

Abstract
Background and purpose
CADASIL is an inherited small-vessel disease responsible for lacunar strokes and cognitive impairment. The disease is caused by highly stereotyped mutations in Notch3, expression of which is highly restricted to vascular smooth muscle cells (vSMC). The underlying vasculopathy is characterized by degeneration of vSMC and the accumulation of granular osmiophilic material (GOM) and Notch3 protein within the cell surface of these cells. In this study we assessed early functional changes related to the expression of mutant Notch3 in resistance arteries.
Methods
Vasomotor function was examined in vitro in arteries from transgenic mice that express a mutant Notch3 in vSMC. Tail artery segments from transgenic and normal wild-type male mice were mounted on small-vessel arteriographs and reactivity to mechanical (flow and pressure) forces and pharmacological stimuli were determined. Mice were studied at 10–11 months of age when vSMC degeneration, GOM deposits and Notch3 accumulation were not yet present.
Results
Passive arterial diameter, contraction to phenylephrine and endothelium-dependent relaxation to acetylcholine were unaffected in transgenic mice. By contrast, flow-induced dilation was significantly decreased and pressure-induced myogenic tone significantly increased in arteries from transgenic mice compared with wild-type mice.
Conclusions
This is the first study providing evidence that mutant Notch3 impairs selectively the response of resistance arteries to flow and pressure. The data suggest an early role of vascular dysfunction in the pathogenic process of the disease.
Keywords: Animals, Arteries, drug effects, physiopathology, CADASIL, etiology, physiopathology, Disease Models, Animal, Male, Mechanotransduction, Cellular, Mice, Mice, Transgenic, Mutation, Phenylephrine, pharmacology, Pressure, Proto-Oncogene Proteins, genetics, Receptors, Cell Surface, genetics, Receptors, Notch, Stress, Mechanical, Vasoconstriction, Vasoconstrictor Agents, pharmacology, Vasodilation
INTRODUCTION
CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) is an inherited small vessel disease characterized by recurrent ischemic strokes, cognitive impairment and premature death1–3. On neuropathological examination, brains show a diffuse myelin loss and multiple small deep infarcts located within the white matter and basal ganglia. These changes are caused by a distinctive arteriopathy characterized by a progressive degeneration of vascular smooth muscle cells (vSMC) and the accumulation of granular osmiophilic material (GOM) within the basement membrane of these cells. Arterial changes are systemic although symptoms are restricted to the central nervous system4–5.
CADASIL is caused by highly stereotyped mutations in the Notch3 receptor, which lead to an odd number of cysteine residues within an Epidermal Growth Factor Repeat (EGFR) of the extracellular domain6–9. Recent works demonstrated that vSMC are the primary targets of the pathogenic process. In human adults, expression of Notch3 is highly restricted to vSMC. In CADASIL, there is an abnormal accumulation of Notch3 at the plasma membrane of vSMC because of an impaired clearance of the receptor10. Consistent with these data, transgenic mice that express a mutant Notch3 in vSMC develop arterial lesions similar to that seen in CADASIL patients11
Functional consequences of vessel changes in the CADASIL pathogenesis process remain to be elucidated. In CADASIL patients, two types of lesions affecting resistance arteries have been described that might affect hemodynamic. First, narrowing of the arterial lumen may reduce the baseline blood flow. Second, degeneration of vSMC may impair the vasomotor function. Analysis of transgenic mice that express a mutant Notch3 revealed a third type of lesions that precede vSMC degeneration, GOM deposits and Notch3 accumulation. These changes are characterized the disruption of anchorage and adhesion of vSMC to neighboring cells as well as cytoskeleton abnormalities of vSMC11. Flow (shear stress) is the main mechanical stimulus activating vascular endothelial cells whereas pressure is the main mechanical stimulus responsible for a sustained vasoconstrictor (myogenic) tone in resistance arteries12–13. We and others have previously shown that vascular mechanotransduction to shear stress and tensile stress relies on the integrity of cytoskeletal proteins and intercellular communications 12–17. On the basis of the aforementioned observations, we hypothesized that these early vascular changes may impair vasomotor function. In the present work, we assessed the vascular reactivity in response to vasoactive agents and mechanical forces in isolated arteries from transgenic mice expressing the Arg90Cys mutant Notch3 protein and wild-type littermates mice. We studied mice at 10 months of age when vSMC cytoskeleton and adhesion defects were present but GOM deposits, Notch3 accumulation and vSMC degeneration were absent. We focused on the tail caudal arteries because alterations have been especially well characterized in these vessels11.
METHODS
Animals
The transgenic mouse model of CADASIL arteriopathy used in this study has been described previously11. In brief, transgenic mice express a mutant human Notch3, carrying the arginine-to cysteine mis-sense mutation at amino acid position 90 in the EGFR n°2, with the expression driven by the SM22a promoter. Two independent transgenic founder lines (TgMa and TgVe) that express a low level of mutant Notch3 (< 50% of murine Notch3) in the tail arteries have been established, with TgVe expressing a higher level than TgMa. Male transgenic mice heterozygous for the transgene and their wild-type littermates were used. All mice were studied at 10–11 months when vSMC degeneration, GOM deposits and Notch3 accumulation were not yet detected.
The procedure followed in the care and euthanasia of the study animals was in accordance with the European Community Standards on the Care and Use of Laboratory Animals (Ministére de l’Agriculture and Préfecture de Paris, authorizations 00577 and 75-071).
Determination of blood pressure
Blood pressure was measured by direct intra-arterial recording. Tygon catheter was inserted into the femoral artery on anesthetized mice with inhaled halothane. Mouse was allowed to recover from anesthesia and held under minimal restraint. Rectal temperature was maintained at 37 °–37.7C. Heart rate and blood pressure were monitored using a 4-Multichannel recorder (Gould RS 3400).
Vascular Function
Mice were anesthetized with an intraperitoneal injection of pentobarbital sodium (75 to 100 mg/kg). Tail caudal arteries were removed and cannulated at both ends in a video-monitored perfusion system (LSI, Burlington, VT) as previously described15,18. Briefly, arteries were bathed in a 5 ml organ bath containing physiological salt solution of the following composition (in mM):135.0, NaCl, 15.0, NaHCO3, 4.6 KC1, 1.5, CaCl2, 1.2, MgSO4, 11.0, glucose, 5.0, N-2-hydroxy-ethylpiperazine-N-2-ethylsulfonic acid. The pH was 7.4. The pO2 was maintained at a value of 160 mmHg and the pCO2 at a value of 37 mmHg. The bath solution was changed continuously at a rate of 4 ml min−1. (pH 7.4, pO2 160 mmHg, pCO2 37 mmHg). Perfusion of the artery was achieved with a similar physiological salt solution. Pressure was controlled by a servo-perfusion system and flow was generated by a peristaltic pump. Flow-induced dilation was studied by increasing flow rate by steps from 0 to 50 μl. min−1. Data were obtained from arterial segments preconstricted with phenylephrine to approximately 50% to 60% of maximum response and with intraluminal pressure set at 75 mmHg as described14. Myogenic tone was determined by increasing intraluminal pressure by steps from 10 to 150 mmHg without intraluminal flow15.
Diameters measured in normal physiological salt solution were considered as diameter under active tone or “active diameter” At the end of each experiment arterial segments were superfused with a Ca2+-free physiological salt solution containing EGTA (2 mM) and sodium nitroprusside (10 μM) and pressure steps were repeated in order to determine the arteries’ passive diameter14.
Results are given in micrometers for artery diameters. Myogenic tone was expressed as active tone (passive diameter - active diameter, in μm) or as the percentage of passive diameter (measured diameter/passive diameter X 100). Flow-induced relaxation was expressed as increase in diameter (μ/m) as a function of shear stress due to flow in each vessel. Shear stress was calculated for each individual segment of artery as previously described13–15 {τ= 4 η · Q/π · r3, where h is viscosity (poise = dyn · s · cm−2), Q is flow (ml/s), and r is radius (cm)}.
In a separate series of arterial segments, we examined dose-dependent contraction of tail arterial segments in response to phenylephrine (10 nmol/L to 1 mmol/L) and relaxation in response to acetylcholine (10 nmol/L to 100 μmol/L) after precontraction with phenylephrine (50% of the maximal contraction) as previously described15. Contraction in response to phenylephrine was expressed as change in diameter (μm) due to phenylephrine. Relaxation in response to acetylcholine was expressed as the percentage of dilation of the active tone (preconstriction)15,16.
Drugs
N-2-hydroxy-ethylpiperazine-N-2-ethylsulfonic acid (HEPES), ethylenbis-(oxyethylenenitrolo) tetra-acetic acid (EGTA), phenylephrine, acetylcholine, were purchased from Sigma (St. Louis, MO). Other reagents were purchased from Prolabo (Paris, France). Drugs were dissolved and diluted in physiological salt solution (HEPES buffer). Concentrations are expressed as final concentration of each drug in the organ bath.
Statistical analysis
Data are expressed as mean ± SEM. Significance of the differences between groups was determined by analysis of variance (ANOVA for consecutive measurements: responses to pressure, flow, phenylephrine and acetylcholine). Means were compared by unpaired t test or Bonferroni’s test. P <0.05 was considered to be statistically significant.
RESULTS
Body weight was not affected by the expression of the mutant protein (41±2 versus 39±2 g, transgenic mice (line TgMa or TgVe) versus wild-type littermate, n= 8 to 12 per group). Mean arterial blood pressure, measured in femoral artery, was not significantly different between transgenic and wild-type mice (108.3 ±4.9, n= 6 in TgMa mice; 105.6±1.8, n=5 in TgVe mice and 111.6±2.7, n=5 in wild-type mice).
Pressure-induced arterial tone
In isolated tail caudal arteries stepwise increases in intraluminal pressure from 0 to 50 mmHg induced an increase in artery diameter, whereas further step increases in pressure induced no more increase or a decrease in diameter reflecting the development of myogenic tone. The difference between passive diameter and the diameter measured under physiological conditions was considered as myogenic tone (Figure 1A). Myogenic tone was significantly increased in arteries from transgenic mice compared with their wild-type littermates (Figure 1A). Myogenic tone, when normalized to the corresponding passive diameters, was also significantly increased in arteries from transgenic mice compared with their wild-type littermates (Figure 1B).
Figure 1. Diameter changes measured in response to pressure in isolated tail caudal arteries.
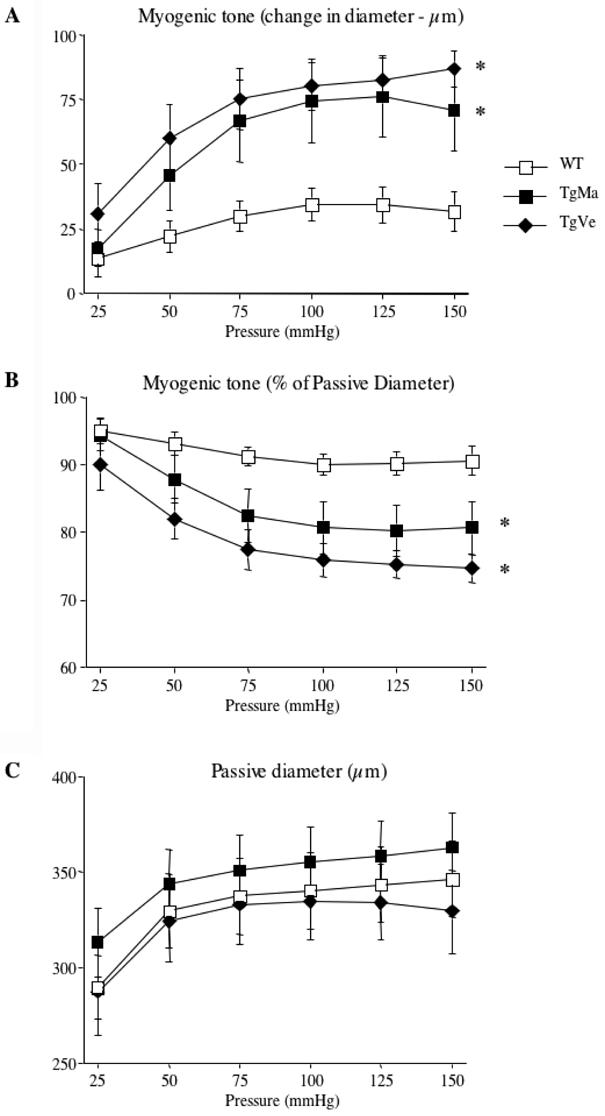
A-Changes in diameter in response to step increases in pressure (myogenic tone) in caudal arteries. Myogenic tone is expressed as active tone (μm).
B- Myogenic tone expressed as percentage of passive diameter.
C- Passive diameter measurements were determined in a Ca2+-free isotonic salt solution containing EGTA (2mmol/L) and in the presence of sodium nitroprusside (10μmol/L) in caudal arteries.
Caudal arteries were isolated from wild-type (n=12) and transgenic mice (n=9 and n=10, lines TgMa and TgVe respectively).
*P <0.001; two-factor ANOVA, transgenic versus wild-type mice.
Isolated tail caudal arteries were also submitted to stepwise increases in intraluminal pressure, in a Ca2+-free physiological salt solution containing EGTA (2 mM) and sodium nitroprusside (10 μM). Under these conditions, arteries responded by a progressive rise in diameter. Internal diameter ranged from 289.6 ± 16.4 μm (pressure, 25 mmHg) to 346.3 ± 20.1 μm (pressure, 150 mmHg) in control mice. Passive arterial diameter curves were unaffected in transgenic mice as compared with their wild-type littermates (Figure 1C).
Flow-induced responses
In arteries submitted to a pressure of 75 mmHg, increasing flow by step induced a progressive arterial dilation. Arterial dilations were quantified as increases in diameter. Flow (shear stress)-induced dilation was significantly reduced in caudal arteries from transgenic mice as compared with arteries from wild-type mice (Figure 2A).
Figure 2. Diameter changes measured in response to flow (shear stress) in isolated tail caudal arteries.
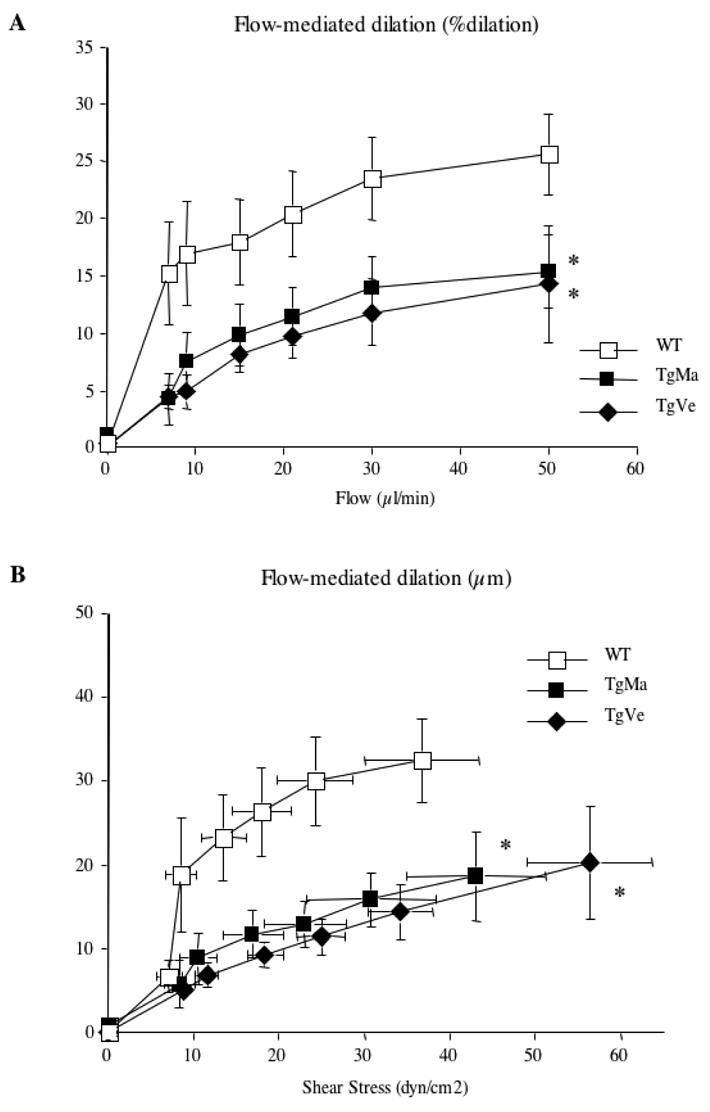
A- Changes in diameter in response to stepwise increases in intraluminal flow were determined in caudal arteries submitted to a pressure of 75mmHg and preconstricted with phenylephrine. Flow-mediated dilation is expressed as percentage dilation of active tone by flow.
B- Flow-induced dilation (in μm change in diameter) is expressed as a function of shear stress. Caudal arteries were isolated from wild-type (n=12) and transgenic mice (n=9 and n=10, lines TgMa and TgVe respectively).
*P <0.001; two-factor ANOVA, transgenic versus wild-type mice.
From the flow and diameter data obtained, wall shear stress was calculated and plotted against the changes in arterial diameter. Figure 2B shows that a given step increase in wall shear stress elicits a significantly attenuated increase in diameter of caudal arteries of transgenic mice compared with those of wild-type mice.
Responses to vasoactive agents
We examined contraction of tail caudal arteries from transgenic and wild-type mice in response to phenylephrine. All arteries contracted in a dose-dependent manner in response to phenylephrine. Vasoconstriction in response to phenylephrine was similar in transgenic and wild type mice (Figure 3A).
Figure 3.
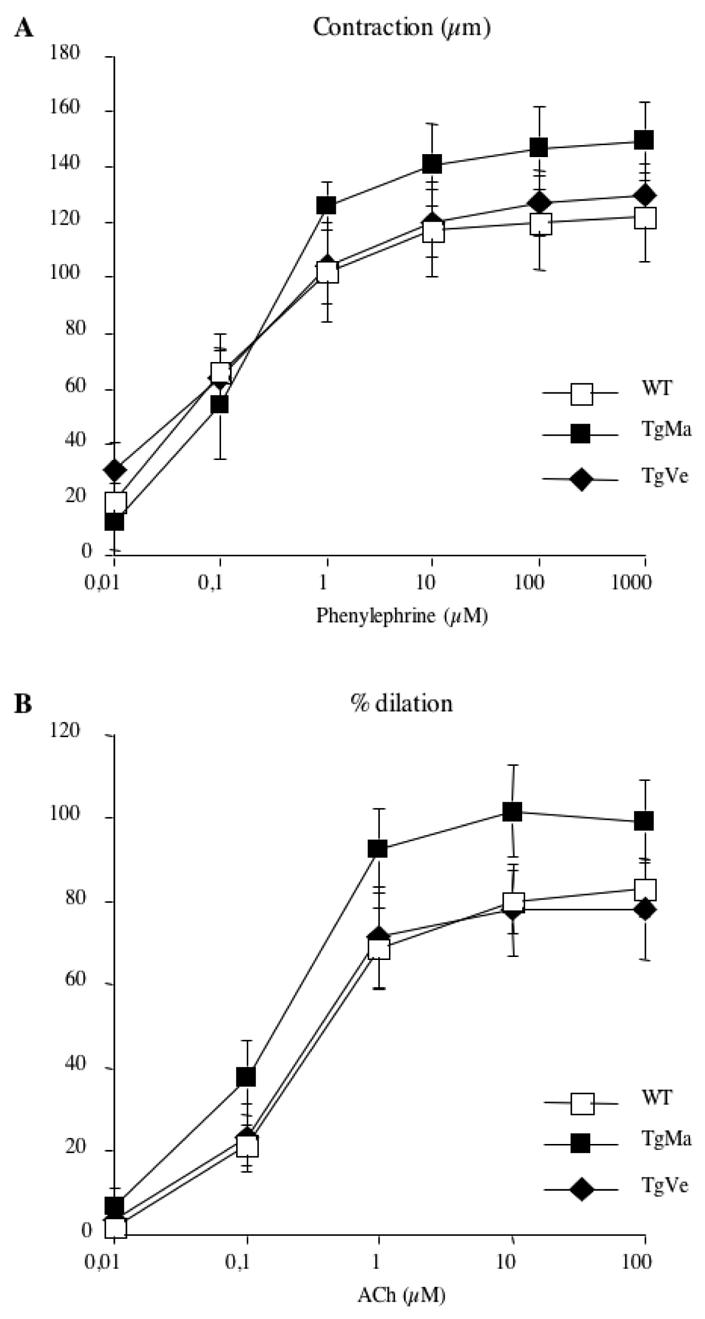
Concentration-dependent-contraction to phenylephrine (A) and concentration-dependent-dilation to acetylcholine (B) in tail caudal arteries isolated from wild-type (n=12) and transgenic mice (n=8 and n=9, lines TgMa and TgVe respectively). No significant difference; 2-factor ANOVA, transgenic versus wild-type mice.
To study vasorelaxation, precontraction was matched in arteries from transgenic and wildtype mice. Acetylcholine induces an endothelium-dependent relaxation. Tail caudal arteries relaxed in a dose-dependent fashion in response to acetylcholine. Responses to acetylcholine were not impaired in transgenic mice as compared with their wild-type littermates (Figure 3B).
DISCUSSION
In this study we showed that transgenic mice expressing a CADASIL mutant Notch3 in vSMC exhibited a significant increase in pressure-induced contraction and a significant decrease in flow-induced dilation in isolated caudal arteries. Vascular dysfunction was detected in 10 month-old transgenic mice, when mutant arteries exhibit disruption of adhesion of vSMC to neighboring endothelial cells and vSMC as well as cytoskeleton abnormalities of vSMC characterized by an increased number of dense plaques and dense bands11. In contrast phenylephrine-induced contraction and acetycholine -induced dilation were unaffected in these transgenic mice, indicating that the defective transduction of mechanical forces did not arise from a global dysfunction of vSMC. Together the data indicate that transgenic mice exhibit a specific defect in the transduction of shear and tensile stress into dilation and constriction respectively. Impaired mechanotransduction with preserved agonist or receptor-induced tone has been previously reported in mice lacking vimentin or dystrophin 14,15. Thus our findings further support the notion that mechanical forces (flow, pressure) and chemical stimuli act, at least in a great part, through different pathways 19–22.
The specific molecular basis for the mechanotransduction defect in Notch3 mutant arteries remains to be elucidated. Both myogenic tone and flow-mediated dilation depend on cytoskeleton integrity and cell to cell force transmission through the cytoskeleton, integrins and the extracellular matrix 12–17. Previous works showed that pressure induced arterial tone involved an initial calcium entry into arteriolar SMC, subsequent activation of signaling pathways (PKC, PLC, Rho-A..) that sensitized the contractile apparatus to calcium and further resulted in the stimulation of actin polymerization in vSMC17,19,20,22. Increased myogenic tone likely arises from a direct deleterious effect of mutant Notch3, expression of which was specifically targeted in vSMC. Ultrastructural analyses are consistent with an increased actin polymerization in SMC of mutant arteries11. In contrast impaired flow-induced dilation, which primarily involves endothelial cells, likely results from an indirect effect of mutant Notch3. Ultrastructural changes of endothelial cells have been detected in mutant arteries11. Additionally, one might speculate that defective myoendothelial communications or even a chronic change in smooth muscle tone could account for this specific endothelial cell dysfunction23. Additional studies are required to address this issue.
Previous studies reported in CADASIL patients an impaired cerebral vasoreactivity, as determined by attenuated vasodilation to inhaled carbon dioxide or acetazolamide and, abnormal vasoconstrictor reactivity in isolated small gluteal arteries24–26. Of note, all three studies included essentially symptomatic patients with a mean age above 45 years, an age where the pathological hallmarks of the CADASIL arteriopathy, including GOM deposits, Notch3 accumulation and vSMC degeneration, are already apparent. Thus these human studies have been difficult to interpret because it was unclear whether impaired vasoreactivity had or not a primary responsibility in the pathogenic process and whether it arose from vascular cells dysfunction or degeneration. In the present study, we demonstrate that vascular mechanotransduction is very early impaired, before the appearance of the pathological hallmarks of CADASIL. Thus, our findings suggest an early role of vascular dysfunction in the pathogenic process of the disease.
Reduced cerebral blood flow has been demonstrated in CADASIL patients using positron emission tomography, magnetic resonance imaging bolus tracking or phase contrast24,27–30. Pressure and flow are two mechanical stimuli that determine the basal vascular tone in resistance arteries and allow for a rapid adaptation to changes in blood flow and pressure. Attenuation in flow-induced dilation might lead to a lesser adaptation to increases in blood flow in organs when a metabolic need requires a higher blood supply. Similarly, increased myogenic tone might lead to a lesser adaptation to decreases in blood pressure. Because there is no organ in the body as dependent as the brain on a continuous blood supply, defective mechanotransduction to shear and tensile stress might reduce cerebral blood flow sufficiently to cause ischemic cell injury.
In conclusion, the present study provides the first evidence that vascular reactivity to the mechanical factors, flow and pressure, is early and selectively impaired in arteries expressing a CADASIL mutant Notch3. Treatments that improve vascular mechanotransduction could be beneficial in Notch3 mutation carriers at risk of developing the CADASIL disease.
Acknowledgments
This work was supported by funds from CADASIL foundation of America, Fédération pour la Recherche sur le Cerveau grant and GIS Maladies Rares grant to AJ, and by funds from Fondation pour la Recherche Médicale to DH. CD is a recipient from Fondation pour la Recherche Médicale.
References
- 1.Tournier-Lasserve E, Joutel A, Melki J, Weissenbach J, Lathrop GM, Chabriat H, Mas JL, Cabanis EA, Baudrimont M, Maciazek J, Bach MA, Bousser MG. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nature Genet. 1993;3:256–259. doi: 10.1038/ng0393-256. [DOI] [PubMed] [Google Scholar]
- 2.Chabriat H, Vahedi K, Iba-Zizen MT, Joutel A, Nibbio A, Nagy TG, Krebs MO, Julien J, Dubois B, Ducrocq X, Levasseur M, Homeyer P, Mas JL, Lyon-Caen O, Tournier-Lasserve E, Bousser MG. Clinical spectrum of CADASIL: a study of 7 families. Lancet. 1995;346:934–939. doi: 10.1016/s0140-6736(95)91557-5. [DOI] [PubMed] [Google Scholar]
- 3.Dichgans M, Mayer M, Uttner I, Bruning R, Muller-Hocker J, Rungger G, Ebke M, Klockgether T, Gasser T. The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol. 1998;44:731–9. doi: 10.1002/ana.410440506. [DOI] [PubMed] [Google Scholar]
- 4.Baudrimont M, Dubas F, Joutel A, Tournier-Lasserve E, Bousser MG. Autosomal Dominant Leukoencephalopathy and Subcortical Ischemic Stroke. A Clinicopathological study. Stroke. 1993;24:122–125. doi: 10.1161/01.str.24.1.122. [DOI] [PubMed] [Google Scholar]
- 5.Ruchoux MM, Guerouaou D, Vandenhaute B, Pruvo JP, Vermersch P, Leys D. Systemic vascular smooth muscle cell impairement in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol. 1995;89:500–512. doi: 10.1007/BF00571504. [DOI] [PubMed] [Google Scholar]
- 6.Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Notch3 mutations in CADASIL, an hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 7.Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssiere C, Cruaud C, Maciazek J, Weissenbach J, Bousser MG, Bach JF, Tournier-Lasserve E. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet. 1997;350:1511–1515. doi: 10.1016/S0140-6736(97)08083-5. [DOI] [PubMed] [Google Scholar]
- 8.Oberstein SA, Ferrari MD, Bakker E, van Gestel J, Kneppers AL, Frants RR, Breuning MH, Haan J. Diagnostic Notch3 sequence analysis in CADASIL: three new mutations in Dutch patients. Dutch CADASIL Research Group. Neurology. 1999;52:1913–5. doi: 10.1212/wnl.52.9.1913. [DOI] [PubMed] [Google Scholar]
- 9.Dichgans M, Ludwig H, Muller-Hocker J, Messerschmidt A, Gasser T. Small in-frame deletions and missense mutations in CADASIL: 3D models predict misfolding of Notch3 EGF-like repeat domains. Eur J Hum Genet. 2000;4:280–5. doi: 10.1038/sj.ejhg.5200460. [DOI] [PubMed] [Google Scholar]
- 10.Joutel A, Andreux F, Gaulis S, Domenga V, Cecillon M, Battail N, Piga N, Chapon F, Godfrain C, Tournier-Lasserve E. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest. 2000;105:597–605. doi: 10.1172/JCI8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruchoux MM, Domenga V, Brulin P, Maciazek J, Limol S, Tournier-Lasserve E, Joutel A. Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am J Pathol. 2003;162:329–42. doi: 10.1016/S0002-9440(10)63824-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis MJ, Hill MA. Signaling Mechanisms Underlying the Vascular Myogenic Response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 14.Henrion D, Terzi F, Matrougui K, Duriez M, Boulanger CM, Colucci-Guyon E, Babinet C, Briand P, Friedlander G, Poitevin P, Levy BI. Impaired flow-induced dilation in mesenteric resistance arteries from mice lacking vimentin. J Clin Invest. 1997;100:2909–14. doi: 10.1172/JCI119840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loufrani L, Matrougui K, Gorny D, Duriez M, Blanc I, Levy BI, Henrion D. Flow (shear stress)-induced endothelium-dependent dilation is altered in mice lacking the gene encoding for dystrophin. Circulation. 2001;13(103 6):864–70. doi: 10.1161/01.cir.103.6.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loufrani L, Matrougui K, Li Z, Levy BI, Lacolley P, Paulin D, Henrion D. Selective microvascular dysfunction in mice lacking the gene encoding for Desmin. FASEB J. 2002;16:117–119. doi: 10.1096/fj.01-0505fje. [DOI] [PubMed] [Google Scholar]
- 17.Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002;16:72–6. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- 18.Halpern W, Osol G, Coy GS. Mechanical behavior of pressurized in vitro prearteriolar vessels determined with a video system. Ann Biomed Eng. 1984;12:463–79. doi: 10.1007/BF02363917. [DOI] [PubMed] [Google Scholar]
- 19.Koller A. Signaling pathways of mechanotransduction in arteriolar endothelium and smooth muscle cells in hypertension. Microcirculation. 2002;9:277–94. doi: 10.1038/sj.mn.7800142. [DOI] [PubMed] [Google Scholar]
- 20.Schubert R, Mulvany MJ. The myogenic response: established facts and attractive hypotheses. Clin Sci (Lond) 1999;96:313–26. [PubMed] [Google Scholar]
- 21.Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284(1):R1–12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- 22.Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca(2+) signaling pathways underlying myogenic reactivity. J Appl Physiol. 2001;91:973–83. doi: 10.1152/jappl.2001.91.2.973. [DOI] [PubMed] [Google Scholar]
- 23.Haefliger JA, Nicod P, Meda P. Contribution of connexins to the function of the vascular wall. Cardiovasc Res. 2004;62(2):345–56. doi: 10.1016/j.cardiores.2003.11.015. Review. [DOI] [PubMed] [Google Scholar]
- 24.Chabriat H, Pappata S, Ostergaard L, Clark CA, Pachot-Clouard M, Vahedi K, Jobert A, Le Bihan D, Bousser MG. Cerebral hemodynamics in CADASIL before and after acetazolamide challenge assessed with MRI bolus tracking. Stroke. 2000;31:1904–12. doi: 10.1161/01.str.31.8.1904. [DOI] [PubMed] [Google Scholar]
- 25.Pfefferkorn T, von Stuckrad-Barre S, Herzog J, Gasser T, Hamann GF, Dichgans M. Reduced cerebrovascular CO(2) reactivity in CADASIL: A transcranial Doppler sonography study. Stroke. 2001;32:17–21. doi: 10.1161/01.str.32.1.17. [DOI] [PubMed] [Google Scholar]
- 26.Hussain MB, Singhal S, Markus HS, Singer DR. Abnormal vasoconstrictor responses to angiotensin II and noradrenaline in isolated small arteries from patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) Stroke. 2004;35:853–8. doi: 10.1161/01.STR.0000120730.54282.A0. [DOI] [PubMed] [Google Scholar]
- 27.Chabriat H, Bousser MG, Pappata S. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: a positron emission tomography study in two affected family members. Stroke. 1995;26:1729–30. [PubMed] [Google Scholar]
- 28.Tuominen S, Juvonen V, Amberla K, Jolma T, Rinne JO, Tuisku S, Kurki T, Marttila R, Poyhonen M, Savontaus ML, Viitanen M, Kalimo H. Phenotype of a homozygous CADASIL patient in comparison to 9 age-matched heterozygous patients with the same R133C Notch3 mutation. Stroke. 2001;32:1767–74. doi: 10.1161/01.str.32.8.1767. [DOI] [PubMed] [Google Scholar]
- 29.van den Boom R, Lesnik Oberstein SA, Spilt A, Behloul F, Ferrari MD, Haan J, Westendorp RG, van Buchem MA. Cerebral hemodynamics and white matter hyperintensities in CADASIL. J Cereb Blood Flow Metab. 2003;23:599–604. doi: 10.1097/01.WCB.0000062341.61367.D3. [DOI] [PubMed] [Google Scholar]
- 30.Tuominen S, Miao Q, Kurki T, Tuisku S, Poyhonen M, Kalimo H, Viitanen M, Sipila HT, Bergman J, Rinne JO. Positron emission tomography examination of cerebral blood flow and glucose metabolism in young CADASIL patients. Stroke. 2004;35:1063–7. doi: 10.1161/01.STR.0000124124.69842.2d. [DOI] [PubMed] [Google Scholar]