

SUMMARY
Actin polymerization in the mammalian cytosol can be locally activated by mechanisms that relieve the autoinhibited state of N-WASP, an initiator of actin assembly, a process that also requires the protein Toca-1. Several pathogenic bacteria, including Shigella, exploit this host feature to infect and disseminate efficiently. The Shigella outer membrane protein IcsA recruits N-WASP, which upon activation at the bacterial surface mediates localized actin polymerization. The molecular role of Toca-1 in N-WASP activation during physiological or pathological actin assembly processes in intact mammalian cells remains unclear. We show that actin tail initiation by S. flexneri requires Toca-1 for the conversion of N-WASP from a closed inactive conformation to an open active one. While N-WASP recruitment is dependent on IcsA, Toca-1 recruitment is instead mediated by S. flexneri type III secretion effectors. Thus, S. flexneri independently hijacks two nodes of the N-WASP actin assembly pathway to initiate localized actin tail assembly.
INTRODUCTION
Actin polymerization in the mammalian cytosol is globally inhibited, but can be locally activated by signals such as the activated form of the small Rho GTPase Cdc42 or phosphatidylinositol 4,5-bisphosphate (PIP2) (Figure 1A). Cdc42 and PIP2 induction of actin polymerization occurs by activating N-WASP, which is otherwise maintained in an inactive autoinhibited conformation in complex with WASP-interacting protein (WIP) (Kim et al., 2000; Martinez-Quiles et al., 2001; Miki et al., 1998). Cdc42 and PIP2 activation of endogenous N-WASP in vitro depend on Toca-1 (“transducer of Cdc42-dependent actin assembly”) (Ho et al., 2004), a member of the pombe Cdc15 homology (PCH) family, which is highly conserved among eukaryotes. While Toca-1 has recently been shown to be involved in the regulation of neurite elongation (Kakimoto et al., 2006), little is known about the molecular role of Toca-1 in activation of N-WASP during physiological actin assembly processes in intact mammalian cells.
Figure 1. Toca-1 Is Required for Efficient Assembly of Actin Tails by Intracellular S. flexneri.
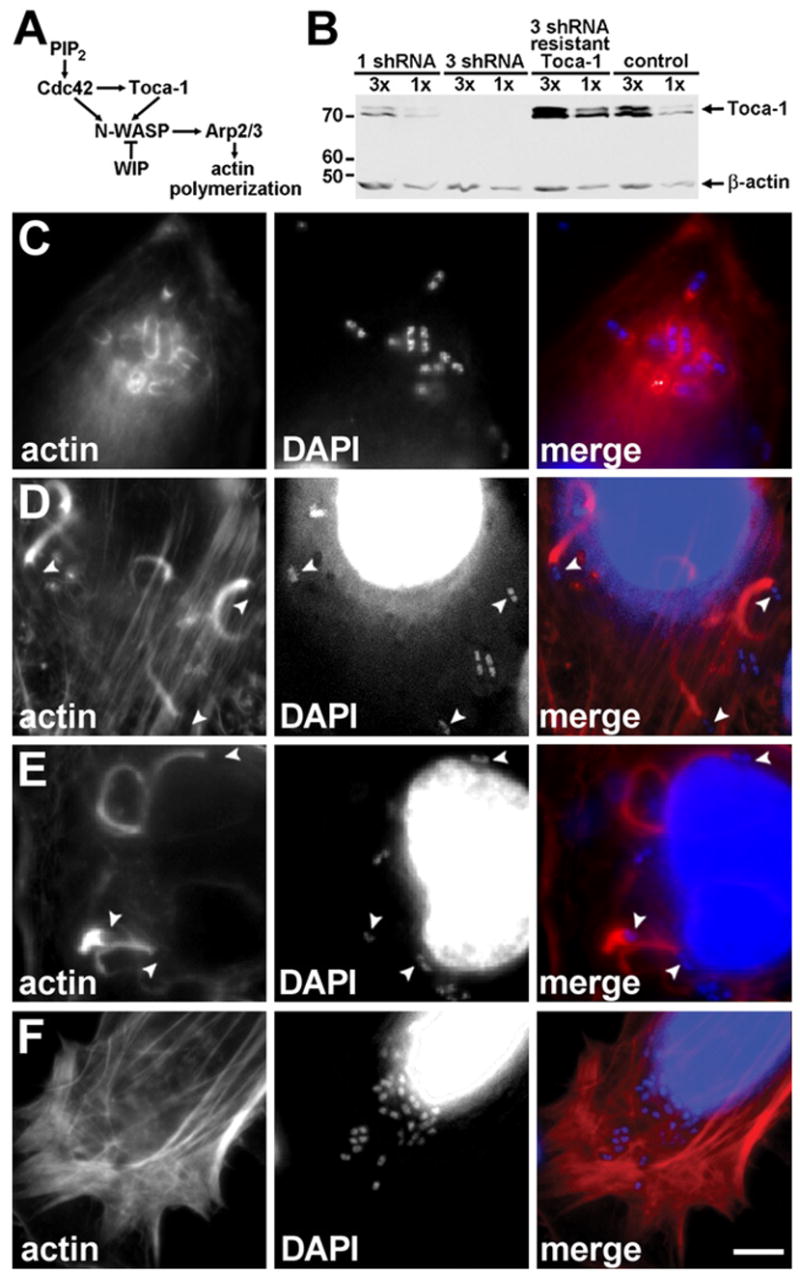
Dramatic reduction in actin tail assembly upon Toca-1 depletion in HeLa cells.
(A) Pathway of endogenous N-WASP activation, adapted from Ho et al. (2004).
(B) Reduction of Toca-1 expression with Toca-1 shRNAi in HeLa cells, using either one or three Toca-1 shRNA constructs. Western blot with relative loading indicated above each lane. β-actin levels confirmed loading. MW markers are in kD.
(C–F) Effect of reduction in Toca-1 on actin tail assembly. Intracellular wild-type S. flexneri in Toca-1-depleted cells (C), mock-depleted cells (D), and Toca-1-depleted cells expressing an RNAi-resistant Toca-1 (E). Cells infected with icsA S. flexneri (F). Fluorescent labeling of polymerized actin (red) and bacterial and cellular DNA with DAPI (blue). Arrowheads, bacteria with normal actin tails. Scale bar: (C)–(F), shown in (F), 10 μm.
Activated N-WASP induces the activation of the Arp2/3 complex, which mediates barbed end actin polymerization and crosslinking of filamentous actin at sites of cytoskeletal rearrangements in cells (Mullins et al., 1998; Welch et al., 1997) (Figure 1A). Several pathogenic bacteria, including Shigella sp. (Bernardini et al., 1989), Listeria monocytogenes (Tilney and Portnoy, 1989), Rickettsia sp. (Teysseire et al., 1992), Mycobacterium marinum (Stamm et al., 2003), and Burkholderia sp. (Kespichayawattana et al., 2000) assemble propulsive actin tails in the cytoplasm of infected mammalian cells by locally activating actin polymerization through the Arp2/3 complex (Egile et al., 1999; Gouin et al., 2004; Jeng et al., 2004; Moreau et al., 2000; Welch et al., 1997). In the case of Shigellae, N-WASP is recruited to intracellular Shigellae by the bacterial outer membrane protein IcsA (VirG) (Suzuki et al., 1998), whereupon N-WASP autoinhibition is overcome (Lommel et al., 2001; Snapper et al., 2001), albeit by mechanisms that have been unclear.
Here we show that Toca-1 is required for the relief of N-WASP autoinhibition during the initiation of actin tail assembly by S. flexneri. In addition, we show that Toca-1 recruitment to the bacterium is independent of IcsA, and instead is dependent on a distinct pathway that involves type III secretion effector proteins. Thus, Shigellae polymerize actin tails by intercepting two discrete nodes of the N-WASP actin assembly pathway using two distinct mechanisms.
RESULTS
Toca-1 Is Required for Efficient Actin Tail Formation
We examined the physiological and molecular function of Toca-1 in mammalian cells infected with S. flexneri. We downregulated Toca-1 expression in HeLa cells by introduction of short-hairpin interfering RNA (shRNA) against Toca-1 mRNA that effectively reduced the expression of endogenous Toca-1 to less than 5% of normal levels (Figure 1B, 3 shRNA). Toca-1 depletion led to a dramatic reduction in actin tail assembly of intracellular S. flexneri (Table 1), frequently resulting in the formation of clusters of intracellular bacteria (Figure 1C), which have also been described for icsA S. flexneri (Bernardini et al., 1989). The reduction in actin tail assembly was rescued by expression of an RNAi-resistant Toca-1 construct (Table 1), indicating that the phenotype was due to effects on Toca-1 levels per se. Similar to icsA S. flexneri, wild-type bacteria in Toca-1-depleted cells did not form normal actin tails; however, notably different from icsA S. flexneri, which do not recruit N-WASP or associate with polymerized actin, wild-type bacteria in Toca-1-depleted cells colocalized with some polymerized actin (Figure 1C versus Figure 1F, and Table 1), suggesting that IcsA was able to recruit N-WASP to these sites and that some of the N-WASP at these sites was active. Although Toca-1 depletion was greater than 95%, it was not complete; therefore, the small amount of actin tail assembly observed in the Toca-1-depleted cells may in part be due to residual Toca-1 present in these cells. Depletion of Toca-1 to 30%–50% of wild-type levels had no significant effect on actin tail assembly (Figure 1B, 1 shRNA, and Table 1), suggesting that low levels of Toca-1 are sufficient to support actin tail assembly.
Figure 3. Toca-1 Recruitment to the Vicinity of Intracellular S. flexneri Is Independent of IcsA and N-WASP.
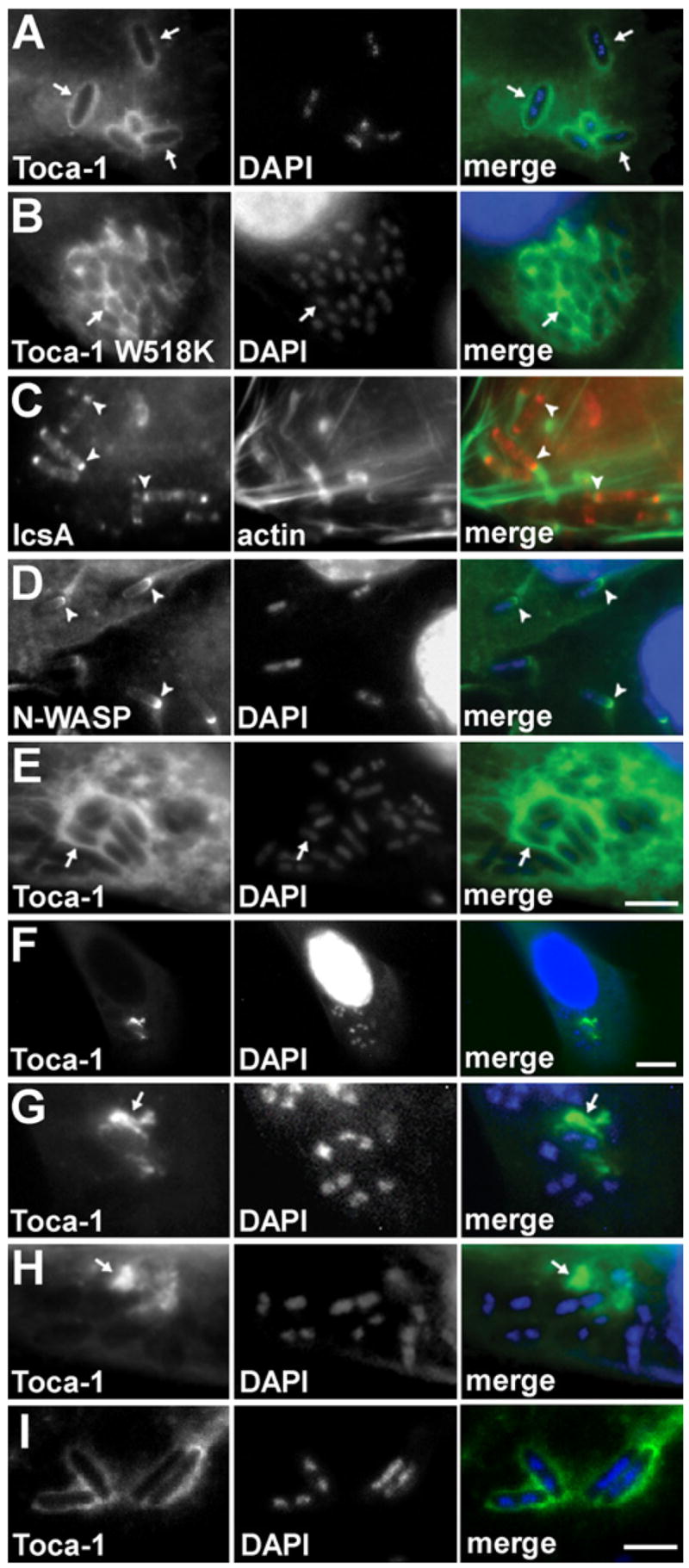
Wild-type (A–D, F, and G), icsA (E and H), or virA (I) S. flexneri with wild-type Toca-1 (A and C–I) or Toca-1 W518K (B) localized around the bacteria inHeLa cells (which are N-WASP+/+, [A]–[E] and [I]) or in N-WASP−/− fibroblast-like cells (F–H). IcsA ([C], arrowheads, red) or N-WASP ([D], GFP-N-WASP, arrowheads, green) localized to one end of the bacteria. Note more diffuse localization of Toca-1 around bacteria in N-WASP−/− cells (F–H). Green, GFP-Toca-1 or GFP-Toca-1 W518K (arrows) (A, B, and E–I), actin (C), or GFP-N-WASP (D). Red, immunofluorescent labeling of IcsA (C). Blue, fluorescent labeling of bacterial and cellular DNA with DAPI. Scale bars: (A)–(E), shown in (E), 5 μm; (F), 15 μm; (G)–(I), shown in (I), 4 μm.
Table 1. S. flexneri Actin Tail Assembly in Cells in Which Toca-1 Has Been Depleted or Overexpressed.
| Transfection Vector | S. flexneri Strain | Bacteria with Well-Formed Actin Tailsa | Bacteria Associated with Actinb | |||
|---|---|---|---|---|---|---|
| % of Bacteria | Mean Length (μm) | % of Bacteria | Mean Length (μm) | |||
| Toca-1 depletionc | three Toca-1 shRNAid | WT | 9.5 ± 1.2g,h | 4.5 ± 0.9l | 71.6 ± 3.0p | 0.6 ± 0.2t,u |
| one Toca-1 shRNAie | WT | 37.5 ± 4.2i | 5.1 ± 0.4m | 79.1 ± 6.4q | 2.1 ± 0.3v | |
| control shRNAi | WT | 41.2 ± 1.9g,i | 5.4 ± 0.2l,m | 74.4 ± 3.3p,q | 2.5 ± 0.1t,v | |
| three Toca-1 shRNAi, PCMV-Toca-1 (resistant)f | WT | 59.9 ± 1.6h | 5.2 ± 0.5 | 78.6 ± 2.5 | 3.3 ± 0.3u | |
| none | icsA | <1 | <0.1 | <1 | <0.1 | |
| Toca-1 overexpressionc | PCMV-Toca-1 | WT | 49.3 ± 5.6j | 5.0 ± 0.3n | 75.3 ± 3.3r | 3.5 ± 0.2w |
| PCMV-Toca-1 W518K | WT | 4.3 ± 1.0k | 6.8 ± 2.23 | 56.4 ± 3.1s | 0.4 ± 0.09x | |
| control plasmid | WT | 19.9 ± 2.3j,k | 5.0 ± 0.1n,o | 72.2 ± 1.7r,s | 1.1 ± 0.09w,x | |
Longer than 0.83 μm (10 pixels).
Associated with detectable polymerized actin, i.e., actin cap or actin tail.
Experimental conditions differ for the Toca-1 depletion and overexpression experiments in that the S. flexneri infection was 1.75 hr for the depletion experiment and 1.5 hr for the overexpression experiment.
>95% depletion of Toca-1.
30%–50% depletion of Toca-1.
RNAi-resistant Toca-1 construct.
p = 0.002.
p = 0.000001.
p = 0.3.
p = 0.0006.
p = 0.0002.
p = 0.2.
p = 0.3.
p = 0.4.
p = 0.1.
p = 0.4.
p = 0.4.
p = 0.1.
p = 0.001.
p = 0.0005.
p = 0.0008.
p = 0.1.
p = 0.00004.
p = 0.0003.
Toca-1 depletion did not affect entry of S. flexneri into host cells (9.0% ± 0.7% of cells were infected following Toca-1 depletion versus 10.9% ± 0.3% of cells following mock depletion). At the experimental time point, 100% of the bacteria examined in both Toca-1-depleted and mock-depleted HeLa cells had escaped endocytic vacuoles, as shown by transmission electron microscopy, whereas in a control infection, only one of eight bacteria of an avirulent S. flexneri strain was able to escape the vacuole following phagocytosis by J774 macrophage-like cells (see Figure S1 available online). Thus, the lack of actin tail formation by wild-type bacteria in Toca-1-depleted cells was not caused by an inability of S. flexneri to escape endocytic vacuoles in the absence of Toca-1.
The length of actin tails associated with intracellular bacteria is a correlate of the efficiency of actin polymerization by the bacteria (Sanger et al., 1992; Theriot et al., 1992). In Toca-1-depleted cells, a large percentage of bacteria accumulated polymerized actin on their surface but did not form actin tails, and the percentage of bacteria that formed normal-appearing actin tails was markedly reduced. We quantified the formation of well-formed tails by determining the frequency and length of tails longer than 10 pixels (0.83 μm). The presence of well-formed tails reflects both the establishment of tails and the efficiency of ongoing polymerization once tails are established. The percentage of bacteria that had well-formed tails was 4-fold decreased in Toca-1-depleted cells versus mock-depleted cells (Table 1, p = 0.002), although the mean length of well-formed tails was similar under the two conditions (Table 1, p = 0.2), suggesting that, although the initiation of actin tail assembly might be defective, ongoing polymerization of established tails was perhaps normal. The observed decrease in efficiency of actin tail assembly by S. flexneri was not due to decreased N-WASP expression or reduced N-WASP recruitment to intracellular bacteria caused by Toca-1 depletion, since both the expression level of N-WASP and the localization pattern of N-WASP on intracellular bacteria were similar in Toca-1-depleted cells and mock-depleted cells (Figure S2). Collectively, these results indicate that Toca-1 is required for efficient actin tail assembly. Moreover, they suggest that a critical role of Toca-1 in actin tail assembly may be at the initiation of actin tail polymerization and that, once established, the subsequent poly-merization process may be independent of Toca-1.
Given these findings, we postulated that an increase in Toca-1 expression would lead to increased efficiency of actin tail assembly by S. flexneri. Indeed, introduction of a vector expressing Toca-1 from the strong CMV promoter significantly increased the percentage of intracellular S. flexneri that had well-formed actin tails (Figure 2, and Table 1, p = 0.0006). Transfected cells expressed approximately 10-fold more Toca-1 protein than mock-transfected cells (Figure 2A). As for Toca-1 depletion, Toca-1 overexpression did not affect the length of well-formed actin tails (Table 1, p = 0.4), again suggesting that a critical role of Toca-1 may be at the step of initiation of actin tail assembly, rather than during ongoing actin tail elongation. The observed efficiency of actin tail formation in control cells was less than that in control cells in the depletion experiment because we reduced the infection time from 1.75 to 1.5 hr, since at later times, S. flexneri infection induced lysis of cells overexpressing Toca-1, likely a result of exuberant actin polymerization in these cells.
Figure 2. Overexpression of Toca-1 Leads to Increased Actin Tail Assembly by S. flexneri.
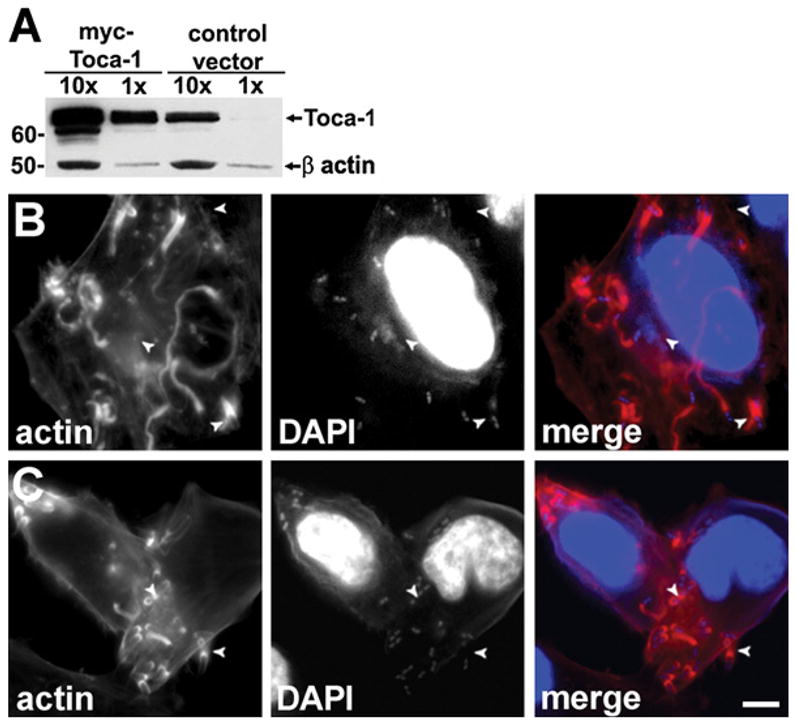
(A) Overexpression of Toca-1 in HeLa cells transfected with PCMV-Toca-1. Western blot with relative loading indicated above each lane. β-actin levels confirmed loading. MW markers are in kD.
(B and C) Effect of Toca-1 overexpression on actin tail assembly. S. flexneri infection of PCMV-Toca-1 transfected (B) or mock-transfected (C) cells. Fluorescent labeling as in Figure 1. Arrowheads, well-formed actin tails. Scale bar: (B) and (C), shown in (C), 10 μm.
Interaction of Toca-1 with N-WASP Is Necessary for Actin Tail Assembly
Toca-1 interacts directly with both N-WASP and Cdc42 (Ho et al., 2004). We tested whether the effect of Toca-1 on bacterial actin tail assembly depended on a direct interaction between Toca-1 and N-WASP, by examining the effect of overexpression of a Toca-1 mutant that does not bind N-WASP as a result of the SH3 domain mutation W518K (Ho et al., 2004). Overexpression of Toca-1 W518K did not lead to the increase in actin tail assembly that was observed in cells overexpressing wild-type Toca-1, consistent with Toca-1 binding to N-WASP being required for its effect on actin tail assembly. Indeed, in cells overexpressing Toca-1 W518K, the percentage of bacteria with well-formed tails was significantly decreased compared to that in either cells over-expressing wild-type Toca-1 or control cells (Table 1). The decrease in actin tail assembly in cells expressing Toca-1 W518K was not due to lack of recruitment of Toca-1 W518K to the bacterial surface, since, like wild-type Toca-1, Toca-1 W518K colocalized to intracellular bacteria (Figures 3A and 3B). The decrease in actin assembly below that observed for control cells is likely due to dominant-negative effects of Toca-1 W518K on bacterial actin assembly; alternatively, it could be due to secondary effects of the mutation on Toca-1 interactions with other proteins that modulate motility.
The Initiation of Bacterial Movement Is Associated with Colocalization of Toca-1
Under each condition, approximately 40% of intracellular bacteria colocalized with Toca-1 (Figure 4A and data not shown). To explore the possibility that Toca-1 might be required for the initiation of actin tail formation, we examined when, during actin tail formation, Toca-1 colocalized with intracellular bacteria. The vast majority of intracellular bacteria (83%) that colocalized with Toca-1 were stationary, as demonstrated by the lack of actin tails on these bacteria. This pattern was consistent with the possibility that Toca-1 might be transiently recruited to the bacterium prior to actin tail formation and that it might then dissociate after actin tail elongation is initiated.
Figure 4. Toca-1 Colocalization with Intracellular S. flexneri Is Associated with the Initiation of Actin-Based Motility.
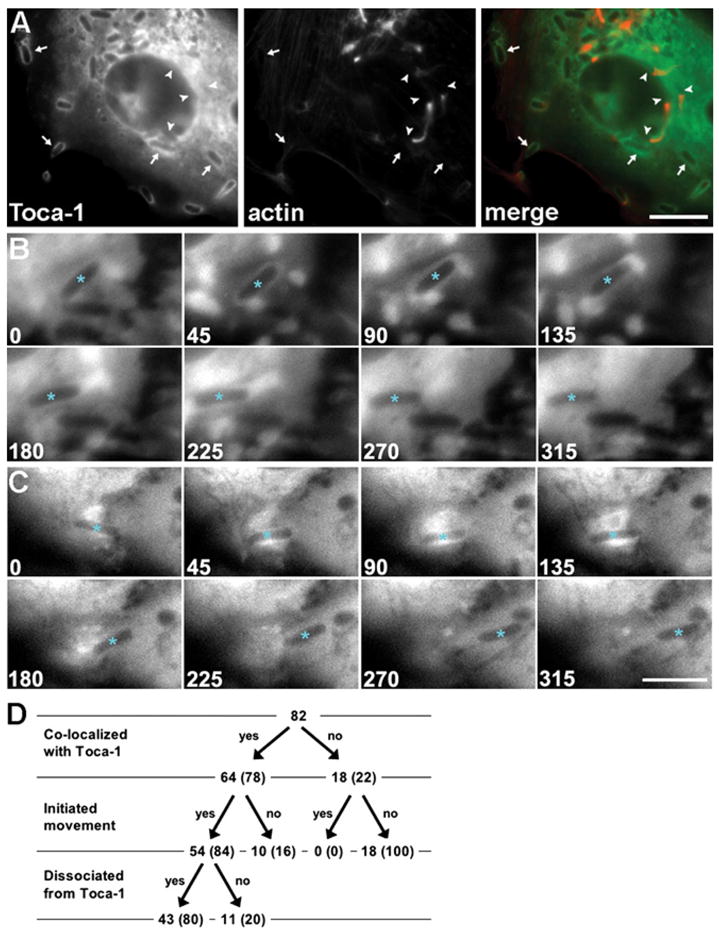
(A) Toca-1 colocalization is predominantly with bacteria that are stationary. GFP-Toca-1 (green) and polymerized actin (red). Arrows, stationary bacteria that colocalize with Toca-1. Arrowheads, bacteria with actin tails that do not colocalize with Toca-1.
(B and C) Temporal sequence of Toca-1 recruitment to bacteria and initiation of actin-based motility in live cells that express GFP-Toca-1. Time-lapse sequences of individual S. flexneri (asterisks) that initiated movement during period of observation. Transient recruitment of Toca-1 to the surface of bacteria at 45–90 s (B) and 45–135 s (C). Scale bars: (A), 10 μm; (B) and (C), shown in
(C), 5 μm.
(D) Flow chart of all bacteria that were initially stationary, tracked for colocalization with Toca-1 and whether actin-based motility was initiated.
We visualized the temporal sequence of Toca-1 recruitment to bacteria relative to initiation of actin tail formation by time-lapse microscopy of S. flexneri in cells expressing GFP-Toca-1. Toca-1 was transiently recruited to the surface of stationary bacteria and dissociated from bacteria as they became motile (Figures 4B and 4C and Movies S1–S3). Frequently, just prior to the initiation of movement, the signal from Toca-1 was observed to transiently form a tight ring or increased signal around the bacterium that then dispersed once the bacterium began to move. Of a total of 104 bacteria visualized in 35 independent series of time-lapse images, 82 (79%) were initially stationary (Figure 4D). Among those that were initially stationary, 64 (78%) colocalized with the GFP-Toca-1 at the onset of the time-lapse sequence and 18 (22%) did not. Of these 18, none (0%) became motile during the course of time-lapse microscopy, whereas 54 (84%) of the 64 bacteria that were initially stationary and colocalized with Toca-1 started moving during the period of observation (5–10 min). Among the bacteria that were initially stationary, initiation of movement was significantly associated with colocalization of Toca-1 (chi-square, p = 0.001). Among the 54 bacteria that colocalized with Toca-1 and started moving, Toca-1 dissociated from 43 (80%) at some point after initiating movement and during the period of observation. The disappearance of the GFP-Toca-1 signal from bacteria that became motile was not due to photobleaching of GFP, since the signals on bacteria that remained stationary continued to be bright throughout the duration of the experiments.
Toca-1 Is Required for the Relief of N-WASP Autoinhibition
Thus, Toca-1 recruitment to stationary bacteria is associated with the initiation of actin tail formation. Moreover, the transience of Toca-1 colocalization with the bacterium suggests that, once actin tail assembly is initiated, Toca-1 is not required for maintenance of actin tail polymerization. These observations are consistent with Toca-1 being required for the activation of N-WASP on the bacterial surface early in the process of actin tail assembly. For actin tail assembly to occur, inactive autoinhibited N-WASP that is recruited to the bacterial surface by S. flexneri IcsA must be converted to an open active conformation. In the case of S. flexneri, the activation of N-WASP occurs independently of Cdc42 (Shibata et al., 2002). Indeed, the effect of Toca-1 depletion on actin tail assembly was unaffected by a dominant-negative or a constitutively active Cdc42 (Table S3). In addition, the effect of Toca-1 depletion on actin tail assembly was unaffected by the absence of WIP, a negative regulator of N-WASP (Figure 1A, and data not shown).
To determine whether Toca-1 is required for relief of N-WASP autoinhibition, we tested whether a constitutively open derivative of N-WASP was independent of Toca-1 in S. flexneri actin tail assembly. We generated a leucine-to-proline mutation at the N-WASP leucine-235, which corresponds to leucine-270 of WASP. The L270P mutation in WASP, which is the cause of human Wiskott-Aldrich syndrome, leads to a constitutively open molecule (Devriendt et al., 2001).
The open conformation N-WASP mutant was independent of Toca-1, as demonstrated by its ability to trigger efficient actin tail formation in Toca-1-depleted cells. Whereas in cells expressing wild-type N-WASP the percentage of bacteria with well-formed actin tails was markedly reduced by depletion of Toca-1 (Table 2, p = 0.0006), in cells expressing N-WASP L235P, the percentage of bacteria with well-formed tails was unaltered by Toca-1 depletion (Table 2, p = 0.07). These results indicate that Toca-1 is involved in the relief of N-WASP autoinhibition and the accompanying conformational change. Once N-WASP is activated, Toca-1 appears to be not required for the subsequent maintenance of N-WASP in the active conformation, as supported by our data that the length of well-formed actin tails is independent of the level of Toca-1 expression and that Toca-1 is transiently recruited to the bacterial surface prior to initiation of movement.
Table 2. S. flexneri Actin Tail Assembly in Cells Expressing Wild-Type or L235P N-WASP with or without Toca-1 Depletion.
| N-WASP Transfection Vector | shRNAi Transfection Vector | Bacteria with Well-Formed Actin Tailsa | Bacteria Associated with Actinb | ||
|---|---|---|---|---|---|
| % of Bacteria | Mean Length (μm) | % of Bacteria | Mean Length (μm) | ||
| WT | Toca-1 | 7.1 ± 2.9c,d | 4.5 ± 0.1f,g | 52.1 ± 6.6m,n | 0.4 ± 0.1p,q |
| control | 35.6 ± 5.3c,e | 4.7 ± 0.7f,h | 59.4 ± 9.4m,o | 1.6 ± 0.4p,r | |
| L235P | Toca-1 | 49.7 ± 7.7d,k | 4.9 ± 0.7g,i | 55.3 ± 3.9n,i | 2.1 ± 0.4q,j |
| control | 41.6 ± 2.2e,k | 5.5 ± 0.8h,i | 62.5 ± 5.5o,i | 2.8 ± 0.6r,j | |
Longer than 0.83 μm (10 pixels).
Associated with detectable polymerized actin, i.e., actin cap or actin tail.
p = 0.0006.
p = 0.00004.
p = 0.02.
p = 0.4.
p = 0.2.
p = 0.1.
p = 0.07.
p = 0.09.
p = 0.07.
p = 0.2.
p = 0.2.
p = 0.3.
p = 0.3.
p = 0.008.
p = 0.001.
p = 0.02.
Like S. flexneri, the intracellular pathogen L. monocytogenes employs actin-based motility to move through cell monolayers. However, actin tail assembly by L. monocytogenes does not require N-WASP, since the L. monocytogenes surface protein ActA functionally mimics N-WASP and is sufficient to activate Arp2/3-mediated actin polymerization (Welch et al., 1997; Gouin et al., 2005; Snapper et al., 2001). Our results indicating that the effect of Toca-1 on S. flexneri actin assembly is via N-WASP suggested that actin tail assembly by L. monocytogenes would be independent of Toca-1. Indeed, analysis of L. monocytogenes-infected cells depleted of Toca-1 shows that this organism assembles actin tails independently of Toca-1 (Table S1).
Toca-1 Recruitment to S. flexneri Is Dependent on Bacterial Type III Secretion
Toca-1 formed tight rings around intracellular S. flexneri (Figure 3A); the distribution of native Toca-1 was identical to that of Toca-1-GFP, and GFP alone did not localize to the bacteria (data not shown). In contrast to the distribution of Toca-1 around the entire bacterium, IcsA is restricted to the bacterial pole (Figure 3C; Goldberg et al., 1993), leading to focal recruitment of N-WASP primarily to one end of the bacterial body (Figure 3D) and suggesting that Toca-1 recruitment may be independent of both IcsA and N-WASP. The recruitment of Toca-1 to intracellular bacteria persisted upon infection with an icsA mutant and expression of the W518K derivative of Toca-1 (Figures 3B and 3E). In cells lacking N-WASP (Figures 3F–3H), Toca-1 was no longer tightly associated with the bacterial surface but was still present in the vicinity of the bacteria, suggesting that both N-WASP and a second diffusible protein contribute to Toca-1 recruitment.
Many S. flexneri virulence proteins that are secreted into the host cytosol during infection are secreted by a type III secretion machinery. Bacterial type III secretion machineries are highly conserved gram-negative bacterial secretion systems that translocate proteins directly from the bacterial cytoplasm into the host cell cytosol during infection (Galan and Wolf-Watz, 2006). Of note, the secretion of IcsA, a bacterial autotransporter, is independent of type III secretion (Brandon et al., 2003). To investigate a possible role for type III secreted effector proteins in recruitment of Toca-1, we examined whether Toca-1 recruitment occurred in the absence of type III secretion. Cells were infected with a S. flexneri derivative that conditionally expresses VirB, the global regulator of type III secretion, under the control of an IPTG promoter (Schuch et al., 1999). Since S. flexneri entry into cells depends on type III secretion, IPTG was included in the media during bacterial entry and was removed or not removed from the culture media for the remainder of the infection. When IPTG was not removed, VirB expression was maintained, and Toca-1 was recruited to intracellular bacteria as it was to wild-type S. flexneri (Figure 5A and Figure S3). In contrast, when IPTG was removed, VirB expression was shut off (Figure S3), and whereas N-WASP recruitment was unaltered, Toca-1 was no longer recruited to intracellular bacteria (Figure 5B and data not shown). As expected by the absence of Toca-1, actin tail assembly by bacteria lacking VirB was markedly reduced (Table S2). The lack of Toca-1 recruitment in the absence of IPTG was not due to an absence of the type III secreted effector protein VirA, which contributes to motility via severing the microtubule cytoskeleton (Yoshida et al., 2006), since a virA mutant recruits Toca-1 to its surface (Figure 3I). These results indicate that expression of type III secretion is necessary for recruitment of Toca-1. Given that the effects of type III secretion are mediated by effector proteins secreted through the type III secretion machinery, these results indicate that Toca-1 recruitment is dependent on type III secreted effectors.
Figure 5. Toca-1 Recruitment to the Surface of Intracellular S. flexneri Is Dependent on Type III Secretion, and Model of Shigella Activation of the N-WASP Actin Assembly Pathway.
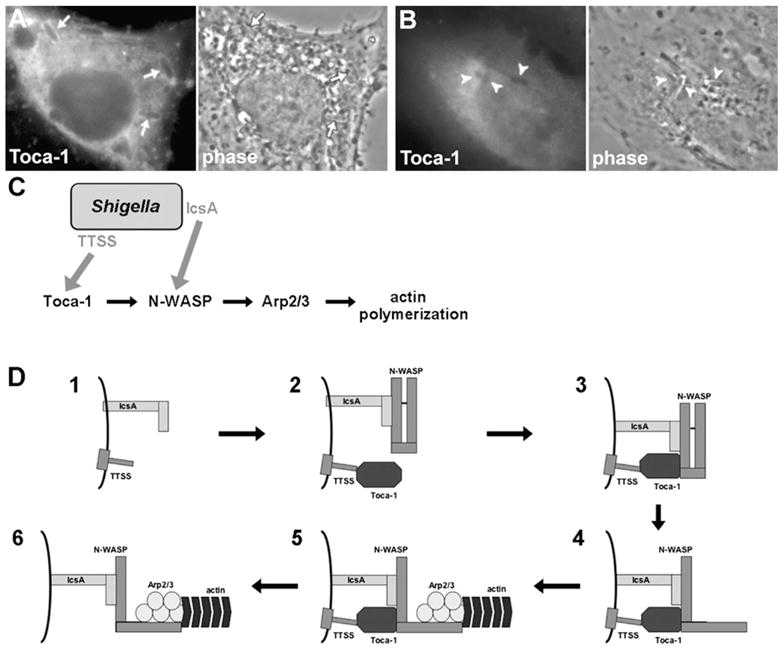
(A and B) Recruitment of Toca-1 to intracellular S. flexneri that express virB ([A], arrows), the master regulator of type III secretion, but not to intracellular S. flexneri that do not express virB ([B], arrowheads). Conditional expression of virB under the control of an IPTG-inducible promoter by addition (A) or absence (B) of IPTG.
(C) Model of Shigella interaction with two discrete nodes of the N-WASP actin assembly pathway using two distinct mechanisms. IcsA recruitment of N-WASP and type III secretion system (TTSS)-dependent recruitment of Toca-1. Shigella factors are indicated with gray text. Gray arrows indicate a role in recruitment of the targeted host factor.
(D) Model of detailed interplay of IcsA-dependent recruitment of N-WASP and TTSS-dependent recruitment of Toca-1. (D1) IcsA and TTSS on the bacterial surface. (D2) IcsA recruits N-WASP, and TTSS-dependent processes recruit Toca-1. (D3) Toca-1 binds N-WASP. (D4) Toca-1 relieves N-WASP autoinhibition, leading to a conformational change in N-WASP. (D5) The active open conformation of N-WASP binds and activates the Arp2/3 complex, leading to localized actin polymerization. (D6) Toca-1 dissociates from the vicinity of the bacterial surface, and N-WASP remains in the active open conformation.
These findings indicate that Shigella has evolved two independent mechanisms of interacting with two discrete nodes of the N-WASP actin assembly pathway. First, the bacterial autotransporter IcsA binds N-WASP at the bacterial pole (Figures 5C and 5D). And second, bacterial type III secreted effector proteins recruit the N-WASP activator Toca-1, with the tight association of Toca-1 to the bacterial surface dependent on the presence of N-WASP as well (Figures 5C and 5D).
DISCUSSION
In cells, N-WASP is maintained in an equilibrium between an inactive autoinhibited conformation and an active open conformation. In resting cells, N-WASP is largely in the inactive autoinhibited conformation. Activation of cellular N-WASP-dependent actin assembly involves recruitment of N-WASP to the plasma membrane and a shift in the N-WASP equilibrium to the active open conformation. The relief of N-WASP autoinhibition involves Cdc42, PIP2, and Toca-1 (Ho et al., 2004; Miki et al., 1996; Rohatgi et al., 1999).
Our data indicate that Shigella recruitment of N-WASP and the subsequent shift in the equilibrium toward the active open conformation occurs in two similarly distinct steps (Figure 5D). IcsA recruits N-WASP to the bacterial surface (Suzuki et al., 1998) and type III secretion effector proteins recruit Toca-1. Then, Toca-1 shifts the N-WASP conformational equilibrium toward active N-WASP, thereby allowing its efficient activation of the downstream effector complex Arp2/3. Thus, in the presence of both Toca-1 and N-WASP, the initiation of actin tail assembly occurs efficiently. In the absence of Toca-1, it appears that only a small amount of N-WASP is in the open conformation, since we observed only a low level of actin polymerization at the bacterial surface.
Once Toca-1 assists in relieving N-WASP autoinhibition, N-WASP appears to be maintained in the open confirmation by Toca-1-independent processes, as supported by our results indicating that Toca-1 is no longer required for actin tail polymerization once movement is initiated (Table 1). In addition, once movement is initiated, Toca-1 appears to dissociate from the bacterial surface (Figure 4). By extension, these findings suggest that, under certain circumstances in cells, Toca-1 may accelerate the conformational change in N-WASP, but may be minimally involved in subsequent steps of actin polymer elongation.
In this way, S. flexneri simultaneously hijacks two distinct nodes of the N-WASP activation pathway (Figure 5D), N-WASP and Toca-1. In cells, N-WASP and Toca-1 are each recruited to the membrane by direct interactions with membrane-anchored activated Cdc42 (Ho et al., 2004), whereupon Cdc42 and Toca-1 together activate N-WASP. S. flexneri has bypassed the cellular requirement for Cdc42 (Shibata et al., 2002) and instead independently recruits N-WASP via IcsA (Suzuki et al., 1998) and Toca-1 via a type III secretion-dependent mechanism. The requirement for both N-WASP and Toca-1 leads to spatial restriction of actin tails to the site where both proteins are present, the bacterial pole. Moreover, this mechanism of independent engagement of two nodes of the cellular N-WASP actin assembly pathway is distinctly different from mechanisms evolved by other intracellular microorganisms that move through the cytosol by actin polymerization.
These data constitute evidence that Toca-1 is required for N-WASP-mediated actin polymerization by a bacterial pathogen in vivo. They also highlight differences between the regulation of endogenous N-WASP and that of recombinant purified N-WASP. Unlike purified N-WASP, endogenous N-WASP is in complex with and inhibited by WIP (Martinez-Quiles et al., 2001), and Cdc42 binding to endogenous N-WASP is not sufficient to activate actin polymerization (Ho et al., 2004); Toca-1 is also required (Ho et al., 2004). In vitro, N-WASP can be activated at the Shigella surface in the absence of Toca-1 (Egile et al., 1999; Goldberg and Theriot, 1995; Kocks et al., 1995; Magdalena and Goldberg, 2002), yet our data indicate that, in vivo, IcsA alone is a poor activator of endogenous N-WASP. Instead, in vivo, Toca-1 is critical for relieving the autoinhibited conformation of N-WASP.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Media
The wild-type S. flexneri strain used in this study is serotype 2a strain 2457T (LaBrec et al., 1964), which contains the large virulence plasmid that is present in all pathogenic Shigellae. The icsA mutant is MBG283, which is 2457T icsA::Ω specR (Ally et al., 2004). The conditional virB mutant is AWY3 (Wing et al., 2004), which is 2457T virB::Tn5, carrying pDSW206-Ptac-virB. pDSW206-Ptac-virB was generated by cloning the fragment containing virB from PLAC-virB (Schuch et al., 1999) into pDSW206 (Weiss et al., 1999). S. flexneri were grown in tryptic soy broth from individual colonies that were red on agar containing Congo red. The wild-type L. monocytogenes strain 10403S was grown in brain heart infusion (BHI) broth or on BHI agar.
Cell Culture
HeLa cells were maintained in minimal essential medium, supplemented with10% fetal bovine serum, under humidified air containing 5% CO2 at 37°C. N-WASP−/− SV fibroblast-like cells (FLCs) (Snapper et al., 2001; gift of Scott B. Snapper) were maintained under the same conditions in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum.
Bacterial Infection of Cells
S. flexneri infection of mammalian cells was performed as described previously (Ally et al., 2004). In brief, exponential phase bacteria grown at 37°C were centrifuged at 700 × g for 10 min onto semiconfluent monolayers of cells that had been seeded onto cover glasses in six-well plates, using a multiplicity of infection (MOI) of 100:1. Bacteria and cells were routinely incubated at 37°C for 30–60 min to allow bacterial entry into cells. Then, gentamicin, which kills extracellular but not intracellular bacteria, was added to the infected monolayers for an additional 60 min. Infection of mammalian cells with L. monocytogenes differed slightly, in that overnight cultures grown at 30°C were back diluted and grown 2 hr at 37°C. Bacteria were then harvested, centrifuged onto cell monolayers, and incubated at 37°C for 5 hr with gentamicin (10 μg/ml) after the initial 1 hr incubation that allowed bacterial entry. For the analysis of bacterial entry, the percentage of cells that were infected in 15 to 20 randomly chosen microscopic fields was determined. The observation of Toca-1 localization with time-lapse microscopy was performed at early times of infection; for these experiments, the total infection time was shortened to 25–35 min. The analysis of whether Toca-1 is involved in the change in conformation of N-WASP was performed in N-WASP−/− cells that had been transfected with either pEGFP-C2-N-WASP or pEGFP-C2-N-WASP-L235P.
For the analysis of the role of bacterial type III secreted effectors in the recruitment of Toca-1, a vector carrying the virB gene under the control of anIPTG-inducible promoter, pDSW206-Ptac-virB, was introduced into a virB mutant of S. flexneri 2457T. IPTG was maintained in the media in which the strain was grown during the hour preceding infection, to induce expression of VirB for bacterial entry, and was removed immediately prior to bacterial contact with cells. Within approximately 25 min of removal of IPTG, expression of VirB ceased, as demonstrated by immunoblotting. Consequently, under the experimental conditions used, VirB was present during entry but not expressed by intracellular bacteria beyond the first 25 min of infection.
Fixation and permeabilization of infected cells were performed as described previously (Ally et al., 2004). Each expression construct was introduced into cells by electroporation of approximately 5 × 106 cells with 25–30 μg of the corresponding plasmid DNA 16 hr prior to infection with S. flexneri. Expression plasmids pCS2-MT-Toca-1, pCS2-EGFP-Toca-1, pCS2-MT-MGD, pCS2-MT-W518K, for the expression under the control of the CMV promoter of Myc-Toca-1, GFP-Toca-1, and Myc-tagged Toca-1 W518K, respectively, were the gift of Henry Ho and Marc Kirschner (Ho et al., 2004). The plasmid pCMV-Myc (BD Biosciences) was used as a control in the Toca-1 overexpression experiments. pEGFP-C2-N-WASP, for the expression of GFP-N-WASP, was the gift of Anna Lyubimova and Scott B. Snapper. pEGFP-C2-N-WASP-L235P was constructed by changing leucine-235 to proline by site-directed mutagenesis of pEGFP-C2-N-WASP using the QuickChange Mutagenesis Kit (Strategene). The N-WASP gene fragment carrying the mutation was then subcloned into the original pEGFP-C2 vector. Using the QuickChange Muta-genesis Kit (Strategene), an RNAi-resistant Toca-1 expression vector was constructed from pCS2-MT-Toca-1 by making two to three silent mutations in each of the three regions of the Toca-1 gene targeted by the shRNA constructs used in this study. The Toca-1 gene fragment carrying these silent mutations was then subcloned into the original pCS2-MT vector. For immunofluorescent labeling of IcsA, a monoclonal antibody to IcsA was used, as described previously (Robbins et al., 2001). Actin filaments were labeled with 200 U/ml Texas red-conjugated or FITC-conjugated phalloidin (Molecular Probes) for 20 min at room temperature. Bacterial and cellular DNA were labeled with 300 nM 4′-6-diamidino-2-phenylindole (DAPI, Molecular Probes) for 5 min at room temperature. Localization of native Toca-1 on bacterial surface was visualized using immunofluorescence with a rabbit polyclonal anti-Toca-1 antibody (gift of M. Kirschner [Ho et al., 2004]). Fixed samples were incubated with anti-Toca-1 antibody at 8 μg/ml for 1 hr at RT, followed by 1 hr of incubation with a FITC-conjugated anti-rabbit secondary antibody at RT.
Toca-1 Depletion and Overexpression
For the depletion of Toca-1, three shRNA vectors that targeted distinct sequences within mammalian TOCA-1 mRNA (V2HS_173540, V2HS_173541 and V2HS_173544, OpenBiosystems) were used in combination. As a control, a nonsilencing pSM2 shRNA vector (OpenBiosystems), which encodes an siRNA sequence not found in the rat or human genome in BLAST search, was used. shRNA and control constructs were introduced into HeLa cells by transfection using Arrest-In transfection reagent (OpenBiosystems). In experiments investigating the effect of partial depletion, only one shRNA vector (V2HS_173544) was used. pCS2-EGFP plasmid DNA, which encodes GFP, or pEGFP-C2-N-WASP, which encodes GFP-N-WASP, was introduced by co-transfection with the shRNA or control vectors at a ratio of 1:20 or 1:10, respectively. Thus, cells that expressed GFP were highly likely to be transfected with the shRNA or control vector. Where appropriate, transfected cells were selected by growth in the presence of 1 μg/ml puromycin (Sigma) for 3–5 days posttransfection prior to infection with S. flexneri or immunoblotting. In Toca-1 “rescue” experiments, Toca-1-depleted cells were transfected with the RNAi-resistant Toca-1 expression vector or control vector 16–20 hr prior to infection with S. flexneri. Transfected cells were identified in fixed samples by immunofluorescent labeling of the Myc tag on Toca-1 using a mouse anti-Myc antibody (Sigma).
Immunoblotting
Cellular proteins were separated by SDS-12.5% PAGE. Equivalent amounts of protein were loaded into each lane by normalizing the harvest volume to cell density and were confirmed by detection of β-actin where appropriate. For western blot analysis, the following antibodies were used: a rabbit polyclonal antibody to Toca-1 (Ho et al., 2004; gift of M. Kirschner), a monoclonal antibody against β-actin (Sigma), a rabbit polyclonal antibody to N-WASP (gift of M. Kirschner), and a rabbit polyclonal antibody to VirB (Beloin et al., 2002; gift of C. Dorman). The signal was detected by enhanced chemiluminescence (Pierce).
Microscopy and Data Analysis
Fluorescent and phase microscopy were performed using a Nikon TE300 or TE2000 microscope with Chroma Technology filters. Images were captured digitally using a black and white Photometrics CoolSNAP HQ charge-coupled device (CCD) camera (Roper Scientific) and IP laboratory software (Scanalytics). Color figures were assembled by separately capturing signals with each of the appropriate filter sets and digitally pseudocoloring the images using Adobe Photoshop software.
Time-lapse microscopy was performed as described previously (Ally et al., 2004). Images were acquired in a blinded fashion. For each data set, measurements were taken for 100 or more bacteria in 20 or more randomly selected infected cells in each of three or four independent experiments. Data analysis was performed using the chi-square test and the Student’s t test.
Samples for transmission electron microscopy were prepared as described previously (Taunton et al., 2000) and viewed on a JEOL 1200 EX microscope. For each experimental condition, eight to ten randomly selected bacteria were analyzed in a blinded fashion for the presence or absence of surrounding vacuolar membranes.
Supplementary Material
Supplemental Data
The Supplemental Data include three supplemental tables, three supplemental figures, and three supplemental movies and can be found with this article online at http://www.cellhostandmicrobe.com/cgi/content/full/3/1/39/DC1/.
Acknowledgments
We thank H.-Y.H. Ho, M.W. Kirschner, A.T. Maurelli, C. Dorman, S.B. Snapper, and C. Lesser for helpful advice and for providing reagents; and R. Lu, J. Heindl, and I. Saran for critical reading of the manuscript. This research was supported by a grant from the National Institute of Allergy and Infectious Diseases (NIH AI52354) to S.B. Snapper and M.B.G.
References
- Ally S, Sauer NJ, Loureiro JJ, Snapper SB, Gertler FB, Goldberg MB. Shigella interactions with the actin cytoskeleton in the absence of Ena/VASP family proteins. Cell Microbiol. 2004;6:355–366. doi: 10.1046/j.1462-5822.2003.00359.x. [DOI] [PubMed] [Google Scholar]
- Beloin C, McKenna S, Dorman CJ. Molecular dissection of VirB, a key regulator of the virulence cascade of Shigella flexneri. J Biol Chem. 2002;277:15333–15344. doi: 10.1074/jbc.M111429200. [DOI] [PubMed] [Google Scholar]
- Bernardini ML, Mounier J, d’Hauteville H, Coquis-Rondon M, Sansonetti PJ. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon LD, Goehring N, Janakiraman A, Yan AW, Wu T, Beckwith J, Goldberg MB. IcsA, a polarly localized autotransporter with an atypical signal peptide, uses the Sec apparatus for secretion, although the Sec apparatus is circumferentially distributed. Mol Microbiol. 2003;50:45–60. doi: 10.1046/j.1365-2958.2003.03674.x. [DOI] [PubMed] [Google Scholar]
- Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Oord JJVD, Verhoef GE, Boogaerts MA, Fryns JP, You D, et al. Constituitively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27:313–317. doi: 10.1038/85886. [DOI] [PubMed] [Google Scholar]
- Egile C, Loisel TP, Laurent V, Li R, Pantaloni D, Sansonetti PJ, Carlier MF. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol. 1999;146:1319–1332. doi: 10.1083/jcb.146.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006;444:567–573. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- Goldberg MB, Barzu O, Parsot C, Sansonetti PJ. Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. J Bacteriol. 1993;175:2189–2196. doi: 10.1128/jb.175.8.2189-2196.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MB, Theriot JA. Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc Natl Acad Sci USA. 1995;92:6572–6576. doi: 10.1073/pnas.92.14.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouin E, Egile C, Dehoux P, Villiers V, Adams J, Gertler F, Li R, Cossart P. The RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature. 2004;427:457–461. doi: 10.1038/nature02318. [DOI] [PubMed] [Google Scholar]
- Gouin E, Welch MD, Cossart P. Actin-based motility of intracellular pathogens. Curr Opin Microbiol. 2005;8:35–45. doi: 10.1016/j.mib.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, Kirschner MW. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Jeng RL, Goley ED, D’Alessio JA, Chaga OY, Svitkina TM, Borisy GG, Heinzen RA, Welch MD. A Rickettsia WASP-like protein activates the Arp2/3 complex and mediates actin-based motility. Cell Microbiol. 2004;6:761–769. doi: 10.1111/j.1462-5822.2004.00402.x. [DOI] [PubMed] [Google Scholar]
- Kakimoto T, Katoh H, Negishi M. Regulation of neuronal morphology by Toca-1, an F-BAR/EFC protein that induces plasma membrane invagination. J Biol Chem. 2006;281:29042–29053. doi: 10.1074/jbc.M604025200. [DOI] [PubMed] [Google Scholar]
- Kespichayawattana W, Rattanachetkul S, Wanun T, Utaisincharoen P, Sirisinha S. Burkholderia pseudomallei induces cell fusion and actin-associated membrane protrusion: A possible mechanism for cell-to-cell spreading. Infect Immun. 2000;68:5377–5384. doi: 10.1128/iai.68.9.5377-5384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature. 2000;404:151–158. doi: 10.1038/35004513. [DOI] [PubMed] [Google Scholar]
- Kocks C, Marchand JB, Gouin E, d’Hauteville H, Sansonetti PJ, Carlier MF, Cossart P. The unrelated surface proteins ActA of Listeria monocytogenes and IcsA of Shigella flexneri are sufficient to confer actin-based motility on Listeria innocua and Escherichia coli, respectively. Mol Microbiol. 1995;18:413–423. doi: 10.1111/j.1365-2958.1995.mmi_18030413.x. [DOI] [PubMed] [Google Scholar]
- LaBrec EH, Schneider H, Magnani TJ, Formal SB. Epithelial cell penetration as an essential step in the pathogenesis of bacillary dysentery. J Bacteriol. 1964;88:1503–1518. doi: 10.1128/jb.88.5.1503-1518.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommel S, Benesch S, Rottner K, Franz T, Wehland J, Kuhn R. Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep. 2001;2:850–857. doi: 10.1093/embo-reports/kve197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdalena J, Goldberg MB. Quantification of Shigella IcsA required for bacterial actin polymerization. Cell Motil Cytoskeleton. 2002;51:187–196. doi: 10.1002/cm.10024. [DOI] [PubMed] [Google Scholar]
- Martinez-Quiles N, Rohatgi R, Anton IM, Medina M, Saville SP, Miki H, Yamaguchi H, Takenawa T, Hartwig JH, Geha RS, Ramesh N. WIP regulates N-WASP-mediated actin polymerization and filopodium formation. Nat Cell Biol. 2001;3:484–491. doi: 10.1038/35074551. [DOI] [PubMed] [Google Scholar]
- Miki H, Miura K, Takenawa T. N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeleton rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. EMBO J. 1996;15:5326–5335. [PMC free article] [PubMed] [Google Scholar]
- Miki H, Sasaki T, Takai Y, Takenawa T. Induction of filopodia formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 1998;391:93–96. doi: 10.1038/34208. [DOI] [PubMed] [Google Scholar]
- Moreau V, Frischknecht F, Reckmann I, Vincentelli R, Rabut G, Stewart D, Way M. A complex of N-WASP and WIP integrates signalling cascades that lead to actin polymerization. Nat Cell Biol. 2000;2:441–448. doi: 10.1038/35017080. [DOI] [PubMed] [Google Scholar]
- Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: Nucleation, high-affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JR, Monack D, McCallum SJ, Vegas A, Pham E, Goldberg MB, Theriot JA. The making of a gradient: IcsA (VirG) polarity in Shigella flexneri. Mol Microbiol. 2001;41:861–872. doi: 10.1046/j.1365-2958.2001.02552.x. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Sanger JM, Sanger JW, Southwick FS. Host cell actin assembly is necessary and likely to provide the propulsive force for intracellular movement of Listeria monocytogenes. Infect Immun. 1992;60:3609–3619. doi: 10.1128/iai.60.9.3609-3619.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch R, Sandlin RC, Maurelli AT. A system for identifying post-invasion functions of invasion genes: Requirements for the Mxi-Spa type III secretion pathway of Shigella flexneri in intercellular dissemination. Mol Microbiol. 1999;34:675–689. doi: 10.1046/j.1365-2958.1999.01627.x. [DOI] [PubMed] [Google Scholar]
- Shibata T, Takeshima F, Chen F, Alt FW, Snapper SB. Cdc42 facilitates invasion but not the actin-based motility of Shigella. Curr Biol. 2002;12:341–345. doi: 10.1016/s0960-9822(02)00689-9. [DOI] [PubMed] [Google Scholar]
- Snapper SB, Takeshima F, Anton I, Liu CH, Thomas S, Nguyen D, Dudley D, Fraser H, Purich D, Klein C, et al. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat Cell Biol. 2001;3:897–904. doi: 10.1038/ncb1001-897. [DOI] [PubMed] [Google Scholar]
- Stamm LM, Morisaki JH, Gao LY, Jeng RL, McDonald KL, Roth R, Takeshita S, Heuser J, Welch MD, Brown EJ. Mycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. J Exp Med. 2003;198:1361–1368. doi: 10.1084/jem.20031072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Miki H, Takenawa T, Sasakawa C. Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 1998;17:2767–2776. doi: 10.1093/emboj/17.10.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taunton J, Rowning BA, Coughlin ML, Wu M, Moon RT, Mitchison TJ, Larabell CA. Actin-dependent propulsion of endosomes and lysosomes by recruitment of N-WASP. J Cell Biol. 2000;148:519–530. doi: 10.1083/jcb.148.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teysseire N, Chiche-Portiche C, Raoult D. Intracellular movements of Rickettsia conorii and R. typhi based on actin polymerization. Res Microbiol. 1992;143:821–829. doi: 10.1016/0923-2508(92)90069-z. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ, Tilney LG, Portnoy DA. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature. 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J Bacteriol. 1999;181:508–520. doi: 10.1128/jb.181.2.508-520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature. 1997;385:265–269. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- Wing HJ, Yan AW, Goldman SR, Goldberg MB. Regulation of IcsP, the outer membrane protease of the Shigella actin tail assembly protein IcsA, by virulence plasmid regulators VirF and VirB. J Bacteriol. 2004;186:699–705. doi: 10.1128/JB.186.3.699-705.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Handa Y, Suzuki T, Ogawa M, Suzuki M, Tamai A, Abe A, Katayama E, Sasakawa C. Microtubule-severing activity of Shigella is pivotal for intercellular spreading. Science. 2006;314:985–989. doi: 10.1126/science.1133174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data
The Supplemental Data include three supplemental tables, three supplemental figures, and three supplemental movies and can be found with this article online at http://www.cellhostandmicrobe.com/cgi/content/full/3/1/39/DC1/.