

Abstract
The antiarrhythmic agent flecainide appears beneficial for painful congenital myotonia and LQT-3/ΔKPQ syndrome. Both diseases manifest small but persistent late Na+ currents in skeletal or cardiac myocytes. Flecainide may therefore block late Na+ currents for its efficacy. To investigate this possibility, we characterized state-dependent block of flecainide in wild-type and inactivation-deficient rNav1.4 muscle Na+ channels (L435W/L437C/A438W) expressed with β1 subunits in Hek293t cells. The flecainide-resting block at −140 mV was weak for wild-type Na+ channels, with an estimated 50% inhibitory concentration (IC50) of 365 μM when the cell was not stimulated for 1,000 s. At 100 μM flecainide, brief monitoring pulses of +30 mV applied at frequencies as low as 1 per 60 s, however, produced an ∼70% use-dependent block of peak Na+ currents. Recovery from this use-dependent block followed an exponential function, with a time constant over 225 s at −140 mV. Inactivated wild-type Na+ channels interacted with flecainide also slowly at −50 mV, with a time constant of 7.9 s. In contrast, flecainide blocked the open state of inactivation-deficient Na+ channels potently as revealed by its rapid time-dependent block of late Na+ currents. The IC50 for flecainide open-channel block at +30 mV was 0.61 μM, right within the therapeutic plasma concentration range; on-rate and off-rate constants were 14.9 μM−1s−1 and 12.2 s−1, respectively. Upon repolarization to −140 mV, flecainide block of inactivation-deficient Na+ channels recovered, with a time constant of 11.2 s, which was ∼20-fold faster than that of wild-type counterparts. We conclude that flecainide directly blocks persistent late Na+ currents with a high affinity. The fast-inactivation gate, probably via its S6 docking site, may further stabilize the flecainide-receptor complex in wild-type Na+ channels.
Keywords: sodium channel, flecainide, persistent sodium currents, state-dependent block, fast inactivation gate
INTRODUCTION
Flecainide is one of class Ic antiarrhythmic drugs taken orally (Roden, 2001). Use of flecainide is limited to patients with supraventricular arrhythmia, as this drug is also proarrhythmic. The primary target of flecainide is the cardiac voltage–gated Na+ channel, which is responsible for the upstroke of cardiac action potentials. Interestingly, the severely affected family member with painful congenital myotonia responded dramatically to flecainide treatment (Rosenfeld et al., 1997). Flecainide has also been found to be effective for patients with Long QT-3 syndromes (e.g., LQT-3/ΔKPQ; Windle et al., 2001). These new therapeutic applications of flecainide give rise to a renewed interest in studying its action on voltage-gated Na+ channels (e.g., Nagatomo et al., 2000; Viswanathan et al., 2001).
The Na+ channel α-subunit isoforms consist of four repeated domains (D1–D4), each with six transmembrane segments (S1–S6). The receptors for flecainide and for traditional local anesthetics (LAs) were found to share common structural determinants at the D4S6 segment of Na+ channel α-subunit (Ragsdale et al., 1996). Like LAs, flecainide appears to have a higher affinity for activated Na+ channels than for resting counterparts, as originally suggested by Anno and Hondeghem (1990) and Nitta et al. (1992). Flecainide also elicits strong use-dependent block during repetitive pulses. This phenotype was examined recently by Liu et al. (2002) using Nav1.5 mutants related to LQT-3 and Brugada syndromes. These authors suggested that flecainide interacts primarily with inactivated states of Na+ channels.
Flecainide apparently blocks persistent late currents more effectively than it blocks peak currents in mutations associated with painful congenital myotonia (Wang et al., 1999) and with LQT-3/ΔKPQ syndrome (Nagatomo et al., 2000). Repetitive pulses elicit additional use-dependent block of peak and late currents by flecainide. The IC50 values of peak and late currents for the wild-type were 127 and 44 μM, respectively, and were 80 and 19 μM, respectively, for LQT-3/ΔKPQ. However, because of their small amplitudes, it was rather difficult to characterize persistent late Na+ currents in these mutants. To increase persistent late currents, Grant et al. (2000) used inactivation-deficient Nav1.5 Na+ channels with mutations at the IFM motif (IFM→QQQ) and found that the use-dependent block of flecainide remains in these mutant channels. The IFM motif is critical for normal fast inactivation and is situated at the cytoplasmic D3-D4 linker, which may function as an inactivation gate (West et al., 1992). The extrapolated flecainide KD value for the noninactivating currents was 27.6 μM. The clinical relevance of these relatively high IC50 and KD values remains unclear, as the therapeutic plasma concentration of flecainide is in a range of 0.4 to 2 μM (Viswanathan et al., 2001).
In this study we sought to address the state-dependent binding of flecainide in wild-type and rNav1.4-L435W/L437C/A438W inactivation-deficient rat-muscle Na+ channels. Residues of L435/L437/A438 are located at the COOH terminus of D1S6, which is different from the IFM locus. We chose this inactivation-deficient rNav1.4 mutant because it expressed well in the Hek293t expression system (Wang et al., 2003), unlike the inactivation-deficient hNav1.5 IFM/QQQ mutant (Grant et al., 2000). Our results demonstrate that flecainide binds rapidly and preferentially with the open state but weakly with the resting state. Once the channel is blocked by flecainide, the inactivation gate may stabilize flecainide binding in situ.
MATERIALS AND METHODS
Site-directed Mutagenesis
We used the QuikChange XL site-directed mutagenesis kit (Stratagene) to create rat skeletal muscle Nav1.4 mutant clones (Wang et al., 2003). To minimize the possibility that unique phenotypes are due to unwanted mutations, we also created independent clones of rNav1.4-L435W/L437C/A438W, rNav1.4-L437C/A438W, and homologous clones in human isoforms (hNav1.4 and hNav1.5). These independent and homologous clones displayed phenotypes similar to those of their rNav1.4 counterparts (Wang et al., 2003). Furthermore, flecainide block in these inactivation-deficient mutant isoforms appeared comparable in our preliminary studies.
Transient Transfection
Human embryonic kidney cells (HEK293t) were grown to ∼50% confluence in DMEM (GIBCO BRL) containing 10% fetal bovine serum (HyClone), 1% penicillin and streptomycin solution (Sigma-Aldrich), 3 mM taurine, and 25 mM HEPES (GIBCO BRL) and then transfected by a calcium phosphate precipitation method (Cannon and Strittmatter, 1993). Transfection of rNav1.4-pcDNA1/Amp or mutant clones (5–10 μg) along with β1 (10–20 μg) and reporter CD8-pih3m (1 μg) was adequate for current recording. Cells were replated 15 h after transfection in 35-mm dishes, maintained at 37°C in a 5% CO2 incubator, and used after 1–4 d. Transfection-positive cells were identified with immunobeads (CD8-Dynabeads).
Whole-cell Voltage Clamp
Whole-cell configuration was used to record Na+ currents (Hamill et al., 1981). Borosilicate micropipettes (Drummond Scientific Company) were pulled with a puller (P-87; Sutter Instrument Co.) and heat polished. Pipette electrodes contained 100 mM NaF, 30 mM NaCl, 10 mM EGTA, and 10 mM HEPES adjusted to pH 7.2 with CsOH. The pipette electrodes had a tip resistance of 0.5–1.0 MΩ. Access resistance was 1–2 MΩ and was further reduced by series resistance compensation. All experiments were performed at room temperature (22–24°C) under a Na+-containing bath solution with 65 mM NaCl, 85 mM choline Cl, 2 mM CaCl2, and 10 mM HEPES adjusted to pH 7.4 with tetramethyl-ammonium hydroxide. Flecainide acetate was purchased from Sigma-Aldrich and dissolved in DMSO solution at 100 mM as stock solution. Final flecainide concentrations were prepared from stock by serial dilution with bath solution. Whole-cell currents were measured by an Axopatch 200B (Axon Instruments, Inc.) or an EPC-7 (List Electronics), filtered at 3 kHz, collected, and analyzed with pClamp8 software (Axon Instruments, Inc.). Holding potential was set at −140 mV. Leak and capacitance were subtracted by the patch clamp device and further by the leak subtraction protocol (P/−4). Voltage error was <4 mV after series resistance compensation. An unpaired Student's t test was used to evaluate estimated parameters (mean ± SEM or fitted value ± SEM of the fit); P values of <0.05 were considered statistically significant.
RESULTS
Families of rNav1.4 Na+ Currents Before and After Flecainide Treatment
Families of superimposed Na+ currents at voltages ranging from −120 to +50 mV were recorded before drug application (Fig. 1 A). Subsequently, flecainide at 30 μM was applied externally and Na+ currents were monitored by test pulses (+30 mV for 5 ms) at a 30-s interval. About 50% of the peak currents were inhibited after flecainide block reached its steady-state, which was usually within 5–7 min. The Na+ current family in 30 μM flecainide was then recorded (Fig. 1 B). The current kinetics remained unchanged, and the conductance/voltage curves remained comparable with or without flecainide (Fig. 1 C).
Figure 1.

Families of superimposed Na+ current traces before and after 30 μM flecainide application. Currents were evoked by 5-ms pulses to voltages ranging from −120 to +50 mV in 10-mV increments and were recorded before (A) and after 30 μM flecainide (B). The inward current evoked by a pulse to −50 mV and the outward current evoked by a pulse to 50 mV are labeled. (C) Conductance was determined from the equation gm = INa/(Em − ENa), where INA is the peak current, Em is the test voltage, and ENa is the estimated reversal potential, and plotted against the corresponding voltage. Plots were fitted with a Boltzmann function. The average midpoint voltage (V0.5) and slope (k) of the function for control Nav1.4 wild-type (circle, n = 5) were (in mV) −30.8 ± 1.1 and 9.9 ± 0.9, respectively, and −30.4 ± 1.4 and 8.2 ± 1.3 for the flecainide treated cell (triangle, n = 4). The holding potential was −140 mV. The β1 subunit was cotransfected with rNav1.4.
Flecainide Block of Na+ Channels at Various Voltages
We applied a voltage-scanning protocol ranging from −180 to −10 mV to determine whether distinct high-affinity binding of flecainide exists in rNav1.4 Na+ channels. This pulse protocol consisted of a conditioning pulse at various voltages, with a 10-s duration intended for drug binding. The 10-s duration was selected because longer duration would elicit greater Na+-channel slow inactivation, particularly at the more positive voltages (Fig. 2 A, open circles). Pulses were applied at 30-s intervals. This protocol was originally used to test if inactivated channels have higher affinities than resting channels for local anesthetics (Wright et al., 1997, 1999). Fig. 2 A (closed circles) shows that flecainide at 30 μM blocks a constant level (∼50%) at hyperpolarizing potentials from −180 to −100 mV. The block increases continuously from −80 to −10 mV when normalized with the control current at each voltage. This voltage dependence of flecainide block in rNav1.4 at the depolarized voltages differed significantly from that of LAs, which reached a constant level around −60 mV (Wright et al., 1997, 1999). This result suggests that flecainide does not have access to the fast-inactivated Na+ channels as readily as traditional LAs. Such a result is consistent with the report by Liu et al. (2003), who found that ionized flecainide (pKa 9.3, 99% charged at pH7.4) gains access to its receptor via an intracellular pathway primarily after channels open.
Figure 2.
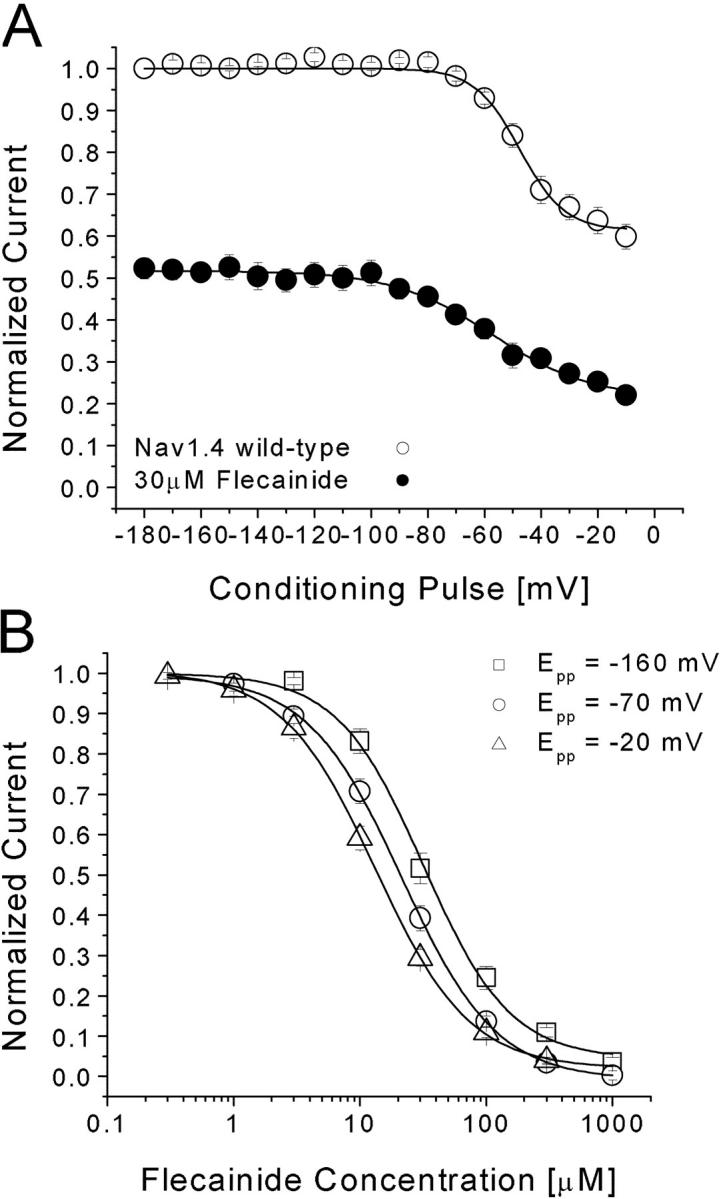
(A) Voltage dependence of flecainide block in rNav1.4 channels. Conditioning prepulses ranging from −180 mV to −10 mV were applied for 10 s. After a 100-ms interval at −140 mV, Na+ currents were evoked by a test pulse at 30 mV for 5-ms. Currents recorded before (open circle, n = 6) and after 30 μM flecainide (closed circle, n = 6) were normalized to the current obtained at the −180 mV conditioning pulse and plotted against the conditioning voltages. Flecainide data were then renormalized at each voltage with respect to the control value without flecainide. Data were fitted with a Boltzmann function (1/[1 + exp((V0.5 − V)/kE)]). The average V0.5 value and kE (slope factor) value for the fitted functions were (in mV) −47.9 ± 1.1 and 8.6 ± 0.9, respectively, for control and −57.3 ± 2.7 and 17.6 ± 2.1, respectively, for flecainide. (B) Dose–response curves for flecainide block. Pulse protocols were the same as in (A) with a conditioning pulse to −20, −70, or −160 mV. The peak amplitudes of Na+ current were measured at various flecainide concentrations, normalized to the peak amplitude of the control, and plotted against drug concentration. Solid lines represent fits to the data with the Hill equation. IC50 values (mean ± SEM) and Hill coefficients (mean ± SEM; in brackets) for Epp = −20 mV (triangle, n = 5), −70 mV (circle, n = 6), and −160 mV (square, n = 5) are 13.3 ± 0.3 μM (1.17 ± 0.03), 21.2 ± 0.4 μM (1.14 ± 0.02), and 31.9 ± 3.0 (1.27 ± 0.12), respectively. Pulses were delivered at 30-s intervals.
To determine the IC50 value directly, we then measured the dose–response curve of flecainide with a 10-s conditioning pulse at −160, −70, and −20 mV. Fig. 2 B shows the results from these measurements at −20, −70, and −160 mV with IC50 values of 13.3 ± 0.3, 21.2 ± 0.4, and 31.9 ± 3.0 μM, respectively. The difference in flecainide affinities at these voltages is less than threefold, confirming the voltage-scanning results shown in Fig. 2 A. In comparison, the difference between the resting and inactivated block is 28-fold for the LA cocaine under identical conditions (250 μM vs. 9 μM; Wright et al., 1997).
Development of and Recovery from Flecainide Block
One possible explanation for the shallow voltage dependence of flecainide block shown in Fig. 2 A is that the inactivated channels interact with the drug rather slowly. This appeared to be the case, as shown in Fig. 3 A, for the development of the inactivated block at −50 mV. We chose −50 mV because steady-state inactivation reached its completion within 100 ms (h∞ = 0; unpublished data). The time constant was measured 7.9 ± 0.7 s. Once developed either at −50 mV or at +30, the recovery from block at 100 μM flecainide was extremely slow, with a time constant of 273 ± 14 s (Fig. 3 B, closed circle) or 225 ± 12 s (Fig. 3 C, closed circle), respectively, as if flecainide was trapped within the channel. Despite our best efforts, we could not ascertain that these recovery time courses reached their plateau as these experiments would take too long to complete. Unexpectedly, the amplitude of the Na+ currents continued to increase during this recovery period. Its amplitude exceeded the current amplitude before the conditioning pulse (∼20% of control; Fig. 3 B) and reached a level that was 78.5 ± 4.9% (n = 9; open triangle) of the control amplitude without flecainide. Thus, block by flecainide under these two conditions recovered nearly to a level ∼80% of the control value if cells were not stimulated for 17 min. These results indicated that the resting block at −140 mV by flecainide is much less than the block we normally monitored at the 30-s interval. The estimated IC50 for the resting block of flecainide in wild-type channels is 365 μM at −140 mV using the Langmuir isotherm. The IC50 results in Fig. 2 B therefore include the background flecainide block, which failed to recover during the 30-s pulse interval.
Figure 3.

Development of and recovery from flecainide block in rNav1.4 wild-type channels by 100 μM flecainide. (A) For the development of inactivated block, the prepulse duration was varied at −50 mV. The peak current was measured at 30 mV after a 100-ms interval at −140 mV, normalized to the initial peak amplitude (t = 0), and then plotted against the prepulse duration. The data were fitted by a single-exponential function. The time constant (and final y0 values) for flecainide block (closed circle, n = 5) was 7.9 ± 0.7 s (20.9 ± 3.2%), respectively. Recovery from 100μM flecainide block at −50 mV for 10 s (B) and at +30 mV for 10 s (C) were measured after a variable recovery interval at −140 mV. A 5-ms test pulse at 30 mV was used to evoke available currents. The recovery time course for control (open circle) and for flecainide (closed circle) were normalized and fitted by the sum of two exponentials. Slow time constants of 273.4 ± 14.3 s and 225.1 ± 12.1 represented the recovery from flecainide block at −50 mV (B, closed circle; n = 5, 72.8%) and at 30 mV (C, closed circle; n = 5, 68.3%), respectively. The flecainide data in B and C were renormalized by the peak current amplitude without drug. After resting for 1,000 s, the peak currents reached to ∼80% of the control level (open triangle).
Background Flecainide Block of Na+ Channels when Stimulated at Low Frequencies
To confirm that background flecainide block indeed occurs during the 30-s pulse interval, we varied the interval duration from 5 to 120 s. Flecainide at 100 μM produced very different degrees of block at intervals of 5, 15, 30, 60, and 120 s (Fig. 4) ; the steady-state block at these frequencies was 87.5%, 82.1%, 81.1%, 70.1%, and 52.5%, respectively. These values are close to the estimated values based on the recovery time course (Fig. 3 C), which yielded 88%, 84%, 78%, 68%, and 54%, respectively. Thus, even with the application of monitoring pulses at very low frequencies, the background block of Na+ currents by flecainide is significant as compared with those by traditional LAs because of the extremely slow recovery of flecainide block (Fig. 3, B and C).
Figure 4.

Use-dependent block by 100 μM flecainide at low stimulus frequencies. Brief test pulses to 30 mV for 5 ms were used to evoke Na+ current at intervals of 5 s (n = 5), 15 s (n = 6), 30 s (n = 5), 60 s (n = 5), and 120 s (n = 5). Peak current of the test pulse was measured and normalized to the initial peak amplitude before flecainide application. Data was plotted against the time after drug exposure. The remaining peak currents (y0) were estimated to be 12.5% (5-s interval), 17.9% (15 s), 18.9% (30 s), 29.9% (60 s), and 47.5% (120 s).
Inactivation-deficient Na+ Current Families Before and After Flecainide Treatment
It is unclear whether flecainide directly blocks open channels, because the channel opens once during depolarization, lasts for ∼0.5 ms, and is then inactivated (Aldrich et al., 1983). To determine direct interactions between flecainide and the open state, we chose to study inactivation-deficient rNav1.4-L435W/L437C/A438W mutant channels (Wang et al., 2003). This mutant channel inactivated minimally during depolarization; as a result, a substantial fraction of peak current was maintained. Fig. 5, A and B , shows superimposed Na+ current families before and after flecainide at 30 μM. A strong time-dependent block of the outward maintained Na+ currents occurred even within the 5-ms test duration. Therefore, this result provides the direct evidence that flecainide binds readily with the open state of the Na+ channel and suggests that this mutant can be used as a tool for detailed studies on flecainide block. The conductance/voltage relationship of inactivation-deficient mutant channels shows that activation and the slope factor were not changed significantly before and after treatment with 30 μM flecainide.
Figure 5.

Inactivation-deficient rNav1.4-L435W/L437C/A438W Na+ currents before and after application of 30 μM flecainide. Superimposed current traces before (A) and after flecainide (B) were evoked by 5-ms pulses ranging from −120 to +50 mV in 10-mV increments. The inward current evoked by a pulse to −50 mV and the outward current evoked by a pulse to +50 mV are labeled. (C) Normalized conductance (gm) plotted versus the amplitude of the 5-ms voltage step. Gm was determined as described in Fig. 1 C. Plots were fitted with a Boltzmann function. The average midpoint voltage (V0.5) and slope (k) of the function in the control solution (open square, n = 5) were −20.2 ± 1.6 and 11.1 ± 1.4, respectively, and −20.2 ± 1.3 and 13.8 ± 1.2 for the flecainide-treated cell (closed square, n = 5).
Dose–response Curve of Flecainide Block in Inactivation-deficient Na+ Channels
We generated persistent late Na+ currents with a test pulse of +30 mV for 140 ms and then measured the time-dependent block of flecainide at various concentrations (Fig. 6 A). We then constructed the dose–response curve of flecainide block of inactivation-deficient Na+ channels (Fig. 6 B). The IC50 values for the open (estimated block at the end of the pulse; open circle) and the resting block at −140 mV (estimated block at the peak current; open triangle) were 0.61 ± 0.07 μM and 306.9 ± 19.3 μM, respectively. In comparison, with a conditioning pulse near activation threshold at −50 mV for 10 s, the IC50 was 4.1 ± 0.1 μM (open square) or about sevenfold less potent than that of the open-channel block. The pulse duration of 10 s was applied since the development of this threshold block had a time constant of 1.38 ± 0.32 s (n = 5). This suggests channel opening is required for the high-affinity block of flecainide. With limited channel opening around activation threshold, the flecainide affinity is not as high as that of the open-channel block. The decay phase of Na+ currents shown in Fig. 6 A after drug applications could be well fitted with a single exponential function. We calculated the inverse of the time constant (τ) and plotted against the corresponding concentration (Fig. 6 C). We estimated the on-rate and off-rate constant of flecainide with the open channel to be 14.9 μM−1s−1 (the slope) and 12.2 s−1 (y-intercept), respectively, as described by O'Leary and Chahine (2002). The calculated dissociation constant (KD) yields 0.82 μM.
Figure 6.

Block of inactivation-deficient Nav1.4 L435W/L437C/A438W channels at various flecainide concentrations. (A) Superimposed Na+ currents evoked by a 140-ms test pulse to 30 mV every 30 s were shown at various flecainide concentrations. Steady-state block at each concentration was achieved within 5 min. (B) Dose–response curves were constructed for open-channel block (relative steady-state block at end of 140 ms test pulse in A; open circle), resting block (relative block of peak current in A; open triangle), and threshold block at −50 mV (open square). All pulses were delivered at 30-s intervals. For threshold block, a conditioning pulse at −50 mV for 10 s followed by a 100-ms interpulse at −140 mV before the test pulse. The peak amplitudes of Na+ currents were measured at various flecainide concentrations, normalized to the peak amplitude of the control, and plotted against drug concentration. Solid lines represent fits to the data with the Hill equation. IC50 values (mean ± SEM) and Hill coefficients (mean ± SEM; in parentheses) are 0.61 ± 0.07 (1.06 ± 0.12) for open-channel block (circle, n = 7), 4.08 ± 0.11 (1.18 ± 0.03) for Epp = −50 mV (square, n = 5), 306.9 ± 19.3 (1.30 ± 0.10) for resting block (triangle, n = 5, renormalized). Renormalization for resting block was performed to correct a small background block (∼10%) by monitoring pulses with 30-s intervals. The decay phase of the Na+ current in A was fitted with a single exponential function, and the corresponding time constant (τ) was inverted and plotted against the corresponding concentration (C). Data were fitted with a linear regression y = 14.9x + 12.16 (solid line). On-rate (kon) corresponded to the slope of the fitted line (14.9 μM−1s−1) and the off-rate (koff) corresponded to the y-intercept (12.16 s−1). The equilibrium dissociation constant was determined by the equation KD = koff/kon and equaled 0.81 μM.
Use-dependent Block of Wild-type and Inactivation-deficient Na+ Currents by Flecainide at 5 Hz
The repetitive pulses of 30 mV at 5 Hz for 60 pulses elicit additional use-dependent block of 100 μM flecainide by ∼45% in wild-type Na+ currents (Fig. 7 A). In comparison with inactivation-deficient mutant channels, the use-dependent block by 10 μM flecainide developed rapidly after the first depolarization (Fig. 7 B, closed circles). It appeared that this rapid phase of the use-dependent block was caused directly by the time-dependent block of the noninactivating current during the pulse (Fig. 6 A, 10 μM). There was also a slow inhibition of peak currents during repetitive pulses in inactivation-deficient channels even without flecainide (Fig. 7 B, open circles); this decrease in peak current amplitude is likely due to the enhanced slow inactivation in this mutant (Wang et al., 2003).
Figure 7.
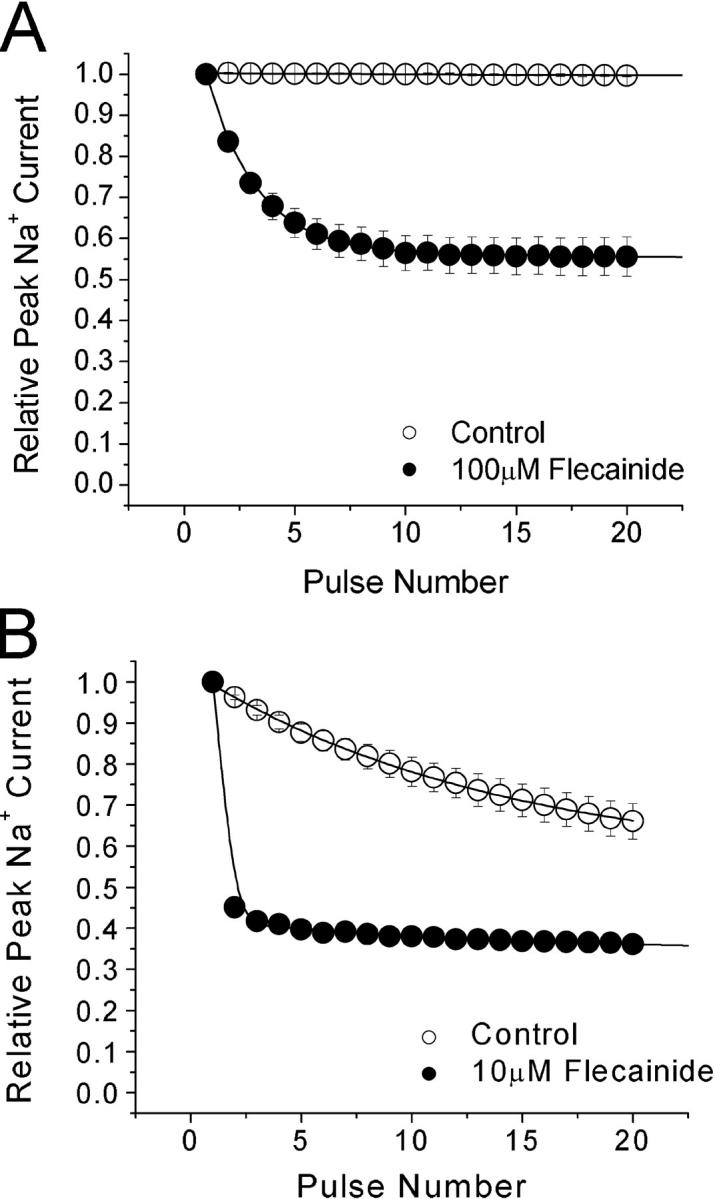
Use-dependent block by flecainide in wild-type and inactivation-deficient mutant channels at a high frequency. Repetitive pulses to +30 mV for 24 ms were delivered at 5 Hz. (A) The wild-type peak amplitude of each dataset was normalized to the first pulse of the set and plotted against the pulse number. In control solution, the pulse protocol did not elicit a use-dependent reduction in current amplitude. Data were best fit by a single exponential function for 100 μM flecainide (closed circle, n = 5) with a time constant of 2.5 ± 0.1 pulses with 54.8 ± 0.1% remaining current. (B) Use-dependent block of Nav1.4 L435W/L437C/A438W by 10 μM flecainide was measured at 5Hz. The time constant for control solution (open circle, n = 6) was 20.4 ± 0.4 pulses. This current reduction was probably caused by the enhanced slow inactivation in these mutant channels. The time constant for 10 μM flecainide (closed circle, n = 5) was too fast to be measured accurately. Steady-state was reached at 33.1 ± 0.1% remaining current.
Recovery from the Open-channel Block of Flecainide in Inactivation-deficient Mutant Channels
We measured the recovery time course of the open-channel block by flecainide once developed in the inactivation-deficient mutant channels. At the activation threshold of −50 mV for 100 ms, few channels were blocked by 10 μM flecainide (Fig. 8 A). In contrast, >90% of inactivation-deficient Na+ channels were blocked by 10 μM flecainide at 30 mV for 100-ms (Fig. 8 B, closed circle). After the open-channel block developed, the current recovered with a time constant of 11.2 s ± 0.7 (n = 5; solid line). In comparison, this recovery time constant of the inactivation-deficient mutant channels is 20-fold faster than that of the wild-type channels (Fig. 3 C). With this recovery time course, flecainide will produce ∼10% background block when the cell is stimulated at 1 per 30 s. In contrast, when the cell is stimulated at 5 Hz, flecainide will produce additional rapid use-dependent block of peak currents, since channels remain blocked during repetitive pulses with a 200-ms interval (5 Hz) (Fig. 7).
Figure 8.

Recovery from the flecainide open-channel block at −140 mV. (A) A conditioning pulse of −50 mV for 100 ms was applied and the recovery of flecainide block was measured with various intervals at −140 mV (inset). Notice that less than 15% of currents recovered slowly in the presence of 10 μM flecainide (closed circle) with a time constant of 13.1 ± 2.7 s (n = 5). (B) With a conditioning pulse of 30 mV for 100 ms, >80% of currents recovered slowly in the presence of 10 μM flecainide (closed circle), with a time constant of 11.2 ± 0.9 s (n = 5; solid line). For comparison purpose with the wild-type, we primarily used this single exponential function in our report. However, this recovery time course could be better fitted with a two-exponential function (dotted line) with τ1 = 3.1 ± 0.7 s (31.0 ± 6.9%) and τ2 = 19.3 ± 3.0 s (55.9 ± 6.6%). The reason for this phenomenon is unclear but may be due to multiple opening modes in inactivation-deficient Na+ channels.
DISCUSSION
We examined the state-dependent block of flecainide in rNav1.4 wild-type and inactivation-deficient L435W/L437C/A438W mutant Na+ channels. There are three novel findings. First, pulse protocols that activate Na+ channels at frequencies even as low as one per 120 s significantly perturb the degree of flecainide block and induce a significant “background” block of Na+ currents. This background block, which developed after Na+ channel activation, has a very slow recovery time course, with a time constant over 225 s at −140 mV. Second, the resting- and open-channel affinities differ by 500-fold in the inactivation-deficient mutant channels (0.61 μM vs. 307 μM, respectively). This IC50 value for flecainide open-channel block is within the therapeutic plasma concentration range of 0.4 to 2.0 μM (Roden, 2001). Hill coefficients of these measurements were near unity, indicating that there is one flecainide receptor per mutant channel. Third, the recovery from the open-channel block by flecainide in inactivation-deficient mutant channels is relatively fast at −140 mV, with a time constant of 11.2 s, or ∼20-fold faster than that in wild-type Na+ channels with intact fast inactivation. The significance of these new findings is discussed as follows.
Resting Block by Flecainide Is Rather Weak
The interaction of flecainide with the resting Na+ channels appears to be weak. Flecainide at 100 μM blocks only 22.5% (n = 9) of peak Na+ currents when the cell is held at −140 mV and is not stimulated in 1,000 s with drug present (Fig. 3, B and C). The calculated IC50 for flecainide block is 365 μM using the Langmuir isotherm. It was time consuming to measure the resting block of flecainide since only one test pulse per ∼17 min could be applied. The true IC50 value for flecainide-resting block could be even larger if the block produced by the monitoring pulse did not recover fully during the time of measurement (at 1,000 s). In contrast, the IC50 for flecainide block of inactivation-deficient mutant Na+ channels can be estimated by the dose–response curve readily from the peak current amplitude (Fig. 6), which yields 307 ± 19 μM for flecainide resting block (n = 5). Thus, the resting flecainide affinity in inactivation-deficient mutant channels is similar to that in wild-type.
Our estimated IC50 values for flecainide-resting block of rNav1.4 wild-type and inactivation-deficient Na+ channels deviate significantly from previous reports. For example, Nagatomo et al. (2000) reported IC50 values of 127 and 80 μM for hH1 wild-type and ΔKPQ mutant channels for flecainide-resting block. Our dose–response curve (Fig. 2 B) shows that the apparent IC50 value at −160 mV is ∼31.9 ± 2.8 μM when measured with a 30-s interval between monitoring pulses. This value is off by 10-fold from that of the flecainide resting affinity (365 μM). The difference arises because <20% of flecainide block produced by monitoring pulses recovered during the 30-s interval (Fig. 3 C). This unusually slow recovery from the use-dependent block of flecainide with a time constant over 225 s (Fig. 3, B and C) differs greatly from that of a long-acting LA, S(−)-bupivacaine, which recovers with a time constant of 4.4 s in cardiac Na+ channels (Valenzuela et al., 1995).
Flecainide Preferentially Blocks the Open State of the Na+ Channel
Repetitive pulses at 5 Hz elicit additional flecainide block of Na+ currents (Fig. 7). This use-dependent phenotype at high frequencies is well documented for flecainide block (Nitta, et al., 1992). Since Na+ channels open only once for ∼0.5 ms during channel activation (Aldrich et al., 1983), it is unclear whether flecainide blocks the open state of the wild-type channel directly. In fact, Liu et al. (2002) recently suggested that use-dependent block requires channel opening but is not likely due to open-channel block. Rather, flecainide appears to interact with inactivated states that follow depolarization-induced channel opening.
Our results from the inactivation-deficient Na+ channels, however, illustrate that use-dependent block is due primarily to open-channel block. Three pieces of evidence support this notion. First, flecainide blocks the late open Na+ channel rapidly and potently (Fig. 5). The IC50 of flecainide block of the open state is 0.61 μM at 30 mV, and its on-rate and off-rate constants are 14.9 μM−1s−1 and 12.2 s−1, respectively. This on-rate value is comparable to those of bupivacaine (Valenzuela et al., 1995; 7 μM−1s−1) and cocaine (O'Leary and Chahine, 2002; 6.8 μM−1s−1). With these on-rate values, both LAs and flecainide will block a limited fraction of peak Na+ currents, as demonstrated by mathematical modeling (Valenzuela et al., 1995), but will in time block most of persistent late Na+ currents even at the low concentration of 3 μM (Fig. 6 A). Second, the time course of flecainide block of the wild-type–inactivated Na+ channel is relatively slow (Fig. 3 A), with a time constant of 7.9 s at −50 mV. This slow rate of binding between flecainide and the inactivated state likely limits the contribution of the inactivated block during repetitive pulses of a short duration (24 ms; Fig. 8 A). Third, there is no apparent high-affinity flecainide block at −50 mV for the inactivated state of Na+ channels (Fig. 2) as normally found for traditional LAs (Wright et al., 1997, 1999). On the basis of these data, we propose that flecainide is primarily an open-channel blocker with minimal interactions with the resting state. However, it remains unclear whether upon flecainide binding the conformation of the open channel can subsequently assume the “inactivated conformation” where the inactivation gate may close but may not bind strongly to its receptor in inactivation-deficient mutant channels.
Does Fast Inactivation Stabilize Flecainide Binding?
As noted above, the recovery from flecainide block after channel activation is very slow at −140 mV for the wild-type, with a time constant >225 s. In contrast, inactivation-deficient mutant channels display a much faster recovery time course of flecainide block, with a time constant of 11.2 s at the holding potential of −140 mV. What causes this difference in the flecainide dissociation rate? It is unlikely that slow inactivation plays a prominent role in this difference, since the inactivation-deficient mutant channels display an enhanced slow inactivation (Wang et al., 2003) and yet flecainide dissociates faster from these channels than it does from the wild-type upon repolarization. One simple interpretation for the slow recovery phenotype in wild-type is that the inactivation gate may stabilize flecainide binding. McPhee et al. (1994)(1995) and Yarov-Yarovoy et al. (2002) previously suggested that the inactivation gate enters the Na+ permeation pathway and interacts with its docking site at S6 COOH termini. Their model also explains the phenotype of our inactivation-deficient Na+ channels with COOH-terminal mutations at D1S6 (L435W/L437C/A438W) (Wang et al., 2003). Such a docking site could be situated adjacent to the flecainide receptor at D4S6 (Ragsdale et al., 1996). If true, the flecainide-receptor complex may be stabilized in situ by the inactivation gate directly. This interpretation seems also consistent with the modulated receptor hypothesis for the complicated action of LAs and antiarrhythmic agents on Na+ channels (Hille, 1977; Hondeghem and Katzung, 1977). It is noteworthy that Vedantham and Cannon (1999) reported no effect of lidocaine on the apparent rate of recovery of lidocaine-blocked Na+ channel from fast inactivation. This phenomenon may be due to differences among various therapeutic Na+ channel blockers in their access and mode of block as suggested by Grant et al. (2000).
Alternatively, the flecainide receptor may be altered significantly by mutations in the inactivation-deficient mutant channel (rNav1.4-L435W/L437C/A438W) and its affinity for flecainide may be reduced drastically so that flecainide dissociated rapidly from its receptor. This possibility is less likely, since the IC50 for the open state in the inactivation-deficient mutant channel is 0.61 μM at 30 mV, suggesting that flecainide binds to its receptor with a high affinity. This potency is far greater than that previously reported for flecainide in wild-type channels. Nitta et al. (1992) reported a KD of 7 μM for the use-dependent block of flecainide at 2.5 Hz. This value still differs ∼10-fold from that for inactivation-deficient mutant channels reported here. The higher apparent KD value of flecainide in wild-type Na+ channels is likely due to their short open time (∼0.5 ms), which limits flecainide access to its receptor. It is unlikely that these high KD values are inherent for the cardiac Na+ channel isoform, since our preliminary results indicate that flecainide also blocks homologous inactivation-deficient hNav1.5 cardiac Na+ channels with similar potency (IC50 = 0.66 μM).
Physiological Significance of Flecainide as an Open-channel Blocker
Flecainide appears to be beneficial for patients with defective Na+ channels in skeletal muscle (Rosenfeld et al., 1997) and in heart (Windle et al., 2001). Many defective mutant channels exhibit persistent late Na+ currents, such as in the cases of LQT-ΔKPQ (Bennett et al., 1995), hyperkalemic periodic paralysis (Cannon and Strittmatter, 1993), or painful congenital myotonia (Wang et al., 1999). In general, the probability of the defective Na+ channel entering the noninactivating mode of gating is <5%. It is noteworthy that persistent late Na+ currents with multiple gating modes are present in the wild-type channels (Patlak and Ortiz, 1985, 1986), although the probability (∼0.1%) is far less than that in defective mutant channels.
Could flecainide block late persistent Na+ currents for its efficacy described above? This is possible since flecainide indeed blocks effectively and rapidly the open state of the inactivation-deficient mutant channels, as shown in this report. The therapeutic plasma concentration of flecainide is in the range of 0.4–2 μM (0.2–1 μg/ml) (Roden, 2001; Viswanathan et al., 2001). A substantial fraction of persistent late currents should be blocked by flecainide at this concentration range, based on an IC50 value of 0.61 μM. Persistent Na+ currents will be particularly susceptible for block because flecainide gains access to its receptor readily during channel opening (Fig. 7 A) and during repetitive pulses (Fig. 8). Hence, it is feasible that flecainide primarily targets persistent late Na+ currents for its therapeutic action but spares most of initial peak currents, which inactivate rapidly. Caution should be taken in extrapolating the IC50 values to the clinical setting, since several factors also could affect the in vivo potency of flecainide (Nagatomo et al., 2000). These include the body temperature, additional β subunits present, posttranslational modifications of Na+ channels, multiple gating modes of late Na+ currents, and flecainide block of other ion channels. Finally, flecainide as an antiarrhythmic agent could become proarrhythmic at a higher pacing frequency and at a higher concentration, as the block of the initial peak currents will accumulate continuously due to its slow recovery. Novel open-channel blockers with a faster recovery time may lessen their proarrhythmic propensity and find broad applications for various pathological conditions that manifest an increase of persistent late Na+ currents in skeletal muscle (Cannon, 1996), in heart (Saint et al., 1992), and in brain (Crill, 1996).
Acknowledgments
This work was supported by National Institutes of Health (GM48090 and HL66076).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: HEK, human embryonic kidney; LA, local anesthetic.
References
- Aldrich, R.W., D.P. Corey, and C.F. Stevens. 1983. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 306:436–441. [DOI] [PubMed] [Google Scholar]
- Anno, T., and L.M. Hondeghem. 1990. Interactions of flecainide with guinea pig cardiac sodium channels. Circ. Res. 66:789–803. [DOI] [PubMed] [Google Scholar]
- Bennett, P.B., K. Yazawa, N. Makita, and A.L. George. 1995. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 376:683–685. [DOI] [PubMed] [Google Scholar]
- Cannon, S.C. 1996. Sodium channel defects in myotonia and periodic paralysis. Annu. Rev. Neurosci. 19:141–164. [DOI] [PubMed] [Google Scholar]
- Cannon, S.C., and S.M. Strittmatter. 1993. Functional expression of sodium channel mutations identified in families with periodic paralysis. Neuron. 10:317–326. [DOI] [PubMed] [Google Scholar]
- Crill, W.E. 1996. Persistent sodium current in mammalian central neurons. Annu. Rev. Physiol. 58:349–362. [DOI] [PubMed] [Google Scholar]
- Grant, A.O., R. Chandra, C. Keller, M. Carboni, and C.F. Starmer. 2000. Block of wild-type and inactivation-deficient cardiac sodium channels IFM/QQQ stably expressed in mammalian cells. Biophys. J. 79:3019–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill, O.P., E. Marty, M.E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1977. Local anesthetics: hydrophilic and hydrophobic pathways for the drug receptor reaction. J. Gen. Physiol. 69:497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem, L.M., and B.G. Katzung. 1977. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim. Biophys. Acta. 472:373–398. [DOI] [PubMed] [Google Scholar]
- Liu, H., J. Atkins, and R.S. Kass. 2003. Common molecular determinants of flecainide and lidocaine block of heart na(+) channels: evidence from experiments with neutral and quaternary flecainide analogues. J. Gen. Physiol. 121:199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H., M. Tateyama, C.E. Clancy, H. Abriel, and R.S. Kass. 2002. Channel opening are necessary but not sufficient for use-dependent block of cardiac Na+ channels by flecainide: evidence from the analysis of disease-linked mutations. J. Gen. Physiol. 120:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee, J.C., D.S. Ragsdale, T. Scheuer, and W.A. Catterall. 1995. A critical role for transmembrane segment IVS6 of the sodium channel alpha subunit in fast inactivation. J. Biol. Chem. 270:12025–12034. [DOI] [PubMed] [Google Scholar]
- McPhee, J.C., D.S. Ragsdale, T. Scheuer, and W.A. Catterall. 1994. A mutation in segment IVS6 disrupts fast inactivation of sodium channels. Proc. Natl. Acad. Sci. USA. 91:12346–12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatomo, T., C.T. January, and J.C. Makielski. 2000. Preferential block of late sodium currents in the LQT3 ΔKPQ mutant by the class I(c) antiarrhythmic flecainide. Mol. Pharmacol. 57:101–107. [PubMed] [Google Scholar]
- Nitta, J., A. Sunami, F. Marumo, and M. Hiraoka. 1992. States and sites of actions of flecainide on guinea-pig cardiac sodium channels. Eur. J. Pharmacol. 214:191–197. [DOI] [PubMed] [Google Scholar]
- O'Leary, M.E., and M. Chahine. 2002. Cocaine binds to a common site on open and inactivated human heart (Nav1.5) sodium channels. J. Physiol. 541:701–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlak, J., and M. Ortiz. 1985. Slow currents through single sodium channels of adult rat heart. J. Gen. Physiol. 86:89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlak, J., and M. Ortiz. 1986. Two modes of gating during late Na+ channel currents in frog sartorius muscle. J. Gen. Physiol. 87:305–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale, D.S., J.C. McPhee, T. Scheuer, and W.A. Catterall. 1996. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. USA. 93:9270–9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden, D.M. 2001. Antiarrhythmic drugs. In Goodman and Gilman's The Pharmacological Basis of Therapeutics. J.G. Hardman, L.E. Limbird, P.B. Molinoff, R.W. Ruddon, and A.G. Gilman, editors. Macmillan Publishing Company, New York. 933–970.
- Rosenfeld, J., K. Sloan-Brown, and A.L. George. 1997. A novel muscle sodium channel mutation causes painful congenital myotonia. Ann. Neurol. 42:811–814. [DOI] [PubMed] [Google Scholar]
- Saint, D.A., Y.-K. Ju, and P.W. Gage. 1992. A persistent sodium current in rat ventricular myocytes. J. Physiol. 453:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela, C., D.J. Snyders, P.B. Bennett, J. Tamargo, and L.M. Hondeghem. 1995. Stereoselective block of cardiac sodium channels by bupivacaine in guinea pig ventricular myocytes. Circulation. 92:3014–3024. [DOI] [PubMed] [Google Scholar]
- Vedantham, V., and S.C. Cannon. 1999. The position of the fast-inactivation gate during lidocaine block of voltage-gated Na+ channels. J. Gen. Physiol. 113:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan, P.C., C.R. Bezzina, A.L. George, D.M. Roden, A.A. Wilde, and J.R. Balser. 2001. Gating-dependent mechanisms for flecainide action in SCN5A-linked arrhythmia syndromes. Circulation. 104:1200–1205. [DOI] [PubMed] [Google Scholar]
- Wang, D.W., D. Van De Carr, P. Ruben, A.L. George, and P.B. Bennett. 1999. Functional consequences of a domain 1/S6 segment sodium channel mutation associated with painful congenital myotonia. FEBS Lett. 448:231–234. [DOI] [PubMed] [Google Scholar]
- Wang, S.-Y., K. Bonner, C. Russell, and G.K. Wang. 2003. Tryptophan scanning of D1S6 and D4S6 C-termini in voltage-gated sodium channels. Biophys. J. 85:911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, J.W., D.E. Patton, T. Scheuer, Y. Wang, A.L. Goldin, and W.A. Catterall. 1992. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc. Natl. Acad. Sci. USA. 89:10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windle, J.R., R.C. Geletka, A.J. Moss, W. Zareba, and D.L. Atkins. 2001. Normalization of ventricular repolarization with flecainide in long QT syndrome patients with SCN5A:deltaKPQ mutation. Ann. Noninvasive Electrocardiol. 6:153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S.N., S.-Y. Wang, R.G. Kallen, and G.K. Wang. 1997. Differences in steady-state inactivation between Na channel isoforms affect local anesthetic binding affinity. Biophys. J. 73:779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S.N., S.-Y. Wang, Y.-F. Xiao, and G.K. Wang. 1999. State-dependent cocaine block of sodium channel isoforms, chimeras, and channels coexpressed with β-subunits. Biophys. J. 76:233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy, V., J.C. McPhee, D. Idsvoog, C. Pate, T. Scheuer, and W.A. Catterall. 2002. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na1 channel a-subunit in voltage-dependent gating and drug block. J. Biol. Chem. 277:35393–35401. [DOI] [PubMed] [Google Scholar]