human stem cells into CML. Med Hypotheses. 2002;59:588–9. doi: 10.1016/s0306-9877(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 76.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–92. [PubMed] [Google Scholar]
- 77.Zhang X, Ren R. Bcr-Abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood. 1998;92:3829–40. [PubMed] [Google Scholar]
- 78.Mutter GL, Ince TA, Baak JP, Kust GA, Zhou XP, Eng C. Molecular identification of latent precancers in histologically normal endometrium. Cancer Res. 2001;61:4311–4. [PubMed] [Google Scholar]
- 79.Crawford YG, Gauthier ML, Joubel A, Mantei K, Kozakiewicz K, Afshari CA, Tlsty TD. Histologically normal human mammary epithelia with silenced p16(INK4a) overexpress COX-2, promoting a premalignant program. Cancer Cell. 2004;5:263–73. doi: 10.1016/s1535-6108(04)00023-6. [DOI] [PubMed] [Google Scholar]
- 80.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–6. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 81.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–7. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 82.Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–52. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cha RS, Thilly WG, Zarbl H. N-nitroso-N-methylurea-induced rat mammary tumors arise from cells with preexisting oncogenic Hras1 gene mutations. Proc Natl Acad Sci U S A. 1994;91:3749–53. doi: 10.1073/pnas.91.9.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, Leffell DJ, Tarone RE, Brash DE. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci U S A. 1996;93:14025–9. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang W, Remenyik E, Zelterman D, Brash DE, Wikonkal NM. Escaping the stem cell compartment: sustained UVB exposure allows p53-mutant keratinocytes to colonize adjacent epidermal proliferating units without incurring additional mutations. Proc Natl Acad Sci U S A. 2001;98:13948–53. doi: 10.1073/pnas.241353198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hainaut P, Pfeifer GP. Patterns of p53 G-->T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22:367–74. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 87.Rodin SN, Rodin AS. Human lung cancer and p53: the interplay between mutagenesis and selection. Proc Natl Acad Sci U S A. 2000;97:12244–9. doi: 10.1073/pnas.180320897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mao L, Lee JS, Kurie JM, Fan YH, Lippman SM, Lee JJ, Ro JY, Broxson A, Yu R, Morice RC, Kemp BL, Khuri FR, Walsh GL, Hittelman WN, Hong WK. Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst. 1997;89:857–62. doi: 10.1093/jnci/89.12.857. [DOI] [PubMed] [Google Scholar]
- 89.Wistuba II, Lam S, Behrens C, Virmani AK, Fong KM, LeRiche J, Samet JM, Srivastava S, Minna JD, Gazdar AF. Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst. 1997;89:1366–73. doi: 10.1093/jnci/89.18.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thilly WG. Have environmental mutagens caused oncomutations in people? Nat Genet. 2003;34:255–9. doi: 10.1038/ng1205. [DOI] [PubMed] [Google Scholar]
- 91.Tomlinson IP, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci U S A. 1996;93:14800–3. doi: 10.1073/pnas.93.25.14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McCullough KD, Coleman WB, Ricketts SL, Wilson JW, Smith GJ, Grisham JW. Plasticity of the neoplastic phenotype in vivo is regulated by epigenetic factors. Proc Natl Acad Sci U S A. 1998;95:15333–8. doi: 10.1073/pnas.95.26.15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Anisimov VN. The relationship between aging and carcinogenesis: a critical appraisal. Crit Rev Oncol Hematol. 2003;45:277–304. doi: 10.1016/s1040-8428(02)00121-x. [DOI] [PubMed] [Google Scholar]
- 94.Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol. 2004;6:168–70. doi: 10.1038/ncb1095. [DOI] [PubMed] [Google Scholar]
- 95.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 97.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:12072–7. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–96. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 100.Pollock PM, Pearson JV, Hayward NK. Compilation of somatic mutations of the CDKN2 gene in human cancers: non-random distribution of base substitutions. Genes Chromosomes Cancer. 1996;15:77–88. doi: 10.1002/(SICI)1098-2264(199602)15:2<77::AID-GCC1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 101.Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Hum Mutat. 2003;21:217–28. doi: 10.1002/humu.10179. [DOI] [PubMed] [Google Scholar]
- 102.Gould SJ, Eldredge N. Punctuated equilibrium comes of age. Nature. 1993;366:223–7. doi: 10.1038/366223a0. [DOI] [PubMed] [Google Scholar]