

Abstract
Polymeric maleimido–quaternary ammonium (QA) compounds have been shown to function as molecular tape measures when covalently tethered to external cysteine residues of a Shaker K+ channel (Blaustein R.O., P.A. Cole, C. Williams, and C. Miller. 2000. Nat. Struct. Biol. 7:309–311). For sufficiently long compounds, the cysteine–maleimide tethering reaction creates a high concentration, at the channel's pore, of a TEA-like moiety that irreversibly blocks current. This paper investigates a striking feature of the maleimide–cysteine tethering kinetics. Strong blockers—those that induce substantial levels (>80%) of irreversible inhibition of current—react with channel cysteines much more rapidly than weak blockers and, when delivered to channels with four cysteine targets, react with multiexponential kinetics. This behavior is shown to arise from the ability of a strong blocker to concentrate its maleimide end near a channel's cysteine target by exploiting the reversible pore-blocking affinity of its QA headgroup.
Keywords: Shaker, tetraethylammonium, block, affinity-label, maleimide
INTRODUCTION
In fulfilling their physiologic roles, voltage-dependent K+ channels accomplish two remarkable feats: they catalyze the rapid and selective passage of potassium ions across the plasma membrane and they regulate this process by opening and closing in response to small changes in transmembrane voltage. To meet these demands, the proteins are equipped with two distinct domains that somehow communicate with each other: a pore-forming unit and a gating module. The entire channel protein consists of four identical subunits, each containing six transmembrane segments (S1–S6). The S5 and S6 segments of each subunit coalesce at the channel's fourfold symmetry axis to form the pore unit and the S1–S4 sequences form the surrounding gating module.
Structure function studies over the past several years have focused predominantly on the pore region, and these labors, together with recent x-ray crystallographic analyses of bacterial potassium channels (Doyle et al., 1998; Jiang and MacKinnon, 2000; Morais-Cabral et al., 2001; Zhou et al., 2001a,b), have deepened our understanding of permeation and selectivity. In contrast, comparatively little is known about the structure of the gating module of these channels. Several groups have started to fill that void by probing the uncharted region using state-dependent chemical modification of cysteine-substituted residues (Yang and Horn, 1995; Larsson et al., 1996; Yusaf et al., 1996; Baker et al., 1998; Gandhi et al., 2000), gating-modifying toxins (Swartz and MacKinnon, 1997a,b; Li-Smerin and Swartz, 2000), scanning mutagenesis (Starace et al., 1997; Monks et al., 1999; Hong and Miller, 2000; Li-Smerin et al., 2000; Starace and Bezanilla, 2001), and spectroscopic rulers (Cha et al., 1999; Glauner et al., 1999).
Another approach to structural mapping was introduced recently (Blaustein et al., 2000). The method uses a series of synthetic maleimido–quaternary ammonium (QA)* compounds of varying length as molecular tape measures. These polymeric molecules possess a QA headgroup resembling the well-known pore blocker TEA, a sulfhydryl-reactive maleimide end capable of tethering the molecule to a cysteine residue, and a flexible linker in between. When a channel containing an external cysteine target is exposed to such a compound, the cysteine–maleimide-tethering reaction creates a cloud of concentrated QA that reaches as far as the extended length of the compound. If the compound is long enough, its QA end will reach the pore and act as a tethered blocker of the channel. Since tethering occurs via a covalent reaction, the cloud of QA cannot be washed away and so a tethered block manifests itself as an irreversible inhibition of current. If the experiment is repeated with shorter compound whose QA end cannot reach the pore, no irreversible block will occur. By varying both the length of the maleimido–QA and the location of the test site, these tethered blockers have been used to determine radial distances of residues in the gating module of the Shaker K+ channel (Blaustein et al., 2000). Since this general approach is potentially useful for structural analysis of other membrane proteins, I was motivated to explore in greater depth the mechanism of action of this new class of compounds.
In the original experiments, an interesting relationship emerged between tether length and extent of irreversible block: at a particular site, longer tethered compounds irreversibly inhibited current to a greater extent than shorter ones (Blaustein et al., 2000). This observation was attributed to the multiple configurations that a longer tethered QA can assume while blocking, whereas a tethered compound whose QA end just reaches the pore can block only if it adopts the highly improbable fully extended state. This entropic view forms the basis for saying that longer compounds have higher effective local QA concentrations near the pore than shorter ones.1 Tethered QA block exhibits another interesting behavior: length-dependent kinetics. Longer compounds react with channel cysteines much more rapidly than do shorter blockers and with a biphasic time course. It is these kinetic phenomena that are explored in detail in this paper. A Shaker K253C mutant was constructed with a cysteine target introduced into the channel in the S1–S2 loop (Fig. 1 A) and the effects of two particular maleimido–QA compounds were studied: Gly5TEA (39 Å), a strong blocker, and Gly3TEA (32 Å), a weak one (Fig. 1 B). The experiments demonstrate that the difference in behavior between these two blockers results from the ability of the strong blocker to act as an affinity label.
Figure 1.

(A) Shaker K253C subunit showing location of target cysteine in the S1–S2 extracellular loop. (B) Structures and extended lengths (measured from the quaternary nitrogen to the distal olefinic carbon of the maleimide) of the two maleimido–QA compounds used.
MATERIALS AND METHODS
Mutagenesis and in vitro Transcription
Shaker B was cloned into pBluescript KS (Stratagene) and contained the point mutation F425G in the channel's external vestibule. Three additional point mutations were made for the purposes of this study: T449F, to increase TEA blocking affinity (MacKinnon and Yellen, 1990; Heginbotham and MacKinnon, 1992; Kavanaugh et al., 1992); K253C, to introduce the sulfhydryl target; and I231W, to speed activation (see below). Mutations were made using PCR-based methods and were confirmed by sequencing through the cloning cassette. Mutant cRNA was transcribed in vitro from linearized plasmids using T7 RNA-polymerase (Promega). With the exception of the “ball-tagging” experiments discussed below, all experiments were performed with channels in which N-type inactivation was removed (Δ6–46) (Schwarz et al., 1988; Hoshi et al., 1990).
Maleimido–QA compounds
Gly5TEA and Gly3TEA were synthesized as described previously (Blaustein et al., 2000). Concentrations of stock solutions of known volume were determined by quantitative amino acid analysis (Yale HHMI/Keck Biopolymer Facility). These solutions were then aliquoted, lyophilized, stored dry at −20°C, and dissolved immediately before use.
Oocyte Preparation, Electrophysiology, and Data Analysis
Xenopus oocytes were surgically removed and, after manual opening of ovarian sacs, were incubated and gently agitated via rotation for 70–80 min in 2 mg/ml collagenase (Worthington) in a Ca2+-free solution containing (mM): 82.5 NaCl, 2 KCl, 1 MgCl2, 5 HEPES, pH 7.5. Defolliculated oocytes (stage V–VI) were injected with 28–51 nL of cRNA (10–100 μg/ml) and stored at 17°C in an ND96-gentamicin solution that contained DTT to prevent cysteine oxidation. The composition was (mM): 2 DTT, 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 10 mM HEPES, pH 7.5 and 0.1 mg/ml gentamicin, and the solution was changed daily. 1–5 d after injection, oocytes were transferred to a homemade low-volume (∼75 μL) chamber built for optimal solution exchange. Oocytes were bathed in a 0.3 mM CaCl2 ND96 solution without DTT or gentamicin; were impaled with glass electrodes (Garner, KG-33) having resistances of 0.3–1.0 MΩ filled with 3 M KCl, 5 mM EGTA, and 10 mM HEPES, pH 7.6; and were examined using a two-electrode voltage clamp (OC-725B, Warner Instruments). Electrical contact to the bath solution was made via 200 mM NaCl agar bridges. After initiation of voltage clamp, oocytes were observed under constant solution flow (3 ml/min) for 1–2 min before addition of Gly5TEA or Gly3TEA to ensure that the steady-state current was drift free. Compounds were added by manually switching the flow to a maleimido–QA-containing syringe, and flow was stopped after 1–2 min. Solution exchange was checked using TEA; at the above flow rate, 90% block was reached within 5 s. The voltage clamp was interfaced to a Digidata 1200 acquisition board (Axon Instruments, Inc.) and a Windows-based PC running pClamp software (v8.0). The standard pulse protocol was a test pulse (50–100 ms duration) to 60 mV from a holding potential of −90 mV, with interpulse intervals ranging from 3–20 s. Currents were filtered at 1 kHz and sampled at 10 kHz. Kinetic data were fitted to single- or double-exponential functions using Origin's (version 6, Microcal Software) implementation of the Levenberg-Marquhart algorithm. The simultaneous fitting of several datasets to Eq. 20, as shown in Fig. 7 , was done by eye using Mathcad 2000 software (Mathsoft).
Figure 7.

Fits of data using Eq. 20. (A) Gly5TEA data of Fig. 6 were fitted to Eq. 20 using the following parameters: B f = 50 μM, B t = 2 mM, K d = 1.47 mM. The intrinsic maleimide reaction rate constant k, and the bound maleimide concentration M t were varied to best simultaneously fit the three datasets, with values of k = 26 M−1s−1 and M t = 10 mM yielding the best fits (by eye). (B) Fit of Gly3TEA data of Fig. 2 C using Eq. 20. The same value for k was used. B t was adjusted to reflect the lower effective concentration of QA headgroup near the pore; a value of 0.24 mM was calculated using the 45% fraction of blocked current with a K d for reversible Gly3TEA block of 1.23 mM (Table I). A value for M t of 1.25 mM was used to maintain a ratio of M t to B t identical to that used for the Gly5TEA data.
Single Cysteine Experiments
To study channels containing a single cysteine target, a “ball-tagging” strategy was employed (MacKinnon, 1991; MacKinnon et al., 1993; Lu and Miller, 1995; Naranjo and Miller, 1996). cRNA encoding a subunit containing a cysteine mutation at position 253 and the N-type inactivation gate was coinjected with cRNA encoding K253 (“wild-type”) inactivation–removed subunits in appropriate ratios so that the inactivating component of current would be composed chiefly of channels containing only one ball-tagged subunit. In the experiments described below, the inactivating fraction (estimated by subtracting the steady-state current from the peak current) ranged from 13 to 23%. At this level, the binomial distribution predicts that at least 91% of the inactivating current will be due to channels containing a single ball-tagged subunit; contamination from channels containing two ball-tagged subunits would amount to <9%, and channels with three or four subunits would account for <1% of the current. The predominance of channels with a single ball-tag was confirmed by measurement of the time constant of inactivation, which ranged from 7–10 ms. One potential problem with ball tagging is that if activation is not sufficiently fast, the peak current can considerably underestimate the true current. Experiments were therefore performed in an I231W background, an S1 mutation previously shown to have an 11 mV left-shifted activation curve and faster activation kinetics (Hong and Miller, 2000).
RESULTS
Shaker K+ channels are reversibly blocked by the TEA-like headgroup of maleimido–QA compounds; at millimolar concentrations these compounds block ∼50% of the current through channels containing an engineered “high affinity” TEA site (Table I) . If such channels also contain a suitably placed cysteine target, the effect of a maleimido–QA is dramatically different. Shaker K253C channels are rapidly and irreversibly inhibited upon exposure to 50 μM Gly5TEA (Fig. 2, A and C , bottom trace). There are several notable features of this inhibition. (a) Gly5TEA is a “strong” blocker that irreversibly inhibits most of the current (84%)—washout does not lead to any recovery of current. (b) This level of irreversible block implies that the effective concentration of tethered QA near the channel's pore is 8 mM. (c) The rate of irreversible inhibition must reflect the rate of the covalent tethering reaction, since the block by the tethered QA headgroup occurs on the submillisecond time domain. (d) The time course of the inhibition is biphasic and well fitted by a double-exponential function. (e) The tethering rate depends on Gly5TEA concentration, with the reciprocals of both the fast and slow time constants proportional to its concentration over a fivefold range (Fig. 3) .
TABLE I.
Reversible Blocking Affinities of Untethered QAs
| Compound | K d |
|---|---|
| mM | |
| TEA | 0.20 ± 0.02 (n = 4) |
| Gly3TEA | 1.23 ± 0.15 (n = 3) |
| Gly5TEA | 1.47 ± 0.17 (n = 3) |
K d values (expressed as mean ± SEM) obtained for the reversible block of Shaker 449F channels by QA derivatives. For each experiment, the concentration of free QA was varied and the degree of block was fitted to a Langmuir function to determine the K d. For TEA block, the same K d was also observed with Shaker 449F channels that carried the K253C mutation.
Figure 2.

Effects of tethered maleimido–QAs on Shaker K253C channels. (A) Gly5TEA. Current traces in response to 75-ms pulses to 60 mV from a holding potential of −90 mV. Dotted line represents zero current. The top trace is before wash-in of 50 μM Gly5TEA; subsequent traces are at 5, 10, 20, 40, 160, and 1,400 s of exposure. (B) Gly3TEA. Currents obtained as above. Top trace is before wash-in of 50 μM Gly3TEA; subsequent traces are at 45, 90, 180, 360, and 1,450 s of exposure. (C) Kinetics of tethered block. Isochronal (at 74 ms) currents from A and B were normalized to their values before maleimido–QA exposure and plotted against time. Arrows indicate exposure periods. Circles, Gly5TEA. Time points are every 5 s. Time course was fitted (solid line) by a double-exponential function with time constants of 25 s (76%) and 268 s (24%). This experiment was performed eight times with mean fast and slow time constants (± SEM) of 28.1 ± 1.6 s and 263 ± 20 s, respectively. Triangles, Gly3TEA. Time points are every 15 s. Time course was fitted by a single-exponential function (solid line) with a time constant of 223 s (mean ± SEM was 227 ± 25 s, n = 7).
Figure 3.

Effect of Gly5TEA concentration on tethering kinetics. (A) The first 750 s of data are shown to illuminate the differences in kinetics. Solid lines represent fits as described below. Circles, 50 μM Gly5TEA. The data are those of Fig. 2 C. Triangles, 25 μM. Time points are every 8 s. The fit is to a double-exponential function with time constants of 52 s (76%) and 398 s (24%). Squares, 10 μM Gly5TEA. Time points are every 15 s. Fit is to a double-exponential function with time constants of 112 s (63%) and 741 s (37%). Inset. Full time course of 10 μM Gly5TEA exposure. (B) Reciprocals of the time constants are proportional to [Gly5TEA]. Experiments were performed using 10 μM (n = 5), 25 μM (n = 6), and 50 μM (n = 8) concentrations of Gly5TEA and fits are to double-exponential functions as above. At each concentration, reciprocals of the fast time constants (circles) were averaged and plotted against concentration; the same was done for the slow time constants (squares). Error bars represent standard errors. Lines are linear least square fits constrained to go through the origin.
The behavior of the shorter Gly3TEA compound stands in stark contrast to that of Gly5TEA in two distinct ways (Fig. 2 C, top trace). Gly3TEA is a much weaker tethered blocker than Gly5TEA, irreversibly blocking only 45% of current. The weaker irreversible inhibition must arise from a substantially lower concentration of its tethered QA headgroup at the channel's pore, since the QA headgroup of Gly3TEA reversibly blocks the pore with similar potency to that of Gly5TEA (Table I). This lower concentration (1 mM vs. 8 mM for Gly5TEA) reflects the fact that the 32 Å Gly3TEA must adopt an extended conformation in order for its QA headgroup to reach the ∼30 Å distance to the pore (Blaustein et al., 2000), in contrast to the longer, less entropically constrained Gly5TEA. Gly3TEA also exhibits markedly different kinetics, reacting much more slowly than Gly5TEA, and with a time course reasonably fit by a single exponential. Configurational entropy naturally accounts for the difference in the degree of irreversible block, but why do the strong and weak blockers react at different rates?
One possibility is that a strong blocker reacts more rapidly because it accelerates its tethering reaction through an affinity label effect initiated by the reversible block of its QA end (Fig. 4) . During these blocking periods, the maleimide end of this bound blocker would be concentrated near its cysteine target—in the same manner that its QA headgroup becomes concentrated at the pore after its maleimide covalently tethers to its cysteine target—and it would then react via a rapid intramolecular reaction. Unlike the case of a typical intramolecular reaction, however, the rate of this tethering reaction would scale with blocker concentration, since it is determined by both the higher maleimide concentration and the proportion of time that the QA end is reversibly blocking; at the concentrations of blockers used (well below the K d for reversible QA block), this occupancy is proportional to concentration. Under this hypothesis, a maleimide therefore reacts with a cysteine in one of two ways: (a) through a process analogous to an intramolecular reaction, with a rate determined by the affinity label effect (Fig. 4, bottom path), or (b) via a standard second-order process whose rate is determined more by the intrinsic cysteine–maleimide rate constant (Fig. 4, top path). Since strong blockers can concentrate their maleimides near their cysteine targets, they should react rapidly via the affinity label path, whereas weak blockers would react predominantly through the latter mechanism.
Figure 4.

Maleimido–QAs react in two ways. Cartoon depicting a cysteine-bearing channel that reacts with a bound maleimido–QA blocker via an affinity-label effect (bottom path) or with a compound free in solution (top path). Shaded circles represent volumes swept out by the compounds when bound.
This affinity label effect also accounts for the biphasic kinetics that strong blockers exhibit. In a channel bearing four equivalent cysteines, the first cysteine reacts rapidly. This reaction surrounds the pore with a high concentration of tethered QA that inhibits pore access by the QA end of a free compound. Subsequent reactions in that channel will proceed chiefly via the slower second path described above, since access to the pore is required for the affinity label effect. A short compound like Gly3TEA does not exhibit a large affinity label effect and so displays little difference in its tethering rate to a channel's four cysteines. The kinetics of its tethering reaction can therefore be approximated by a single exponential.
This kinetic mechanism leads to a number of other predictions. (a) Channels containing a single cysteine target should react with single-exponential kinetics. (b) These channels should exhibit an affinity label effect when reacted with strong blockers, but the time constant for their reaction will be fourfold larger than the fast time constant seen with channels containing four cysteines. (c) TEA should act as a competitive inhibitor and decrease the tethering rate of a strong blocker.
The first two predictions were tested by mixing inactivating cysteine-bearing subunits with noninactivating cysteineless subunits (see materials and methods), and exposing the resultant single-cysteine channels to Gly5TEA. With this approach, single-target channels are represented by the inactivating component of current, and the tethering kinetics are assayed by tracking the disappearance of this inactivating fraction (Fig. 5) . In the presence of a strong blocker like Gly5TEA, the tethering kinetics are well described by a single time constant (Fig. 5 B), an observation consistent with the above kinetic mechanism, as biphasic tethering kinetics require multiple cysteine targets. Since these channels offer only one-fourth the number of tethering sites as channels containing four cysteine targets, they will tether at one-fourth the rate of the fast rate seen with these latter four-target channels (compare Fig. 5 B with Fig. 2 C).
Figure 5.

Kinetics of single-cysteine–containing channels using a ball-tagging approach. Subunits containing both a K253C mutation and the N-type inactivation sequence were mixed with subunits containing neither, and resultant channels were exposed to 50 μM Gly5TEA. (A) Current traces in response to 100 ms pulses to +60 mV from a holding potential of –90 mV. Dotted line represents zero current. The top trace is before wash-in of 50 μM Gly5TEA; subsequent traces are at 30, 90, 210, and 630 s of exposure. (B) Inactivating fraction of current (peak minus steady-state) was normalized to that before Gly5TEA exposure and plotted against time. Time points are every 10s. Time course was fitted by a single-exponential function (solid line) with time constant of 108 s (mean ± SEM was 97 ± 3 s, n = 7).
The third prediction—that TEA will slow the tethering reaction for a strong blocker—was tested by examining the tethering kinetics at two different TEA concentrations: 0.2 mM, representing 50% occupancy by TEA, and 1.25 mM, representing 86% occupancy. Shaker 253C channels were exposed to TEA at the desired concentration, and were subsequently exposed to 50 μM Gly5TEA. To best compare the kinetics in the absence and presence of TEA, reductions in current were all normalized to a 0–1 scale and plotted on the same set of axes (Fig. 6) . Although TEA's effects are analyzed in greater detail in the discussion, their qualitative nature is evident in the figure. As TEA's occupancy at the pore increases, the occupancy of the QA end of Gly5TEA decreases. Since the latter initiates the affinity label effect, the tethering rate slows. At the higher occupancy, the affinity label effect is largely reduced and biphasic kinetics are no longer obvious.
Figure 6.

Effect of TEA on Gly5TEA tethering kinetics. 50 μM Gly5TEA was used for all experiments. Currents were scaled to a 0–1 range of inhibition. Circles, no TEA (same data as Fig. 2). Triangles, 0.2 mM TEA (occupancy of 0.5). Squares, 1.25 mM (occupancy of 0.86). Occupancies were calculated using a measured K d for TEA block of 0.2 mM. The time courses shown are representative of experiments repeated at least six times at each TEA concentration.
DISCUSSION
The notion of exploiting a compound's affinity to enhance its covalent attachment to a target molecule is not new. The term “affinity label” was coined by (Wofsy et al., 1962) almost 40 yr ago to describe the general method, developed by several groups concurrently, for covalently labeling the active sites of antibodies and enzymes, or for improving the activity of antineoplastic antimetabolites (for review see Singer, 1967). The approach was introduced to the ion channel field when Karlin showed that certain maleimide compounds acted as affinity labels of the reduced form of the Electrophorus acetylcholine receptor (Karlin and Winnik, 1968; Karlin, 1969). Compounds bearing a trimethylammonium group reacted several orders of magnitude faster than N-ethylmaleimide or maleimides containing a tertiary amine group, an effect mitigated by the presence of a competing QA such as hexamethonium (Karlin and Winnik, 1968). These bifunctional alkylating agents ranged in length from 9–12 Å, and were able to span the distance from the QA binding site in the receptor to the neighboring cysteine target. In the present study I show that this effect can be extended to the covalent modification of sites considerably farther away (∼30 Å) if sufficiently long compounds are employed.
The arguments presented in the results section to explain the kinetic data were qualitative. Although a detailed kinetic analysis is given below, the essence of the affinity label effect can be appreciated from a simplified treatment. Consider two situations: a strong blocker that, after reaction with all four cysteines on a channel, irreversibly blocks 95% of the current; and a weak blocker that blocks only 50% of the current after it has fully reacted. We assume that the QA headgroup of each compound blocks with a K d of 1 mM and that we expose the channels to these compounds at a concentration B f of 10 μM. In the case of the strong blocker, we can estimate the concentration of tethered QA near the pore to be ∼20 mM; if each of the four tethered QAs acts independently and additively, the local concentration due to each QA headgroup, B t, would be 5 mM. For the weak blocker, the total local QA concentration will be 1 mM, with each QA contributing 0.25 mM, or 20-fold less than the strong blocker. For the sake of this analysis, we also assume that during the time its QA is bound to the pore, a compound's maleimide group will be concentrated near its cysteine target to the same extent that a single QA is concentrated near the pore when it is tethered. Therefore, for the strong blocker, the effective maleimide concentration will be 5 mM multiplied by the proportion of time that its QA headgroup is bound to the pore. For both blockers, this occupancy will be 10 μM/(10 μM + 1 mM) or 1/100. The strong blocker will concentrate its maleimide to 5 mM × 0.01 = 0.05 mM, which is five times higher than its free concentration in solution. The weak blocker, however, will only achieve a concentration of 0.25 mM × 0.01, or 2.5 μM, an increase of only 25% above its free concentration! The rate of the first tethering reaction is then the unenhanced tethering rate plus the accelerated rate from the affinity label effect:
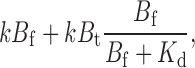 |
where k is the intrinsic second order rate constant of the maleimide–cysteine tethering reaction. Since B f << K d, this is approximately equal to
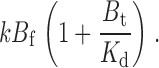 |
The magnitude of the affinity label effect will therefore be determined by B t/K d.
The analysis can be made more rigorous by considering all of the possible kinetic states in which the tetrameric channel, with its four cysteine targets, can exist (Scheme I) . The goal of such an endeavor is not to determine precisely all of the various rates, but rather to show that a kinetic model derived from the affinity label hypothesis can reasonably reproduce the data. As above, k represents the intrinsic second order rate constant of the maleimide–cysteine tethering reaction, B f is the concentration of free maleimido–QA blocker, K d is the dissociation constant for pore block of the QA end of both a free and tethered blocker, and B t is the local concentration of a single tethered blocker at the pore. Furthermore, we assume that: (a) unlike in the example above, a compound's maleimide might not necessarily achieve a concentration near its cysteine target that is identical to its tethered QA concentration, so we explicitly define that quantity as M t; (b) the second order rate constant for the maleimide–cysteine tethering reaction will be the same for both a free maleimido–QA and a maleimide whose QA is bound to the pore; (c) we can extrapolate from the measured K d for pore block of a free maleimido–QA (Table I) to that of a tethered QA; (d) k, the intrinsic maleimide–cysteine reaction rate, is the same for Gly5TEA and Gly3TEA, and; (e) each tethered QA contributes equally and additively to the total effective QA concentration at the channel's pore.
SCHEME I.

All possible states and reaction pathways for a tetrameric channel bearing four equivalent cysteine targets. The M at the end of the curvy line represents an unreacted maleimide group and an encircled plus sign represents an ammonium group. k, B t, B f, K d, M t are as defined in the text. The vertical double arrows represent rapid equilibria between blocked and unblocked states. Horizontal single-headed arrows represent irreversible tethering reactions in which channels react with free maleimido–QAs; downward-pointing single-headed arrows represent reactions with bound blockers. For a compound exhibiting a large affinity label effect, the reaction represented by the thicker downward and rightward-pointing arrow will dominate the tethering kinetics.
In this kinetic model (Scheme I), the first column contains channels that have no cysteines reacted; the second column channels with only one reacted cysteine, and so on. In the second row are unblocked channels that can be reversibly blocked either by the QA end of a free blocker to generate the channels in the first row, or by a tethered QA to yield the channels occupying the third row. The vertical double-headed arrows connecting states in the same column represent rapid equilibria between blocked and unblocked states. The horizontal single-headed arrows represent irreversible tethering reactions in which channels react with free maleimido–QAs. Since there are four cysteines available to react, these tethering reactions will occur at rates that are in a 4:3:2:1 ratio. The downward-pointing single-headed arrows represent reactions with bound blockers. Although the individual rate constants for these reactions also demonstrate a 4:3:2:1 ratio, the actual reaction rates will be quite different, since the concentration of reactant changes in each column. For compounds exhibiting large affinity label effects, the reaction represented by the thick (leftmost) downward-pointing arrow will dominate the tethering kinetics. For subsequent reactions, the proportion of channels in the first row will be very small, since channels with more than one tethered blocker will mostly be blocked.
The kinetics of reversible block by either a tethered or free QA compound are several orders of magnitude faster than the covalent maleimide–tethering reaction. Channels within a given column can therefore be thought of as interconverting via a rapid equilibrium and so can be treated as a single kinetic state. This greatly simplifies the kinetic scheme to one with five states, C 0–C 4 (Scheme II) , where C 0 represents channels with no cysteines reacted, C 1 represents channels in which one cysteine has reacted, and k 1, k 2, k 3, and k 4 represent the pseudo first-order rate constants of the irreversible tethering reactions.
SCHEME II.

The ks are readily determined from Scheme I by considering the tethering rate of each distinct state and the fraction of channels in that state. Thus, we have that
 |
(1) |
 |
(2) |
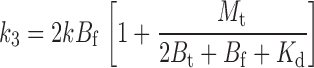 |
(3) |
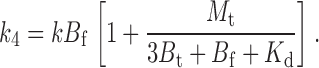 |
(4) |
Determining the time dependence of the current requires solving for each C as a function of time and multiplying that C(t) by the fraction of channels that are not blocked by either a free or a tethered QA. The relevant differential rate equations for Scheme II are:
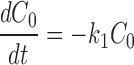 |
(5) |
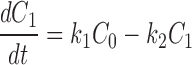 |
(6) |
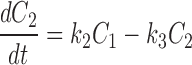 |
(7) |
 |
(8) |
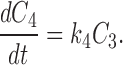 |
(9) |
The solution to these simultaneous equations is (see appendix)
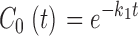 |
(10) |
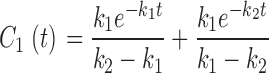 |
(11) |
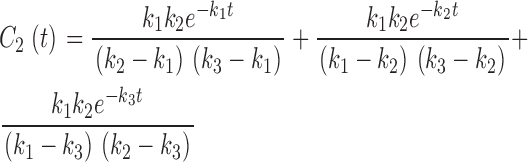 |
(12) |
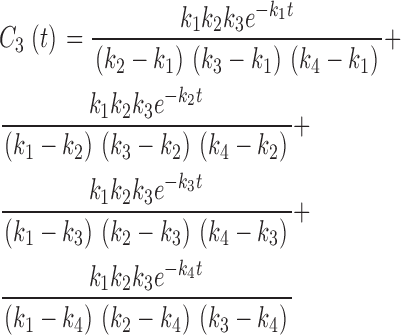 |
(13) |
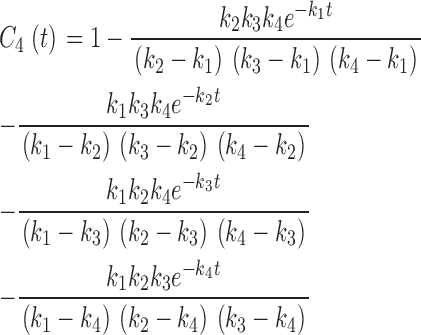 |
(14) |
f i, the fraction of unblocked channels in each of the C i states, is
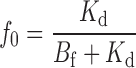 |
(15) |
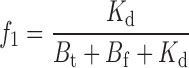 |
(16) |
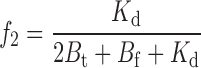 |
(17) |
 |
(18) |
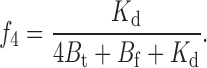 |
(19) |
The total current will then be
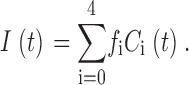 |
(20) |
The K d for Gly5TEA was experimentally determined to be 1.47 mM; this value is significantly higher than that for TEA (see Table I), a not surprising observation considering differences in steric and orientation factors. Although Gly5TEA's free concentration (B f) in these experiments ranged from 10 to 50 μM, the focus here will be on data generated using 50 μM. How is B t determined? Although it would seem logical to attempt to extract B t from the amount of block seen when channels containing a single cysteine target are exposed to a maleimido–QA (as in Fig. 5), technical limitations associated with the ball-tagging approach preclude such determination.2 Instead, we estimate B t as in the above example. The effective local concentration of QA at the pore for the Gly5TEA experiment shown in Fig. 2 C (84% block) was calculated to be 8 mM; we therefore estimate B t to be 1/4 that concentration, or 2 mM.
The two remaining free parameters are k and M t, with a further constraint on k coming from the experiments performed at different TEA concentrations. The affinity label effect should decrease with increasing concentrations of TEA; in the limit of infinite [TEA] we expect the rate of Gly5TEA tethering to be governed by the rate constants of Eqs. 1–4 using a value of 0 for M t. Modifying the above kinetic model to include the effect of TEA makes explicit the functional dependence of the rate constants, and of Eqs. 15–19 on [TEA]. This requires the addition of another set of states communicating with those of the second row of Scheme I, so that channels in this second row can now be blocked reversibly by either a free TEA molecule, a free maleimido–QA, or a tethered QA. If r = [TEA]/Kd TEA, then Eqs. 1–4 and 15–19 can be rewritten as
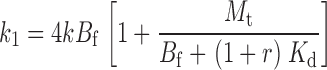 |
(21) |
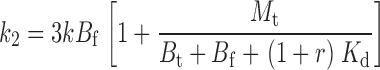 |
(22) |
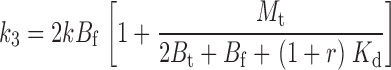 |
(23) |
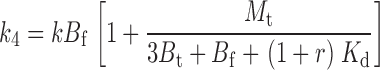 |
(24) |
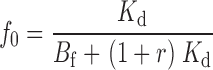 |
(25) |
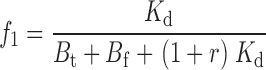 |
(26) |
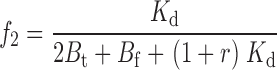 |
(27) |
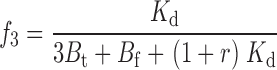 |
(28) |
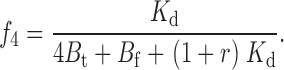 |
(29) |
As expected, when [TEA] = 0, r = 0, and Eqs. 21–24 and 25–29 reduce to 1–4 and 10–14. The three tethering reactions whose kinetics are shown in Fig. 6—one in the absence of TEA (r = 0), one with [TEA] = 0.2 mM (r = 1.0), and the third at 1.25 mM [TEA] (r = 6.2)—were simultaneously fitted to Eq. 20 using the three values of r (a measured value of 0.2 mM was used for the Kd of TEA). Since the kinetics of Gly5TEA in the presence of high [TEA], and the slow phase of the reaction in the absence of TEA are each determined more by the intrinsic tethering rate than by M t, it is not surprising that fits to these data were most sensitive to changes in k. In contrast, the fast phase of the Gly5TEA reaction in the absence of TEA, which predominantly reflects the first of the four cysteine reactions, is sensitive to both M t and k. As seen in Fig. 7 A, the data are nicely fit by Eq. 20 with k = 26 M−1 s−1 and M t = 10 mM.
The data cannot be reasonably fitted by Eq. 20 if M t is constrained to be equal to B t, and there are a number of reasons why this is not surprising. (a) Although the basic mechanism underlying the increased concentration of a tethered QA near the pore is the same as that underlying the increased concentration near a cysteine of the maleimide end of a bound maleimido–QA, there is no a priori reason why these concentrations should be identical. (b) These concentration terms encompass the respective geometries of the maleimide–cysteine and QA–pore interactions, and these are likely to be different. (c) Although a single dissociation constant (K d) was used for both a free and tethered QA, it is unlikely that the QA–pore interaction will be the same for each; actual differences in K ds are essentially incorporated into the B t term. (d) Likewise, the same second-order rate constant (k) was used for the reaction of a cysteine with a free maleimido–QA as for a maleimide transiently tethered via its bound QA headgroup; differences in rate constants for the two situations will be incorporated into the M t term. In light of all of these factors, fitting the data with values of M t and B t that differ by a factor of ∼5 is not unreasonable.
The affinity label hypothesis explains why a strong blocker tethers with biphasic kinetics. It also predicts that TEA will slow the tethering of a strong blocker by acting as a competitive inhibitor and that it will predominantly affect the faster phase of the tethering reaction. These effects are seen experimentally and they can be quantified using the kinetic model. Doing so requires calculation of the weights associated with the four predicted pseudo-first-order rate constants (k 1–k 4), and this is accomplished by rearranging Eq. 20 to generate an equation that contains the sum of four exponentials and a constant term. In the absence of TEA (Fig. 7 A, circles), the fit to Eq. 20 generates a fast time constant of 25 s—∼8-fold faster than that predicted in the absence of any affinity label effect—with a weight of 56%. The three slower constants—66, 134, and 324 s—are weighted similarly to each other at 19%, 13%, and 12%, respectively, and it is therefore not surprising that the time course described by the kinetic parameters exhibits a fast phase with a time constant of ∼25 s, and a single slow phase with a time constant whose value lies somewhere in the range of the three slow time constants. The actual Gly5TEA data are well fit by a double-exponential function with time constants of 25 s (76%) and 268 s (24%). In the presence of 0.2 mM TEA (Fig. 7 A, triangles), the time constants are 44 s (33%), 84 s (23%), 156 s (21%), and 359 s (23%). At 1.25 mM TEA (Fig. 7 A, squares), six times its K d, the biphasic nature of the tethering kinetics is blurred. The fastest time constant has slowed to 99 s, but its weight is now only 4%; although the other three time constants do exhibit some slowing, more notable is the shift in their weights in favor of the slower components—142 s (13%), 227 s (28%), and 477 s (56%).
Is the observed behavior of a weak blocker like Gly3TEA consistent with this kinetic model? Eq. 20 was used to simulate the time course of Gly3TEA tethering using a value for the intrinsic maleimide–cysteine rate constant identical to that used above. B t was adjusted to reflect the lower effective concentration of QA headgroup near the pore; a value of 0.24 mM was calculated from the 45% fraction of blocked current and the K d for reversible Gly3TEA block of 1.23 mM (Table I). A value of 1.25 mM was used for M t to preserve the ratio of M t to B t used with Gly5TEA. The curve generated by these parameters provides an excellent fit of the Gly3TEA data (Fig. 7 B). As was seen with Gly5TEA with 1.25 mM TEA present, Eq. 20 generates a sum of four exponentials whose time constants and weights—97 s (5%), 141 s (13%), 225 s (28%), and 473 s (55%)—conspire to produce a curve that appears single exponential.
How does the intrinsic second order rate constant of 26 M−1 s−1 predicted by Eq. 20 compare with that measured with free cysteine or with other cysteine-containing proteins? Reaction rates depend on both pH and the pK of the thiol, since maleimides react with the thiolate moiety (Gorin et al., 1966; Bednar, 1990); at pH 7.0 the second order rate constant for the reaction of N-ethylmaleimide with a free cysteine is on the order of 103 M−1 s−1 (Gorin et al., 1966). Reactions at this pH with various water-soluble cysteine-containing proteins, however, yield rate constants ranging from this value to 5 orders of magnitude lower (Franklin and Leslie, 1968; Felberg and Hollocher, 1972; Bednar, 1990). Steric factors can play a large role, as is evident when denaturation of a protein speeds the reaction to rates comparable to free cysteine (Franklin and Leslie, 1968; Bednar, 1990), but electrostatics, hydrophobic interactions, and hydrogen bonding also likely contribute. Single cysteines substituted at residues in a solvent-exposed loop of the aspartate receptor, a membrane-spanning sensory transducer protein, react with maleimides with rate constants of ∼15 M−1 s−1 at pH 7.0 (Falke et al., 1988), a value comparable to that obtained in Shaker with maleimido–QA's at pH 7.5.
The kinetics of the compounds studied here are complex—even when applied to a well-characterized channel—and the impetus to probe their behavior in depth stems from the belief that understanding such mechanistic underpinnings is critical, particularly if the general approach holds promise for the study of other channels or receptors. The above experiments demonstrate that two requirements must be met for a compound to tether with biphasic kinetics: it must act as an affinity label for its target molecule and this molecule must contain multiple tethering sites. Shaker's tetrameric structure was not in question here; in fact the presence of its four tethering sites formed the basis of a kinetic model that helped confirm the central hypotheses of this paper. Although these tethered blockers were originally designed as tape measures, this study suggests another use for these compounds—analysis of their tethering kinetics to a molecule of unknown architecture has the potential to provide information about its subunit stoichiometry.
Acknowledgments
I thank Chris Miller, Merritt Maduke, and Dan Cox for numerous helpful discussions and for critically reviewing the manuscript. I am also grateful to Dan Cox for sharing with me his exact solution of Eqs. 5–9 of the text, and to Larry Sirovich for helpful mathematical discussions.
This work was supported by National Institutes of Health grant HL-03462 and by a Tufts-New England Medical Center New Investigator Award.
APPENDIX
We consider the linear kinetic scheme of N forward reactions taking place among N + 1 reactants whose concentrations are governed by the set of coupled differential equations A1.
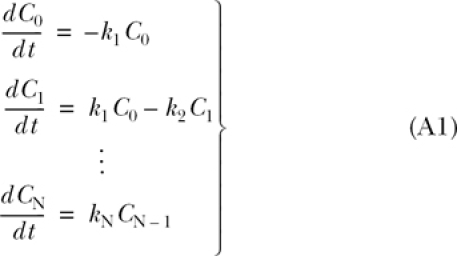 |
(A1) |
Figure .

Several methods can be used to solve this system of equations; the following solution is essentially that of Bateman (1910) who was inspired by the importance of these equations in Rutherford's analysis of radioactive decay. It represents one of the first uses of integral transforms to solve differential equations.
For n = 1, 2 … N, we define c n(x) as the Laplace transform of C n(t)
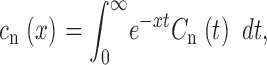 |
(A2) |
and using integration by parts, we generate the Laplace transform of 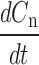
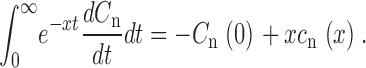 |
(A3) |
Taking the Laplace transform of both sides of Eq. A1 and applying Eqs. A2 and A3, we have that
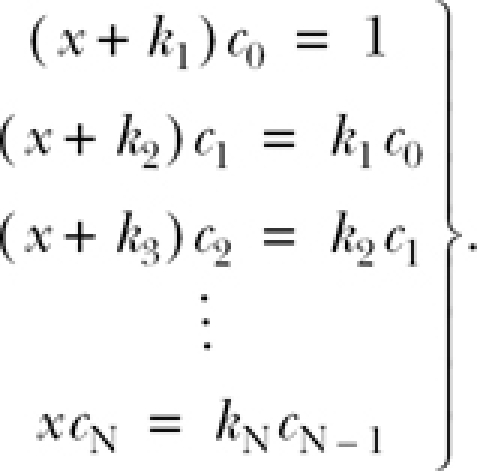 |
(A4) |
If the system is initially in state C 0, then C 0(0) = 1, and C 1(0) … C N(0) are all zero. Substituting these values into Eq. A4 and rearranging yields
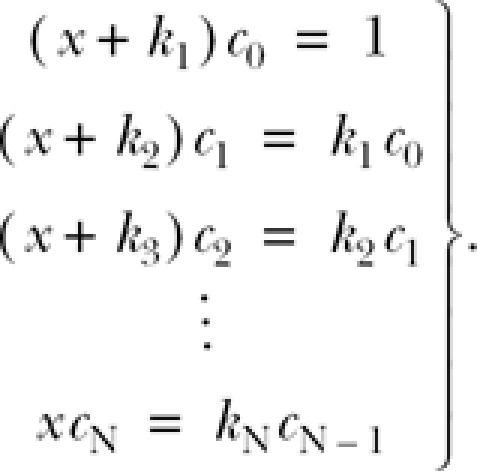 |
Sequentially solving these recursive equations for each c n, we have that
 |
(A5) |
where c n is the nth term, for n < N, and c N represents the final term (note its slightly different form). Using partial fraction decomposition (see Sirovich [1988] for a general discussion of this approach), we can express c n in a manner that will make it more amenable to inverse transformation:
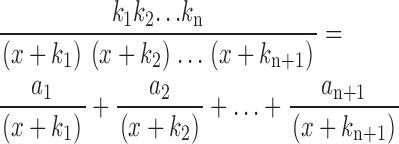 |
(A6) |
To determine a 1 we multiply both sides of Eq. A6 by (x + k 1), set x = −k 1, and solve; a 2 … a n + 1 are obtained similarly. (Note that for each c n there is a distinct set of a's.) We therefore have that
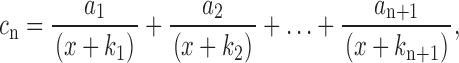 |
(A7) |
where
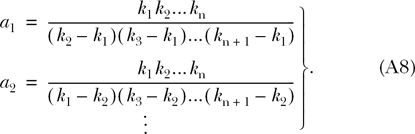 |
(A8) |
Similarly, the final term, c N, can be written as
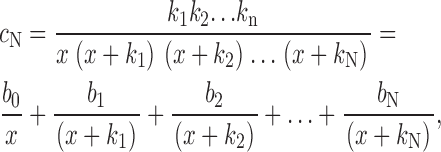 |
(A9) |
where
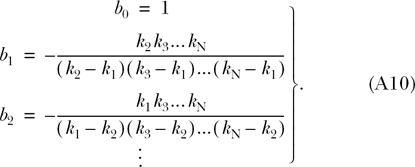 |
(A10) |
Using the fact that
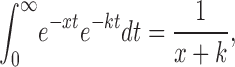 |
as well as the linearity of the Laplace transform operator, we take the inverse transform of Eq. A5, apply Eqs. A7–A10, and solve for the C n(t)'s and the final C N(t) (Eq. A11). When N = 4, these equations yield Eqs. 10–14 of the text.
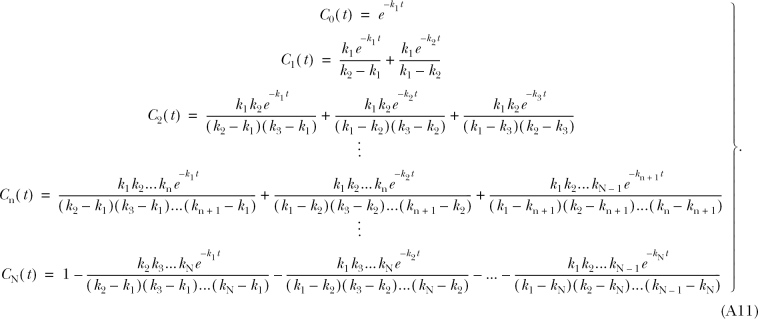 |
(A11) |
Footnotes
This increase in concentration with increasing length may not seem intuitive; however, the length dependence of tethered QA concentration at the pore involves a complex interplay between the number of configurational states a tethered compound can block in, and the volume element that the compound sweeps out. Polymer statistical theory predicts that a length will eventually be reached for which the increase in volume element overcomes the increase in the number of states. At this point the length dependence will reverse, and longer compounds will actually lead to a lower concentration of QA at the pore. I have not yet seen this occur with the range of compounds that I have tried (up to 45 Å in length).
The peak current minus the steady-state current underestimates the true inactivating component because channels begin inactivating immediately after depolarization. This error is magnified when the inactivating fraction becomes blocked. Although simulations (not depicted) reveal that this does not significantly affect kinetic measurements, it does preclude accurate calculation of the extent of the tethering reaction.
References
- Baker, O.S., H.P. Larsson, L.M. Mannuzzu, and E.Y. Isacoff. 1998. Three transmembrane conformations and sequence-dependent displacement of the S4 domain in Shaker K+ channel gating. Neuron. 20:1283–1294. [DOI] [PubMed] [Google Scholar]
- Bateman, H. 1910. The solution of a system of differential equations occurring in the theory of radio-active transformations. Proc. Camb. Phil. Soc. 15:423-427. [Google Scholar]
- Bednar, R.A. 1990. Reactivity and pH dependence of thiol conjugation to N-ethylmaleimide: detection of a conformational change in chalcone isomerase. Biochemistry. 29:3684–3690. [DOI] [PubMed] [Google Scholar]
- Blaustein, R.O., P.A. Cole, C. Williams, and C. Miller. 2000. Tethered blockers as molecular ‘tape measures’ for a voltage-gated K+ channel. Nat. Struct. Biol. 7:309–311. [DOI] [PubMed] [Google Scholar]
- Cha, A., P.C. Ruben, A.L. George, Jr., E. Fujimoto, and F. Bezanilla. 1999. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 22:73–87. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J.H. Morais-Cabral, A. Pfuetzner, J.M. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–76. [DOI] [PubMed] [Google Scholar]
- Falke, J.J., A.F. Dernburg, D.A. Sternberg, N. Zalkin, D.L. Milligan, and D.E. Koshland, Jr. 1988. Structure of a bacterial sensory receptor. A site-directed sulfhydryl study. J. Biol. Chem. 263:14850–14858. [PubMed] [Google Scholar]
- Felberg, N.T., and T.C. Hollocher. 1972. Inactivation of succinate dehydrogenase by N-ethylmaleimide. Stoichiometry and chemistry. J. Biol. Chem. 247:4539–4542. [PubMed] [Google Scholar]
- Franklin, J.G., and J. Leslie. 1968. The kinetics of the reaction of N-ethylmaleimide with denatured beta-lactoglobulin and ovalbumin. Biochim. Biophys. Acta. 160:333–339. [DOI] [PubMed] [Google Scholar]
- Gandhi, C.S., E. Loots, and E.Y. Isacoff. 2000. Reconstructing voltage sensor-pore interaction from a fluorescence scan of a voltage-gated K+ channel. Neuron. 27:585–595. [DOI] [PubMed] [Google Scholar]
- Glauner, K.S., L.M. Mannuzzu, C.S. Gandhi, and E.Y. Isacoff. 1999. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 402:813–817. [DOI] [PubMed] [Google Scholar]
- Gorin, G., P.A. Martic, and G. Doughty. 1966. Kinetics of the reaction of N-ethylmaleimide with cysteine and some congeners. Arch. Biochem. Biophys. 115:593–597. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., and R. MacKinnon. 1992. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 8:483–491. [DOI] [PubMed] [Google Scholar]
- Hong, K.H., and C. Miller. 2000. The lipid–protein interface of a Shaker K+ channel. J. Gen. Physiol. 115:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi, T., W.N. Zagotta, and R.W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., and R. MacKinnon. 2000. The barium site in a potassium channel by x-ray crystallography. J. Gen. Physiol. 115:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin, A. 1969. Chemical modification of the active site of the acetylcholine receptor. J. Gen. Physiol. 54:245s–261s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin, A., and M. Winnik. 1968. Reduction and specific alkylation of the receptor for acetylcholine. Proc. Natl. Acad. Sci. USA. 60:668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh, M.P., R.S. Hurst, J. Yakel, M.D. Varnum, J.P. Adelman, and R.A. North. 1992. Multiple subunits of a voltage-dependent potassium channel contribute to the binding site for tetraethylammonium. Neuron. 8:493–497. [DOI] [PubMed] [Google Scholar]
- Larsson, H.P., O.S. Baker, D.S. Dhillon, and E.Y. Isacoff. 1996. Transmembrane movement of the Shaker K+ channel S4. Neuron. 16:387–397. [DOI] [PubMed] [Google Scholar]
- Li-Smerin, Y., D.H. Hackos, and K.J. Swartz. 2000. Alpha-helical structural elements within the voltage-sensing domains of a K+ channel. J. Gen. Physiol. 115:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin, Y., and K.J. Swartz. 2000. Localization and molecular determinants of the Hanatoxin receptors on the voltage-sensing domains of a K+ channel. J. Gen. Physiol. 115:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Q., and C. Miller. 1995. Silver as a probe of pore-forming residues in a potassium channel. Science. 268:304–307. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R. 1991. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 500:232–235. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., R.W. Aldrich, and A.W. Lee. 1993. Functional stoichiometry of Shaker potassium channel inactivation. Science. 262:757–759. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., and G. Yellen. 1990. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 250:276–279. [DOI] [PubMed] [Google Scholar]
- Monks, S.A., D.J. Needleman, and C. Miller. 1999. Helical structure and packing orientation of the S2 segment in the Shaker K+ channel. J. Gen. Physiol. 113:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais-Cabral, J.H., Y. Zhou, and R. MacKinnon. 2001. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 414:37–42. [DOI] [PubMed] [Google Scholar]
- Naranjo, D., and C. Miller. 1996. A strongly interacting pair of residues on the contact surface of charybdotoxin and a Shaker K+ channel. Neuron. 16:123–130. [DOI] [PubMed] [Google Scholar]
- Schwarz, T.L., B.L. Tempel, D.M. Papazian, Y.-N. Jan, and L.-Y. Jan. 1988. Multiple potassium-channel components are produced by alternative splicing at the Shaker locus in Drosophila. Nature. 331:137–142. [DOI] [PubMed] [Google Scholar]
- Singer, S.J. 1967. Advances in Protein Chemistry. Vol. 22. Academic Press, New York and London.
- Sirovich, L. 1988. Introduction to Applied Mathematics. Springer-Verlag, New York.
- Starace, D.M., and F. Bezanilla. 2001. Histidine scanning mutagenesis of basic residues of the S4 segment of the Shaker K+ channel. J. Gen. Physiol. 117:469–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace, D.M., E. Stefani, and F. Bezanilla. 1997. Voltage-dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron. 19:1319–1327. [DOI] [PubMed] [Google Scholar]
- Swartz, K.J., and R. MacKinnon. 1997. a. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 18:665–673. [DOI] [PubMed] [Google Scholar]
- Swartz, K.J., and R. MacKinnon. 1997. b. Mapping the receptor site for Hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron. 18:675–682. [DOI] [PubMed] [Google Scholar]
- Wofsy, L., H. Metzger, and S.J. Singer. 1962. Affinity labeling–a general method for labeling the active sites of antibody and enzyme molecules. Biochemistry. 1:1031–1039. [DOI] [PubMed] [Google Scholar]
- Yang, N., and R. Horn. 1995. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 15:213–218. [DOI] [PubMed] [Google Scholar]
- Yusaf, S.P., D. Wray, and A. Sivaprasadarao. 1996. Measurement of the movement of the S4 segment during the activation of a voltage-gated potassium channel. Pflugers Arch. 433:91–97. [DOI] [PubMed] [Google Scholar]
- Zhou, M., J.H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. a. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., J.H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. b. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]