

Abstract
The human mitochondrial peptide deformylase (HsPDF) provides a potential new target for broadly acting antiproliferative agents. To identify novel nonpeptidomimetic and nonhydroxamic acid–based inhibitors of HsPDF, the authors have developed a high-throughput screening (HTS) strategy using a fluorescence polarization (FP)–based binding assay as the primary assay for screening chemical libraries, followed by an enzymatic-based assay to confirm hits, prior to characterization of their antiproliferative activity against established tumor cell lines. The authors present the results and performance of the established strategy tested in a pilot screen of 2880 compounds and the identification of the 1st inhibitors. Two common scaffolds were identified within the hits. Furthermore, cytotoxicity studies revealed that most of the confirmed hits have antiproliferative activity. These findings demonstrate that the designed strategy can identify novel functional inhibitors and provide a powerful alternative to the use of functional assays in HTS and support the hypothesis that HsPDF inhibitors may constitute a new class of antiproliferative agent.
Keywords: human peptide deformylase, high-throughput screening, fluorescence polarization, antiproliferative agents
INTRODUCTION
Protein synthesis in prokaryotes of eukaryotic proteins is initiated with and in a small subset an N-formylated methionine. In bacteria, the N-formyl group is subsequently removed from most of the peptides by peptide deformylase (PDF). This activity is followed by further processing of the N-terminal methionine by methionine aminopeptidase and other enzymes to yield mature proteins. PDF has been shown to be essential to bacterial growth.1,2 Until recently, PDF was thought to be absent from eukaryotic systems, making it an attractive target for the development of new antibiotics.3 However, genome database searches have revealed eukaryotic PDF-like sequences in parasites, plants, and mammals,4 and more recently, the human mitochondrial peptide deformylase (HsPDF) has been cloned and characterized in our laboratory as well as by others.5–7
Actinonin, first described as a naturally occurring antibiotic by Gordon and coworkers,8 was later found to be a potent bacterial PDF inhibitor.9 In recent studies, we have shown that actinonin is also a potent inhibitor of HsPDF.5 Furthermore, we found that HsPDF inhibition by actinonin and actinonin analogs, or by specific small interfering RNA knockdown of expression, is associated with selective antiproliferative properties in cancer cells.10 Therefore, HsPDF may constitute an attractive target for the development of a small-molecule therapeutic agent.7,10
These observations led us to screen for novel nonpeptidomimetic and nonhydroxamic acid–based inhibitors of HsPDF as potentially new antiproliferative agents. Efforts elsewhere have been concentrated on the discovery of bacterial PDF inhibitors.11,12 Actinonin, which inhibits aminopeptidase-N/CD13,13 is among a few potent HsPDF inhibitors currently known.7 Therefore, we decided to develop and adapt robust assays and integrate them into a sequential strategy to help us discover novel HsPDF inhibitors. Several families of bacterial PDF inhibitors have been identified through the screening of chemical libraries.12 Among them, 2 well-described PDF inhibitors (actinonin and BB-3497) were identified directly or indirectly by screening.9,11 However, nearly all bacterial PDF inhibitors currently in development share a common scaffold based on peptidomimetic backbone linked to a chelating moiety.12 Indeed, many of the reported screens were performed on small, rationally designed libraries, often focused on chelator-based compounds.9,11,12 We sought here to identify structurally diverse HsPDF inhibitors, structurally different from known bacterial PDF inhibitors, so that they would potentially be specific for HsPDF.
In most of the reported screens for bacterial PDF inhibitors, 2 different assays were employed. A coupled assay consists of monitoring the release of formate from a peptide substrate for PDF through a reduction reaction involving formate dehydrogenase. The readout is performed by following the production of NADH, which absorbs at 340 nm.14 A fluorescence intensity (fluorescamine; FLUO) assay relies on the use of fluorescamine to yield a fluorogenic derivative after deformylation of the N-terminal amine of a peptide substrate for PDF. Fluorescamine is intrinsically nonfluorescent but reacts with exposed primary aliphatic amines to yield highly fluorescent derivatives (λex max = 381 nm, λem max = 470 nm). This fluorophore has been widely studied and its use optimized,15 as it allows the detection of primary amines with high sensitivity. These 2 assays were successful in identifying potent bacterial PDF inhibitors.9,11 However, both assays present drawbacks that limit their use for large high-throughput screening (HTS) campaigns. 1) The coupled assay is not very sensitive because of its reliance on absorbance readout. 2) Coupled assays identify inhibitors of both the target enzyme and the reporter enzyme. Therefore, a secondary screen must be performed to identify the inhibitors of the target enzyme. 3) Both the coupled assay and the FLUO assay rely on the detection of compounds that absorb at wavelengths less than 400 nm. As many chemicals absorb in this range of wavelengths, these assays are prone to identify false positives. 4) Reagent stability is an important concern in large screens relying on enzymatic activity, as a stable signal is required during the entire length of the screen. A loss of enzymatic activity over time can jeopardize the integrity of a full screening campaign. In our case, this fact was especially important to consider because of the lability of PDF.16, 7
In light of the limitations associated with the methods described above, we sought to develop an assay compatible with the HTS for HsPDF inhibitors. Fluorescence polarization (FP) constitutes a powerful technique that is compatible with the requirements of HTS.18,19 1) It allows the development of assays that are homogeneous in that they do not necessitate a separation step and most often require the addition of only a single reagent. 2) This technology is therefore amenable to miniaturization and automation. 3) FP reagents are stable, and the readout is mostly independent of intensity. These properties make FP a technique of choice for large-scale automated assays.18,19
In this article, we describe the development of an FP binding assay for the screening of HsPDF inhibitors, amenable to the screening of large chemical libraries. We also report the miniaturization of the FLUO assay. The FLUO assay, despite not being amenable to large-scale screens for the above-mentioned reasons, is being used in this study as part of our strategy to assess the inhibitory activity of positives obtained using this novel FP binding assay for HsPDF. We present here the results of this novel screening strategy and report the identification of the 1st nonpeptidomimetics and nonhydroxamic acid–based small-molecule inhibitors of HsPDF. Most of them exhibited antiproliferative activity against several well-established tumor cell lines.
MATERIALS AND METHODS
Chemicals
The probe SKI-267088 was synthesized at the Sloan-Kettering Organic Synthesis Core Facility. Its molecular weight was confirmed by mass spectrometry analysis performed at the Sloan-Kettering Analytical Pharmacology Core Facility (Fig. 1). The peptide N-formyl-Met-Ala-Ser was purchased from Bachem Bioscience Inc. (King of Prussia, PA). The peptide N-formyl-Met-Ala-His-Ala was purchased from Biopeptide Co. Inc. (San Diego, CA). Fluorescamine, actinonin, and L-alanine were obtained from Sigma Chemical Co. (St. Louis, MO).
FIG. 1.

Mass spectrometry (MS) analysis of the probe SKI-267088. (A) MS scan of SKI-267088. (B) Product ion of m/z 1054, showing a major fragment of m/z 770. (C) Precursor scan of 770, showing the parent ion of m/z 1054.
Expression and purification of MBP-HsPDF
A 63–amino acid N-terminally truncated HsPDF was cloned as a fusion with maltose-binding protein (MBP), with MBP at the N-terminus. HsPDF was cloned into the vector pIADL-16,20 which contains the coding sequence for MBP expressed under the control of the T7 promoter. HsPDF was subcloned into pIADL-16 from a restriction digest of a pET-29b vector containing HsPDF.5 Ligation of the NdeI/XhoI restriction fragment from pET-29b into pIADL-16 resulted in a vector (pIADL-16-MBP-HsPDF) coding for an MBP-HsPDF fusion with a C-terminal 6His-tag. BL21 (DE3) competent cells were transformed with pIADL-16-MBP-HsPDF and the plasmid GroES/EL, which codes for a chaperone. Transformed bacteria were inoculated in 100 mL of LB media containing ampicillin and kanamycin at 50 μg/mL and 10 μg/mL, respectively, and grown overnight at 37 °C. The overnight culture was diluted 1:50 in 4 L of fresh LB with the same concentration of antibiotics described above. These cells were allowed to grow at 37 °C for approximately 2 h, until the optical density at 600 nm reached 0.4 to 0.8. Induction was started by addition of IPTG at a final concentration of 0.1 mM and 0.1 mM CoCl2, followed by incubation at 20 °C for 5 h. Cells were harvested by centrifuging at 5000 rpm for 30 min. The cell pellet was resuspended in a 50 mL final volume of buffer (50 mM HEPES pH 7.5, 50 mM NaCl, 0.1 mM CoCl2) and lysed by passing 2 times through a French press. The cell lysate was centrifuged at 15,000g for 45 min. The supernatant was applied to a column packed with 15 mL of amylose-resin slurry (New England Biolabs, Ipswich, MA). MBP-HsPDF was eluted with a maltose gradient starting with lysis buffer without maltose to reach a final concentration of 10 mM maltose in the same buffer. Fractions containing MBP-HsPDF, determined through sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), were pooled and the protein concentration measured using the Dc protein assay (Bio-rad, Hercules, CA). MBP-HsPDF purity was assessed by SDS-PAGE and GelCode Blue staining (Pierce, Rockford, IL). A total of 20 to 30 mg of MBP-HsPDF was obtained per liter of cell culture.
FP binding assay
Compounds or high/low controls were added to the wells at a volume of 2 μL. Low controls for this assay consisted of actinonin at a final concentration of 100 μM in 1% DMSO (v/v). High controls consisted of 1% DMSO (v/v). MBP-HsPDF was diluted in the assay buffer (25 mM HEPES, 50 mM NaCl, 0.005% Tween 20, pH 7.5), and 10 μL was added to the 384-well microplates (low-volume, round-bottom, NBS-treated plates; Corning, Corning, NY) to achieve a final concentration of 1 μM. After the addition of MBP-HsPDF to the tested compounds, the 384-well microplates were preincubated for 1 h at room temperature. Then, 8 μL of the probe SKI-267088 in solution in assay buffer was added to the wells at a final concentration of 5 nM. After 1 h incubation at room temperature, the FP was read using the Amersham (Buckinghamshire, UK) LEADseeker™ Multimodality Imaging System equipped with Cy3 excitation/emission filters and Cy3 FP epi-mirror. Quench tests were performed in duplicate by measuring the FP of wells containing the probe, before and after addition of the compounds at 100 μM. Compounds inducing a variation of FP greater than 20% were flagged as optically active compounds.
FLUO assay
Compounds or high/low controls were added to the wells at a volume of 2 μL. Low controls for this assay consisted of actinonin at a final concentration of 100 μM in 1% DMSO. High controls consisted of 1% DMSO (v/v). MBP-HsPDF was diluted in the assay buffer (25 mM HEPES, 50 mM NaCl, 0.005% Tween 20, pH 7.5), and 10 μL of this solution was added to the wells of the 384-well microplates (low-volume, round-bottom, NBS-treated plates; Corning) at a final concentration of 1 μM. After 1 h incubation at room temperature, 10 μL of the substrate peptide fMAHA diluted in the assay buffer was added to the wells at a final concentration of 0.5 mM. The deformylation reaction mixture was incubated for 1 h at room temperature. A separate set of plates containing 3 μL of fluorescamine at 1 mg/mL in 100% DMSO was prepared for the labeling step. Then, 17 μL of the reaction mixture from the original set of plates was transferred to the plates containing the fluorescamine solution for the labeling step. The readout was performed on a Perkin Elmer (Waltham, MA) VICTOR3 V™ Multilabel counter, using an excitation wavelength of 355 nm and an emission wavelength of 460 nm. Quench tests were performed in duplicate by measuring the fluorescence of wells containing L-alanine labeled with fluorescamine as a surrogate for the fluorescamine-labeled deformylated substrate, before and after addition of the compounds at 100 μM. Compounds inducing a variation of fluorescence greater than 20% were flagged as optically active compounds.
Cytotoxicity assay
The cell lines K562 (human chronic myelogenous leukemia), NCEB-1 (human non-Hodgkin’s lymphoma), HL-60 (human acute promyelocytic leukemia), Jurkat (human acute T-cell leukemia), and HEK293 (human embryonic kidney) were obtained from ATCC (Manassas, VA) and cultured following ATCC recommendations. The cell line HL-60/RV+ (a P-glycoprotein–overexpressing multidrug-resistant HL-60 variant selected by continuous exposure to vincristine) has been described elsewhere.21 The assay used for the cytotoxicity studies is based on the dye resazurin, commercially sold as Alamar Blue.22 Cells were grown in 45 μL medium for 24 h before addition of the compounds in 5 μL of 1% DMSO (v/v). After a total of 60 h incubation, 5 μL Alamar Blue was added. The cells were then incubated for another 16 h, and the fluorescence intensity was read on the Amersham LEADseeker™ Multimodality Imaging System equipped with Cy3 excitation and excitation filters and FLINT epi-mirror. Quench tests were performed in duplicate by measuring the fluorescence of wells with cells grown in the same assay conditions, before and after addition of the compounds at 100 μM. Compounds inducing a variation of fluorescence greater than 20% were flagged as optically active compounds.
Dose-response studies
In each assay, the signal inhibition induced by the compounds was expressed as a percentage compared to high and low controls located on the same plate, as defined as percentage inhibition = (high control average – read value)/(high control average – low control average) × 100. The dose response was assessed in duplicate using 12-point doubling dilutions with a 100-μM compound concentration as the upper limit. The dose-response curve for each set of data was fitted seperately, and the 2 IC50 values obtained were averaged. For compounds having an IC50 less than 1 μM, the dose-response study was repeated using dilutions starting at 10 μM for a more accurate determination of the IC50 value.
Statistical analysis
The Z′ factor was used to assess assay performance. The Z′ factor constitutes a dimensionless parameter that ranges from 1 (infinite separation) to <0. It is defined as 1 – Z′ = (3σc+ +3σc–)/|μc+ – μc–|, where σc+, σc–, μc+, and μc– are the standard deviations (σ) and averages (μ) of the high (c+) and low (c–) controls.23
For the scatter plot analysis of the FLUO screen data, the percentage inhibition was calculated after log-transforming the signal intensities. For the scatter plot comparing the hits identified in the FLUO and in the FP screening campaigns, the percentage inhibition was calculated as the average of the 2 validation sets for the corresponding assay.
Chemical libraries
The library used for the pilot screen combines approximately 3000 chemicals obtained commercially from Prestwick (Washington, DC) and MicroSource (Gaylordsville, CT). Actinonin was part of the MicroSource library. The MicroSource library contains 2000 biologically active and structurally diverse compounds from known drugs, experimental bioactives, and pure natural products. The library includes a reference collection of 160 synthetic and natural toxic substances (inhibitors of DNA/RNA synthesis, protein synthesis, cellular respiration, and membrane integrity); a collection of 80 compounds representing classical and experimental pesticides, herbicides, and endocrine disruptors; and a unique collection of 720 natural products and their derivatives. The collection includes simple and complex oxygen-containing heterocycles, alkaloids, sequiterpenes, diterpenes, pentercyclic triterpenes, sterols, and many other diverse representatives. The Prestwick Chemical Library is a unique collection of 880 high-purity chemical compounds (all off patent) carefully selected for structural diversity, a broad spectrum covering several therapeutic areas (from neuropsychiatry to cardiology, immunology, anti-inflammatory, analgesia, and more), known safety, and bioavailability in humans. More than 85% of its compounds are marketed drugs.
Liquid-dispensing systems
Three different liquid-dispensing devices were used in this study. Compounds were plated at a volume of 2 μL for the enzymatic assays and added to the cytotoxicity assay plates in 5 μL using a TPS-384 total pipetting solution (Apricot Designs, Monrovia, CA). For the dispensing of MBP-HsPDF in 10 μL, a Multidrop 384 (Thermo Electron Corporation, Waltham, MA) was used. The addition of Alamar Blue (5 μL) was performed using a Flexdrop IV (Perkin Elmer).
Automation system and screening data management
The assays were performed on a fully automated linear track robotic platform (CRS F3 Robot System; Thermo Electron, Canada) using several integrated peripherals for plate handling, liquid dispensing, and fluorescence detection. Screening data files from the Amersham LEADseeker™ Multimodality Imaging System or from a Perkin Elmer VICTOR3 V™ Multilabel counter were loaded into the HTS Core Screening Data Management System, a custom-built suite of modules for compound registration, plating, and data management that is powered by ChemAxon Cheminformatic tools (ChemAxon, Budapest, Hungary). Data were analyzed for compounds exhibiting 30% inhibition or greater, and the summary of the identified positives was exported as SD files for further analysis and reporting.
RESULTS AND DISCUSSION
Development of a high-throughput assay to screen for inhibitors of HsPDF
For the purpose of developing the FP assay for HsPDF, we expressed HsPDF as an MBP fusion protein. This approach presents several advantages. First, a target protein of higher molecular weight allows better sensitivity in FP binding assays. Indeed, the observed differential in polarization upon binding of the fast-rotating, small-molecular-weight probe to the slow-rotating, larger protein is higher. A 2nd advantage of the MBP portion is that it facilitates the purification of the fusion protein, and MBP-HsPDF can therefore be produced in the amounts required for screening of a large number of compounds. A 63–amino acid N-terminally truncated HsPDF was cloned as a fusion with MBP. MBP-HsPDF was produced as the cobalt form and purified. As a result of the fusion, the overall size of the protein increased to 64 kDa, compared to 21 kDa for the native protein (Fig. 2).
FIG. 2.
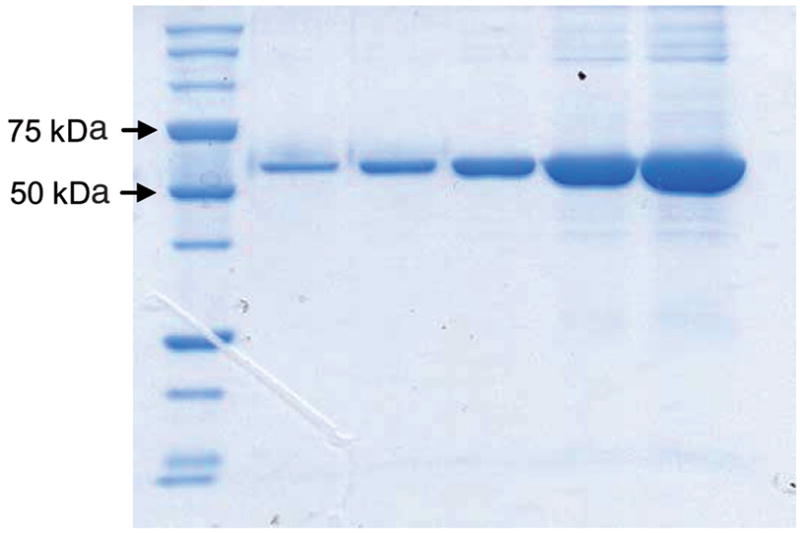
Electrophoretic analysis of purified maltose-binding protein human mitochondrial peptide deformylase stained by Coomassie Blue.
The next step in the conception of the FP assay for HsPDF consisted of designing a reporter probe. For the selection of the binding moiety, we took adavantage of the fact that actinonin is a potent inhibitor of HsPDF.5 We based our probe on the similarly potent derivative of actinonin SKI-267087 previously described as actinonin derivative 11111110 (Fig. 3) and more amenable to chemistry. For the choice of the fluorescent moiety, we took into consideration the fact that a lot of compounds interfere optically either directly or indirectly with typical fluorescent tracers such as fluorescein.24 Therefore, we opted to use the Amersham Cy3b dye, a red-shifted dye with an excitation wavelength of 525 nm and emission wavelength of 580 nm. Besides its excellent fluorescence properties, the fluorophore Cy3b was also chosen for its good water solubility, pH insensitivity, and easy amenability to derivatization.25 The binding moiety was linked to the fluorophore Cy3b through a 6-carbon (C6) linker to form the reporter probe SKI-267088 (Fig. 3). The probe was able to bind to MBP-HsPDF and to induce an increase in FP as the concentration of MBP-HsPDF increased (Fig. 4A). To assess the binding specificity of the probe SKI-267088, we tested its binding to bovine serum albumin (BSA) and to the cysteine protease cathepsin B. No increase in FP was observed when the probe was incubated with increasing concentrations of BSA or cathepsin B (Fig. 4A). This result suggests that the probe behaves much like its parent compound actinonin and that the addition of the Cy3B moiety and of the linker does not induce any nonspecific binding to other proteins. We miniaturized the assay for a 384-well format in a final volume of 20 μL. We set the probe concentration at 5 nM and the HsPDF concentration at 1 μM based on the binding experiment presented in Figure 4A. Under these conditions, enough protein to bind approximately 80% of the probe is provided. This allows for maximal separation of the signal from the background and for maximal sensitivity.19
FIG. 3.

Chemical structure of actinonin; actinonin analogs SKI-267087, SKI-274447, and SKI-274448; and the actinonin-based fluorescence polarization probe SKI-267088.
FIG. 4.
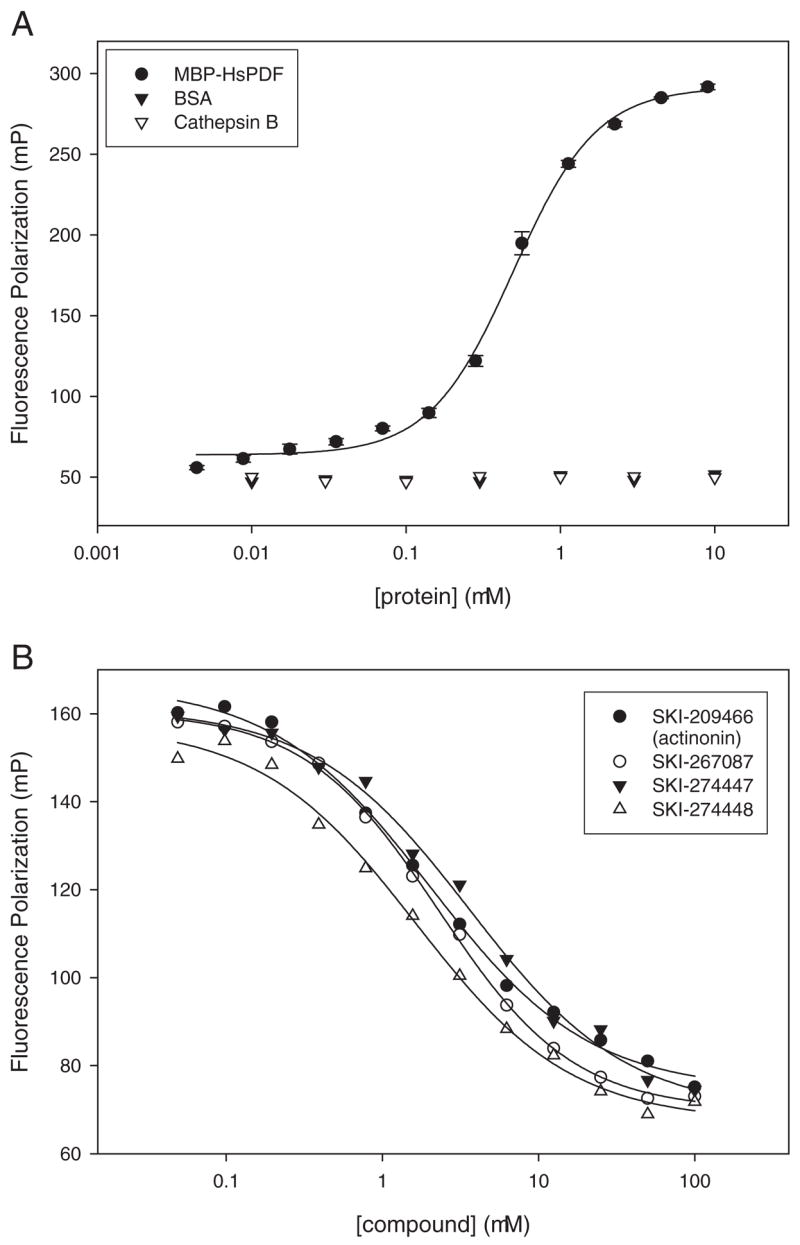
(A) Binding of the actinonin-based fluorescence polarization probe SKI-267088 to maltose-binding protein human mitochondrial peptide deformylase (MBP-HsPDF) and test of the nonspecific binding of the probe SKI-267088 to bovine serum albumin (BSA; and cathep-sin B. The error bars represent plus or minus 1 standard deviation from triplicate measurements. (B) Displacement of the probe SKI-267088 from HsPDF by actinonin (IC50 = 2.3 μM) and the actinonin analogs SKI-267087 (IC50 = 2.1 μM), SKI-274447 (IC50 = 3.9 μM), and SKI-274448 (IC50 = 1.4 μM).
As a proof of principle for the assay’s ability to identify potential inhibitors that would prevent binding of the reporter probe to HsPDF, we measured the dose-response curve of actinonin using 12-point doubling dilutions with 100 μM as the upper limit and in the presence of 1% DMSO (v/v). We found that preincubation of the enzyme with actinonin for an hour before adding the probe was necessary to observe full inhibition of binding (data not shown). Because other inhibitors might also require a preincubation time with the enzyme because of their mechanism of action, we chose to include this step in the assay protocol for maximum sensitivity. As expected, the fluorescence polarization decreased when MBP-HsPDF was preincubated with increasing concentrations of actinonin (Fig. 4B). The calculated IC50 of actinonin in this assay was 2.3 μM. Similar dose-response profiles were observed with the actinonin analogs SKI-267087 (IC50 = 2.1 μM), SKI-274447 (IC50 = 3.9 μM), and SKI-274448 (IC50 μ = 1.4 M; Fig. 4B), indicating that the tracer binds to the active site of the enzyme, and its binding can be prevented upon addition of an increasing concentration of inhibitors.
To further evaluate assay robustness, we performed a control run of 1152 data points for each of the high-control (1% DMSO [v/v]) and low-control wells (100 μM actinonin in 1% DMSO [v/v]). The assay performance was evaluated by measuring the coefficients of variation (CVs) and the Z′ factor.23 The Z′ factor has been widely accepted by the HTS community for the evaluation of assay quality and performance. We found that both high- and low-control data points showed very little variability, with CVs in the 4% range, a signal-to-noise ratio of 3:1 (signal window of 140 mP), and a calculated Z′ value of 0.76, results that are indicative of an excellent assay performance and robustness (Fig. 5A).
FIG. 5.

(A) Fluorescence polarization (FP) assay control run. (B) Scatter plot of the duplicate values of percentage inhibition for each compound in the FP pilot screen. (C) Z′ factor distribution during the pilot screen. (D) Fluorescence intensity (FLUO) assay control run.
Screening for inhibitors of HsPDF using the FP assay
The developed assay was used in a high-throughput pilot screen of a library of 2880 commercially available compounds. The collection includes known drugs and is composed of synthetic as well as natural compounds. The library was plated in nine 384-well plates, with columns 13 and 14 left empty for the addition of high- and low-control wells, allowing monitoring of assay performance during the automated screening run. The pilot screen was carried out exactly the same way the full-scale HTS will be performed (e.g., same robotic platform and readers). The pilot screen was performed on 2 separate days against the same set of compounds to assess day-to-day variability. The compound screening concentration was 10 μM in 1% DMSO (v/v). This validation step allowed us to obtain field data on assay performance as well as an estimate of the initial hit rate and an overall assessment of compound optical interference. Most important, we could evaluate the assay reproducibility by comparing the 2 data sets to identify the number of outliers that hit on one day but not on the other. The results were plotted as a scatter plot (Fig. 5B). The Z′ value for each plate was calculated and shows the reproducibility of the assay between plates (Fig. 5C). The overall Z′ factor of 0.8 is indicative of the excellent robustness of the assay during fully automated screening.
At an inhibition threshold of 30%, we observed a very good reproducibility between the 2 screening days, reflected by a correlation factor of 0.85. An initial positive hit rate of 1.3% yielded 38 positives (Fig. 5B). Among these positives was actinonin, which is present in the library; this internal control demonstrates that the developed FP assay is able to identify HsPDF inhibitors among several thousand compounds and is amenable to screening larger chemical libraries.
Miniaturization of a functional assay for HsPDF
We miniaturized the fluorescamine functional assay previously described for bacterial PDF11 to 20 μL in 384-well microplates. We originally developed the assay using HsPDF expressed as a polyhistidine fusion protein. Subsequent studies with MBP-HsPDF showed that the MBP portion did not significantly alter the signal, and MBP-HsPDF was subsequently used for the FLUO assay. Previous studies in our laboratory had shown that fMAS and fMAHA were the best substrates for HsPDF among those tested (data not shown). We measured the signal-to-noise ratio for various concentrations of both substrates and for various concentrations of enzyme and established the following as optimal conditions for the assay: The substrate was fMAHA and used at 0.5 mM, and the HsPDF concentration was 1 μM (Fig. 6). During the optimization of the FLUO assay, we observed a great variability of the high-control signal (data not shown). This was presumably due to the mixing of a small volume (3 μL) of fluorescamine in 100% DMSO into a larger aqueous volume (17 μL). For this reason, we modified the assay so that the reaction mixture (17 μL of aqueous) after the deformylation step (Fig. 7) was added to plates already containing fluorescamine (3 μL in 100% DMSO) for the labeling step, in an effort to enhance the rapidity and efficiency of mixing. This modification to the protocol helped to minimize the variability of the high signal (data not shown). However, this extra step makes the FLUO assay heterogeneous in nature, excluding its use for large-scale screening campaigns.
FIG. 6.
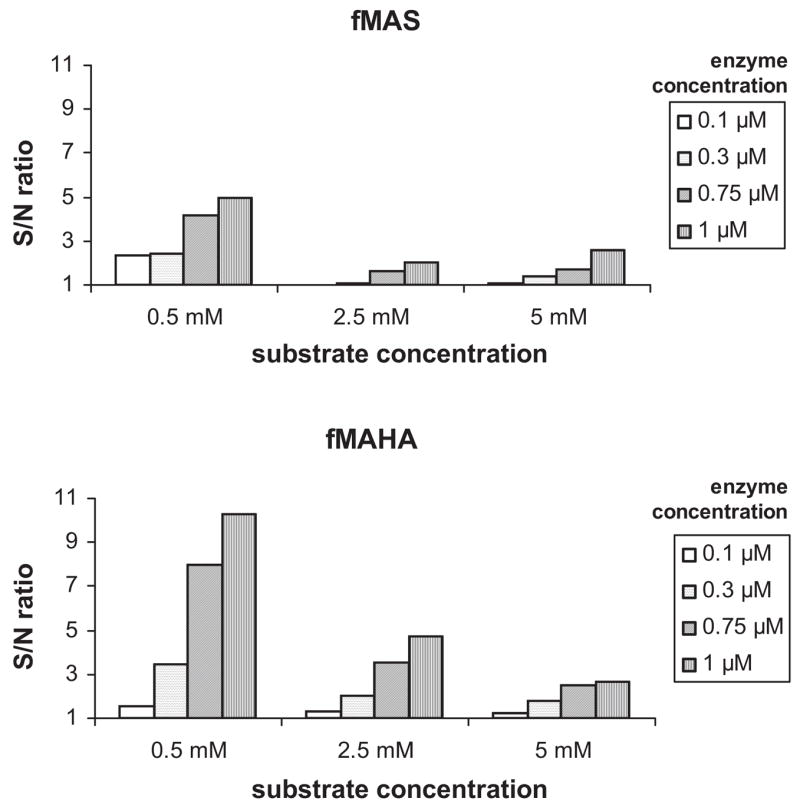
Optimization of the enzyme and substrate concentration for the fluorescamine assay with 2 different substrates.
FIG. 7.

Principle of the fluorescamine assay.
To evaluate assay performance of the miniaturized assay, we performed a control run of 1152 data points for each of the high-control (1% DMSO [v/v]) and low-control wells (100 μM actinonin in 1% DMSO [v/v]). The assay performance was evaluated by measuring the CVs and the Z′ factor. We found that both high- and low-control data points showed high variability, with CVs in the 14% to 16% range and with a calculated Z′ value of 0.41; this result is indicative of a somewhat poor performance and robustness for this miniaturized assay for HTS (Fig. 5D). Furthermore, the drift in the high-control signal between plates that occurred in the control run (Fig. 5D) indicates that a larger variability of the high signal would likely be observed when screening a higher number of plates. This poor performance may be attributed to the fact that fluorescamine is soluble only in organic solvents; acetonitrile has traditionally been the solvent of choice. However, acetonitrile is not suitable for our automated pipettors because of its surface tension and evaporation properties. DMF is incompatible with the use of fluorescamine.15 Therefore, we used DMSO as the carrier suitable to automated liquid handling. Despite its being less robust than the FP assay, our optimization efforts allowed us to use the miniaturized FLUO assay to reliably perform confirmation studies on the 38 positives obtained in the FP assay from a pilot library of 2880 compounds. Although a larger number of positives could easily be identified from screening larger chemical libraries, we propose that the performance limits of the FLUO assay used in an automated fashion make it more of a confirmatory assay rather than a primary one in a sequential screening strategy using the FP assay as the primary screening assay.
Confirming hits in the FP and the FLUO assays
The 38 positives obtained from the FP assay were submitted to further evaluation. They were first evaluated in a series of confirmatory tests to exclude any false positives as part of our positive to hit confirmation work flow (Fig. 8). Insoluble aggregates can disrupt biological activity through nonspecific interactions.26 Therefore, assessing the poor solubility of a compound in the assay conditions is important for identifying potentially false positives. Laser nephelometry has been shown to be a reliable and sensitive technique for the measurement of solubility in the 384-well plate format.27 Despite the use of detergent in our assay buffer, 4 positives were found to have solubility limits less than 100 μM as measured by laser nephelometry (Fig. 9A). This low solubility possibly interfered with the subsequent dose-response studies; these compounds were therefore not selected for further evaluation. Optically active compounds can potentially interfere with the fluorescence-based technologies used in this study. As mentioned earlier, fluorescamine fluorescence is prone to optical interference. Also, despite our choice of the Cy3 dye as a low interference–prone fluorescent probe, chemical libraries may still contain optically active compounds that could interfere in the FP assay. Therefore, we performed quench tests for each assay to ensure that we selected as confirmed hits only those positives that do not optically interfere with the detection. Six compounds were found to interfere in the FP assay and 5 in the FLUO assay (Fig. 9B). Therefore, they were flagged as false positives and not considered for further evaluation. Next, we assessed the dose response for the 25 remaining positives in both the FP and the FLUO assay using 12-point doubling dilutions, with a 100-μM compound concentration as the upper limit. Fifteen compounds in the FP assay and 5 compounds in the FLUO assay were found to exhibit dose-dependent inhibition, with their apparent IC50 values ranging from 1.5 μM to 55 μM (Table 1). Dose-response curves in the FP assay are shown for a selection of the hits (Fig. 10). Interestingly, the IC50 of actinonin was approximately 4 times lower in the FLUO assay than in the FP assay (Table 1). This discrepancy is probably explained by the fact that the FLUO assay is a catalytic assay compared to the FP assay, which monitors binding. If only a fraction of the expressed HsPDF enzymes coordinate a catalytic metal ion in their active site, a lower concentration in actinonin might be required to chelate this metal and inhibit the functional HsPDF enzymes than to displace the probe from all HsPDF enzymes. On the other hand, all the other compounds that we identified were more potent in the FP assay than in the functional assay. This may suggest a different mechanism of action for these compounds, possibly allosteric inhibition. Further studies would be required to characterize the mode of action of the newly discovered HsPDF inhibitors.
FIG. 8.

Flow chart of the high-throughput assay strategy employed in this study. SAR = structure-activity relationship; cmpd = compound; inhib = inhibitor.
FIG. 9.

(A) Structure of 4 compounds identified by laser nephelometry as having a solubility limit <100 μM in the fluorescence polarization (FP) assay conditions. (B) Structure of 8 compounds inducing optical interference in the FP or in the fluorescamine (FLUO) assay. The ratio of fluorescence intensity before and after addition of the compound in the assay conditions at a final concentration of 100 μM is indicated.
Table 1.
Summary of the Confirmed Hits
| IC50 (μM)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SKI ID | Name | Scaffold | K562 | NCEB-1 | HL-60 | Jurkat | RV+ | HEK293 | FP | FLUO |
| 209325 | Hematoxylin | A | — | — | 60 | 4.7 | 59 | — | 2.7 | — |
| 209412 | 2′,2′-Bisepigallocatechin monogallate | A | — | — | — | 3.0 | — | — | 4.1 | 54 |
| 209466 | Actinonin | — | — | 46 | 22 | — | 28 | 5.5 | 1.5 | |
| 209469 | Pyrogallin | B | — | — | — | 0.24 | — | — | 5.7 | 46 |
| 209343 | Theaflavin | A/B | — | — | — | 53 | — | — | 6.1 | — |
| 209459 | Theaflavin monogallates | A/B | — | — | — | 1.3 | — | — | 7.1 | — |
| 209468 | Purpurogallin | B | — | — | — | 11 | — | — | 8.8 | — |
| 210479 | Quinalizarin | — | — | — | — | — | — | 8.9 | — | |
| 210893 | Hematein | A | — | — | — | 2.3 | — | — | 12 | — |
| 209399 | 7,8-Dihydroxyflavone | — | — | 52 | 7.5 | — | 28 | 17 | — | |
| 209392 | Irigenol | — | — | — | 3.2 | — | — | 17 | 61 | |
| 218293 | Epitheaflavin monogallate | A/B | — | — | — | 3.4 | — | — | 29 | — |
| 211431 | 3-Methoxycatechol | — | — | 7.0 | 5.5 | 11 | 28 | 31 | — | |
| 209415 | Epigallocatechin 3,5-digallate | A | — | — | — | 0.57 | — | — | 38 | — |
| 211412 | Chloranil | — | — | — | 15 | — | 19 | 48 | — | |
| 211062 | Thiram | 53 | — | 0.03 | 3.2 | 0.3 | 0.81 | — | 43 | |
The IC50 (μM) for each compound in the fluorescence polarization (FP) assay, fluorescamine (FLUO) assay, and cytotoxicity studies against a panel of cell lines is summarized. — = IC50 >100 μM.
FIG. 10.
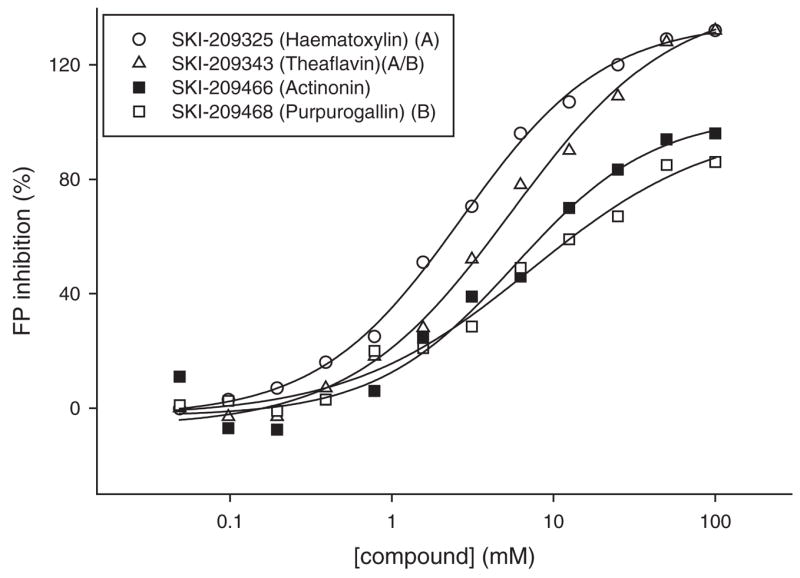
Dose response in the fluorescence polarization (FP) binding assay of selected compounds representative of each scaffold, compared to the reference inhibitor actinonin. The IC50s for each compound were hematoxylin, 2.7 μM; theaflavin, 6.1 μM; actinonin, 5.5 μM; and purpurogallin, 8.8 μM.
Two common scaffolds among the confirmed hits were identified, and 9 of the 16 confirmed compounds could be grouped into 2 distinct chemical scaffolds designated as A and B. Some compound structures belonging to both scaffolds are identified in this article as A/B (Fig. 11A). Importantly, 1 representative of each scaffold was active in both the FP and the FLUO assay. These compounds are structurally diverse compared to most known PDF inhibitors, which are mostly peptidomimetics with a chelating moiety at 1 end of the molecule.12 Our screening strategy was therefore successful in that the FP screen allowed the discovery of novel chemical scaffolds for the development of HsPDF inhibitors. The 2 identified scaffolds and the structure of their different members provide important preliminary structure-activity relationship data for the future investigation of the hits.
FIG. 11.

(A) Scaffolds identified within the screening hits. (B) Structures of selected representatives of each scaffold.
Biological evaluation of the confirmed hits
We then assessed the cytotoxicity of the confirmed hits from the FP screen in a panel of 6 established human tumor cell lines (K562, NCEB-1, HL-60, HL60/RV+, Jurkat, HEK293). In this study, we used the Alamar Blue assay, based on the reduction of the dye resazurin by living cells.22 Fluorescence is proportional to the number of metabolically active cells. A quench test was performed to identify potential false positives, but no confirmed hit interfered in the assay. Fifteen of the 16 confirmed hits were found to be cytotoxic toward at least 1 tumor cell line. The IC50s for each cell line are summarized in Table 1. This important observation supports the hypothesis that HsPDF inhibitors may constitute a novel class of antiproliferative agents. However, it is possible that some of the confirmed hits kill cells through a mechanism of action unrelated to HsPDF inhibition. Further studies would be required to understand the mechanism of killing. The most potent hit had an IC50 in the 30-nM range toward the HL-60 cell line (SKI-211062, 28 nM; Table 1). Interestingly, 3 confirmed hits were found to be cyto-toxic toward the highly multidrug-resistant (MDR) cell line RV+. The most potent compound toward this cell line (SKI-211062) had an IC50 of 300 nM (Table 1). The mechanism of action of this compound is worth investigating, as many antiproliferative drugs are significantly less active against MDR cell lines.
Several members of scaffold A (Fig. 11A, B) were found to have cytotoxic activity toward several tumor cell lines, including the MDR cell line RV+ (Table 1). Among the cytotoxic hits, hematoxylin (SKI-209325) belongs to a family of compounds (brazilins) that was previously reported to have potent antimicrobial activity against a number of bacteria, including methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, and MDR Burkholderia cepacia.28 Also belonging to scaffold A are several epicatechins and theaflavins that were identified as positives in the FP screen. One of them (SKI-209412) was confirmed as an HsPDF inhibitor in the FLUO assay as well. These flavonoids from tea extracts are mostly known for their antioxidative properties.29 They have also been reported to interact with specific signal transduction pathways resulting in anticancer activity.30 HsPDF inhibition could constitute a novel mechanism of action explaining their antiproliferative effects. Members of scaffold B are potent HsPDF inhibitors in the FP assay (Fig. 10, Table 1) and are cytotoxic toward at least 1 tumor cell line. No previous report of biological activity for the members of scaffold B has been published.
Our strategy therefore proved successful, in that an FP binding primary screen identified confirmed HsPDF inhibitors in both the FP and the FLUO assay, and a large majority of the confirmed hits are active in vitro against tumor cell lines.
Sensitivity of the FP and the FLUO assays in identifying hits
A concern about FP binding assays lies in their ability to identify inhibitors that displace the fluorescent probe from the target protein but not necessarily inhibit its enzymatic function. Therefore, a primary screen based on an FP binding assay could potentially be less efficient than a functional assay at identifying functional inhibitors. However, our study shows that we could identify the known HsPDF inhibitor actinonin among thousands of compounds in a pilot screen using an FP binding assay. Four other functional inhibitors of HsPDF were identified (Table 1). Moreover, 9 confirmed hits were found to belong to 2 distinct chemical scaffolds, and 1 representative of each scaffold was later confirmed as a functional HsPDF inhibitor (Table 1). This important observation demonstrates that the 2 classes of compounds that we identified not only bind to HsPDF but also have the potential to interfere with its enzymatic activity. We therefore demonstrated that performing a primary screen using the FP assay to identify positives and confirming their activities using both the FP and the FLUO assay constitutes a powerful strategy for the discovery of HsPDF inhibitors.
Applications of this HTS strategy
This article reports the successful development and use of an FP binding assay based on the PDF inhibitor actinonin and the miniaturization of the functional assay to be used as a confirmatory assay, together creating a sequential screening strategy for screening inhibitors of HsPDF. During the development of the 2 assays, it became apparent that only the FP binding assay was amenable to HTS because of the high signal variability inherent to the use of fluorescamine in automated conditions. Because the FP binding assay could potentially identify compounds that bind to the target enzyme but do not inhibit its deformylase function, we sought to confirm the positives using the adapted FLUO assay and the FP assay. Our goal was to determine whether a primary screen using the HsPDF FP binding assay could identify compounds that also inhibit HsPDF in the FLUO assay. We determined that the FP assay was efficient at identifying functional inhibitors. This strategy allowed us to identify 2 chemical scaffolds that constitute a basis for the development of the 1st nonpeptidomimetic inhibitors of HsPDF. Interestingly, all the identified compounds belonging to the 2 scaffolds were active in vitro toward at least 1 tumor cell line out of a panel of 6. This important observation supports the hypothesis that HsPDF inhibitors may constitute a novel class of antiproliferative agents. This study therefore demonstrates that a dual approach consisting of performing an FP binding primary screen, followed by a confirmation using a functional assay, can provide a powerful alternative strategy when a functional assay is not amenable to large-scale HTS.
SUPPLEMENTARY EXPERIMENTAL PROCEDURES
Mass spectrometry analysis of the probe SKI-267088
The sample was diluted with methanol to a final concentration of approximately 10 μM. An Applied Biosystem Sciex API 4000 was used for the mass spectrometry analysis. A mass survey scan was performed in the range of m/z 950 to 1150; covering the expected mass range under a constant infusion flow, the flow rate was 10 μL/min for the sample and 200 μL/min for delivering solvents (0.1% formic acid/100% acetonitrile = 1/1, v/v), respectively. During standard electrospray ionization in a positive mode, source voltage was optimized at 2000 with a vaporizer temperature of 300; N2 flow rate was 30 L/min for gas 1 and 5 L/min for gas 2. The major peak of m/z 1054 (M + H+) was further identified by its product ion and precursor ion scans.
Acknowledgments
We thank M. Lee, V. Jindal, and the members of the HTS Core facility for their support. This work was financially supported by grant R21 NS057008 to H.D., Mr. William H. Goodwin, and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the William Randolph Hearst Fund in Experimental Therapeutics, and the MSKCC Experimental Therapeutics Center.
References
- 1.Mazel D, Pochet S, Marliere P. Genetic characterization of polypeptide deformylase, a distinctive enzyme of eubacterial translation. EMBO J. 1994;13:914–923. doi: 10.1002/j.1460-2075.1994.tb06335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meinnel T, Blanquet S. Characterization of the Thermus thermophilus locus encoding peptide deformylase and methionyl-tRNA(fMet) formyl-transferase. J Bacteriol. 1994;176:7387–7390. doi: 10.1128/jb.176.23.7387-7390.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meinnel T, Patiny L, Ragusa S, Blanquet S. Design and synthesis of substrate analogue inhibitors of peptide deformylase. Biochemistry. 1999;38:4287–4295. doi: 10.1021/bi982622r. [DOI] [PubMed] [Google Scholar]
- 4.Giglione C, Serero A, Pierre M, Boisson B, Meinnel T. Identification of eukaryotic peptide deformylases reveals universality of N-terminal protein processing mechanisms. EMBO J. 2000;19:5916–5929. doi: 10.1093/emboj/19.21.5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MD, Antczak C, Li Y, Sirotnak FM, Bornmann WG, Scheinberg DA. A new human peptide deformylase inhibitable by actinonin. Biochem Biophys Res Commun. 2003;312:309–315. doi: 10.1016/j.bbrc.2003.10.123. [DOI] [PubMed] [Google Scholar]
- 6.Serero A, Giglione C, Sardini A, Martinez-Sanz J, Meinnel T. An unusual peptide deformylase features in the human mitochondrial N-terminal methionine excision pathway. J Biol Chem. 2003;278:52953–52963. doi: 10.1074/jbc.M309770200. [DOI] [PubMed] [Google Scholar]
- 7.Leeds JA, Dean CR. Peptide deformylase as an antibacterial target: a critical assessment. Curr Opin Pharmacol. 2006;6:445–452. doi: 10.1016/j.coph.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Gordon JJ, Kelly BK, Miller GA. Actinonin: an antibiotic substance produced by an actinomycete. Nature. 1962;195:701–702. doi: 10.1038/195701b0. [DOI] [PubMed] [Google Scholar]
- 9.Chen DZ, Patel DV, Hackbarth CJ, Wang W, Dreyer G, Young DC, et al. Actinonin, a naturally occurring antibacterial agent, is a potent deformylase inhibitor. Biochemistry. 2000;39:1256–1262. doi: 10.1021/bi992245y. [DOI] [PubMed] [Google Scholar]
- 10.Lee MD, She Y, Soskis MJ, Borella CP, Gardner JR, Hayes PA, et al. Human mitochondrial peptide deformylase, a new anticancer target of actinonin-based antibiotics. J Clin Invest. 2004;114:1107–1116. doi: 10.1172/JCI22269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clements JM, Beckett RP, Brown A, Catlin G, Lobell M, Palan S, et al. Antibiotic activity and characterization of BB-3497, a novel peptide deformylase inhibitor. Antimicrob Agents Chemother. 2001;45:563–570. doi: 10.1128/AAC.45.2.563-570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain R, Chen D, White RJ, Patel DV, Yuan Z. Bacterial peptide deformylase inhibitors: a new class of antibacterial agents. Curr Med Chem. 2005;12:1607–1621. doi: 10.2174/0929867054367194. [DOI] [PubMed] [Google Scholar]
- 13.Bauvois B, Dauzonne D. Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: chemistry, biological evaluations, and therapeutic prospects. Med Res Rev. 2006;26:88–130. doi: 10.1002/med.20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lazennec C, Meinnel T. Formate dehydrogenase-coupled spectrophotometric assay of peptide deformylase. Anal Biochem. 1997;244:180–182. doi: 10.1006/abio.1996.9910. [DOI] [PubMed] [Google Scholar]
- 15.Bantan-Polak T, Kassai M, Grant KB. A comparison of fluorescamine and naphthalene-2,3-dicarboxaldehyde fluorogenic reagents for microplate-based detection of amino acids. Anal Biochem. 2001;297:128–136. doi: 10.1006/abio.2001.5338. [DOI] [PubMed] [Google Scholar]
- 16.Adams JM. On the release of the formyl group from nascent protein. J Mol Biol. 1968;33:571–589. doi: 10.1016/0022-2836(68)90307-0. [DOI] [PubMed] [Google Scholar]
- 17.Livingston DM, Leder P. Deformylation and protein biosynthesis. Biochemistry. 1969;8:435–443. doi: 10.1021/bi00829a059. [DOI] [PubMed] [Google Scholar]
- 18.Nasir MS, Jolley ME. Fluorescence polarization: an analytical tool for immunoassay and drug discovery. Comb Chem High Throughput Screen. 1999;2:177–190. [PubMed] [Google Scholar]
- 19.Burke TJ, Loniello KR, Beebe JA, Ervin KM. Development and application of fluorescence polarization assays in drug discovery. Comb Chem High Throughput Screen. 2003;6:183–194. doi: 10.2174/138620703106298365. [DOI] [PubMed] [Google Scholar]
- 20.McCafferty DG, Lessard IA, Walsh CT. Mutational analysis of potential zinc-binding residues in the active site of the enterococcal D-Ala-D-Ala dipeptidase VanX. Biochemistry. 1997;36:10498–10505. doi: 10.1021/bi970543u. [DOI] [PubMed] [Google Scholar]
- 21.Weisburg JH, Roepe PD, Dzekunov S, Scheinberg DA. Intracellular pH and multidrug resistance regulate complement-mediated cytotoxicity of nucleated human cells. J Biol Chem. 1999;274:10877–10888. doi: 10.1074/jbc.274.16.10877. [DOI] [PubMed] [Google Scholar]
- 22.O’Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high-throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 24.Turek-Etienne TC, Small EC, Soh SC, Xin TA, Gaitonde PV, Barrabee EB, et al. Evaluation of fluorescent compound interference in 4 fluorescence polarization assays: 2 kinases, 1 protease, and 1 phosphatase. J Biomol Screen. 2003;8:176–184. doi: 10.1177/1087057103252304. [DOI] [PubMed] [Google Scholar]
- 25.Cooper M, Ebner A, Briggs M, Burrows M, Gardner N, Richardson R, et al. Cy3B: improving the performance of cyanine dyes. J Fluoresc. 2004;14:145–150. doi: 10.1023/b:jofl.0000016286.62641.59. [DOI] [PubMed] [Google Scholar]
- 26.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 27.Bevan CD, Lloyd RS. A high-throughput screening method for the determination of aqueous drug solubility using laser nephelometry in microtiter plates. Anal Chem. 2000;72:1781–1787. doi: 10.1021/ac9912247. [DOI] [PubMed] [Google Scholar]
- 28.Xu HX, Lee SF. The antibacterial principle of Caesalpina sappan. Phytother Res. 2004;18:647–651. doi: 10.1002/ptr.1524. [DOI] [PubMed] [Google Scholar]
- 29.Luczaj W, Skrzydlewska E. Antioxidative properties of black tea. Prev Med. 2005;40:910–918. doi: 10.1016/j.ypmed.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 30.Park AM, Dong Z. Signal transduction pathways: targets for green and black tea polyphenols. J Biochem Mol Biol. 2003;36:66–77. [PubMed] [Google Scholar]