

Abstract
The onset of the adaptive immune response to Mycobacterium tuberculosis is delayed compared with that of other infections or immunization, and allows the bacterial population in the lungs to expand markedly during the preimmune phase of infection. We used adoptive transfer of M. tuberculosis Ag85B-specific CD4+ T cells to determine that the delayed adaptive response is caused by a delay in initial activation of CD4+ T cells, which occurs earliest in the local lung-draining mediastinal lymph node. We also found that initial activation of Ag85B-specific T cells depends on production of antigen by bacteria in the lymph node, despite the presence of 100-fold more bacteria in the lungs. Although dendritic cells have been found to transport M. tuberculosis from the lungs to the local lymph node, airway administration of LPS did not accelerate transport of bacteria to the lymph node and did not accelerate activation of Ag85B-specific T cells. These results indicate that delayed initial activation of CD4+ T cells in tuberculosis is caused by the presence of the bacteria in a compartment that cannot be mobilized from the lungs to the lymph node, where initial T cell activation occurs.
Protective immunity to tuberculosis depends on CD4+ T lymphocytes in humans and in mice (1, 2), but adaptive immune responses are unable to eradicate Mycobacterium tuberculosis or to provide sterile immunity. One characteristic of the adaptive immune response to tuberculosis is the long interval required for its development compared with the response to immunization or to other infections. In humans, development of adaptive immunity to tuberculosis, which is measured as a response to a tuberculin skin test, requires up to 5–6 wk after infection (3, 4), whereas in mice, the earliest antigen-specific CD4+ T cell responses require a minimum of 12 d after aerosol infection (5). Although these intervals are much longer than those required for development of adaptive immune responses to other pathogens such as Salmonella enterica, Listeria monocytogenes, Francisella tularensis, Influenza virus, Leishmania major, or Plasmodium yoelii sporozoites (6–12), the mechanisms that account for the delay in adaptive immune responses in tuberculosis are poorly understood. Indeed, the mechanisms that underlie the initiation of adaptive immune responses to M. tuberculosis are themselves poorly understood. One prior study revealed that antigen-specific CD4+ T lymphocytes did not appear in the lung-draining mediastinal lymph node until after M. tuberculosis had disseminated to that lymph node, indicating that, whereas M. tuberculosis is a lung pathogen residing in antigen-presenting cells, the adaptive immune response to M. tuberculosis antigens is initiated in the local lymph node (5). That study also found that bacteria and T cell recall responses appear earlier in the mediastinal lymph nodes of C57BL/6 than in C3H/HeJ mice infected with M. tuberculosis, but it did not address the mechanisms of dissemination of the bacteria from the lungs to the mediastinal lymph node.
We recently reported that, in addition to macrophages, dendritic cells are infected with M. tuberculosis with high frequency in the lungs and mediastinal lymph node of mice, and we reported evidence that dendritic cells transport M. tuberculosis from the lungs to the mediastinal lymph node (13). In these studies, we used adoptive transfer of CD4+ T cells that express a transgenic T cell antigen receptor specific for a peptide from M. tuberculosis Antigen 85B to characterize the spatial and temporal determinants of initiation of the adaptive immune response to M. tuberculosis. We found that activation of CD4+ T cells in the mediastinal lymph node is the limiting step in development of the adaptive immune response, and that after initial proliferation in the lymph node, effector CD4+ T cells traffic rapidly to the lungs. We also found that initial CD4+ T cell responses depend on the presence of bacteria in the mediastinal lymph node, and that before dissemination in the lymph node, the bacteria are in a compartment in the lungs that cannot be mobilized to the lymph node by an additional proinflammatory stimulus. These findings indicate that initiation of an adaptive immune response to M. tuberculosis depends on transport of live bacteria from the lungs to the mediastinal lymph node, and that M. tuberculosis may delay this process to expand the bacterial population in the lungs and to evade immune effector mechanisms and establish chronic infection.
RESULTS
The adaptive immune response to M. tuberculosis in the lungs is delayed and has limited efficacy
In wild-type C57BL/6 mice infected by the aerosol route, M. tuberculosis grows progressively in the lungs for ∼17–19 d (Fig. 1). Coincident with the appearance of a detectable adaptive immune response, the bacterial population stops expanding, and reaches a stable plateau. IFNγ, which is primarily produced by CD4+ T cells, is one measure of adaptive immunity to M. tuberculosis, and its expression in the lungs coincides with the appearance of CD4+ and CD8+ T lymphocytes (not depicted) and with control of bacterial growth (Fig. 1). During the time required for appearance of an adaptive immune response to M. tuberculosis in the lungs, the bacterial population expanded >20,000-fold, and although the adaptive immune response is capable of arresting progressive growth of the bacteria, it does not lead to apparent bacterial killing.
Figure 1.

Timing of the appearance of adaptive immunity compared with bacterial growth in the lungs of M. tuberculosis–infected C57BL/6 mice. C57BL/6 mice were infected by the aerosol route with 50 cfu of M. tuberculosis (H37Rv). At designated time points, the lungs were removed and assayed for bacteria by plating and for IFNγ gene expression by real-time RT-qPCR. Four mice were assayed at each time point. Error bars represent the SEM.
The long interval between infection and appearance of adaptive immunity to M. tuberculosis could be attributable to delayed priming of naive antigen-specific T lymphocytes, or to delayed trafficking of effector T lymphocytes to the site of infection. To determine the contributions of these two steps to the time required for appearance of M. tuberculosis–specific T lymphocytes in the lungs, we used CD4+ T lymphocytes from transgenic mice that express a T-cell antigen receptor, termed P25TCR-Tg, which is specific for peptide 25 (aa 240–254) of M. tuberculosis Antigen 85B.
P25TCR-Tg CD4+ T cells recognize Antigen 85B produced by M. tuberculosis
We first assessed the ability of P25TCR-Tg CD4+ T cells to respond to antigen presented by cultured cells. Splenocytes from P25TCR-Tg mice stimulated with peptide antigen (peptide 25) and whole Antigen 85B (Ag85B) responded in a dose-dependent manner with maximal responses achieved at 10 μg/ml for both peptide 25 and native Ag85 isolated from M. tuberculosis (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20071367/DC1). In addition, naive P25TCR-Tg/Rag1−/− CD4+ T cells responded to Ag85B expressed by M. tuberculosis in cultured macrophages by producing IFNγ and proliferating (Fig. S1). These results confirm that P25TCR-Tg CD4+ T cells are capable of sensitive detection of Ag85B produced by live, intracellular M. tuberculosis.
Proliferation of Ag85B-specific CD4+ T cells begins in the mediastinal lymph node, and is delayed until 11 d after infection
To characterize the time and location of M. tuberculosis Ag85B-specific T cell priming after M. tuberculosis infection in vivo, we adoptively transferred CFSE-labeled P25TCR-Tg CD4+ T cells into CD45.1 congenic wild-type mice (14). After adoptive transfer, the P25TCR-Tg CD4+ T cells trafficked to secondary lymphoid tissues and represented an equal percentage (1.3%) of the CD4+ T cells in all of the secondary lymphoid tissues examined, as well as in the lungs and blood (unpublished data). When examined 7 or 11 d after aerosol infection with M. tuberculosis (∼100 cfu/mouse), a single population of CFSEbright P25TCR-Tg CD4+ T cells was detected in the lungs and mediastinal lymph node, indicating that none of the cells had proliferated (Fig. 2 A). 14 d after infection, a small percentage of the P25TCR-Tg CD4+ T cells had proliferated in the mediastinal lymph node, but not in other tissues examined (lung, spleen, and inguinal lymph node). Because some P25TCR-Tg CD4+ T cells had undergone up to 7 cycles of replication, we estimated that proliferation had begun (but was not completed) on day 11, assuming ∼10.6 h per cell cycle (15). This was confirmed in a subsequent experiment in which ∼4% of the P25TCR-Tg CD4+ T cells in 5 mice were in the first 2 cycles of proliferation in the mediastinal lymph node on day 12 after infection (unpublished data). By day 17 of infection, the absolute number of P25TCR-Tg CD4+ T cells in the mediastinal lymph node reached a peak after expanding 110-fold, and accounted for 8% of the CD4+ T cells in the mediastinal lymph node.
Figure 2.

Proliferation of P25TCR-Tg CD4+ T cells occurs in the mediastinal lymph node 11 d after infection. (A) CFSE proliferation profile of P25TCR-Tg CD4+ T cells in the mediastinal lymph node and lungs over the course of the infection with M. tuberculosis. Plots are representative of five mice at each time point. Data from days 7, 14, 17, and 21 are from one experiment; day 11 results are from a separate experiment, using the same bacterial inoculum and number of adoptively transferred cells. (B) Total number of P25TCR-Tg CD4+ T cells in the mediastinal lymph node (solid line) and lung (dashed line). Replicates were averaged, and the error bars represent the SEM of five mice per time point. (C) Comparison of the number of total CD4+ T cells and the P25TCR-Tg CD4+ T cells in the lungs with time. Error bars represent the mean ± the SD.
M. tuberculosis Ag85B-specific CD4+ T cells traffic to the lungs after proliferating in the mediastinal lymph node
We also assessed Ag85B-specific T cell proliferation in the lungs. Few (2 × 105) CD4+ T cells were found in the lung before infection and during the first week of infection, of which 1.03% were P25TCR-Tg CD4+ cells. After 14 d of infection, when T cell proliferation was detectable in the mediastinal lymph node, none of the P25TCR-Tg CD4+ T cells had proliferated in the lungs (Fig. 2 A). By day 17 after infection, when a high proportion of the P25TCR-Tg CD4+ T cells were actively proliferating in the mediastinal lymph node, P25TCR-Tg CD4+ T cells in the lung were either CFSEhigh or CFSElow, with few cells of intermediate CFSE intensity. The lack of cells in the first 3 cycles of proliferation in the lungs at those time points indicates that the CFSE low-to-negative cells had migrated to the lungs after undergoing several cycles of proliferation in the lymph node. This observation is consistent with the regulation of T cell egress from the lymph node by sphingosine-1-phosphate and its receptor after initial T cell activation (16). Further evidence that Ag85B-specific CD4+ T cells are recruited to the lungs after proliferating in the mediastinal lymph node was provided by the observation that the total number of P25TCR-Tg CD4+ T cells in the lungs increases coincident with a decrease in the number of P25TCR-Tg CD4+ T cells in the mediastinal lymph node (Fig. 2 B). The observation that the number of P25TCR-Tg CD4+ T cells in the lung does not equal the number produced in the mediastinal lymph node is likely caused by the death of a large number of the cells that initially proliferated in the mediastinal lymph node (17). These observations with P25TCR-Tg CD4+ T cells are likely to be representative of overall M. tuberculosis antigen-specific CD4+ T cell responses, as the increases in P25TCR-Tg CD4+ T cells in the lung mirror the overall increase in CD4+ T cells in the lung (Fig. 2 C).
We considered the possibility that the delayed initial activation of P25TCR-Tg CD4+ T cells was a consequence of the number of cells administered because adoptive transfer of large numbers of TCR transgenic CD4+ T cells influences their extent of expansion (18), differentiation (19), and survival (20). However, adoptive transfer of lower numbers (in 10-fold dilutions to as low as 103 cells/mouse) of P25TCR-Tg CD4+ T cells did not result in a detectable decrease in the time required for their initial activation after M. tuberculosis infection (unpublished data). We also considered the possibility that the long interval required for activation of P25TCR-Tg CD4+ T cells was an intrinsic property of the cells, or was the consequence of the route of antigen delivery. Therefore, we examined the proliferation of P25TCR-Tg CD4+ T cells after administration of recombinant Ag85B by the respiratory and subcutaneous routes. This revealed that adoptively transferred P25TCR-Tg CD4+ T cells could respond by proliferating extensively in vivo within 3 d of immunization, irrespective of the route of antigen delivery (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20071367/DC1). These findings indicate that the delayed initial activation of P25TCR-Tg CD4+ cells in vivo after M. tuberculosis infection is not attributable to an artifact of the experimental system.
P25TCR-Tg CD4+ T cell responses are specific for Antigen 85B
To characterize the specificity of P25TCR-Tg CD4+ T cells in vivo, we infected mice with an Antigen 85B–null strain of M. tuberculosis (Ag85BKO-Mtb). After aerosol infection, the Ag85BKO-Mtb strain grew in the lungs and disseminated to the mediastinal lymph node at a rate equivalent to that of wild-type M. tuberculosis H37Rv (Fig. 3 A). Nevertheless, P25TCR-Tg CD4+ T cell proliferation was not detectable in the mediastinal lymph node or in the lung, even as late as 27 d after infection (Fig. 3 B). The results demonstrate that all the P25TCR-Tg CD4+ T cell proliferation detected in vivo was Antigen 85B specific. Additionally, the lack of any response in the absence of antigen implies a lack of expansion of bystander CD4+ T cells during the initial phase of M. tuberculosis infection.
Figure 3.

P25TCR-Tg CD4+ T cell responses are specific for M. tuberculosis Antigen 85B. (A) CFSE-labeled CD4+ P25TCR-Tg CD4+ T cells were adoptively transferred into C57BL/6J mice, and after 24 h, mice were infected with either wild-type M. tuberculosis (Wt-Mtb, solid lines) or Antigen 85BKO M. tuberculosis (Ag85BKO-Mtb, dashed lines with open symbols) by the aerosol route. The initial inoculum for both strains was ∼100 bacteria/mouse. Total bacterial load was assessed over the first 28 d of infection in the lungs (circles) and the mediastinal lymph node (squares). At each time point, two Ag85BKO-Mtb–infected mice were assessed with a difference of <0.5 log10. Error bars for WT-Mtb represent the mean ± the SD of five mice. The dashed line represents the limit of detection for the cfu assay (18.75 cfu/mouse). The mediastinal lymph node cfu of all the mice at day 7 were below the limit of detection. (B) CFSE proliferation profile of P25TCR-Tg CD4+ T cells in the mediastinal lymph node.
Analysis of T cell activation markers and IFNγ production
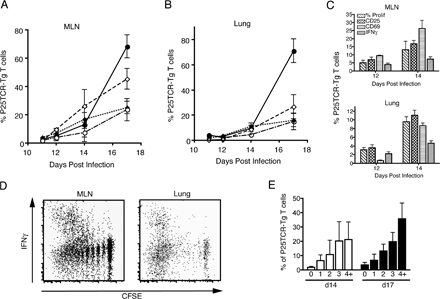
We also considered the possibility that M. tuberculosis–specific CD4+ T cells might become partially activated earlier in infection, but that there is an abnormally long lag period between initial activation and proliferation. We therefore assessed expression of the early activation marker CD69, as well as CD25 expression on the surface of P25TCR-Tg CD4+ T cells during the course of infection. CD69 expression on P25TCR-Tg CD4+ T cells was first observed on day 12, which is approximately coincident with the beginning of measurable proliferation in the mediastinal lymph node, although 5% more of the P25TCR-Tg CD4+ T cells had up-regulated CD69 than had begun to proliferate, indicating that CD69 is up-regulated before proliferation begins (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20071367/DC1). The percentage of CD25+ P25TCR-Tg CD4+ T cells increased concurrent with the onset of proliferation in the mediastinal lymph node (Fig. S3). In addition, a small percentage (4%) of the adoptively transferred cells were competent to produce IFNγ by day 14 of infection; the percentage of IFNγ-producing P25TCR-Tg cells increased further between 14 and 17 d after infection, and was highest in cells that had undergone 4 or more cycles of cell division (Fig. S3).
Analysis of CD69 and CD25 expression on P25TCR-Tg CD4+ cells in the lungs revealed that neither of these markers were expressed before the appearance of cells that had previously proliferated (Fig. S3), further supporting the conclusion that initial CD4+ T cell activation occurs before recruitment to the lungs. Likewise, cells that were competent for IFNγ production did not appear in the lungs until the arrival of P25TCR-Tg CD4+ T cells that had previously proliferated (4 or more cycles) outside the lungs.
Delayed activation of M. tuberculosis–specific CD4+ T cells is not solely caused by a low bacterial inoculum
Because infection with M. tuberculosis was routinely established with a small bacterial inoculum (∼100 cfu), it is possible that one determinant of the delay in initiation of adaptive immune responses is the abundance of antigen, especially because M. tuberculosis replicates slowly. To assess this possibility, we infected four groups of mice by the aerosol route with a range of bacterial inocula between 15 and 700 cfu per mouse and assessed proliferation of adoptively transferred P25TCR-Tg CD4+ T cells during the first 24 d of infection. In the lungs, each inoculum resulted in a distinct bacterial burden over the course of the infection, with a 2 log10 difference between the highest and lowest dose at each time point (Fig. 4 A). Varying the inoculum also resulted in a 2 log10 difference in bacteria in the mediastinal lymph node in mice that received the highest and lowest inocula (Fig. 4 B). This revealed that decreasing the initial inoculum below 30 cfu/mouse led to an additional 3 d delay in P25TCR-Tg CD4+ T cell proliferation, whereas increasing the inoculum to 700 cfu/mouse did not lead to earlier proliferation, but caused a slight increase in the fraction of the cells that proliferated by day 14 (Fig. 4 B). These results indicate that, whereas initial activation of Ag85B-specific CD4+ T cells is partially sensitive to the number of bacteria administered, the delay between initial infection and the earliest activation of CD4+ T cells could not be attributed solely to the low number of bacteria used to initiate infection.
Figure 4.

Dependence of initiation of proliferation on the inoculum of M. tuberculosis. Bacterial cfu in the lung (A) and mediastinal lymph node (B) over the course of the infection after inocula of <15, 30, 100, or 700 bacteria per mouse. (B) Histograms are representative of P25TCR-Tg CD4+ T cell proliferation in the mediastinal lymph node 14 (left column) or 17 (right column) d after infection in mice that received the designated inocula. (C) Correlation of mediastinal lymph node bacterial burden and proliferation of P25TCR-Tg CD4+ T cells. Data are from three independent experiments, and include data from mice that received distinct inocula and that were harvested on various days after infection. (D) Correlation of lung and mediastinal lymph node cfu and P25TCR-Tg CD4+ T cell proliferation. Six mice with similar lung cfu and dissimilar mediastinal lymph node cfu are highlighted. Inset table shows the log10 lung cfu (Lung), log10 mediastinal lymph node cfu (LN), and the percentage of P25TCR-Tg CD4+ T cells that had undergone one or more cycles of proliferation in the mediastinal lymph node for each of the highlighted mice.
Initial activation of M. tuberculosis–specific CD4+ T cells is determined by the number of bacteria in the mediastinal lymph node
Because naive T lymphocytes traffic to the lungs and other peripheral tissues with low frequency, it was not surprising that activation of M. tuberculosis–specific T cells occurred earliest in the mediastinal lymph node. To determine whether the source of antigen for initial stimulation of Ag85B-specific CD4+ T cells is in the lungs or in the mediastinal lymph node (which have ∼100-fold fewer bacteria than the lungs), we tested the hypothesis that stimulation of CD4+ T cells depends on the number of bacteria in the lymph node by examining the relationship between the number of bacteria in the mediastinal lymph node and proliferation of adoptively transferred P25TCR-Tg cells. We pooled the results of three independent experiments and plotted the log10 mediastinal lymph node cfu versus the percentage of P25TCR-Tg CD4+ T cells that had proliferated in the mediastinal lymph node for each mouse. The resulting plot revealed that no proliferation occurred when there were fewer than ∼1,500 cfu in the mediastinal lymph node, even when there were as many as 5 × 104 cfu in the lungs. However, once a threshold number of bacteria (1,500–3,000 per lymph node) were present in the lymph node, there was a linear correlation between cfu and the percentage of P25TCR-Tg CD4+ T cells that proliferated (r2 = 0.77; Fig. 4 C).
When lung cfu were plotted against the mediastinal lymph node cfu, there was little correlation between the lung cfu and the lymph node cfu in individual mice early in infection, when lung cfu were <5 log10 (Fig. 4 D). When we analyzed a subgroup of mice with lung bacterial loads in the narrow range between 4.33 and 4.71 log10 cfu, individual mice exhibited a broad range (1.24–4.7 log10) in the number of bacteria in the mediastinal lymph node. In these mice, proliferation of P25TCR-Tg CD4+ T cells also exhibited a broad variation (1.2–40%), and correlated closely with the number of bacteria in the lymph node and not in the lung. Thus, we conclude that the timing of CD4+ T cell activation is tightly linked to the number of bacteria in the mediastinal lymph node.
Delayed bacterial dissemination to the mediastinal lymph results in delayed T cell activation
We recently reported that plt mice, which lack expression of the CCR7 ligands CCL19 and CCL21ser, exhibit defective trafficking of M. tuberculosis from the lungs to the mediastinal lymph node (13). If initiation of CD4+ T cell responses to M. tuberculosis depends on the number of bacteria expressing antigen in the lymph node, then plt mice should exhibit delayed activation of M. tuberculosis–specific CD4+ T cells in the mediastinal lymph node. To test this hypothesis, we adoptively transferred CFSE-labeled P25TCR-Tg CD4+ T cells into M. tuberculosis–infected wild-type and plt mice and assessed the timing of T cell proliferation. 14 d after infection, when antigen-specific CD4+ T cell proliferation was detected in wild-type mice, 50% fewer P25TCR-Tg CD4+ T cells had started to divide in plt mice at the same time point (Fig. 5 A). The reduced T cell proliferation in plt mice was accompanied by a 40% reduction in the number of bacteria in the mediastinal lymph node compared with wild-type mice at day 14 (Fig. 5 B). Overall, the CD4+ T cell responses were delayed by ∼3 d in plt mice.
Figure 5.

Delayed dissemination of M. tuberculosis in plt mice results in further delayed T cell activation. (A) Proliferation of CFSE-labeled P25TCR-Tg CD4+ T cells after adoptive transfer to M. tuberculosis–infected wild-type C57BL/6J or plt mice. Results shown are the percentage of P25TCR-Tg CD4+ T cells that had started to divide, to correct for the partial defect in the number of adoptively transferred cells that trafficked to the mediastinal lymph node of plt mice. To detect P25TCR-Tg CD4+ T cells on days 10 and 14, it was necessary to pool lymph nodes from 5 mice, therefore statistical analysis was not done on those samples. Error bars on days 19 and 21 represent the mean ± the SD of 3 C57BL/6J mice and 5 plt mice. *, P < 0.05 by Student's t test. The percentage of CD69+ P25TCR-Tg CD4+ T cells was also fourfold lower in plt mice compared with wild-type at day 14 (not depicted). (B) Bar graph displaying the percentage of P25TCR-Tg CD4+ T cells that had divided and the cfu at days 14 and 19 for the plt mice as the percentage of wild-type levels.
Ag85B-specific T cell proliferation occurs in nondraining lymph nodes and spleen if sufficient bacteria are present in the tissue
To further test the hypothesis that activation of M. tuberculosis–specific CD4+ T cells requires production of antigen by bacteria in the lymph node, rather than occurring by transfer of soluble antigen synthesized by bacteria in the lungs, we assessed proliferation of P25TCR-Tg CD4+ T cells in the inguinal lymph node, which does not receive lymphatic drainage from the lungs. This revealed that activation and proliferation of Ag85B-specific CD4+ T cells could be initiated in the inguinal lymph node after aerosol infection with M. tuberculosis (Fig. 6 A). The timing of P25TCR-Tg CD4+ T cell proliferation in the inguinal lymph node followed responses in the mediastinal lymph node by 7–10 d, and varied more from mouse to mouse than did responses in the mediastinal lymph node. Nevertheless, proliferative responses in the inguinal lymph node correlated closely with the number of bacteria present in the lymph node (Fig. 6, B and C). Additionally, we found that lymph nodes in diverse anatomical locations could be sites of proliferation of P25TCR-Tg CD4+ T cells, if they contained sufficient numbers of viable M. tuberculosis (unpublished data). The initiation of proliferation of M. tuberculosis–specific CD4+ T cells in lymph nodes that contain viable M. tuberculosis, but do not drain the lungs, further supports the conclusion that CD4+ T cell activation requires synthesis of antigens in the local lymph node, and that production of M. tuberculosis antigens in the lungs plays little, if any, role in initiating responses of naive T lymphocytes.
Figure 6.

M. tuberculosis disseminates and activates P25TCR-Tg CD4+ T cells in lymphoid tissues that do not drain the lungs. (A) Bacterial load in the mediastinal lymph node (MLN), spleen, and inguinal lymph node (ILN) with time. The dashed line represents the limit of detection (18.75 cfu/mouse). Error bars represent the SD. (B) The percentage of P25TCR-Tg CD4+ T cells that have undergone at least 1 cycle of proliferation in the mediastinal lymph node (MLN), spleen, and inguinal lymph node (ILN). Error bars represent the SEM. (C) CFSE histograms form the inguinal lymph node at days 17 and 21 after infection; each histogram represents one mouse. Values in the top left corner represent the number of cfu found in the inguinal lymph node of that mouse.
In the spleen, we found that on day 14 of infection there were >10-fold fewer bacteria in the spleen compared with the mediastinal lymph node (Fig. 6 B). The difference in bacterial load narrowed over the next 2 wk of the infection. Proliferation of P25TCR-Tg CD4+ T cells occurred in the spleen, but followed T cell responses in the mediastinal lymph node by ∼3 d (Fig. 6 A). At the point when P25TCR-Tg CD4+ T cell proliferation occurred in the spleen (day 17), the bacterial load in the spleen had reached approximately the same level as in the mediastinal lymph node when initial proliferation was detected.
Addition of a proinflammatory stimulus does not accelerate the T cell response to live M. tuberculosis
The observations that the initial adaptive immune response to an M. tuberculosis is delayed, that activation of M. tuberculosis–specific CD4+ T cells depends on bacterial production of antigen in the lymph node, and that the number of bacteria in the lymph node is only ∼1% of that in the lung suggest that M. tuberculosis might impair maturation and/or trafficking of dendritic cells from the lungs to the lymph node. To test this hypothesis, we administered LPS intranasally (i.n.) to mice that had been infected with wild-type M. tuberculosis 10 d earlier. The administration of LPS resulted in a 12.4-fold increase in the number of myeloid dendritic cells (CD11chiCD11bhi) in the lungs, and in a 12-fold increase in myeloid dendritic cells recruited to the mediastinal lymph node (Fig. 7 A). The increase in the number of myeloid dendritic cells was accompanied by their increased maturation, as assessed by surface expression of MHC class II (Fig. 7 B). However, despite the increased number of dendritic cells in the lungs and mediastinal lymph node, there was no increase in the number of bacteria transported to the mediastinal lymph node from the lungs (Fig. 7 C), and there was no increase in proliferation of P25TCR-Tg CD4+ T cells in the mediastinal lymph node after treatment with LPS (Fig. 7 D), unless recombinant Ag85B was coadministered with LPS (Fig. 7 E).
Figure 7.

Airway administration of LPS does not increase translocation of M. tuberculosis from the lungs to the mediastinal lymph node, and does not accelerate P25TCR-Tg CD4+ T cell responses. Mice that were either uninfected or infected 10 d earlier with wild-type M. tuberculosis received 5 × 106 CFSE-labeled P25TCR-Tg CD4+ T cells 4 d before harvest. 24 h later, i.e., 3 d before harvest, mice were given sterile PBS or 1 μg LPS with or without 50 ng of recombinant Antigen 85B (rAg85B) by the intranasal route. (A) The number of myeloid dendritic cell in the mediastinal lymph node. (B) Surface MHC class II mean fluorescence intensity (MFI) on mediastinal lymph node myeloid dendritic cell. (C) Cfu in the lung and mediastinal lymph node (MLN) in mice infected with wild-type M. tuberculosis that either received PBS or LPS i.n. (D) Change in the number of P25TCR-Tg CD4+ T cells caused by proliferation as assessed in the mediastinal lymph node at day 14 after infection (4 d after LPS or control). Histograms show day 14 CFSE proliferation profiles of representative mice given either PBS (D, left) or LPS (D, right) i.n. (E) Comparison of the number of P25TCR-Tg CD4+ T cells in the mediastinal lymph node of uninfected or M. tuberculosis–infected mice that received either PBS or rAg85B + LPS i.n. Histograms represent the proliferation profile of the CFSE-labeled P25TCR-Tg cells in uninfected + rAg85B + LPS (left) and infected + rAg85B + LPS (right). The number of cells was determined by multiplying the percentage as determined by flow cytometry by the total number of cells in the lymph node as assessed by trypan exclusion. Three mice were examined per group, and the error bars represent the SD. Statistics were done using Student t test. *, P < 0.05; **, P < 0.01. ns, not significant.
DISCUSSION
The adaptive cellular immune response to M. tuberculosis is only partially efficacious, as it can restrict progressive growth of the bacteria, but rarely if ever eradicates them. In our studies, we used adoptive transfer of cells from transgenic mice whose CD4+ T lymphocytes express an antigen receptor specific for a peptide from M. tuberculosis Antigen 85B to characterize the initial events in the adaptive immune response to M. tuberculosis. In accord with studies that used ex vivo T cell restimulation to characterize the location and timing of CD4+ T cell responses to M. tuberculosis, we found that the earliest detectable responses of adoptively transferred Ag85B-specific CD4+ T cells occur in the lung-draining mediastinal lymph node, are delayed until 10–12 d after infection, and are preceded by the appearance of live M. tuberculosis in the mediastinal lymph node. In addition, we found strong evidence that activation of Ag85B-specific CD4+ T cells depends on the number of M. tuberculosis in the lymph node rather than in the lungs. We also found that transfer of M. tuberculosis from the lungs to the lymph node could not be accelerated by pulmonary administration of LPS, indicating that early in infection, the bacteria are in a compartment that is not competent to migrate, suggesting that slow growth of M. tuberculosis in the lungs does not fully account for delayed T cell activation. Collectively, these results indicate that during early stages of infection in vivo, M. tuberculosis occupies one or more compartments that do not promote antigen presentation to naive CD4+ T cells, and this allows dramatic expansion of the bacterial population in the lungs. We propose that this allows the large bacterial population to resist effector mechanisms of the adaptive immune response, and allows M. tuberculosis to persist in the lungs.
Antigen 85B is an abundant secreted protein of M. tuberculosis that represents >20% of the protein in culture filtrates, and is expressed and secreted by M. tuberculosis within host cells (21, 22). As a secreted protein and frequent target of immune responses in humans (23) and mice, Ag85B has been considered to be a potential candidate vaccine antigen, and a recombinant BCG strain that overproduces Ag85B is currently being prepared for human clinical trials. Because Ag85B is secreted, has been observed in extraphagosomal vesicles of macrophages (22), and expression has been detected in the lungs early during infection (24), we expected that production and secretion of Ag85B by M. tuberculosis in the lungs would be sufficient to deliver soluble antigen to activate Ag85B-specific CD4+ T cells in the local draining lymph node. If this were the case, Ag85B-specific T cell responses in the lymph node should directly correlate with the number of bacteria in the lungs. Instead, we found that although the number of bacteria in the lungs increases progressively during the first 2–3 wk of infection, Ag85B-specific CD4+ T cell responses were not observed until a threshold number of viable bacteria (∼1,500–3,000) appeared in the mediastinal lymph node. Moreover, although the number of bacteria in the lymph node in most mice was closely related to (and ∼100-fold less than) the number of bacteria in the lungs, analysis of several mice with discordant bacterial burdens in the lungs and lymph node revealed that responses of Ag85B-specific CD4+ T cells correlated with the number of bacteria in the mediastinal lymph node, and not with the number of bacteria in the lungs. Additional evidence that production of Ag85B by live bacteria in the lymph node is essential to initiate T cell responses in the context of M. tuberculosis infection was provided by analysis of Ag85B-specific CD4+ T cell responses in lymph nodes that do not drain the lungs. This analysis revealed that Ag85B-specific CD4+ T cell responses occurred in peripheral (nonlung draining) lymph nodes as long as a sufficient number of live bacteria were present in the same lymph node. These results indicate that, even for an abundant secreted protein antigen, stimulation of CD4+ T cells requires short-range interactions between M. tuberculosis–infected cells and antigen-specific T cells.
We considered the possibility that the immune response to M. tuberculosis Ag85B is aberrant or otherwise unrepresentative of M. tuberculosis antigens. However, we found that the temporal and quantitative characteristics of expansion of adoptively transferred Ag85B-specific CD4+ T cells paralleled that of total CD4+ T cell responses during the course of M. tuberculosis infection (Fig. 2). Moreover, our results correspond closely to those obtained by ex vivo restimulation with an M. tuberculosis lysate (5).
The observation that initial Ag85B-specific CD4+ T cell responses require the presence of live bacteria in the lymph node suggests that the time required for M. tuberculosis to enter a compartment that is competent for transport to the local lymph node may account for the delay in initiation of the adaptive immune response in tuberculosis. Although slow replication of M. tuberculosis could be assumed to account for the delayed cellular immune response, our data imply that slow bacterial growth cannot fully explain the temporal characteristics of the immune response. If a simple model of slow growth accounted for insufficient antigen for CD4+ T cell activation, the time required to initiate the immune response should be reduced by a higher inoculum of bacteria. For example, increasing the inoculum from 100 to 700 bacteria/mouse in the initial inoculum should have reduced the time dependence of Ag85B-specific CD4+ T cell activation by ∼2.6 bacterial doubling times (71 h, based on the growth curve shown in Fig. 1). Instead, we found no difference in the time required to initiate proliferation of Ag85B-specific CD4+ T cells in mice infected with 100 or 700 bacteria, which implies that one or more factors other than a low initial inoculum contribute to the long interval preceding Ag85B-specific CD4+ T cell responses.
We recently reported that, after aerosol infection of mice with M. tuberculosis, myeloid dendritic cells become infected in the lungs, and represent the predominant cells that contain M. tuberculosis in the mediastinal lymph node (13). Moreover, we found that plt mice exhibited a parallel decrease in dendritic cell recruitment and dissemination of M. tuberculosis to the mediastinal lymph node. These results provide evidence that myeloid dendritic cells become infected in the lungs and transport live M. tuberculosis to the local lymph node. Together with the observation that plt mice also show an increased delay in activation of Ag85B-specific CD4+ T cells (Fig. 5), these results suggest that myeloid dendritic cell transport of M. tuberculosis to the local lymph node is a critical determinant of the initiation of the adaptive immune response in tuberculosis.
Because transport of M. tuberculosis to the mediastinal lymph node was not apparent until 11–12 d after infection, we tested the hypothesis that administration of lipopolysaccharide to M. tuberculosis–infected mice would accelerate migration of infected dendritic cells from the lungs to the mediastinal lymph node, and thereby accelerate activation of Ag85B-specific CD4+ T cells. Although intranasal administration of lipopolysaccharide did result in recruitment of additional macrophages and dendritic cells to the lungs of infected mice, and resulted in recruitment of additional dendritic cells to the mediastinal lymph node, this did not result in an increase in the number of bacteria transported to the lymph node, and did not result in increased activation of Ag85B-specific CD4+ T cell responses. These results imply that during the early stage of M. tuberculosis infection, the bacteria are in a cellular compartment that is unable to migrate from the lungs to the lymph node. Whether the bacteria are in cells that intrinsically lack the ability to migrate, or whether they inhibit migration of dendritic cells from the lungs to the lymph node, will require further study.
The findings reported here have several implications for understanding the biology of tuberculosis. In particular, they indicate that even though the lungs are the site of infection with M. tuberculosis, the cellular immune response is not initiated in the lungs. This is consistent with the well-characterized trafficking patterns of naive T lymphocytes that circulate through peripheral nonlymphoid organs with low frequency (25, 26). Therefore, even though a large number of bacteria are present and a large quantity of antigen is produced by bacteria in the lungs (24), initiation of the adaptive immune response requires transport of bacteria from the lungs to the local draining lymph node or other secondary lymphoid tissues. Second, the observation that the bacterial population in the lungs expands 10,000–100,000-fold between the time of initial infection and appearance of the adaptive immune response in the lungs implies that the effector mechanisms of the adaptive immune response confront a large bacterial burden when they do appear in the lungs. Because M. tuberculosis has been found to inhibit cellular responses to IFNγ (27–30) and MHC class II antigen presentation (31–33), the existence of such a large bacterial population in the lungs may interfere with recognition by and/or effector functions of CD4+ T cells, and thereby inhibit the ability of the adaptive immune response to eradicate M. tuberculosis from the lungs. Third, these findings may provide a basis for further analysis of the differential outcomes in humans infected with M. tuberculosis. Although a high proportion of humans that encounter M. tuberculosis develop immune responses that contain the infection as latent tuberculosis, ∼5% progress to active, symptomatic infection within 2 yr of initial infection (34). Although identifiable factors such as HIV infection account for some cases of progressive infection (35), most are unexplained. The findings reported here from studies of mice suggest that variations in transport of bacteria from the lungs to the local lymph node and initiation of the adaptive immune response may underlie some of the differential outcomes of M. tuberculosis infection in humans.
MATERIALS AND METHODS
Mice.
P25TCR-Tg mice, whose CD4+ T cells express a transgenic T-cell antigen receptor that recognizes peptide 25 (aa 240–254) of M. tuberculosis Antigen 85B bound to I-Ab was prepared on a C57BL/6 background, as previously described (36). Mice for experiments were bred in the New York University School of Medicine animal facility. Genotypes of all mice were confirmed by PCR testing of tail genomic DNA. CD45.1 mice were either bred in the New York University School of Medicine animal facility or purchased from Taconic Farms, Inc. All animal experiments were done in accordance with procedures approved by the New York University School of Medicine Institutional Animal Care and Use Committee.
Generation and characterization of Antigen 85B–deficient M. tuberculosis.
Antigen 85B (Rv1886c)–deficient M. tuberculosis mutant was created with the conditionally replicating mycobacteriophages, as previously described (37). The upper and lower allelic exchange substrates were PCR amplified from H37Rv genomic DNA and cloned into pCR2.1-TOPO (Invitrogen). The allelic exchange substrates were sequenced and directionally cloned into pYUB854. After transduction of H37Rv and plating on Middlebrook 7H9 agar supplemented with albumin-dextrose-catalase, several colonies were picked and screened for the absence of Antigen 85B mRNA by real-time RT-PCR. Antigen 85B–deficient clones (Ag85BKO-Mtb) were also examined by Western blot to confirm the absence of Antigen 85B protein using a monoclonal antibody against the Antigen 85B complex.
Preparation and characterization of recombinant Antigen 85B.
Recombinant Antigen 85B was produced using the expression plasmid pMRLB47.Rv1886c obtained through the National Institutes of Health TB Vaccine Testing and Research Materials Contract (NIAID N01AO040091). Escherichia coli BL21(DE3)pLysS (Novagen) was transformed with the expression plasmid. Bacteria were grown in Lurie-Bertani broth at 37°C until OD600 = 0.5, and then the culture was shifted to 30°C in the presence of 0.5 mM IPTG. Lysis of E. coli and extraction of recombinant protein was done using BugBusters reagent and His-Bind kit (Novagen) according to the manufacturer's instructions, followed by dialysis in PBS and quantitation using BCA Protein Assay (Thermo Fisher Scientific). Protein purity was assessed by SDS-PAGE and Bio-Safe Coomassie staining (Bio-Rad Laboratories). Endotoxin was removed using EndoClean reagent (BioVintage). Recombinant Antigen 85B was considered endotoxin-free when it was determined to contain <0.1 EU/ml as assessed by PyroGene Recombinant Factor C Endotoxin Detection System (Cambrex).
In vitro T cell stimulation assays.
BM-derived macrophages (BMDMs) from C57BL/6 mice were cultured as previously described (38). After 6–7 d of culture, cells were replated at 105 cells/well in flat bottom 96-well plates and treated for 24 h with recombinant mouse IFNγ (20 ng/ml; BD Biosciences). For infection of cultured cells, M. tuberculosis H37Rv was grown to an OD580 of 0.5–1 in 7H9 broth supplemented with albumin-dextrose-catalase enrichment. A suspension of single bacteria was generated by pelleting the bacteria, resuspending in macrophage culture media, and allowing the bacteria to flow by gravity through a 5-μm syringe filter (Millipore) to remove clumps. Bacteria were enumerated by counting a dilution in a Petroff-Hausser chamber and confirmed by serial dilution and growth on 7H11 agar. BMDMs were incubated with bacteria for 24 h in BMDM growth media. Growth media was removed and extracellular bacteria were removed by washing with PBS. P25TCR-Tg CD4+ T cells were added at 2 × 105 cells/well in complete media (RPMI, 10% FCS, l-glutamine, nonessential amino acids, sodium pyruvate, Hepes, and β-mercaptoethanol) with or without peptide 25 (custom synthesis by Invitrogen and EZ Biolabs), and cells were incubated for 1–5 d. Supernatants of triplicate wells were pooled and assayed for IFNγ by ELISA (BD Biosciences) and read on a Bio Tek EXL800 plate reader, or proliferation of CFSE-labeled P25TCR-Tg/Rag1−/− CD4+ T cells was assessed by flow cytometry.
P25TCR-Tg CD4+ T cell isolation and labeling.
P25TCR-Tg mice between 8–16 wk of age were killed according to approved laboratory animal procedures. Lymph nodes and spleen were aseptically removed, and tissues were disrupted by forcing them through a 70-μm cell strainer (BD Biosciences) in RPMI, 5% FCS, 10 mM Hepes. Red blood cells were removed using ACK lysis buffer (155 mM NH3Cl, 10 mM KHCO3, and 88 μM EDTA). Live cells were counted in a hemacytometer using trypan blue exclusion. CD4+ T cells were magnetically isolated using a CD4+ T Cell Isolation kit and an AutoMACS (Miltenyi Biotech). CD4+ T cell purity was routinely >90% as assessed by flow cytometry. For proliferation assays, CD4+ T cells were labeled with CFSE by resuspending the P25TCT-Tg CD4+ cells at a density of 107 cells/ml in room temperature PBS containing 1 μM CFSE (CFDA-SE; Invitrogen). Cells were incubated with CFSE for 7 min at 37°C. Labeling was stopped with an excess of FCS and the cells were washed three times with RPMI with 10% FCS.
Adoptive transfer and infection.
CD45.1 mice routinely received 3–5 × 106 CFSE-labeled CD4+ P25TCR-Tg CD4+ T cells (CD45.2) by tail vein injection in 100 μl of sterile PBS. After 24 h, mice were infected by the aerosol route using an Inhalation Exposure Unit (Glas-Col), as previously described (13). The infectious dose was confirmed by killing 4–5 mice within 24 h, and then removing the lungs, forcing them through a 70-μm cell strainer (BD Biosciences), and plating the homogenate on 7H11 agar. Plates were incubated at 37°C, and colonies were counted 14–21 d later.
Tissue processing and flow cytometry.
At designated time points, 3–5 infected mice in each group were killed, and tissues were used to prepare single-cell suspensions, as previously described (13), with the exception that enzyme digestion was omitted when tissues were used only for T cell isolation. 5 × 106 cells were stained with anti-CD4, anti-CD45.2, and other surface marker antibodies at a density of 1.5 × 107 cells/ml in FACS buffer (PBS, 1% FCS, 0.1% sodium azide, and 1 mM EDTA) and incubated at 4°C for 20–30 min. Cells were washed and fixed in 1% PFA overnight at 4°C. Data were acquired on either a FACSCalibur or LSRII (BD Biosciences). Flow cytometry antibodies were purchased from BD Biosciences or BioLegend. The number of cells of a specific phenotype was determined by taking the percentage of that cell type determined by flow cytometry multiplied by the number of total cells. “Percent proliferating” was defined as the percentage of all P25TCR-Tg CD4+ T cells that had undergone at least one cycle of replication. “Percent divided” was defined as the percentage of the original number of P25TCR-Tg CD4+ T cells that have begun to proliferate assuming no cell death, calculated by FlowJo (Tree Star, Inc.) proliferation platform.
Detection of IFNγ-producing cells by intracellular cytokine staining.
Single-cell suspensions of lung and lymph node were prepared as in the previous section. Cells were plated at 106 cells per well in a round-bottom 96-well tissue culture plate (Corning) in complete media. 5–7 wells of cells were plated for each mouse and incubated with or without 10 μg of peptide 25 per well for 4–6 h in the presence of Brefeldin A at 37°C with 5% CO2. Cells were stained for the surface markers CD4 and CD45.2 for 30 min at 4°C, followed by fixation for 2 h at 4°C in 2% PFA. IFNγ was detected using Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's instructions. Data were acquired on an LSR-II flow cytometer using FACSDiva Software (BD Biosciences) and analyzed using FlowJo software.
Determination of bacterial load.
At every time point, each tissue from each mouse was assessed for bacterial load by taking an aliquot of the total tissue homogenate before any washing. The aliquot was serial diluted and plated on 7H11 agar. Plates were incubated at 37°C and colonies were counted 14–21 d later. Total colony forming units were determined based on the total volume of tissue homogenate.
Intranasal and subcutaneous immunization with recombinant Antigen 85B.
M. tuberculosis–infected or uninfected mice were anesthetized with a mixture of ketamine and xylazine administered i.p., and given sterile PBS with or without 50 ng of endotoxin-free recombinant Antigen 85B in 30–50 μl of sterile PBS, together with 1 μg of LPS for intranasal immunization or in an emulsion of TiterMax adjuvant (Sigma-Aldrich) for subcutaneous immunization. 3 d after immunization, tissues were harvested and single-cell suspensions were analyzed by flow cytometry and plated for cfu, as described above.
Online supplemental material.
An analysis of the in vitro response of P25TCR-Tg T cells to Antigen 85B, peptide 25, and M. tuberculosis–infected cells can be found in Fig. S1. Fig. S2 shows the P25TCR-Tg T cell responses to intranasal and subcutaneous immunization with Ag85B, demonstrating that the transgenic T cells do not display inherently delayed responsiveness. The expression patterns of CD69, CD25, and IFNγ on P25TCR-Tg T cells over the course of the infection in the lung and mediastinal lymph node are depicted in Fig. S3. In addition, Fig. S3 shows the expression of IFNγ relative to the number of cycles of P25TCR-Tg proliferation in the lung and mediastinal lymph node. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20071367/DC1.
Supplemental Material
Acknowledgments
We thank Dan Littman and members of his laboratory for the use of their LSR-II flow cytometer, and Dr. Mark Eggena for providing training in techniques used for T cell adoptive transfer techniques.
Supported by National Institutes of Health R01-AI051242, and by Strategic cooperation to control emerging and reemerging infections funded by the Special Coordination Funds for Promoting Science and Technology of the Ministry of Education, Culture, Sports, Science and Technology.
The authors have no conflicting financial interests.
Abbreviation used: i.n., intranasally.
References
- 1.Mogues, T., M.E. Goodrich, L. Ryan, R. LaCourse, and R.J. North. 2001. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 193:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perlman, D.C., W.M. El-Sadr, E.T. Nelson, J.P. Matts, E.E. Telzak, N. Salomon, K. Chirgwin, and R. Hafner. 1997. Variation of chest radiographic patterns in pulmonary tuberculosis by degree of human immunodeficiency virus-related immunosuppression. Clin. Infect. Dis. 25:242–246. [DOI] [PubMed] [Google Scholar]
- 3.Poulsen, A. 1950. Some clinical features of tuberculosis I. Incubation period. Acta Tuberc. Pneumol. Scand. 24:311–346. [PubMed] [Google Scholar]
- 4.Wallgren, A. 1948. The time-table of tuberculosis. Tubercle. 29:245–251. [DOI] [PubMed] [Google Scholar]
- 5.Chackerian, A.A., J.M. Alt, T.V. Perera, C.C. Dascher, and S.M. Behar. 2002. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect. Immun. 70:4501–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baron, S.D., R. Singh, and D.W. Metzger. 2007. Inactivated Francisella tularensis live vaccine strain protects against respiratory tularemia by intranasal vaccination in an immunoglobulin A-dependent fashion. Infect. Immun. 75:2152–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakravarty, S., I.A. Cockburn, S. Kuk, M.G. Overstreet, J.B. Sacci, and F. Zavala. 2007. CD8(+) T lymphocytes protective against malaria liver stages are primed in skin-draining lymph nodes. Nat. Med. 13:1035–1041. [DOI] [PubMed] [Google Scholar]
- 8.Kursar, M., K. Bonhagen, A. Kohler, T. Kamradt, S.H.E. Kaufmann, and H.-W. Mittrucker. 2002. Organ-specific CD4+ T cell response during Listeria monocytogenes infection. J. Immunol. 168:6382–6387. [DOI] [PubMed] [Google Scholar]
- 9.Lira, R., M. Doherty, G. Modi, and D. Sacks. 2000. Evolution of lesion formation, parasitic load, immune response, and reservoir potential in c57bl/6 mice following high- and low-dose challenge with Leishmania major. Infect. Immun. 68:5176–5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McSorley, S.J., S. Asch, M. Costalonga, R.L. Reinhardt, and M.K. Jenkins. 2002. Tracking Salmonella-specific CD4 T cells in vivo reveals a local mucosal response to a disseminated infection. Immunity. 16:365–377. [DOI] [PubMed] [Google Scholar]
- 11.Moskophidis, D., and D. Kioussis. 1998. Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J. Exp. Med. 188:223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srinivasan, A., J. Foley, R. Ravindran, and S.J. McSorley. 2004. Low-dose Salmonella infection evades activation of flagellin-specific CD4 T cells. J. Immunol. 173:4091–4099. [DOI] [PubMed] [Google Scholar]
- 13.Wolf, A.J., B. Linas, G.J. Trevejo-Nunez, E. Kincaid, T. Tamura, K. Takatsu, and J.D. Ernst. 2007. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 179:2509–2519. [DOI] [PubMed] [Google Scholar]
- 14.Parish, C.R. 1999. Fluorescent dyes for lymphocyte migration and proliferation studies. Immunol. Cell Biol. 77:499–508. [DOI] [PubMed] [Google Scholar]
- 15.Gudmundsdottir, H., A.D. Wells, and L.A. Turka. 1999. Dynamics and requirements of T cell clonal expansion in vivo at the single-cell level: effector function is linked to proliferative capacity. J. Immunol. 162:5212–5223. [PubMed] [Google Scholar]
- 16.Matloubian, M., C.G. Lo, G. Cinamon, M.J. Lesneski, Y. Xu, V. Brinkmann, M.L. Allende, R.L. Proia, and J.G. Cyster. 2004. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 427:355–360. [DOI] [PubMed] [Google Scholar]
- 17.Ahmed, R., and D. Gray. 1996. Immunological memory and protective immunity: understanding their relation. Science. 272:54–60. [DOI] [PubMed] [Google Scholar]
- 18.Rivera, A., G. Ro, H.L. Van Epps, T. Simpson, I. Leiner, D.B. Sant'Angelo, and E.G.P. Am. 2006. Innate immune activation and CD4+ T cell priming during respiratory fungal infection. Immunity. 25:665–675. [DOI] [PubMed] [Google Scholar]
- 19.Foulds, K.E., and H. Shen. 2006. Clonal competition inhibits the proliferation and differentiation of adoptively transferred TCR transgenic CD4 T cells in response to infection. J. Immunol. 176:3037–3043. [DOI] [PubMed] [Google Scholar]
- 20.Hataye, J., J.J. Moon, A. Khoruts, C. Reilly, and M.K. Jenkins. 2006. Naive and memory CD4+ T cell survival controlled by clonal abundance. Science. 312:114–116. [DOI] [PubMed] [Google Scholar]
- 21.Lee, B.Y., and M.A. Horwitz. 1995. Identification of macrophage and stress-induced proteins of Mycobacterium tuberculosis. J. Clin. Invest. 96:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harth, G., B.Y. Lee, J. Wang, D.L. Clemens, and M.A. Horwitz. 1996. Novel insights into the genetics, biochemistry, and immunocytochemistry of the 30-kilodalton major extracellular protein of Mycobacterium tuberculosis. Infect. Immun. 64:3038–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Havlir, D.V., R.S. Wallis, W.H. Boom, T.M. Daniel, K. Chervenak, and J.J. Ellner. 1991. Human immune response to Mycobacterium tuberculosis antigens. Infect. Immun. 59:665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi, L., Y.-J. Jung, S. Tyagi, M.L. Gennaro, and R.J. North. 2003. Expression of Th1-mediated immunity in mouse lungs induces a Mycobacterium tuberculosis transcription pattern characteristic of nonreplicating persistence. Proc. Natl. Acad. Sci. USA. 100:241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mora, J.R., and U.H. von Andrian. 2006. T-cell homing specificity and plasticity: new concepts and future challenges. Trends Immunol. 27:235–243. [DOI] [PubMed] [Google Scholar]
- 26.Westermann, J., E.M. Ehlers, M.S. Exton, M. Kaiser, and U. Bode. 2001. Migration of naive, effector and memory T cells: implications for the regulation of immune responses. Immunol. Rev. 184:20–37. [DOI] [PubMed] [Google Scholar]
- 27.Ting, L.-M., A.C. Kim, A. Cattamanchi, and J.D. Ernst. 1999. Mycobacterium tuberculosis Inhibits IFN-{gamma } transcriptional responses without inhibiting activation of STAT1. J. Immunol. 163:3898–3906. [PubMed] [Google Scholar]
- 28.Kincaid, E.Z., and J.D. Ernst. 2003. Mycobacterium tuberculosis exerts gene-selective inhibition of transcriptional responses to IFN-{gamma } without inhibiting STAT1 function. J. Immunol. 171:2042–2049. [DOI] [PubMed] [Google Scholar]
- 29.Fortune, S.M., A. Solache, A. Jaeger, P.J. Hill, J.T. Belisle, B.R. Bloom, E.J. Rubin, and J.D. Ernst. 2004. Mycobacterium tuberculosis inhibits macrophage responses to IFN-{gamma } through myeloid differentiation factor 88-dependent and -independent mechanisms. J. Immunol. 172:6272–6280. [DOI] [PubMed] [Google Scholar]
- 30.Arko-Mensah, J., E. Julian, M. Singh, and C. Fernandez. 2007. TLR2 but not TLR4 signalling is critically involved in the inhibition of IFN-gamma-induced killing of mycobacteria by murine macrophages. Scand. J. Immunol. 65:148–157. [DOI] [PubMed] [Google Scholar]
- 31.Noss, E.H., C.V. Harding, and W.H. Boom. 2000. Mycobacterium tuberculosis Inhibits MHC Class II antigen processing in murine bone marrow macrophages. Cell. Immunol. 201:63–74. [DOI] [PubMed] [Google Scholar]
- 32.Noss, E.H., R.K. Pai, T.J. Sellati, J.D. Radolf, J. Belisle, D.T. Golenbock, W.H. Boom, and C.V. Harding. 2001. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. J. Immunol. 167:910–918. [DOI] [PubMed] [Google Scholar]
- 33.Fulton, S.A., S.M. Reba, R.K. Pai, M. Pennini, M. Torres, C.V. Harding, and W.H. Boom. 2004. Inhibition of major histocompatibility complex ii expression and antigen processing in murine alveolar macrophages by mycobacterium bovis BCG and the 19-kilodalton mycobacterial lipoprotein. Infect. Immun. 72:2101–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Comstock, G.W., V.T. Livesay, and S.F. Woolpert. 1974. The prognosis of positive tuberculin reation in childhood and adolescence. Am. J. Epidemiol. 99:131–138. [DOI] [PubMed] [Google Scholar]
- 35.Daley, C.L., P.M. Small, G.F. Schecter, G.K. Schoolnik, R.A. McAdam, W.R. Jacobs, and P.C. Hopewell. 1992. An outbreak of tuberculosis with accelerated progression among persons infected with the human immunodeficiency virus. An analysis using restriction-fragment-length polymorphisms. N. Engl. J. Med. 326:231–235. [DOI] [PubMed] [Google Scholar]
- 36.Tamura, T., H. Ariga, T. Kinashi, S. Uehara, T. Kikuchi, M. Nakada, T. Tokunaga, W. Xu, A. Kariyone, T. Saito, et al. 2004. The role of antigenic peptide in CD4+ T helper phenotype development in a T cell receptor transgenic model. Int. Immunol. 16:1691–1699. [DOI] [PubMed] [Google Scholar]
- 37.Bardarov, S., S. Bardarov Jr., M.S. Pavelka Jr., V. Sambandamurthy, M. Larsen, J. Tufariello, J. Chan, G. Hatfull, and W.R. Jacobs Jr. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology. 148:3007–3017. [DOI] [PubMed] [Google Scholar]
- 38.Banaiee, N., E.Z. Kincaid, U. Buchwald, W.R. Jacobs Jr., and J.D. Ernst. 2006. Potent inhibition of macrophage responses to IFN-{gamma } by live virulent Mycobacterium tuberculosis is independent of mature mycobacterial lipoproteins but dependent on TLR2. J. Immunol. 176:3019–3027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}