

Abstract
The tumor suppressor p53 is important for inhibiting the development of breast carcinomas. However, little is known about the effects of increased p53 activity on mammary gland development. Therefore, the effect of p53 dosage on mammary gland development was examined by utilizing the p53+/m mouse, a p53 mutant which exhibits increased wildtype p53 activity, increased tumor resistance, a shortened longevity, and a variety of accelerated aging phenotypes. Here we report that p53+/m virgin mice exhibit a defect in mammary gland ductal morphogenesis. Transplants of mammary epithelium into p53+/m recipient mice demonstrate decreased outgrowth of wildtype and p53+/m donor epithelium, suggesting systemic or stromal alterations in the p53+/m mouse. Supporting these data, p53+/m mice display decreased levels of serum IGF-1 and reduced IGF-1 signaling in virgin glands. The induction of pregnancy or treatment of p53+/m mice with estrogen, progesterone, estrogen and progesterone in combination, or IGF-1 stimulates ductal outgrowth, rescuing the p53+/m mammary phenotype. Serial mammary epithelium transplants demonstrate that p53+/m epithelium exhibits decreased transplant capabilities, suggesting early stem cell exhaustion. These data indicate that appropriate levels of p53 activity are important in regulating mammary gland ductal morphogenesis, in part through regulation of the IGF-1 pathway.
Keywords: p53, mammary gland, mouse, ductal morphogenesis, IGF-1
Introduction
In response to cellular stress, the tumor suppressor p53 plays an important role in maintaining genome integrity and inhibiting both cancer formation and progression by regulating the expression of genes responsible for mediating DNA repair, cell cycle arrest, apoptosis, and senescence (Bunz et al., 1998; Chao et al., 2000; Vogelstein et al., 2000). Dysfunctional p53 signaling pathways or p53 mutations are present in over 80% of all human cancers, including breast cancer (Lozano and Elledge, 2000). Proper function and regulation of p53 are important to prevent the development of human breast cancer, as mutations in p53 are found in 30–40% of spontaneous breast carcinomas and female Li-Fraumeni syndrome patients who carry a germline mutation in one p53 allele have an increased incidence of early-onset breast cancer (Akashi et al., 1998; Coles et al., 1992; Moll et al., 1992). Further work to understand the role of p53 in mammary gland development may provide insight into mechanisms that regulate malignant transformation as well as normal mammary epithelial cell growth.
The loss of p53 has been reported to have little effect on mammary gland development, as p53 null mammary glands were initially shown to be morphologically and functionally comparable to wildtype glands (Donehower et al., 1992; Jacks et al., 1994; Kuperwasser et al., 2000; Purdie et al., 1994). However, loss of p53 in a BALB/c background causes a transient delay in involution, indicating that p53 is expressed by the mammary epithelium during early involution, where it mediates apoptosis (Jerry et al., 1998). In the quiescent virgin gland, p53 mRNA levels are high in the mammary epithelium, however the virgin gland exhibits a decreased p53 response to DNA damage, characterized by cytoplasmic sequestration of p53 (Kuperwasser et al., 2000; Minter et al., 2002). p53 activation is highest during early and mid-pregnancy, correlating with an increase in proliferation and steroid hormones (Becker et al., 2005; Minter et al., 2002). Interestingly, treatment with pregnancy-associated hormones allows nuclear accumulation of p53 following irradiation in virgin glands (Kuperwasser et al., 2000). Limited hormonal exposure of the mammary gland during postpubertal development, through pregnancy or hormonal treatment, has a protective effect against breast cancer in humans and rodents, which is dependent on the presence of wildtype p53 (Medina et al., 2003). These studies show that p53 activity is dependent upon hormonal status and varies throughout development.
Recent studies have identified the ability of truncated forms of p53 to enhance the activity of full-length wildtype p53, including a mouse model developed in our laboratory, the p53+/m mouse (Tyner et al., 2002). The p53+/m mice express one wildtype p53 allele and a truncated C-terminal p53 mutant allele, referred to as the “m” allele, which consists of p53 exons 7–11, with an Arg to Trp mutation at codon 245 (Tyner et al., 2002). p53+/m mice are highly resistant to tumors, yet display a reduction in median lifespan and exhibit a variety of accelerated aging phenotypes (Tyner et al., 2002). The p53+/m mice also exhibit alterations in hematopoietic stem cell (HSC) number, proliferation, and engraftment capabilities with age, suggesting that altered p53 levels affect stem cell functionality (Dumble et al., 2007; Gatza et al., 2007). In response to γ-irradiation, p53+/m tissues exhibit increased wildtype p53 protein levels, indicating that the m protein can stabilize wildtype p53 (Tyner et al., 2002; Moore et al., 2007). The m allele also increases the activity of wildtype p53, leading to increased transactivation of the p53 target gene p21 and increased resistance to oncogenic transformation in MEFs (Tyner et al., 2002). Although the m protein is not a natural isoform of p53, it is similar to several naturally occurring p53 isoforms, p44 and Δ133p53 (Bourdon et al., 2005). In addition, the similarities between the p53+/m and p44 mouse models indicate that the p53+/m mouse is biologically significant as a model of increased p53 activity (Maier et al., 2004; Tyner et al., 2002).
Since p53 levels play an important role in protection against mammary carcinogenesis, we investigated the effect of increased p53 activity on mammary gland development and stem cell dynamics by utilizing the p53+/m mouse model. Mammary gland stem cells are necessary for mammary gland development and functionality, and alterations in stem cell proliferative capabilities may lead to defects in the gland (Shackleton et al., 2006; Stingl et al., 2006; Woodward et al., 2005). Additionally, IGF-1, which plays a critical role in mediating mammary gland development, has also been implicated in aging (Holzenberger et al., 2003). p53 is known to transcriptionally repress expression of the IGF-1 receptor (IGF-1R) and transcriptionally upregulate IGF-1 signaling inhibitors IGF-BP3 and PTEN, demonstrating a direct role in p53-mediated regulation of the IGF-1 pathway (Buckbinder et al., 1995; Stambolic et al., 2001; Werner et al., 1996). Recently, several mouse models with accelerated aging phenotypes have been shown to exhibit alterations in IGF-1 signaling (Maier et al., 2004; Niedernhofer et al., 2006; Shukla et al., 2006). Collectively, these studies suggest that the p53+/m mouse may exhibit altered IGF-1 signaling, which could affect mammary gland development.
Here we report that p53+/m virgin mice exhibit a defect in mammary gland ductal morphogenesis. Transplants of mammary epithelium into p53+/m recipient mice demonstrate a significant reduction of WT and p53+/m donor epithelium outgrowth, suggesting systemic or stromal alterations in p53+/m mice. In fact, p53+/m mice exhibit a decrease in serum IGF-1 levels and attenuated IGF-1 signaling in the virgin gland. The p53+/m mammary phenotype is rescued by induction of pregnancy, treatment with estrogen, progesterone, estrogen and progesterone in combination, or LR3-IGF-1. Collectively, these data demonstrate that alterations in p53 activity can have a profound effect in the regulation of mammary gland development.
Materials and Methods
Mice
The mice used in this study were previously reported by our laboratory (Donehower et al., 1992; Tyner et al., 2002), except the TTR-IGF-1 mice (Liao et al., 2006). p53 wildtype (WT) and p53+/m mice backcrossed seven generations into C57BL/6 and p53+/− mice backcrossed twelve generations into C57BL/6 were used for all experiments. The WT and p53+/m mice were generated by crossing WT mice to p53+/m mice and were genotyped by PCR as previously described (Dumble et al., 2007). p53+/− mice were produced by heterozygous crossings and were genotyped by Southern Blot analysis as previously described (Donehower et al., 1992). TTR-IGF-1 heterozygous mice were crossed to BC7-p53+/m mice to generate TTR-IGF-1/+; p53+/m mice and were genotyped by PCR as previously described (Dumble et al., 2007; Liao et al., 2006).
All mice were bred and maintained in a specific pathogen free animal facility at Baylor College of Medicine. The three week old C57BL/6 female recipient mice used in the serial mammary epithelial transplantation experiments were purchased from the Baylor College of Medicine Transgenic Mouse Barrier Facility.
Whole Mount Preparation
Mammary gland tissue was isolated and whole mounts prepared as previously described (Medina et al., 2002). The number four inguinal mammary glands were harvested from WT, p53+/−, and p53+/m mice at 8 week virgin, 12 week virgin, 24 week virgin, mid-pregnancy (day 12), and D10 involution (10 days post weaning) and fixed in 10% neutral buffered formalin for at least 24 hours. Whole mounts were examined for ductal outgrowth under a dissecting microscope (Olympus SZX12).
In vivo BrdU Incorporation
Two hours prior to sacrifice, mice were treated with 7 mg/mL bromodeoxyuridine (BrdU) cell proliferation reagent (Sigma Aldrich, St. Louis, MO, USA) at 0.01 ml/gm of body weight. The number four inguinal mammary glands were harvested from WT, p53+/−, and p53+/m mice at 8 week virgin, 12 week virgin, mid-pregnancy (day 12), and D10 involution and fixed in 4% paraformaldehyde for two hours followed by 70% ethanol. Fixed tissues were paraffin-embedded and sectioned. Immunostaining for BrdU incorporation was performed using the BrdU In-Situ Detection Kit (BD Pharmingen, San Diego, CA, USA). For each sample, 500 ductal epithelial cells were counted over multiple fields and scored as positive or negative for BrdU incorporation. The BrdU labeling index was determined by calculating the average of the percent BrdU positive cells from three separate glands.
TUNEL Assay
The number four mammary glands were harvested from WT, p53+/−, and p53+/m mice at 8 week virgin, 12 week virgin, mid-pregnancy (day 12), and D10 involution and fixed in 4% paraformaldehyde for two hours followed by 70% ethanol. Fixed tissues were paraffin embedded and sectioned. Immunostaining for apoptosis was performed using the In Situ Apoptosis Detection Kit (Trevigen, Helgerman, CT, USA). For each sample, 500 cells were counted over multiple fields and scored as TUNEL positive or negative. The percent apoptosis was determined by calculating the average of the percent TUNEL positive cells from three separate glands.
Serial Mammary Epithelial Transplants
Serial mammary transplants were performed as previously described (Medina et al., 2002). Briefly, the number four inguinal glands of five three week old female WT C57BL/6 recipients were cleared by removal of the duct containing fat pad below the primary lymph node and proximal to the nipple. A 1mm2 piece of tissue from the number four mammary gland of an eight week old female WT, p53+/−, or p53+/m donor mouse was transplanted into the cleared glands of each of the five WT C57BL/6 recipients. Eight weeks post transplant, the outgrowths were retransplanted into five WT recipients and the glands were harvested, fixed in 4% paraformaldehyde for two hours, transferred to 70% ethanol, and then paraffin embedded or examined by whole mount. Serial transplants were continued until the outgrowths failed to fill 10% of the mammary fat pad.
Mammary Epithelium Transplants into p53+/m Recipients
The mammary glands of three week old WT or p53+/m recipients were cleared as previously described. Mammary epithelium from an eight week WT donor was transplanted into the left gland, while tissue from an eight week p53+/m donor was transplanted into the right mammary gland of each recipient mouse. Transplants were harvested four weeks post transplant and whole mounted as previously described. Examination of outgrowth was performed by blinded assessment of whole mounts.
Hormone Treatment
10–12 week WT, p53+/−, and p53+/m virgin mice were subcutaneously implanted in the sub scapular region with Silastic brand capsules (Dow Corning, Midland, MI, USA) containing either 50 μg estrogen (Sigma Aldrich, St. Louis, MO, USA), 20 mg progesterone (Sigma Aldrich, St. Louis, MO, USA), or 50 μg estrogen and 20 mg progesterone. The number four inguinal mammary glands were removed at day zero (untreated) and day 14, fixed in 10% neutral buffered formalin and analyzed for ductal outgrowth by whole mount analysis.
IGF-1 Serum Analysis
Blood was collected retro-orbitally from anesthetized (Isoflurane) 8 week WT, p53+/−, and p53+/m virgin mice using heparinized capillary tubes (Fisher Scientific, Pittsburgh, PA, USA) and serum was collected by centrifugation. IGF-1 serum levels were analyzed by an enzyme-immuno-assay kit (Immunodiagnostic Systems, Ltd, Boldon Business Park, Bolden, Tyne & Wear, NE, USA) according to the manufacturer’s instructions.
Western Blotting
Mammary gland extracts were prepared from 8 week virgin mice or 5 week WT and p53+/m virgin mice untreated or treated once with 100 μl of 0.25 μg/μl LR3-IGF-1 recombinant analog (Sigma Aldrich, St. Louis, MO, USA) in 0.9% saline + 10 mg/ml BSA. Tissue was harvested one hour post treatment. Mammary gland extracts were prepared by pulverizing snap-frozen tissue followed by lysis in RIPA buffer (Pierce, Rockford, IL, USA) plus a protease inhibitor cocktail (Roche, Applied Science, Mannheim, Germany) and phosphatase inhibitors (Pierce, Rockford, IL, USA), and cleared by centrifugation. Protein concentrations were determined using the BCA protein quantification assay (Pierce, Rockford, IL, USA). Extracts were electrophoresed on a 3–8% Tris-Acetate gel (Invitrogen, Carlsbad, CA, USA) or 4–12% Bis-Tris gel (Invitrogen, Carlsbad, CA, USA) and transferred to PVDF membrane (Invitrogen, Carlsbad, CA, USA). Membranes were blocked in 5% Milk-TBS-0.1% Tween followed by incubation with IRS-1 (Upstate Lake Placid, NY, USA), AKT (Cell Signaling, Danvers, MA, USA), pAKT (Ser473) (Cell Signaling, Danvers, MA, USA), IGF-1R (Santa Cruz Biotechnology, Santa Cruz, CA, USA), pIGF-1R (Tyr1135/1136) (Cell Signaling, Danvers, MA, USA), p21 (F-5) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p53 (R19) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and Actin (Lab Vision, Freemont, CA, USA). Peroxidase-conjugated bovine anti-rabbit (Santa Cruz Biotech, Santa Cruz, CA, USA) and anti-mouse secondary antibodies (Santa Cruz Biotech, Santa Cruz, CA, USA) were used at 1:2000 and signal was detected with Supersignal West Pico Solutions (Pierce, Rockford IL, USA). Membranes were stripped and reprobed whenever possible. Protein levels were determined by immunoblot analysis followed by densitometric analysis (GE Molecular Dynamics Personal Densitometer SI) utilizing ImageQuaNT software (GE Molecular Dynamics).
Treatment of mice with LR3-IGF-1
6 week WT, p53+/−, and p53+/m virgin mice were intraperitoneally (IP) injected twice daily with 100 μl of 0.25 μg/μl LR3-IGF-1 recombinant analog (Sigma Aldrich, St. Louis, MO, USA) in 0.9% saline + 10 mg/ml BSA for 5 days. Control mice were IP injected twice daily with sterile 0.9% saline + 10 mg/ml BSA. Mammary glands were harvested, fixed in 10% neutral buffered formalin and whole mounted as previously described. Examination of ductal outgrowth was performed by assessment of whole mounts using a dissecting microscope (Olympus SZX12).
Analysis of IGF-1 mRNA levels
Total RNA was extracted from 12 week virgin WT, p53+/−, and p53+/m mammary glands using the RNeasy Lipid Tissue Mini Kit per the manufacturer’s instructions (Qiagen, Valencia, CA, USA). 1 mu;g of RNA was reverse transcribed using the SuperScript First Strand Synthesis System for RT PCR (Invitrogen, Carlsbad, CA, USA). Each PCR reaction contained 1 μg of cDNA along with IGF-1 primers: IGF-1F 5′ GGA CCA GAG ACC CTT TGC GGG G 3′ and IGF-1R 5′ AAC AGA ACT TAG GAC GAG GG 3′ or β-actinF 5′ AAC AGA ACT TAG GAC GAG GG 3′ and β-actinR 5′ GGA AAC CAG GTT GTC AGT C 3′ and was run for 35 cycles. These primers generate a 267 bp IGF-1 product and a 209 bp β-actin product, respectively. IGF-1 expression was normalized to β-actin levels using KODAK Digital Science 1D Software.
Results
p53+/m Mice Exhibit Delayed Ductal Morphogenesis
Mammary gland morphology was examined during developmental stages 8, 12, and 24 week virgin, mid-pregnancy (day 12), and involution (day 10) in WT, p53+/−, and p53+/m mice by whole mount analysis. Consistent with previous reports, p53+/−mammary glands were comparable to wildtype and no phenotypic alterations were observed at any developmental stages (data not shown) (Donehower et al., 1992; Jacks et al., 1994; Purdie et al., 1994). In contrast, mammary glands from 8 week old p53+/m virgin mice show reduced ductal outgrowth, with minimal ductal growth past the primary lymph node (Fig. 1A). To confirm this delay in ductal morphogenesis, we examined p53+/m virgin mice at 12 and 24 weeks of age and found that these mice also exhibit decreased ductal outgrowth (Fig. 1A). Despite decreased ductal outgrowth, the limited number of ducts in the p53+/m glands exhibit normal gross and cellular morphology when examined by whole mount analysis and by hematoxylin and eosin (H&E) staining (data not shown). Surprisingly, by mid-pregnancy (day 12) the glands of p53+/m mice were comparable to those of both WT and p53+/− mice, demonstrating ductal outgrowth encompassing the entire fat pad as well as normal formation of alveolar buds (Fig. 1B). No morphological differences in involution (day 10) were noted between WT, p53+/−, or p53+/m glands (Fig. 1C). Furthermore, it was observed that female p53+/m mice were capable of nursing their pups and supporting them until weaning, suggesting that p53+/m mammary glands are functionally analogous to wildtype glands post-partum. This suggests that the pregnancy-mediated increase in steroid hormones may be required to induce ductal outgrowth in the p53+/m mammary gland.
Figure 1. Representative whole mounts of WT and p53+/m mammary glands demonstrate the effects of altered p53 dosage on ductal morphogenesis.
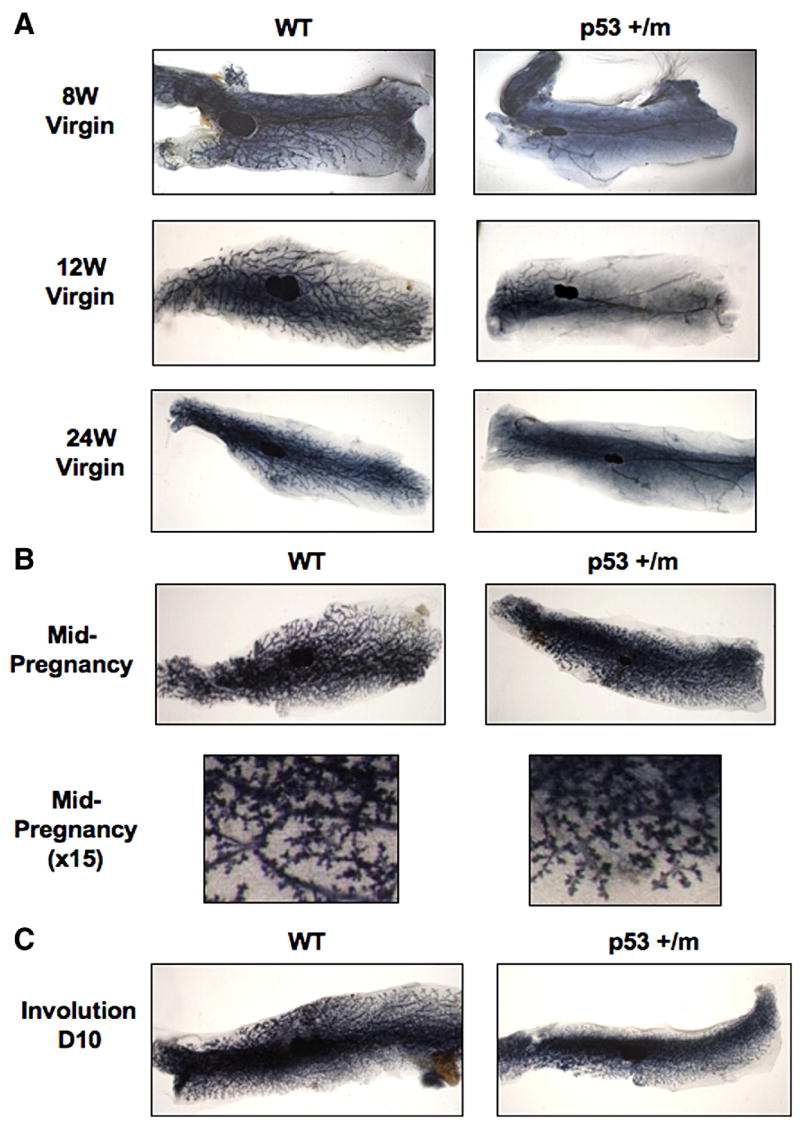
WT and p53+/m mammary glands at eight, 12, and 24 week old virgin (A), mid-pregnancy (day 12) (B), and day 10 involution (C). All images are shown at 3.5X or 15X magnification.
Rates of Cell Proliferation and Apoptosis During Development
The p53+/m mice were previously shown to exhibit increased levels of p53 protein in response to irradiation in the kidney (Tyner et al., 2002). Although p53 protein levels were not significantly increased in the mammary glands of p53+/m virgin mice, these glands exhibit an increase in p21 levels in response to irradiation, indicative of an increase in p53 activity (Supplementary Fig. 1). The increased p53 activity in the p53+/m mouse suggests that the decreased ductal outgrowth observed in these mice could be due to alterations in proliferation and/or apoptosis, which are important during ductal morphogenesis, successive cycles of pregnancy, and critical during involution (Richert et al., 2000). An in vivo BrdU proliferation assay was performed to determine if levels of proliferation were altered in the p53+/m mammary gland. Proliferation was examined in WT, p53+/−, and p53+/m mice at developmental stages 8 and 12 week virgin, mid-pregnancy (day 12), and D10 involution. No proliferation was observed in the limited number of ductal epithelial cells found in the 8 and 12 week p53+/m virgin glands, however by mid-pregnancy (day 12) and during involution, the p53+/m glands exhibited rates of cell proliferation comparable to both WT and p53+/− glands (Fig. 2A, B). These results further suggest that pregnancy is required to induce epithelial cell proliferation and ductal outgrowth in the p53+/m gland.
Figure 2. Rates of cell proliferation and apoptosis in WT, p53+/−, and p53+/m mammary glands during development.
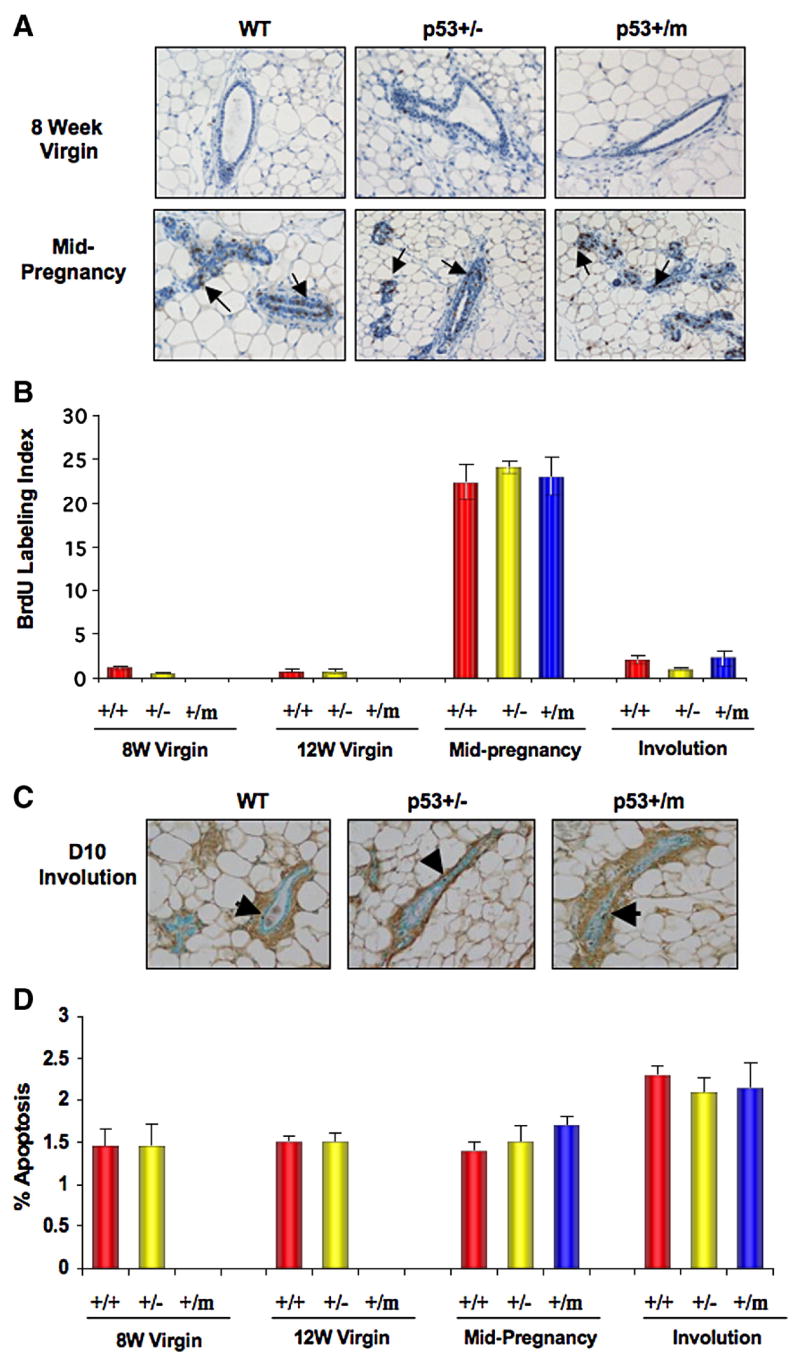
(A) Representative WT, p53+/−, and p53+/m mammary gland sections stained for BrdU incorporation at 8 week virgin and mid-pregnancy (day 12) stages of development. Arrows indicate BrdU labeled cells. (B) Rate of cell proliferation at various developmental stages presented as mean percent of BrdU Labeling Index (± s.e.m) in a bar graph. (n=3 for all groups.) (C) Representative WT, p53+/−, and p53+/m mammary gland sections TUNEL stained at D10 involution. Arrows indicate TUNEL stained apoptotic cells. (D) Quantitative analysis of the mean percent apoptosis (± s.e.m) in WT, p53+/−, and p53+/m mammary glands at various development stages. (n=3 for all groups.) All images are shown at 20X magnification.
A TUNEL assay was performed to examine levels of apoptosis in WT, p53+/−, and p53+/m mice during various developmental stages. Consistent with our previous results, limited numbers of ductal epithelial cells were observed in the p53+/m 8 and 12 week virgin glands. Virgin glands showed comparable low levels of apoptosis across all genotypes and no significant increase in apoptosis was detected in these glands by TUNEL assay (Fig. 2C, D). Moreover, we did not observe alterations in apoptotic levels between genotypes at mid-pregnancy (day 12) or D10 involution and no differences in morphology were seen at D10 involution (Figs 1D, 2C, 2D). These data demonstrate that the m allele does not increase apoptosis in the mammary gland, so apoptosis does not play a role in the ductal defect seen in the p53+/m virgin gland.
Mammary Epithelial Transplants into p53+/m Recipients Exhibit Reduced Ductal Outgrowth
The delayed ductal outgrowth observed in the virgin p53+/m gland suggests that mammary gland stem proliferation and/or differentiation may be inhibited by increased p53 activity (Fig. 1). Therefore, we compared the replicative lifespan of WT, p53+/−, and p53+/m mammary gland stem cells in vivo by performing serial mammary epithelium transplants into wildtype recipient mice. The p53+/m transplants exhibited a slightly reduced regenerative capacity compared to their WT counterparts, suggesting that cell autonomous effects of augmented p53 activity in the p53+/m mammary stem cells may have a marginal effect on stem cell regeneration (Supplementary Table 1). Interestingly, p53+/m transplants were capable of growing to fill the entire mammary fat pad by eight weeks post-transplant, suggesting a systemic effect on mammary gland development in the p53+/m mice (Fig. 3A).
Figure 3. Mammary epithelium transplants demonstrate that p53 dosage alters the systemic environment.

(A) Representative whole mounts of WT and p53+/m serial mammary epithelial transplants into wildtype recipient mice (transplant number one) demonstrate complete ductal outgrowth in p53+/m transplants. (B) Representative whole mounts of WT and p53+/m mammary epithelium transplants into p53+/m recipient mice four weeks post transplant demonstrate reduced ductal outgrowth of both WT and p53+/m transplants into p53+/m recipient mice. Each recipient mouse was transplanted with WT mammary epithelium in the left mammary gland and p53+/m mammary epithelium in the right gland. All images are shown at 3.5X magnification.
The complete ductal outgrowth observed in the serial transplantation of p53+/m epithelium into WT recipients, in contrast to the limited ductal outgrowth seen in virgin p53+/m glands, suggests that systemic or stromal alterations in the p53+/m mice may play a major role in the p53+/m mammary defect. The mammary stroma has been reported to play an important role in regulating development and ductal morphogenesis in the mammary gland (Walden et al., 1998). Proliferation signals mediated through epithelial-stromal crosstalk such as IGF-1, estrogen, and progesterone are critical for normal development to occur (Ruan et al., 1999; Ruan et al., 2005). To determine if systemic or stromal alterations were affecting ductal outgrowth in the p53+/m mice, mammary epithelial transplants were performed with p53+/m recipient mice. Mammary glands from WT or p53+/m virgin recipient mice were cleared and transplanted with 8 week virgin WT donor epithelium in the left gland and 8 week virgin p53+/m donor epithelium in the right gland to allow for a comparison of outgrowth. Whole mount analysis of the transplanted glands revealed that the majority of WT and p53+/m transplants into WT recipient mice showed robust ductal outgrowth, with all outgrowth filling greater than 50% of the fat pad by four weeks post transplant (Fig. 3B, Table 1). However, it was noted that fewer p53+/m transplants demonstrated outgrowth compared to WT transplants (Table 1). Interestingly, transplantation of either WT or p53+/m mammary epithelium into p53+/m recipient mice exhibited significantly reduced ductal outgrowth, with a mean percentage of fat pad filled of less than 20% in the majority of WT transplants and only 6% ductal outgrowth in the p53+/m transplants (Fig. 3B, Table 1). In addition, fewer WT and p53+/m transplants into p53+/m recipients demonstrated outgrowth (Table 1). These data suggest that the systemic increase in p53 activity and/or other systemic alterations in the p53+/m mice contribute to the developmental delay observed in the p53+/m mammary gland.
Table 1.
Mammary epithelial transplants of WT and p53+/m epithelium into p53+/m recipient mice.
| Recipient | Donor | # Outgrowths / # Recipients | Average % Outgrowth |
|---|---|---|---|
| P53+/+ | p53+/+ | 12/14* | 80 ± 6** |
| P53+/+ | p53+/m | 8/14 | 73 ± 11a |
| P53+/m | p53+/+ | 7/12 | 19 ± 6b |
| P53+/m | p53+/m | 6/12 | 6 ± 2c,d |
Proportion of outgrowths observed in recipient mice. Each recipient was transplanted with wildtype epithelium in the left gland and p53+/m epithelium in the right gland. Glands were harvested four weeks post transplant and examined for outgrowth by whole mount analysis. n=12–14 recipients, n=2 donors per genotype.
Values represent the mean percentage of ductal outgrowth ± s.e.m.
p < 0.6 compared to WT donor transplanted into WT recipient
p < 2.88 x 10−6 compared to WT donor transplanted into +/m recipient
p < 0.0002 compared to +/m donor transplanted into WT recipient
p < 0.06 compared to WT donor transplanted into +/m recipient
Steroid Hormone Treatment of p53+/m mice Rescues Mammary Gland Development
Examination of p53+/m whole mounts during development revealed that by mid-pregnancy (day 12), the p53+/m mammary gland exhibited complete ductal outgrowth with normal development of alveolar structures comparable to WT glands (Fig. 1B). These results suggest that increased levels of steroid hormones during pregnancy may be required to allow ductal outgrowth in the presence of increased p53 levels. To determine whether hormonal treatment is capable of stimulating mammary gland development in virgin p53+/m mice, mice were treated with estrogen, progesterone, or a combination of estrogen and progesterone for 14 days. Whole mount examination of mammary glands demonstrate that treatment with estrogen alone, progesterone alone, or estrogen and progesterone in combination rescues the ductal defect in virgin p53+/m mice, stimulating ductal outgrowth past the primary lymph node (Fig. 4). These data confirm that alterations in hormone levels induced by pregnancy are capable of stimulating ductal outgrowth in the p53+/m mice.
Figure 4. Treatment of p53+/m mice with steroid hormones restores ductal outgrowth.

Representative whole mounts of mammary glands from WT and p53+/m mice untreated or treated with estrogen (E), progesterone (P), or estrogen and progesterone (E+P) in combination for 14 days. (n=3–4).
p53+/m Mice Exhibit Decreased Levels of Serum IGF-1 and Attenuated IGF-1 Signaling
The decreased outgrowth observed in mammary epithelium transplants into p53+/m recipient mice, in addition to the stimulation of ductal outgrowth by hormonal treatment, suggests that the increased p53 activity in the p53+/m mice leads to alterations in stromal or systemic signaling which may play a role in the decreased ductal growth. Estrogen and progesterone are both known to enhance IGF-1 activity during ductal morphogenesis (Ruan et al., 2005). IGF-1, which is produced primarily in the stroma and mediates the proliferation signals of growth hormone in an autocrine fashion through stroma-epithelial crosstalk, is known to play a critical role in ductal morphogenesis (Ruan et al., 1999). In addition, serum IGF-1 may affect the mammary gland in an endocrine manner (Liu et al., 1999; Ruan et al., 1999). Interestingly, IGF-1 null mice exhibit impaired mammary gland development characterized by decreased numbers of terminal end buds and ducts as well as a decrease in the percentage of fat pad occupied by ductal outgrowth (Ruan et al., 1999). Since these phenotypes are comparable to those seen in the p53+/m mammary gland and p53 itself has been shown to play an integral role in regulating IGF-1 signaling through transcriptional repression of the IGF-1R (Werner et al., 1996), levels of IGF-1 were examined in the p53+/m mouse. Serum levels of IGF-1 were found to be significantly decreased (34%) in 8 week virgin p53+/m mice when compared to WT mice of the same age (Fig. 5A). While the decrease in circulating IGF-1 levels may contribute to the p53+/m mammary phenotype, it has been reported that paracrine levels of IGF-1 may play a more important role in mammary ductal morphogenesis than circulating levels of IGF-1 (Richards et al., 2004). Therefore, RT-PCR was performed to examine in situ levels of IGF-1 mRNA in the mammary gland and levels of IGF-1 signaling proteins were examined by Western blot analysis. p53+/m virgin glands exhibit reduced levels of insulin-receptor substrate 1 (IRS-1), IGF-1R, pIGF-R, AKT, and pAKT proteins, demonstrating reduced IGF-1 signaling (Fig. 5B), although no difference was observed in levels of IGF-1 mRNA in p53+/m virgin mammary glands when compared to WT glands (Supplementary Fig. 2). These data suggest that decreased IGF-1 serum levels lead to decreased IGF-1 signaling in the p53+/m virgin mammary gland which may contribute to the mammary phenotype.
Figure 5. Altered IGF-1 contributes to the mammary phenotype of the p53+/m mice.

(A) Quantitative comparison of mean IGF-1 serum levels (± s.e.m) of 8 week virgin WT, p53+/−, and p53+/m mice. (n=9). * p < 0.003 compared to WT. (B) Western blot analysis of IRS-1, AKT, pAKT, pIGF-1R, and IGF-1R, shows decreased IGF-1 signaling in p53+/m 5 week virgin mammary glands compared to wildtype virgin mammary glands. Treatment with LR3-IGF-1 rescues the attenuation of IGF-1 signaling in p53+/m glands. Values below each lane indicate relative protein levels after normalization to actin levels. (C) Representative whole mounts of 6 week virgin WT and p53+/m mammary glands from mice treated twice daily with LR3-IGF-1 for 5 days. Control mice were treated twice daily with 0.9% saline plus 10 mg/ml BSA. (n=4). All images are shown at 3.5X magnification. (D) Representative whole mounts of 8 week virgin WT, p53+/m, TTR-IGF-1/+, and TTR-IGF-1/+;p53+/m mammary glands. (n=3–4). All images are shown at 3.5X magnification.
Increased IGF-1 Serum Levels Rescue Mammary Gland Development in p53+/m Mice
To determine if the decrease in serum IGF-1 contributes to the decreased ductal outgrowth, p53+/m mice were treated twice daily with LR3-IGF-1 for five days. Examination of whole mounts following LR3-IGF-1 treatment demonstrated a rescue of ductal morphogenesis, characterized by increased ductal outgrowth past the primary lymph node, with a significant increase in the mean percentage of fat pad filled by ductal outgrowth (Fig. 5C, Supplementary Fig. 3). LR3-IGF-1 treatment of p53+/m mice also restored levels of IGF-1 signaling, with levels of IRS-1, IGF-1R, pIGF-R, pAKT, and AKT proteins comparable to wildtype post treatment (Fig. 5B).
Additionally, p53+/m mice were crossed to TTR-IGF-1/+ mice, which contain three copies of an IGF-1 transgene under the transthyretin promoter, which allows for liver specific overexpression of IGF-1, resulting in a 50–60% increase in serum IGF-1 levels (Liao et al., 2006). To determine if the increased serum IGF-1 levels in the TTR-IGF-1/+ mice could rescue the mammary phenotype of the p53+/m mice, 8 week virgin glands from TTR-IGF-1/+ ;p53+/m mice were examined by whole mount. TTR-IGF-1/+ ;p53+/m virgin glands exhibit an increase in ductal outgrowth past the primary lymph node (Fig. 5D), suggesting that the increased serum IGF-1 levels due to the presence of the TTR-IGF-1 transgenes is sufficient to rescue the p53+/m mammary gland defect. These data confirm that decreased levels of serum IGF-1 and decreased IGF-1 signaling contribute to the p53+/m mammary phenotype.
Discussion
The data reported here demonstrate that the increased wildtype p53 activity observed in the p53+/m mouse leads to a defect in mammary gland ductal morphogenesis, which is characterized by minimal ductal outgrowth beyond the primary lymph node in virgin mice. Previous studies have reported that p53 typically remains cytoplasmically sequestered in virgin glands, demonstrating that p53 activity is tightly regulated in the quiescent virgin gland (Kuperwasser et al., 2000; Minter et al., 2002). We have previously shown that the m allele increases both p53 stability and activity (Tyner et al., 2002; Moore et al., 2007). Additionally, the m allele drives nuclear localization of p53 even in the absence of stress, which may contribute to the increased p53 activity observed in the presence of the m allele (Moore et al., 2007). Although no significant increase in p53 protein levels was observed in the p53+/m virgin mammary gland, there is an increase in levels of p21 in response to irradiation, suggesting an increase in p53 activity in the p53+/m mammary gland (Supplementary Fig. 1). An increase in p53 activity could result in altered apoptotic levels. However, no increase in apoptosis was observed at any developmental stage, demonstrating that apoptosis does not play a role in the mammary phenotype of the p53+/m mouse. Increases in p53 activity in other tissues in the p53+/m mice, as previously observed in the kidney (Tyner et al., 2002), could cause systemic effects that alter mammary gland development, such as the observed decrease in serum IGF-1 levels. In fact, preliminary data suggests that there is a reduced ratio of IGF-1/IGFBP-3 mRNA levels in p53+/m liver (M. Pollak, unpublished data).
The ability of p53+/m serial mammary epithelial transplants to exhibit complete ductal outgrowth in a WT fat pad suggested that systemic or stromal alterations in the p53+/m mice, such as loss of IGF-1 signaling, contribute to the phenotype. In fact, p53+/m mice exhibit a reduction in IGF-1 serum levels that may contribute to the decreased proliferation observed in the p53+/m virgin gland. This suggests that p53 may be able to directly or indirectly regulate IGF-1 levels, although the mechanism remains unknown at this time. Interestingly, two progeroid mouse models, the DNA repair deficient XPF-ERCC1−/− mouse and the Csbm/m/Xpa −/− mouse, exhibit a reduced lifespan and chronic genotoxic stress which leads to suppression of the GH/IGF-1 somatotroph axis (Niedernhofer et al., 2006; 2006; van der Pluijm et al., 2006). It is possible that the increased p53 activity in the p53+/m mice may potentially replicate some aspects of the chronic genotoxic stress observed in these mice, as p53 is directly activated by genotoxic stress. The increased p53 activity in the p53+/m mice could induce a chronic augmented stress response, which may lead to the downregulation of the IGF-1 somatotrophic axis.
It was previously reported that paracrine levels of IGF-1 may play a more important role in mammary ductal morphogenesis than circulating levels of IGF-1 (Richards et al., 2004). However, the rescue of the p53+/m mammary phenotype by systemic treatment with LR3-IGF-1 and by crossing to the TTR-IGF-1/+ mice suggests that serum levels of IGF-1 significantly contribute to mammary gland development. Although RT-PCR analysis showed no alterations in IGF-1 mRNA levels in the p53+/m mammary gland itself, these glands exhibit a decrease in levels of IGF-1 signaling proteins, including the IGF-1R, a p53 target gene. p53 is known to transcriptionally repress expression of the IGF-1R and transcriptionally activate expression of IGF-1 signaling inhibitors IGF-BP3 and PTEN, demonstrating a direct role in p53-mediated regulation of the IGF-1 pathway (Buckbinder et al., 1995; Stambolic et al., 2001; Werner et al., 1996). In fact, disruption of the IGF-1R gene has been shown to reduce levels of proliferation in terminal end buds (Bonnette et al., 2001). These data suggest that the increased p53 activity in the p53+/m mouse attenuates IGF-1 signaling.
Strikingly, the p53+/m mice are also highly tumor resistant, which may be due in part to reduced levels of circulating IGF-1, as an increase in IGF-1 and IGF-1R levels have been shown to correlate with an increased risk of tumor development (Hankinson et al., 1998). In contrast to the p53+/m model, some of the premature aging mouse models which have been shown to exhibit increased IGF-1 levels also display increased incidence of tumorigenesis (Shukla et al., 2006; Xu et al., 2001).
Surprisingly, we saw a complete rescue of the p53+/m mammary gland phenotype with the induction of pregnancy. This rescue may be due to increased levels of estrogen and progesterone which are known to cause secondary changes in growth factor expression (Sternlicht et al., 2006). Interestingly, there is evidence that pregnancy can upregulate IGF signaling (Bonnette et al., 2001), suggesting that pregnancy may compensate for the reduction in circulating IGF-1 levels and decreased IGF-1 signaling observed in the p53+/m mice. In fact, both progesterone and estrogen have been shown to synergize with and enhance IGF-1 mediated ductal morphogenesis (Ruan et al., 2005). Supporting this, our data demonstrate that treatment with estrogen, progesterone, estrogen plus progesterone, or LR3-IGF-1 stimulates ductal outgrowth in the p53+/m gland. While, we can not rule out a p53-mediated reduction in estrogen or progesterone, we observed no alterations in fecundity or fertility in p53+/m mice, suggesting that alterations in estrogen and/or progesterone do not contribute to the mammary phenotypes. These data suggest that p53-mediated regulation of the IGF-1 signaling pathway may contribute to mammary gland development and support a model in which pregnancy-induced proliferation signals compensate for the increased p53 activity in the p53+/m mouse by stimulating or synergizing with IGF-1 signaling.
The decreased ductal outgrowth observed in transplantation of wildtype and p53+/m mammary epithelium into p53+/m recipients suggests that serum and/or stromal factors may also play a major role in the extrinsic regulation of mammary stem cell proliferation (Fig. 3B, Table 1). One of these extrinsic factors is likely to be IGF-1, as there is growing evidence that multiple stem cell types are functionally stimulated by IGF-1 (Bendall et al., 2007; Musaro et al., 2004). Intrinsic cell autonomous effects of enhanced p53 in the mammary gland stem cells may have only a minor role, as p53+/m mammary gland stem cells exhibited a marginal reduction in serial transplantation capacity compared to their WT counterparts (Supplementary Table 1). However, additional studies will be required to further clarify the role of p53 in regulation of the mammary gland stem cell.
The data presented here suggest that increased p53 activity can dramatically affect mammary gland development, in part through regulation of IGF-1 signaling. The increased p53 activity present in p53+/m mice during mammary gland ductal morphogenesis contributes to decreased proliferation in virgin glands, decreased levels of serum IGF-1, and decreased IGF-1 signaling in p53+/m virgin glands. However, other effects of enhanced p53 on estrogen and progesterone receptor signaling could also be playing an important role in this model. Collectively it is believed that these effects of increased p53 activity lead to the decreased ductal outgrowth observed in the p53+/m virgin glands. Our work solidifies the link between p53 and hormonal regulation, demonstrating that hormonal stimulation is able to overcome the proliferative block induced by increased p53 activity. These data suggest that the regulation of p53 activity is important for mammary gland development. However, further work is needed to fully elucidate the effects of altering p53 levels.
Supplementary Material
Western blot analysis of p53 and p21 in 8 week virgin WT and p53+/m mice shows increased levels of p21 in response to irradiation in p53+/m glands compared to WT glands. Values below each lane indicate relative protein levels after normalization to actin levels.
(A) RT-PCR analysis of IGF-1 mRNA levels in 12 week virgin WT, p53+/−, and p53+/m mammary glands. (B) Quantitative analysis of RT-PCR analysis of IGF-1 mRNA levels shows mean level of IGF-1 normalized to mean level of β-actin (± s.e.m). (n=4). * p < 0.3 compared to WT.
(A) Graph of the mean percentage of fat pad filled (FPF) in control and LR3-IGF-1 treated mice (± s.e.m). * p < 9.75 x 10−5 compared to WT control; ** p < 0.009 compared to p53+/m control. Mice were treated twice daily with 100μl of 0.25 μg/μl LR3-IGF-1 in 0.9% saline + 10 mg/ml BSA for 5 days. Control mice were treated twice a day with sterile 0.9% saline + 10 mg/ml BSA for 5 days. (n=4).
Acknowledgments
We thank Lynette Moore, George Hinkal, and Michael Gatza for helpful discussions. This work was supported by a grant from the National Cancer Institute to A. V. L. (PO1CA30195) and a grant from the National Institute for Aging to L.A.D. (R01AG19693). C.G. was supported, in part, by National Institute of Health training grant #T32 CA09197.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Akashi M, Koeffler HP. Li-Fraumeni syndrome and the role of the p53 tumor suppressor gene in cancer susceptibility. Clin Obstet Gynecol. 1998;41:172–199. doi: 10.1097/00003081-199803000-00024. [DOI] [PubMed] [Google Scholar]
- Becker KA, Lu S, Dickinson ES, Dunphy KA, Mathews L, Schneider SS, Jerry DJ. Estrogen and progesterone regulate radiation-induced p53 activity in mammary epithelium through TGF-[beta]-dependent pathways. Oncogene. 2005;24:6345–6353. doi: 10.1038/sj.onc.1208787. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, Werbowetski-Ogilvie T, Ramos-Mejia V, Rouleau A, Yang J, Bosse M, Lajoie G, Bhatia M. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature advanced online publication. 2007 doi: 10.1038/nature06027. [DOI] [PubMed] [Google Scholar]
- Bonnette SG, Hadsell DL. Targeted Disruption of the IGF-I Receptor Gene Decreases Cellular Proliferation in Mammary Terminal End Buds. Endocrinology. 2001;142:4937–4945. doi: 10.1210/endo.142.11.8500. [DOI] [PubMed] [Google Scholar]
- Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–2137. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger B, Kley N. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 1995;377:646–649. doi: 10.1038/377646a0. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Chao C, Saito S, Anderson CW, Appella E, Xu Y. Phosphorylation of murine p53 at ser-18 regulates the p53 responses to DNA damage. Proc Natl Acad Sci U S A. 2000;97:11936–11941. doi: 10.1073/pnas.220252297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles C, Condie A, Chetty U, Michael Steel C, John Evans H, Prosser J. p53 Mutations in Breast Cancer. Cancer Res. 1992;52:5291–5298. [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, Allan B. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Dumble M, Moore L, Chambers SM, Geiger H, Van Zant G, Goodell MA, Donehower LA. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736–1742. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatza C, Moore L, Dumble M, Donehower LA. Tumor suppressor dosage regulates stem cell dynamics during aging. Cell Cycle. 2007;6:52–55. doi: 10.4161/cc.6.1.3667. [DOI] [PubMed] [Google Scholar]
- Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, Rosner B, Speizer FE, Pollak M. Circulating concentrations of insulin-like growth factor I and risk of breast cancer. The Lancet. 1998;351:1393–1396. doi: 10.1016/S0140-6736(97)10384-1. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Current Biology. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Jerry DJ, Kuperwasser C, Downing SR, Pinkas J, He C, Dickinson E, Marconi S, Naber SP. Delayed involution of the mammary epithelium in BALB/c-p53null mice. Oncogene. 1998;17:2305–2312. doi: 10.1038/sj.onc.1202157. [DOI] [PubMed] [Google Scholar]
- Kuperwasser C, Pinkas J, Hurlbut GD, Naber SP, Jerry DJ. Cytoplasmic Sequestration and Functional Repression of p53 in the Mammary Epithelium Is Reversed by Hormonal Treatment. Cancer Res. 2000;60:2723–2729. [PubMed] [Google Scholar]
- Liao L, Dearth RK, Zhou S, Britton OL, Lee AV, Xu J. Liver-Specific Overexpression of the Insulin-like Growth Factor-I Enhances Somatic Growth and Partially Prevents the Effects of Growth Hormone Deficiency. Endocrinology. 2006;147:3877–3888. doi: 10.1210/en.2005-1537. [DOI] [PubMed] [Google Scholar]
- Liu JL, LeRoith D. Insulin-Like Growth Factor I Is Essential for Postnatal Growth in Response to Growth Hormone. Endocrinology. 1999;140:5178–5184. doi: 10.1210/endo.140.11.7151. [DOI] [PubMed] [Google Scholar]
- Lozano G, Elledge SJ. p53 sends nucleotides to repair DNA. Nature. 2000;404:24–25. doi: 10.1038/35003670. [DOI] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306–319. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D, Kittrell FS, Shephard A, Stephens LC, Jiang C, Lu, Allred DC, McCarthy M, Ullrich RL. Biological and genetic properties of the p53 null preneoplastic mammary epithelium. FASEB J. 2002;16:881–883. doi: 10.1096/fj.01-0885fje. [DOI] [PubMed] [Google Scholar]
- Medina D, Kittrell FS. p53 Function Is Required for Hormone-Mediated Protection of Mouse Mammary Tumorigenesis. Cancer Res. 2003;63:6140–6143. [PubMed] [Google Scholar]
- Minter LM, Dickinson ES, Naber SP, Jerry DJ. Epithelial cell cycling predicts p53 responsiveness to {gamma}-irradiation during post-natal mammary gland development. Development. 2002;129:2997–3008. doi: 10.1242/dev.129.12.2997. [DOI] [PubMed] [Google Scholar]
- Moll UM, Riou G, Levine AJ. Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc Natl Acad Sci U S A. 1992;89:7262–7266. doi: 10.1073/pnas.89.15.7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore L, Lu X, Ghebranious N, Tyner S, Donehower LA. Aging-associated truncated form of p53 interacts with wild-type p53 and alters p53 stability, localization, and activity. Mech Aging Dev. 2007 doi: 10.1016/j.mad.2007.10.011. (in revision). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musaro A, Giacinti C, Borsellino G, Dobrowolny G, Pelosi L, Cairns L, Ottolenghi S, Cossu G, Bernardi G, Battistini L, Molinaro M, Rosenthal N. Stem cell-mediated muscle regeneration is enhanced by local isoform of insulin-like growth factor 1. PNAS. 2004;101:1206–1210. doi: 10.1073/pnas.0303792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GTJ, Meinecke P, Kleijer WJ, Vijg J, Jaspers NGJ, Hoeijmakers JHJ. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- Purdie CA, Harrison DJ, Peter A, Dobbie L, White S, Howie SE, Salter DM, Bird CC, Wyllie AH, Hooper ML. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene. 1994;9:603–609. [PubMed] [Google Scholar]
- Richards RG, Klotz DM, Walker MP, DiAugustine RP. Mammary Gland Branching Morphogenesis Is Diminished in Mice with a Deficiency of Insulin-like Growth Factor-I (IGF-I), But Not in Mice with a Liver-Specific Deletion of IGF-I. Endocrinology. 2004;145:3106–3110. doi: 10.1210/en.2003-1112. [DOI] [PubMed] [Google Scholar]
- Richert MM, Schwertfeger KL, Ryder JW, Anderson SM. An atlas of mouse mammary gland development. J Mammary Gland Biol Neoplasia. 2000;5:227–241. doi: 10.1023/a:1026499523505. [DOI] [PubMed] [Google Scholar]
- Ruan W, Kleinberg DL. Insulin-Like Growth Factor I Is Essential for Terminal End Bud Formation and Ductal Morphogenesis during Mammary Development. Endocrinology. 1999;140:5075–5081. doi: 10.1210/endo.140.11.7095. [DOI] [PubMed] [Google Scholar]
- Ruan W, Monaco ME, Kleinberg DL. Progesterone Stimulates Mammary Gland Ductal Morphogenesis by Synergizing with and Enhancing Insulin-Like Growth Factor-I Action. Endocrinology. 2005;146:1170–1178. doi: 10.1210/en.2004-1360. [DOI] [PubMed] [Google Scholar]
- Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- Shukla V, Coumoul X, Cao L, Wang RH, Xiao C, Xu X, Ando S, Yakar S, LeRoith D, Deng C. Absence of the Full-Length Breast Cancer-Associated Gene-1 Leads to Increased Expression of Insulin-Like Growth Factor Signaling Axis Members. Cancer Res. 2006;66:7151–7157. doi: 10.1158/0008-5472.CAN-05-4570. [DOI] [PubMed] [Google Scholar]
- Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. Regulation of PTEN Transcription by p53. Molecular Cell. 2001;8:317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Kouros-Mehr H, Lu P, Werb Z. Hormonal and local control of mammary branching morphogenesis. Differentiation. 2006;74:365–381. doi: 10.1111/j.1432-0436.2006.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu XB, Soron G, Cooper B, Brayton C, Park SH, Thompson T, Karsenty G, Bradley A, Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- van der Pluijm I, Garinis GA, Brandt RM, Gorgels TG, Wijnhoven SW, Diderich KE, de Wit J, Mitchell JR, van Oostrom C, Beems R, Niedernhofer LJ, Velasco S, Friedberg EC, Tanaka K, van Steeg H, Hoeijmakers JH, van der Horst GT. Impaired genome maintenance suppresses the growth hormone--insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 2006;5:e2. doi: 10.1371/journal.pbio.0050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Walden PD, Ruan W, Feldman M, Kleinberg DL. Evidence That the Mammary Fat Pad Mediates the Action of Growth Hormone in Mammary Gland Development. Endocrinology. 1998;139:659–662. doi: 10.1210/endo.139.2.5718. [DOI] [PubMed] [Google Scholar]
- Werner H, Karnieli E, Rauscher FJ, III, LeRoith D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptorágene. PNAS. 1996;93:8318–8323. doi: 10.1073/pnas.93.16.8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward WA, Chen MS, Behbod F, Rosen JM. On mammary stem cells. J Cell Sci. 2005;118:3585–3594. doi: 10.1242/jcs.02532. [DOI] [PubMed] [Google Scholar]
- Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA, Harris CC, Den CX. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001;28:266–271. doi: 10.1038/90108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Western blot analysis of p53 and p21 in 8 week virgin WT and p53+/m mice shows increased levels of p21 in response to irradiation in p53+/m glands compared to WT glands. Values below each lane indicate relative protein levels after normalization to actin levels.
(A) RT-PCR analysis of IGF-1 mRNA levels in 12 week virgin WT, p53+/−, and p53+/m mammary glands. (B) Quantitative analysis of RT-PCR analysis of IGF-1 mRNA levels shows mean level of IGF-1 normalized to mean level of β-actin (± s.e.m). (n=4). * p < 0.3 compared to WT.
(A) Graph of the mean percentage of fat pad filled (FPF) in control and LR3-IGF-1 treated mice (± s.e.m). * p < 9.75 x 10−5 compared to WT control; ** p < 0.009 compared to p53+/m control. Mice were treated twice daily with 100μl of 0.25 μg/μl LR3-IGF-1 in 0.9% saline + 10 mg/ml BSA for 5 days. Control mice were treated twice a day with sterile 0.9% saline + 10 mg/ml BSA for 5 days. (n=4).