

Abstract
CFTR, the protein defective in cystic fibrosis, functions as a Cl− channel regulated by cAMP-dependent protein kinase (PKA). CFTR is also an ATPase, comprising two nucleotide-binding domains (NBDs) thought to bind and hydrolyze ATP. In hydrolyzable nucleoside triphosphates, PKA-phosphorylated CFTR channels open into bursts, lasting on the order of a second, from closed (interburst) intervals of a second or more. To investigate nucleotide interactions underlying channel gating, we examined photolabeling by [α32P]8-N3ATP or [γ32P]8-N3ATP of intact CFTR channels expressed in HEK293T cells or Xenopus oocytes. We also exploited split CFTR channels to distinguish photolabeling at NBD1 from that at NBD2. To examine simple binding of nucleotide in the absence of hydrolysis and gating reactions, we photolabeled after incubation at 0°C with no washing. Nucleotide interactions under gating conditions were probed by photolabeling after incubation at 30°C, with extensive washing, also at 30°C. Phosphorylation of CFTR by PKA only slightly influenced photolabeling after either protocol. Strikingly, at 30°C nucleotide remained tightly bound at NBD1 for many minutes, in the form of nonhydrolyzed nucleoside triphosphate. As nucleotide-dependent gating of CFTR channels occurred on the time scale of seconds under comparable conditions, this suggests that the nucleotide interactions, including hydrolysis, that time CFTR channel opening and closing occur predominantly at NBD2. Vanadate also appeared to act at NBD2, presumably interrupting its hydrolytic cycle, and markedly delayed termination of channel open bursts. Vanadate somewhat increased the magnitude, but did not alter the rate, of the slow loss of nucleotide tightly bound at NBD1. Kinetic analysis of channel gating in Mg8-N3ATP or MgATP reveals that the rate-limiting step for CFTR channel opening at saturating [nucleotide] follows nucleotide binding to both NBDs. We propose that ATP remains tightly bound or occluded at CFTR's NBD1 for long periods, that binding of ATP at NBD2 leads to channel opening wherupon its hydrolysis prompts channel closing, and that phosphorylation acts like an automobile clutch that engages the NBD events to drive gating of the transmembrane ion pore.
Keywords: ABC transporters, photolabeling, 8-azido nucleotides, ATP binding and hydrolysis, orthovanadate
INTRODUCTION
The CFTR Cl− ion channel is encoded by the gene found mutated in cystic fibrosis patients (Riordan et al., 1989). CFTR (ABCC7) belongs to subfamily C in the large family of ABC (ATP-binding cassette) proteins. Other subfamily C members include the multidrug resistance–related protein, MRP1, believed to transport cytotoxic substances out of cells, and the sulfonylurea receptors, such as SUR1, that regulate ATP-sensitive inwardly-rectifying K+ channels with which they closely associate. Full-length CFTR, MRP1, and SUR1 proteins each incorporate two nucleotide binding domains (NBDs) anticipated to bind and hydrolyze ATP to drive the conformational changes that underlie their regulatory or transport function. Hydrolysis of ATP has been demonstrated directly for CFTR (Li et al., 1996; Ramjeesingh et al., 1999) and MRP1 (Chang et al., 1998), and inferred for SUR1 (e.g., Matsuo et al., 1999), but in none of these cases is the link between nucleotide binding and hydrolysis and ABC protein function well understood.
In principle, insights into functional mechanisms of these ABC proteins can be gained by examining interactions of their NBDs with the photoactivatable nucleotides [α32P]8-N3ATP and [γ32P]8-N3ATP in parallel with the functional consequences of those interactions. This approach was exploited in studies (Urbatsch et al., 1995a,b) of the ABC-B subfamily member, multidrug resistance protein, MDR (also called P-glycoprotein, P-gp), that analyzed inhibition of ATP hydrolysis by the inorganic phosphate analogue orthovanadate (Vi). In other ATPases, Vi was shown to bind tightly in the catalytic site in place of released phosphate to form an extremely stable complex with the other hydrolysis product, ADP, and thereby prevent further cycles of hydrolysis (e.g., Lindquist et al., 1973). In P-gp, cross-linking of Vi-trapped [α32P]8-N3ADP by UV light revealed that ATPase activity was fully inhibited when only a single NBD in each P-gp was photolabeled, but that labeling was found more-or-less equally in NH2- and COOH-terminal NBDs (Urbatsch et al., 1995a,b; but cf. Hrycyna et al., 1999). This suggested that the two NBDs randomly hydrolyzed ATP, but displayed obligatory catalytic cooperativity (Senior et al., 1995; Urbatsch et al., 1995b) such that inhibition of ATP hydrolysis at one catalytic site prevented even a single hydrolysis event at the other (Urbatsch et al., 1998). The implied functional similarity between the two NBDs corresponds to their substantial sequence identity, and is corroborated by the finding that mutant P-gp with a second copy of NBD1 in place of NBD2 could still extrude drug from cells (Hrycyna et al., 1999).
In contrast, in the ABC-C family members, MRP1, SUR1, and CFTR, the two NBDs diverge not only structurally (e.g., only 27% identity between NBD1 and NBD2 sequences in CFTR), but also functionally. Thus, analysis of photolabeling measurements with [α32P]8-N3ATP with and without Vi suggested that ATP hydrolysis in MRP1 (Chang et al., 1998) occurred predominantly at NBD2 (Gao et al., 2000; Hou et al., 2000; Nagata et al., 2000). Also, in SUR1, although both NBDs were photolabeled after incubation with [α32P] 8-N3ATP, only NBD1 (and not NBD2) was photolabeled by [γ32P]8-N3ATP (Ueda et al., 1997; Matsuo et al., 1999), suggesting that hydrolysis likely occurs at NBD2 but not NBD1. Further mechanistic interpretations have been hampered, however, because transport assays for MRP1 are largely limited to steady-state population measurements (e.g., Gao et al., 2000; Hou et al., 2000), and functional assays for SUR1 are complicated by the likely octameric nature of the entire SUR1–K+ channel complex (Shyng and Nichols, 1997).
CFTR, on the other hand, comprises a Cl− ion channel whose activity is regulated by events at its NBDs (e.g., Gadsby and Nairn, 1999; Sheppard and Welsh, 1999), so that electrophysiological measurements afford high-resolution assays of function, even at the single-channel level. In photolabeling studies, nucleotide at NBD1 in CFTR was found to survive washes with ice-cold buffer before photocrosslinking (Szabó et al., 1999; Aleksandrov et al., 2001), and labeling with [γ32P]8-N3ATP suggested that the surviving nucleotide could include nonhydrolyzed ATP (Aleksandrov et al., 2002). Because NBD2 in CFTR could be labeled by [α32P]8-N3ATP, but not by [γ32P]8-N3ATP (Aleksandrov et al., 2002), it appeared that, as suggested for the other ABC-C proteins, NBD2 of CFTR hydrolyzes ATP. However, the relationships, if any, between these nucleotide interactions and gating of CFTR channels remain unexplained.
In this work, we attempted to correlate measurements of nucleotide labeling of wild-type and mutant CFTR under nonhydrolytic (0°C) and hydrolytic (30°C) conditions, with or without Mg2+ or Vi, with measurements of CFTR channel activity. Because CFTR channel gating requires phosphorylation by PKA (e.g., Gadsby and Nairn, 1999; Sheppard and Welsh, 1999), we also examined the influence of phosphorylation on CFTR photolabeling. We found that the nucleotide remained at NBD1 in the form of nonhydrolyzed nucleoside triphosphate for several minutes at a temperature at which CFTR channels open and close every second or two. This suggests that the nucleotide association and dissociation events, together with any ATP hydrolysis, that underlie CFTR channel gating occur predominantly at NBD2. We found that Vi delayed the termination of open bursts, presumably by disrupting the hydrolytic cycle at NBD2, and somewhat increased the magnitude, without altering the rate, of the slow nucleotide loss from NBD1. Phosphorylation of CFTR by PKA had little effect on either binding or retention of nucleotide at NBD1. Moreover, by comparing channel gating kinetics during activation by Mg8-N3ATP or MgATP, we conclude that the slow step that at saturating [nucleotide] limits the rate of CFTR channel opening to a burst follows nucleotide binding to both NBDs.
MATERIALS AND METHODS
Molecular Biology
pIRES-CFTR and pIRES-Flag-CFTR were constructed by subcloning CFTR or Flag-CFTR from pGEMHE-CFTR or pGEMHE-Flag-CFTR (Chan et al., 2000) into the EcoRV site of the pIRES vector (Clontech Laboratories, Inc.). We have previously described pGEMHE-CFTR, pGEMHE-Flag-3–835 (Chan et al., 2000), and pGEMHE-K464A (Vergani et al., 2003). pGEMHE-Flag-837–1480 (with Flag sequence inserted in the extracellular loop between transmembrane helices 7 and 8; cf. Howard et al., 1995) was a gift from Dr. R. Frizzell (University of Pittsburgh School of Medicine, Pittsburgh, PA).
CFTR Expression and Preparation of Membranes
HEK293T cells were maintained in DMEM medium (GIBCO) supplemented with 10% Fetal bovine serum (GIBCO BRL). Cells plated in 100-mm dishes were transfected with 2 μg of pIRES-Flag-CFTR using Effectene transfection reagent (QIAGEN), and were harvested 2 d later for membrane preparation. To obtain membranes, cells scraped from two dishes were resuspended in 1 ml of ice-cold lysis buffer containing 10 mM HEPES (pH 7.5), 5 mM EDTA, 50 mM NaCl, 1 mg/ml BSA, and protease inhibitor cocktail (Calbiochem; final concentrations, 1 mM AEBSF HCl, 300 nM aprotinin, 2 μM E-64, 2 μM leupeptin hemisulfate), and disrupted by sonication. Xenopus laevis stage V-VI oocytes were isolated as described (Chan et al., 2000), injected with 25 ng of each cRNA (in 50 nl), and incubated at 18°C for 3 d. For membrane preparation, ∼200 oocytes were resuspended in 1 ml of lysis buffer and homogenized at 4°C. Membrane suspensions from HEK293T cells or oocytes were centrifuged twice at 3,000 g for 10 min at 4°C to remove debris, and then 2 ml of lysis buffer was added to the supernatant and the membranes pelleted at 173,600 g for 1 h at 4°C. The pelleted membranes were washed with 3 ml of modified lysis buffer (with 10% glycerol and no BSA), centrifuged again, and then resuspended in 0.2 ml modified lysis buffer and stored at −80°C. Protein concentration was measured with bicinchoninic acid (Pierce Chemical Co.).
Immunoprecipitation and Protein Detection
Immunoprecipitation with anti-Flag M2-agarose beads (affinity gel; Sigma-Aldrich) was essentially as described (Picciotto et al., 1992). Membranes were dissolved in 300 μl solubilization buffer (25 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM EDTA, 2.5% nonidet P-40, 0.5% SDS). After centrifugation at 15,000 g for 10 min, 40 μl of antibody beads was added to the supernatant, and the mixture rocked overnight at 4°C. The samples were spun at 15,000 g for 10 min and washed with 500 μl of a high-salt buffer (10 mM Tris-HCl, pH 7.5, 500 mM NaCl, 0.5% nonidet P-40, 0.05% SDS), then isotonic buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% nonidet P-40, 0.05% SDS), and finally NaCl-free buffer (10 mM Tris-HCl, pH 7.5, 0.05% SDS). The immunoprecipitated proteins were released from the beads by resuspension in 50 μl of 2.5× Laemmli buffer (2.5% SDS) and centrifugation for 10 min at 15,000 g. The proteins in the supernatant were separated (150–200 μg per lane for immunoblots, 300–400 μg for 32P detection) on 6% or 8% SDS-PAGE gels, which were dried and exposed for 32P detection, or transferred onto nitrocellulose membranes using a semidry transfer cell (Bio-Rad Laboratories, Inc.) for blotting. Protein bands containing the R domain were detected with polyclonal anti-R domain antibody (Picciotto et al., 1992) and bands containing the C terminus with polyclonal anti-COOH-terminal antibody α-1468 (Marino et al., 1991; a gift from Dr. J.A. Cohn, Duke University Medical Center, Durham, NC), with horseradish peroxidase–conjugated goat anti–rabbit as secondary antibody. Protein bands were visualized with an ECL kit (Amersham Biosciences). Radiolabeled protein was visualized and quantified with a Storm phosphorimager and Imagequant 5.0 software.
In vitro Phosphorylation
Membranes were resuspended in 100 μl phosphorylation buffer, containing 50 mM HEPES, 1 mM EGTA, 10 mM MgCl2, and 1 μM microcystin, with 50 μM MgATP. PKA catalytic subunit from bovine heart (Kaczmarek et al., 1980) was added to a final concentration of ∼500 nM (20 μg/ml), and the reaction was incubated for 20 min at 30°C (unless otherwise indicated) and stopped by addition of 900 μl ice-cold wash buffer (40 mM Tris-HCl, 0.1 mM EGTA, 1 μM microcystin). After centrifugation at 15,000 g for 20 min at 4°C, the membranes were washed a second time with 1 ml of wash buffer.
[α32P]8-N3ATP and [γ32P]8-N3ATP Photolabeling of CFTR at 0°C
Stored HEK293T cell, or oocyte, membranes were centrifuged at 15,000 g at 4°C for 20 min, or 1 h, respectively, and the membrane pellet was resuspended in 100 μl ice-cold reaction buffer containing 50 mM HEPES, 1 mM EGTA, 10 mM MgCl2, 1 μM microcystin, 5 μM PKA inhibitor (PKI). In Mg2+-free buffer, 1 mM EDTA replaced the 10 mM MgCl2. For binding competition experiments, membrane aliquots (50 μl, ∼400 μg protein) were incubated on ice with various concentrations of each competing nucleotide together with 5 μM [α32P]8-N3ATP (triethylammonium salt, specific activity = 20 mCi/μmol; ICN Biomedicals, Inc.). The nucleotides were added from stock solutions: 200 mM MgATP (pH 7.4 with NMG), 93 mM TrisATP (pH 7.3 with NMG), 200 mM MgADP (pH 7.0 with NMG). The samples were incubated on ice for 5 min to allow equilibration of binding, and were then cross-linked by UV irradiation (254 nm; Spectroline) for 10 min on ice without any intervening washing step. After washing with 900 μl ice-cold wash buffer to remove noncross-linked nucleotide, the membranes were centrifuged at 15,000 g for 20 min at 4°C, solubilized by resuspension in 150 μl 1% SDS, and CFTR then immunoprecipitated (after addition of 150 μl of 2× solubilization buffer, with ingredients as listed above in section on immunoprecipitation, but without SDS). The same protocol assayed binding of [γ32P]8-N3ATP (triethylammonium salt, specific activity = 19.8 mCi/μmol; ICN Biomedicals, Inc.) at 0°C.
[α32P]8-N3ATP and [γ32P]8-N3ATP Photolabeling of CFTR at 30°C
Na orthovanadate (Vi) was prepared as a 200 mM aqueous solution (pH >10) and kept frozen at −80°C; this stock solution was boiled for 15 min and cooled to room temperature before each use. To assay photolabeling with [α32P]8-N3ATP under conditions that support hydrolysis, membranes (300–400 μg of protein) were resuspended in 100 μl reaction buffer with or without 1 mM Vi (and with or without Mg2+), as specified, and incubated for 15 min at 30°C. After this incubation, before cross-linking, the membranes were washed once with 900 μl ice-cold wash buffer (composition specified above, under in vitro phosphorylation), pelleted at 15,000 g for 20 min at 4°C, and washed a second time with 1,000 μl ice-cold wash buffer; then, before repeating the centrifugation, the membranes were subjected to a 0–15-min (usually 5 min) postincubation period at 30°C (compare Ueda et al., 1999) still in the nucleotide-free wash buffer. After centrifugation, the membrane pellet was resuspended in 47.5 μl cold buffer and the samples were irradiated with UV for 10 min on ice. Finally, 2.5 μl 20% SDS was added to solubilize the membranes and, after 10 min at room temperature, a further 100 μl 1% SDS was added and CFTR then immunoprecipitated. Photolabeling with [γ32P]8-N3ATP followed the same protocol, but controls without UV exposure were included to assess covalent incorporation of γ32P into CFTR via phosphorylation by endogenous membrane-associated kinases (compare Hou et al., 2000).
Electrophysiology
HEK293T cells were transfected with pIRES-CFTR directly on poly-L-lysine-coated glass coverslips. Oocytes were injected with 0.1–10 ng of cRNA transcribed from pGEMHE-CFTR (WT or K464A). Starting 12 h after transfection or cRNA injection, HEK293T cells or oocytes were transferred to a Petri dish containing standard (designed to minimize cation channel currents) bath solution (in mM): 138 NMG, 2 MgCl2, 5 HEPES, 0.5 EGTA, 134 sulfamic acid (pH 7.1 with sulfamic acid). 2–7-MΩ fire-polished pipettes contained pipette solution with (in mM): 138 NMG, 2 MgCl2, 5 HEPES, 136 HCl (pH 7.4 with HCl). After seal formation (>100 GΩ), an inside-out patch was excised and moved to a chamber where its cytosolic face was continuously superfused (0.2–0.6 ml/min) with bath solution supplemented, as indicated, with 300 nM PKA and/or nucleotides, at room temperature (21–26°C). Na28-N3ATP (ICN Biomedicals, Inc.) was kept as a 100 mM stock solution (pH 7.0 with NMG) in bath solution plus equimolar Mg-sulfamate. Solutions were switched by computer-driven valves (General Valve): exchange time (<1 s) was checked by the decay of endogenous Ca2+-activated Cl− currents after brief exposure to 2 mM Ca2+ (sulfamate). Outward CFTR channel currents were measured at +40 or +50 mV using an Axopatch 200B amplifier (Axon Instruments, Inc.), filtered at 50 Hz (8-pole Bessel, Frequency Devices), digitized at 1 kHz via a Digidata 1200 interface (Axon Instruments, Inc.), and saved to a PC hard disk with Clampex 7 acquisition software (Axon Instruments, Inc.).
Data Analysis
Data analysis was as described (Chan et al., 2000; Csanády et al., 2000; Vergani et al., 2003). CFTR channels open from long closures into open bursts (average duration, τb) interrupted by brief flickery closures (Haws et al., 1992), and dwell-time analyses of single-channel records reveal, within the bandwidth of our measurements, one population of open states (average duration ≈ mean interval between flickery closures) but two distinct populations of closed states. The durations of the brief, intraburst flickery closures and of individual openings within bursts are not influenced by [MgATP] (e.g., Vergani et al., 2003), but the duration (τib) of the long-lived interburst closures declines as [MgATP] is increased (Gunderson and Kopito, 1994; Winter et al., 1994). Here we compare the influence of [MgATP] with that of [Mg8-N3ATP] on CFTR channel gating kinetics in the presence or absence of Vi, using three kinds of measurements. We estimated absolute gating rates in patches with a known small number (≤8) of active channels, we determined rates relative to a standard condition when the number of active channels was uncertain, and we measured rates of macroscopic current relaxation in patches with large numbers of active channels.
For kinetic analysis, digitized segments of records in which individual channel opening and closing events could be discerned were baseline subtracted (to remove slow drifts and small, <0.5 pA, changes in the magnitude of the seal current accompanying solution exchange), and idealized using half-amplitude threshold crossing. The resulting events lists were used to generate dwell-time distributions at all conductance levels (0, 1, 2, 3… channels open) that were then simultaneously fitted with a maximum likelihood algorithm (Csanády, 2000) to determine rate constants (rAB: average number of transitions from state A to state B occurring per unit of dwell time in state A, measured in s−1). Likelihood of the three-state “C-O-CF” scheme.
was optimized with respect to the rate constants indicated: “C” represents the long interburst closed state, “O” the open state, and “CF” the short flickery closed state. The rates of channel opening to a burst (often simply called channel opening rate) and closing from a burst (often called channel closing rate) are given directly by the simple rate constants rCO and rOC (where 1/τib = rCO; 1/τb = rOC/{1 + (rOF/rFO)}). An artificial dead time of 5–6.5 ms (larger than filter dead time of ∼3.6 ms) was imposed to implement a correction for events missed due to the limited bandwidth (Csanády, 2000).
During exposure to saturating 5 mM MgATP in the presence of PKA, channel open probability, Po, was usually sufficiently high (∼0.3) that in patches with few channels the observed maximum number of simultaneously open channels could be shown by statistical tests (Csanády et al., 2000) to reasonably approximate the true number of active channels. In patches with ≤8 simultaneously open channels under those conditions, we could then also estimate absolute opening and closing rates in 5 mM MgATP after withdrawal of PKA: rCO = 0.27 ± 0.03 s−1, n = 18, rOC = 3.28 ± 0.21 s−1, n = 18 (Vergani et al., 2003). As 2 mM MgATP and 2 mM Mg8-N3ATP are both saturating concentrations (see Fig. 1, below), in each of 17 patches with few channels we directly compared opening and closing rates of the same channels (hence with the same phosphorylation status) under these two conditions, bracketing the measurements where possible: the ratios of these rates then yielded, via the absolute rates determined at saturating [MgATP], absolute gating rates at saturating [Mg8-N3ATP]. To collect enough gating events for analysis at lower [nucleotide], when Po is lower, we used patches with more channels. Due to practical limits imposed by computer-processing times (Csanády, 2000), the maximum likelihood fitting programs do not allow the number, N, of active channels to exceed 8. So we analyzed records only from patches with maximum conductance levels of ≤8 open channels under all test and reference conditions used, and we assumed N = 8 for the maximum likelihood fit. In those cases, we extracted not absolute values for rCO and hence τib, but values relative to those in the same channels in the same patch during bracketing exposures to a reference condition (e.g., 2 mM [Mg8-N3ATP]; see Fig. 1, below). These normalized values (which should be relatively insensitive to N) were then scaled by the absolute rates at saturating nucleotide (described above) to yield the data in Fig. 1, B and C (below).
Figure 1.

The 8-azido group slows CFTR Cl− channel opening and closing. (A) Outward currents (at +40 mV) activated by nucleotides indicated beneath traces in patches excised from oocytes expressing WT CFTR, after prior exposure to PKA plus 5 mM MgATP. (B and C) Mean rates of channel opening to a burst (rCO, B) and of closing from a burst (rOC, C) vs. [MgATP] (filled circles) or [Mg8-N3ATP] (red triangles), from analysis of records as in A (lower trace). For each test concentration (e.g., 15 μM Mg8-N3ATP, in A), kinetic parameters were normalized to the average of the values obtained from the same channels during the bracketing exposures to saturating nucleotide (e.g., 2 mM Mg8-N3ATP, in A). These ratios were then scaled by our estimates of absolute opening and closing rates at saturating [nucleotide] (see materials and methods): for MgATP, rCO(5 mM MgATP) = 0.27 ± 0.03 s−1 and rOC(5 mM MgATP)= 3.3 ± 0.2 s−1 (n = 18; Vergani et al., 2003). For Mg8-N3ATP we multiplied those rates by the factors (0.42 for rCO, and 0.19 for rOC) determined by direct comparison (as in A, top trace), assuming that 2 and 5 mM MgATP are both saturating concentrations. Curves in B show Michelis-Menten fits, with parameters (mean ± SEM; 2 ≤ n ≤ 7) K0.5 = 55 ± 5 μM and 11 ± 3 μM and Vmax = 1.02 ± 0.02 and 1.01 ± 0.04, for MgATP and Mg8-N3ATP, respectively. The high apparent affinity for Mg8-N3ATP is directly evident in Fig. 1 A, bottom trace: thus, rCO at 15 μM Mg8-N3ATP averaged 60 ± 11% (n = 7) of rCO at saturating, 2 mM, Mg8-N3ATP, whereas rCO at 15 μM MgATP was only 17 ± 1% (n = 8) of rCO at saturating [MgATP] (Fig. 1 B).
The marked prolongation of burst duration caused by Vi made it difficult to collect enough events for the kinetic analyses just described. To estimate τb under those conditions, we analyzed macroscopic current decay reflecting closure of large numbers of channels after sudden nucleotide removal. Records were digitally refiltered at 10 Hz, and then fitted with single or double exponential decay functions by nonlinear least squares (Sigmaplot; Jandel Scientific). Unless otherwise noted, data are given as mean ± SEM (n), where n represents the number of observations.
RESULTS
Mg8-N3ATP Supports CFTR Cl− Channel Gating but with Altered Single-channel Kinetics
Use of 8-azido–substituted nucleotides as surrogates rests on the assumption that the substitution does not grossly affect nucleotide interactions with the subject protein. To this end, Mg8-N3ATP was found to support substrate transport by MRP1 with comparable maximal rate and apparent affinity (10–50 μM) as for ATP (Gao et al., 2000; Hou et al., 2000), and 1 mM Mg8-N3ATP activated macroscopic Cl− current in excised patches containing PKA-phosphorylated CFTR channels (Travis et al., 1993). The records in Fig. 1 A confirm that Mg8-N3ATP supports activity of phosphorylated wild-type (WT) CFTR channels expressed in Xenopus oocytes, but they also show that the rates of channel opening to a burst (rCO, see materials and methods), and of closing from a burst (rOC), are both lower during exposure to 2 mM Mg8-N3ATP than they are in 2 mM MgATP. Kinetic analysis (Csanády, 2000) showed that, at the saturating concentration of 2 mM, the 8-N3 group reduced the maximal rates of opening to a burst ∼2 fold (rCO2mM 8-N3ATP/rCO2mM ATP = 0.42 ± 0.04, n = 17), and of closing from a burst ∼5 fold (in 2 mM MgATP, rOC = 3.83 ± 0.25 s−1; in 2 mM Mg8-N3ATP, rOC = 0.74 ± 0.09 s−1; n = 17). For both MgATP and Mg8-N3ATP, opening rate was a saturable function of concentration (Fig. 1 B), but closing rate was approximately constant (Fig. 1 C). The opening rates were half-maximal at 55 ± 5 μM MgATP (Fig. 1 B, black circles) or 11 ± 3 μM Mg8-N3ATP (Fig. 1 B, red triangles).
Binding of [32P]8-N3ATP to CFTR at 0°C
We examined simple binding of 8-N3ATP to CFTR at 0°C, a temperature anticipated to largely prevent hydrolysis. To facilitate immunoprecipitation, we used CFTR with an NH2-terminal Flag epitope, which in Xenopus oocytes differed from WT CFTR only in its ∼3-fold lower maximal opening rate (Chan et al., 2000). We incubated membranes from HEK293T cells expressing Flag-CFTR on ice with varying concentrations of [α32P]8-N3ATP (in the presence of 10 mM Mg2+) for 5 min, and then subjected them to UV irradiation on ice, with no intervening wash. Autoradiography of the photolabeled CFTR after immunoprecipitation and SDS-PAGE showed that, in agreement with others (Travis et al., 1993; Aleksandrov et al., 2002), the labeling saturated at low μM [α32P]8-N3ATP (≤50 μM; e.g., Fig. 8 B, below).
Figure 8.

Mutation K464A in CFTR impairs occlusion at 30°C, but not binding at 0°C, at low [nucleotide], and disrupts Vi-induced stabilization of open burst state. (A, left) Membranes of oocytes expressing WT or K464A CFTR (without Flag tags) were run on SDS-PAGE gels, transferred to nitrocellulose membranes, and blotted with anti–R-domain antibody. WT CFTR was expressed at least twice as well as K464A CFTR in this batch of oocytes (arrow marks mature fully-glycosylated CFTR; lower, sharper band is core-glycosylated CFTR). (Right) Autoradiogram showing nucleotide occlusion in the same membranes as at left, after incubation at 30°C for 15 min with 5 μM [α32P]8-N3ATP ± 1 mM Vi, washing including 5-min postincubation at 30°C, irradiation with UV, solubilization, and SDS-PAGE. Twice as much membrane was used for K464A samples as for WT samples. (B, left) Immunoblots of membranes from Flag-WT– and Flag-K464A–expressing oocytes (the Flag tags facilitated immunoprecipitation to enhanced signal-to-noise ratio, as membranes were not washed before photocrosslinking) blotted with anti–R-domain antibody as in A. The membranes contained about one third more WT than mutant K464A protein. (Right) Autoradiogram showing photolabeling of CFTR in the same membranes as at left after incubation with 5 or 50 μM [α32P]8-N3ATP for 5 min on ice, followed by UV irradiation, immunoprecipitation, SDS-PAGE, and autoradiography. Approximately 30% more membrane was used for Flag-K464A samples as for Flag-WT samples. (C) Macroscopic current in an oocyte patch containing hundreds of K464A CFTR channels. Current decays with comparable biexponential time course (superimposed fit lines largely obscured by data) after activation by 5 mM MgATP plus 300 nM PKA with 5 mM Vi (for trace shown: af = 23 pA, as = 7 pA, τf = 0.7 s, τs = 5.5 s) or without it (first decay af = 29 pA; as = 31 pA, τf = 0.5 s, τs = 1.4 s, second decay af = 57 pA; as = 5 pA, τf = 0.6 s, τs = 2.1 s). Solution exchange time <1 s. Bars beneath trace offset to signal different solution compositions. (D and E) Mean fit parameters for the time constant (D) and fractional amplitude (E) of the slow component of current decay after activation by 5 mM MgATP plus 300 nM PKA without (white bars) or with 5 mM Vi (black bars) for WT (n = 19) or K464A (n = 10).
Relative binding affinities at 0°C were determined by including various concentrations of unlabeled competing nucleotide during the 5-min incubation with 5 μM Mg[α32P]8-N3ATP before photocrosslinking. For example, the labeling by 5 μM Mg[α32P]8-N3ATP was reduced to about half by 10–50 μM unlabeled (cold) MgATP (Fig. 2 A). Assuming a Kd of 5 μM for Mg8-N3ATP binding (see Fig. 8 B, below) and simple competition between Mg8-N3ATP and other Mg-nucleotide complexes, least-squares fits yielded Ki values of 1 ± 1 μM for MgADP, 6 ± 2 mM for MgAMPPNP, and 16 ± 4 μM for MgATP (Fig. 2 C). In separate experiments, we found that omission of Mg2+ (and addition of 1 mM EDTA) reduced photolabeling by 5 μM [α32P]8-N3ATP only to 77 ± 9% (n = 4) of the level found with Mg2+. By including this constraint (i.e., for Mg2+-free [α32P]8-N3ATP, Kd = 8 μM) in the fit, we estimate a Ki for Mg2+-free (Tris) ATP of 69 ± 20 μM (Fig. 2 C).
Figure 2.

[α32P]8-N3ATP and [γ32P]8-N3ATP binding to Flag-CFTR in HEK293T cell membranes and inhibition by nucleotides. (A) Autoradiogram showing competition by cold MgATP for binding of 5 μM [α32P]8-N3ATP to CFTR at 0°C, followed by UV irradiation, washing, immunoprecipitation, and SDS-PAGE. (B) CFTR incubated with 5 μM [γ32P]8-N3ATP at 0°C was photolabeled only after UV irradiation, indicating lack of phosphorylation by endogenous kinases under these conditions. (C) Quantification of competition for binding of 5 μM [α32P]8-N3ATP by unlabeled MgATP (as in A), MgADP, or MgAMPPNP (all in Mg2+-containing buffer), or by unlabeled TrisATP (in Mg2+-free buffer). Photolabeling was normalized to the signal obtained in the absence of competing nucleotide (note broken abscissa), and the data (error bars show ±SD) fitted assuming simple competition and a Kd of 5 μM for Mg8-N3ATP, or 8 μM for free 8-N3ATP. Ki values from the fits were 1 ± 1 μM for MgADP (filled triangles, n = 2), 6 ± 2 μM for MgAMPPNP (empty squares, n = 4), 16 ± 4 μM for MgATP (filled circles, n = 4), and 69 ± 20 μM for TrisATP (empty circles, n = 3). Given the finite duration of UV irradiation, these apparent binding affinities might be less accurate than the ratios of the values obtained for the different nucleotides.
UV irradiation after incubation with 5 μM Mg [γ32P]8-N3ATP on ice resulted in a comparable level of photolabeling of CFTR (Fig. 2 B) to that seen with 5 μM Mg[α32P]8-N3ATP (Fig. 2 A).
Binding of Low Concentrations of 8-N3 Nucleotide at 0°C Occurs Predominantly at NBD1
To examine the relative labeling of the two NBDs, we coexpressed in Xenopus oocytes two Flag-tagged halves of CFTR: the NH2-terminal half, Flag-3–835, containing NBD1, and the COOH-terminal half, Flag-837–1480, containing NBD2. Gating of full-length CFTR with an extracellular Flag epitope, as in Flag-837–1480, is reportedly identical to that of WT CFTR (Schultz et al., 1997). We previously characterized in detail gating of Flag-3–835 + 837–1480 split CFTR (with a single Flag tag) in oocyte patches: in the presence of MgATP, the channels showed measurable phosphorylation-independent activity that was enhanced ∼5-fold by PKA (Csanády et al., 2000). The same pattern of gating and enhancement by PKA was found for the doubly-tagged split channel, Flag-3–835 + Flag-837–1480 CFTR, in oocyte patches (not depicted). Binding of 5 or 50 μM [α32P]8-N3ATP to these doubly-tagged split channels in oocyte membranes at 0°C yielded a prominent band in the autoradiogram from injected (+), but not from noninjected (−), oocytes at the level expected for the ∼96-kD Flag-3–835 half channel that harbors NBD1 (Fig. 3 A, arrow). The parallel immunoblot with anti–R-domain antibody (Fig. 3 B) confirmed this assignment; the smaller (≤90 kD), presumably truncated, band recognized by the anti–R-domain antibody (Fig. 3 B; see also Figs. 5 A and 6 C, below) was weakly labeled in the autoradiogram. In contrast, no comparably prominent signals that would indicate strong labeling of NBD2 by [α32P]8-N3ATP were seen in the autoradiogram (Fig. 3 A), i.e., at positions corresponding to the bands recognized by the anti–COOH-terminal CFTR antibody (Fig. 3 B). We cannot rule out that relatively weak labeling of NBD2-containing fragments may be partly obscured by the faint nonspecific labeling evident in uninjected oocytes (Fig. 3 A, left lane). Nevertheless, the results suggest that in this assay of simple binding at 0°C low micromolar concentrations of nucleotide label mostly NBD1, and hence that the relative nucleotide affinities assayed in Fig. 2 also largely reflect nucleotide interactions at NBD1.
Figure 3.

Binding of [α32P]8-N3ATP to split CFTR channels. (A) Membranes from oocytes coexpressing Flag-3–835 CFTR and Flag-837–1480 CFTR (+), or noninjected controls (−), incubated with 5 or 50 μM [α32P]8-N3ATP at 0°C were irradiated with UV before washing, immunoprecipitation, and SDS-PAGE. Arrow indicates photolabeled band in the autoradiogram corresponding to CFTR half containing NBD1. (B) Immunoblots of proteins immunoprecipitated and run in same SDS-PAGE gel used for A, before transfer to nitrocellulose and blotting with anti-R domain antibody (left) that recognizes Flag-3–835-containing NBD1, or with anti–COOH-terminal antibody (right) that recognizes Flag-837–1480-containing NBD2.
Figure 5.

(A) In split CFTR, tightly bound [α32P]8-azido nucleotide predominantly labels NBD1. (Left) Autoradiogram from membranes of oocytes coexpressing Flag-3–835 and Flag-837–1480 (+), or of noninjected oocytes (−) incubated for 15 min at 30°C with 5 μM [α32P]8-N3ATP and 10 mM Mg2+ ± 1 mM Vi, subjected to the standard wash procedure including 5-min postincubation at 30°C, irradiated with UV, immunoprecipitated, and subjected to SDS-PAGE. Arrow indicates photolabeled band corresponding to CFTR half containing NBD1. (Right) Proteins from the same membranes were immunoprecipitated and blotted with either anti–R-domain antibody, which recognizes the half of CFTR containing NBD1 (left two lanes), or with anti–COOH-terminal antibody, which recognizes the half containing NBD2 (right two lanes). (B) Extremely slow release of [α32P]8-azido nucleotide during nucleotide-free postincubation at 30°C before UV irradiation. Initial incubation of membranes from HEK293T cells expressing Flag-CFTR was with 5 μM [α32P]8-N3ATP for 15 min at 30°C with (+Vi) or without (−Vi) 1 mM Vi. After postincubation for time indicated, immunoprecipitation, and SDS-PAGE, the resulting autoradiogram signals were normalized to that obtained with Vi but without postincubation (postincubation time = 0) in each experiment (n = 6 for 0- and 5-min points; n = 2 for 1.5- and 15-min points; error bars give ± SD). Curves show simultaneous least-squares fits to both datasets with single time constant, yielding τ = 15 ± 2 min.
With Mg8-N3ATP, as with MgATP, Vi Markedly Prolongs the Open Burst State of CFTR Channels
Before using Vi in photolabeling studies, we examined its influence on CFTR channel gating in MgATP and in Mg8-N3ATP. After activating CFTR channels in large oocyte patches with PKA, MgATP, and Vi, sudden nucleotide withdrawal caused rapid closure of about half the channels, with a time constant, τf = 1.1 ± 0.3 s (n = 6), identical to that of most current decay on MgATP removal in the absence of Vi (Fig. 4 C, left; τf = 1.1 ± 0.2 s; n = 6), and similar to the mean burst duration at saturating [MgATP] in PKA (τburst ∼700 ms; Csanády et al., 2000; Vergani et al., 2003): this fast component presumably represents closing of channels that had not interacted with Vi. The other half of the open channels (fractional amplitude, as/(as + af) = 0.48 ± 0.06; n = 6; Fig. 4 E, black bar) closed slowly (τs = 31.5 ± 3.8 s; n = 6; Fig. 4 D, black bar) reflecting marked stabilization of their open burst state by Vi (Fig. 4, A and C; Baukrowitz et al., 1994; Gunderson and Kopito, 1994). On average, the fraction of its open time a single channel spends in short (or long) bursts equals the fraction of the population of open channels in short (or long) bursts at any instant. At the instant of MgATP removal, the ratio of these population fractions is given by the ratio of the fast/slow fractional amplitudes, af/as (0.52/0.48 ≈ 1), which thus equals the ratio nτf/mτs, where n and m are the mean numbers of short (τf ≈ 1 s) and long (τs ≈ 30 s) bursts, respectively, entered by each channel in any given (sufficiently long) time interval. So, the ratio n/m, ∼30 here, indicates that 1 in ∼31 openings resulted in a Vi-stabilized open burst.
Figure 4.
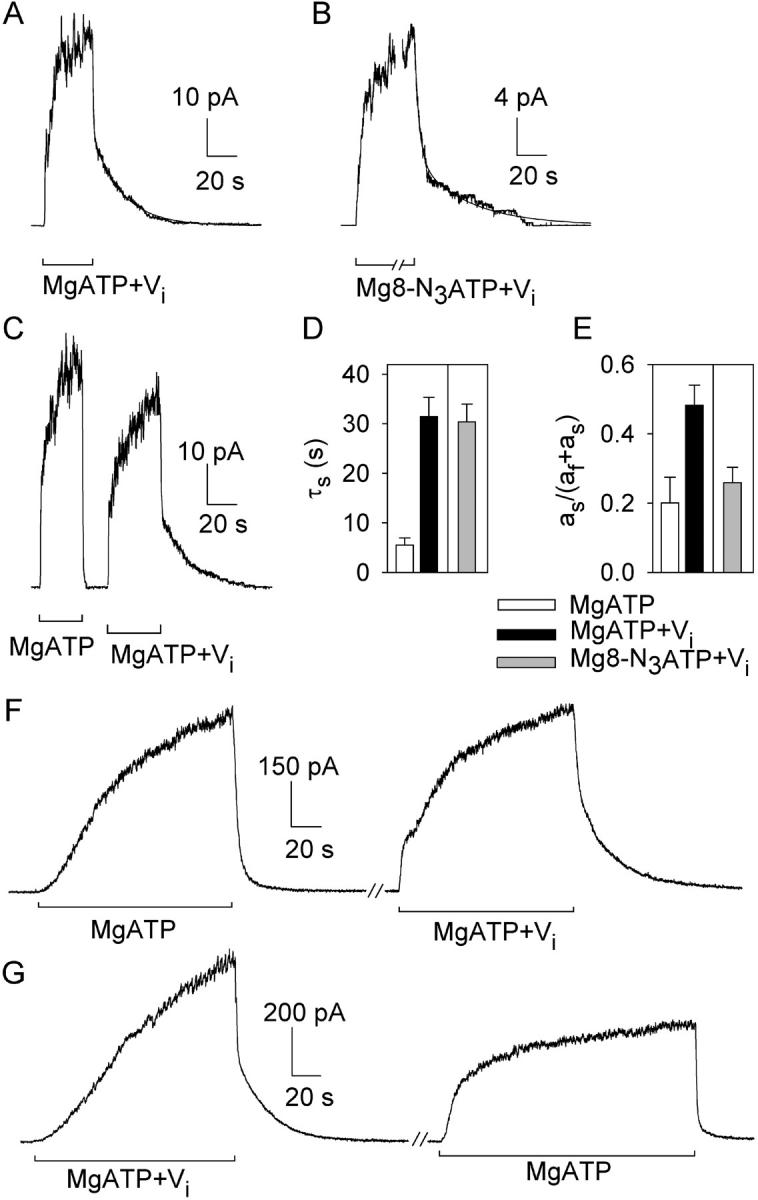
Vi locks open WT CFTR channels activated by PKA and either MgATP or Mg8-N3ATP. (A–C) Macroscopic current, in patches containing hundreds of channels excised from oocytes expressing WT CFTR, relaxed with double exponential time course (smooth fit lines, largely obscured by data) after removal of nucleotides. Nucleotides (0.5 mM in A and B; 5 mM in C) and Vi were applied in the presence of 300 nM PKA. Fit parameters for trace in A are, af = 25 pA, as = 24 pA, τf = 0.5 s, τs = 20 s; for trace in B, af = 14 pA, as = 7 pA, τf = 3 s, τs = 35 s; for first decay in C, af = 50 pA, τf = 0.5 s; second decay in C, af = 27 pA, as = 18 pA, τf = 0.6 s, τs = 21 s. Solution exchange took <1 s, estimated from current decay after brief activation of endogenous Ca2+-activated Cl− channels. (D and E) Summary of mean (±SEM, n = 6) time constants (D) and fractional amplitudes (E) of slow components of current decay after activation by PKA and 0.5 mM MgATP without (white bars), or with (black bars) 5 mM Vi, or by PKA and 0.5 mM Mg8-N3ATP with 5 mM Vi (gray bars). (F and G) Macroscopic current records from inside-out patches excised from HEK293T cells expressing WT CFTR; MgATP (5 mM) and Vi (5 mM) applications were in the presence of 300 nM PKA; the breaks indicate omission of 30 s (F) and 100 s (G) of record. (In C, F, and G, offset bars beneath traces signal different solution compositions.)
When Mg8-N3ATP was used to open the channels (Fig. 4 B), the slow time constant reflecting the Vi-stabilized open state was unaltered (τs = 30.4 ± 3.6 s; n = 6; Fig. 4 D, gray bar), though the steady-state fraction of such locked-open channels was somewhat reduced (as/(as + af) = 0.26 ± 0.04, n = 6; Fig. 4 E, gray bar). The fast time constant after Mg8-N3ATP, τf = 3.0 ± 0.3 s (n = 6), like the mean burst duration (Fig. 1, A and C), was larger than that with MgATP, supporting our interpretation that the fast component reflects normal closure of channels not locked by Vi. This larger τf can account for the observed smaller as/(as+af) if, in Mg8-N3ATP too, 1 (m = 1) of 31 channel openings becomes an ∼30-s Vi-locked burst and the other 30 of 31 (n = 30) openings are normal (for Mg8-N3ATP plus PKA) ∼3-s bursts: i.e., af/as ≈ (30 × 3 s)/(1 × 30 s) ≈ 0.75/0.25. So, the 8-N3 group slows “normal” channel closing, but appears to alter neither the stability of the Vi-locked open-burst state nor the frequency of entry into that state.
Vi similarly affected CFTR expressed in HEK293T cells (Fig. 4, F and G). On nucleotide withdrawal after channel activation with PKA + MgATP, >90% of the current relaxed rapidly (τf = 1.9 ± 0.5 s; n = 4); but, after activation with PKA + MgATP + Vi, 39 ± 4% (n = 4) of the current relaxed slowly (τs = 22.0 ± 2.6 s; n = 4), reflecting channel closing from Vi-stabilized open bursts, whereas the rest decayed rapidly (τf = 1.9 ± 0.6 s; n = 4) like channels not exposed to Vi. This large slow component was seen only after exposure to Vi, regardless of whether that exposure occurred after (Fig. 4 F) or before (Fig. 4 G) the control activation without Vi.
Tightly Bound [α32P]8-N3 Nucleotide after Incubation at 30°C Photolabels Predominantly NBD1
To assay protracted nucleotide interactions with the NBDs, it was essential to wash away freely exchangeable nucleotide before photocrosslinking. After incubation with [α32P]8-N3ATP at 30°C, Flag-CFTR–containing HEK293T-cell membranes were diluted 10-fold into ice-cold nucleotide-free buffer, spun down at 4°C, then resuspended in more of the same buffer and subjected to a 5-min postwash incubation at 30°C (compare Ueda et al., 1999) before UV irradiation on ice to cross-link tightly bound nucleotide, followed by immunoprecipitation, SDS-PAGE, and autoradiography. We found, like Szabó et al. (1999) (but cf. Aleksandrov et al., 2002), that there was little tight binding without Mg2+: only 11 ± 3% (n = 2) in the absence of Vi and 20 ± 7% in its presence, of that with both Vi and Mg2+. The presence of Mg2+ alone increased labeling to 70 ± 16% of that seen when Vi was also added (cf. Szabó et al., 1999; Aleksandrov et al., 2001, 2002). Varying the concentration of [α32P]8-N3ATP, in the presence of Mg2+ and 1 mM Vi, showed that the 5 μM [α32P]8-N3ATP used in most experiments was not saturating, and that half-maximal tight binding (or occlusion) of nucleotide at 30°C occurred at ∼10 μM [α32P]8-N3ATP (unpublished data), the concentration at which channel opening rate at room temperature was also half maximal (Fig. 1 B). Additional controls, with 5 μM [α32P]8-N3ATP, Mg2+, and Vi, demonstrated that the standard 15-min incubation and 10-min irradiation times yielded robust signals that appeared relatively insensitive to even ∼2-fold reductions in those times (unpublished data).
When split CFTR channels (Flag-3–835 + Flag-837–1480) in oocyte membranes were incubated at 30°C with 5 μM [α32P]8-N3ATP and Mg2+, with or without Vi, followed by the two washes and 5-min 30°C postincubation just described, the tightly-bound 8-N3 nucleotide predominantly photolabeled NBD1 (compare Szabó et al., 1999; Aleksandrov et al., 2001, 2002). The autoradiogram (Fig. 5 A, left) showed strong labeling, enhanced by Vi, of a single band near ∼90 kD, not seen in uninjected oocyte membranes (right lane). Comparison with the immunoblots (Fig. 5 A, right) shows that the photolabeled band corresponds to the principal band recognized by the anti–R-domain antibody (epitope near NBD1), and that none of the bands recognized by the anti–COOH-terminal antibody (Fig. 5 A, right; epitope near NBD2) was strongly photolabeled (including, importantly, the broad band that corresponds to fully glycosylated protein; Chan et al., 2000), indicating that the occluded [α32P]8-N3 nucleotide was virtually exclusively cross-linked to NBD1.
Dwell Time of 8-N3 Nucleotide Occluded at NBD1 with or without Vi
To learn just how long the nucleotide remains associated with NBD1 at a temperature that supports channel gating, we used Flag-CFTR expressed in HEK293T cells and varied the postincubation period at 30°C from 0 to 15 min before photocrosslinking (Fig. 5 B). Three results are clear. First, the standard 5-min postincubation released less than half the label found with no postincubation (just two cold washes), regardless of whether the initial incubation included Vi (Fig. 5 B). Second, the initial presence of Vi increased tightly bound nucleotide on average by <50% for all postincubation times up to 5 min (Fig. 5 B). Third, nucleotide loss during postincubation at 30°C could be fitted with the same exponential time course (τ ∼15 min) whether or not Vi had been present during the initial incubation (fitted curves, Fig. 5 B).
Is the Nucleotide Tightly Bound at NBD1 8-N3ADP, or 8-N3ATP?
To learn whether the nucleotide occluded at NBD1 had been hydrolyzed to N3ADP during the postincubation at 30°C, we incubated Flag-CFTR from HEK293T cells with 5 μM [γ32P]8-N3ATP instead of [α32P]8-N3ATP, at 30°C in the presence of Vi, subjected the membranes to the standard two ice-cold washes with intervening warm 5-min postincubation, and then incubated samples with or without UV irradiation (Fig. 6 A). A low level of covalent incorporation of [γ32P] into CFTR was seen without UV irradiation. We attribute this to phosphorylation of CFTR by endogenous kinases, as it was observed with [γ32P]ATP as well as [γ32P]8-N3ATP and was enhanced by Vi, an inhibitor of protein phosphatases (unpublished data), but was absent at 0°C (Fig. 2 B, above). A similar phosphorylation was found with MRP1 (Hou et al., 2000). Nonetheless, UV irradiation markedly increased the label incorporated into CFTR (Fig. 6 A, left lane). That UV-dependent component of labeling with [γ32P]8-N3ATP represents photocrosslinking of nonhydrolyzed N3ATP, which must have survived intact throughout the initial 15-min incubation at 30°C, and the subsequent cold washes and 5-min postincubation at 30°C. So we may conclude that some of the tightly bound nucleotide remained in the form of ATP, but what fraction?
Figure 6.

Tightly bound 8-N3 nucleotide at NBD1 is largely ATP. (A) Autoradiogram showing labeling of CFTR by [γ32P]8-N3ATP without UV (reflecting protein phosphorylation; right) and after UV irradiation (reflecting both photocrosslinking and phosphorylation; left). Prephosphorylated (with PKA; see Fig. 7) HEK293T membranes containing Flag-CFTR were incubated for 15 min at 30°C with 5 μM [γ32P]8-N3ATP plus 1 mM Vi, subjected to usual washes plus 5-min nucleotide-free postincubation at 30°C, exposed to UV (+) or kept in the dark (−), immunoprecipitated, run on SDS-PAGE, and exposed. (B and C) Both [γ32P]8-N3ATP and [α32P]8-N3ATP predominantly photolabel split CFTR (arrow) at NBD1. (B) Autoradiogram from (not prephosphorylated) membranes of oocytes coexpressing Flag-3–835 and Flag-837–1480 (+) or from noninjected oocytes (−), incubated for 15 min at 30°C with 1 mM Vi and either 5 μM [α32P]8-N3ATP (left two lanes) or 5 μM [γ32P]8-N3ATP (right three lanes), followed by washes and 5-min postincubation at 30°C and then exposure to UV light (+) or darkness (−), before immunoprecipitation and SDS-PAGE. (C) Immunoprecipitates from the same membranes were immunoblotted with anti–R-domain antibody (left) or anti–COOH-terminal antibody (right).
To answer this, we examined radiolabeling of membranes from oocytes expressing split CFTR, Flag-3–835 plus Flag-837–1480, after incubation with 5 μM [γ32P]8-N3ATP or 5 μM [α32P]8-N3ATP, this time without Vi, followed by a 5-min postincubation at 30°C (Fig. 6 B). Comparison with the immunoblots (Fig. 6 C) confirmed that both [α32P]8-N3ATP and [γ32P]8-N3ATP labeled essentially only the NH2-terminal half of CFTR, and that the labeling with [γ32P]8-N3ATP was stronger with than without UV irradiation (Fig. 6 B, right); the latter presumably reflected R-domain phosphorylation by native kinases. The UV-dependent component of [γ32P]8-N3ATP labeling, estimated by subtracting the signal without from that with UV exposure, had an intensity comparable (∼60% in this case) to that of the labeling by [α32P]8-N3ATP (entirely dependent on UV irradiation; Fig. 7 B). As the specific activities of the [α32P]- and [γ32P]-labeled nucleotides were identical (within 1%), this finding establishes that most of the nucleotide remaining at NBD1 after a 5-min postincubation at 30°C in nucleotide-free buffer is nonhydrolyzed N3ATP. We conclude that, even at 30°C, NBD1 in CFTR hydrolyzes ATP little, if at all.
Figure 7.

Little influence of phosphorylation with PKA on [α32P]8-N3ATP binding at 0°C (A) or tight binding/occlusion at 30°C (B and C). (A) Before the binding assay, HEK293T membranes expressing Flag-CFTR were incubated with (+PKA) or without (−PKA) PKA, washed twice with ice-cold nucleotide-free (and PKA-free) buffer, and then incubated for 5 min on ice with 0.5, 1, or 5 μM [α32P]8-N3ATP before UV irradiation without washes, immunoprecipitation, and SDS-PAGE to yield the autoradiogram shown. (B) Membranes were prephosphorylated with PKA (+) or not (−) and washed as in A, then incubated at 30°C for 15 min with 5 μM PKI and 5 μM [α32P]8-N3ATP ± 1 mM Vi (as indicated), washed as usual including a 5-min postincubation at 30°C, and then UV irradiated (except lane marked “−”) before immunoprecipitation, SDS-PAGE, and autoradiography. The right lane is a control using the same membranes but prephosphorylated with PKA and [γ32P]ATP, then washed and incubated with Vi for 15 min at 30°C like the other samples, but without [α32P]8-N3ATP, before washing, immunoprecipitation, SDS-PAGE, and autoradiography: the PKA-mediated phosphorylation of CFTR was retained throughout the incubation period with 8-N3 nucleotide. (C) PKA was included (+) or not (−) in the 10-min incubation of HEK293T-cell membranes expressing Flag-CFTR with 5 μM [α32P]8-N3ATP ± 1 mM Vi (as indicated) at 25°C, after which the membranes were washed as usual including a 5-min postincubation at 25°C, and then UV irradiated before immunoprecipitation, SDS-PAGE, and autoradiography.
Influence of Prephosphorylation of CFTR on Nucleotide Binding at 0°C and on Occlusion at 30°C
We first phosphorylated Flag-CFTR in HEK293T cell membranes by incubating them for 20 min at 30°C with cold (nonradiolabeled) MgATP, with or without PKA catalytic subunit; a control sample included [γ32P]ATP with PKA to monitor CFTR phosphorylation. All samples were washed with ice-cold nucleotide-free buffer and pelleted twice, to remove PKA and/or ATP, before they were incubated with [α32P]8-N3ATP at 0°C to assess binding or at 30°C to assess occlusion. In vitro phosphorylation appeared (Fig. 7 A) to slightly diminish nucleotide binding at 0°C (on average by ∼15%), consistent with the negligible effect reported by Travis et al. (1993). Prephosphorylation also consistently reduced occlusion of [α32P]8-N3ATP at 30°C, to 80 ± 7% (n = 9) of control with Vi, and to 75 ± 15% (n = 6) without it (Fig. 7 B), i.e., regardless of the small augmentation caused by the Vi itself. The strong signal in the right lane of Fig. 7 B confirms that CFTR remained phosphorylated throughout all the incubations. This reduction in [α32P]8-N3ATP occlusion seems unlikely to be entirely attributable to residual partial occupancy of NBD1 by unlabeled ATP that was bound during the prephosphorylation and remained bound during the subsequent washes. Inclusion of PKA during a 10-min incubation with [α32P]8-N3ATP at 25°C also reduced occlusion, by about one third (to 67%) in the presence of Vi (Fig. 7 C; compare lane 1 to lane 3), and by almost one quarter (to 78%) in its absence (Fig. 7 C; compare lane 2 to lane 4), consistent with the above results. We conclude that the strong facilitatory effect of PKA on CFTR channel gating is not due to enhanced binding or occlusion of nucleotide at NBD1.
The K464A Mutation Little Influences Binding at 0°C, but Diminishes Occlusion at 30°C, at Low [Nucleotide]
Because the mutation K464A at the Walker A lysine in NBD1 impairs CFTR maturation in mammalian cells (e.g., Aleksandrov et al., 2001), we compared nucleotide interactions with WT and K464A CFTR expressed in oocytes, in which we also compared electrophysiological responses. Despite loading twice as much membrane protein in the K464A lanes as in the WT lanes (Fig. 8 A, right) to compensate for diminished K464A expression (Fig. 8 A, left), negligible occlusion of [α32P]8-N3ATP at 30°C was observed for K464A CFTR, either with or without Vi. In contrast, simple binding of 5 or 50 μM [α32P]8-N3ATP, assayed by photolabeling in oocyte membranes at 0°C, seemed little different for Flag-K464A than for Flag-WT CFTR (Fig. 8 B), even though that binding occurs predominantly at NBD1 (Fig. 3, above), the structurally altered catalytic site. The fact that opening of K464A CFTR channels is impaired at low μM [MgATP] (Vergani et al., 2003) suggests that channel opening and nucleotide occlusion at NBD1 share a common step that occurs after the simple association with nucleotide.
The K464A Mutation Virtually Eliminates the Vi-dependent Slowing of CFTR Channel Closing
Unlike the slow decline of roughly half of the WT CFTR current (Fig. 4, above), the macroscopic current flowing through K464A channels activated by PKA, MgATP, and Vi decayed relatively rapidly upon nucleotide withdrawal (Fig. 8 C). The current relaxation after exposure to Vi had a double exponential time course (smooth fit line, Fig. 8 C, center): most of the current decayed rapidly (τfast = 0.9 ± 0.1 s, n = 10) with a time constant like that of closing (τb = 620 ± 58 ms, n = 24; Vergani et al., 2003) of K464A channels exposed to just PKA and 5 mM MgATP, indicating that the majority of K464A CFTR channels did not respond to the presence of Vi. The smaller current component after Vi (as/(as + af) = 0.32 ± 0.06, n = 10; Fig. 8 E, black bar) decayed somewhat more slowly (τs = 5.2 ± 0.5 s, n = 10; Fig. 8 D, black bar), but still ∼5 times faster than the slower component of WT CFTR channel current after exposure to Vi (compare black bars, Fig. 8 D).
DISCUSSION
Our measurements of interactions of 8-N3–labeled nucleotides with CFTR confirm and substantially extend results from previous studies (Travis et al., 1993; Szabó et al., 1999; Aleksandrov et al., 2001, 2002). By comparing the kinetics of these nucleotide interactions with the kinetics of CFTR channel gating, we are able to derive new mechanistic insights. We find that nucleotide binds with relatively high affinity to NBD1 with or without Mg2+, before becoming occluded at NBD1 in a Mg2+-requiring reaction; once occluded, the nucleotide dissociates with a time constant of ∼15 min at 30°C, whether or not Vi was present during the initial incubation. As this dissociation is orders of magnitude slower than channel opening and closing events, we conclude that the nucleotide remains bound at NBD1 throughout many CFTR channel gating cycles. This means that the nucleotide-dependent events that control the timing of normal channel gating (on the time scale of seconds) occur at NBD2. In addition, we find that the nucleotide remaining for several minutes at NBD1 at 30°C is largely ATP, not ADP, which leads us to conclude that NBD1 is incapable of rapidly hydrolyzing ATP. K464A CFTR, mutated at the Walker A lysine in NBD1, binds but does not occlude micromolar concentrations of 8-N3ATP, in accord with our conclusion from analyses of channel gating that the mutation lowers the apparent affinity for nucleotide interactions required for the transition to the channel open-burst state. Our finding that phosphorylation of CFTR by PKA little alters nucleotide binding or occlusion suggests that phosphorylation might regulate not events at the NBDs, but transduction of those events to channel gates elsewhere. By comparing CFTR channel gating kinetics in Mg8-N3ATP and in MgATP, we find that the slow step that rate limits channel opening at saturating [nucleotide] follows nucleotide binding to both NBDs.
Nucleotide Occlusion at NBD1 at 30°C, and CFTR Channel Gating
That [α32P]8-N3 nucleotide bound at NBD1, but not at NBD2, could survive washing with ice-cold buffer before photocrosslinking with UV light was recently reported (Szabó et al., 1999; Aleksandrov et al., 2001, 2002), but the dwell time of nucleotide at NBD1 at temperatures capable of supporting CFTR channel gating was unknown. We show here that, after incubation at 30°C in the presence of Mg2+ (with or without Vi), 8-N3 nucleotide not only remains at NBD1 throughout two washes with ice-cold buffer, but dissociates very slowly during subsequent postincubation at 30°C, with a time constant of ∼15 min (Fig. 5 B). 8-N3 nucleotide similarly became occluded at 37°C, regardless of the presence of Mg2+ (or of Vi), in NBD1 (and not in NBD2) of the ABC-C family relative SUR1 (Ueda et al., 1997), and that nucleotide also dissociated slowly during postincubation at 37°C (Ueda et al., 1999).
We also find that, on average, CFTR channels exposed to MgATP or Mg8-N3ATP switch back and forth between the closed interburst state and the open burst state at least once every few seconds at room temperature (Fig. 1), and gating is even faster at 30°C (Mathews et al., 1998; Aleksandrov and Riordan, 1998; Csanády et al., 2000). Together, these results imply that nucleotide remains tightly bound at NBD1 throughout many gating cycles. The nucleotide-dependent events that time CFTR channel opening and closing must therefore occur at NBD2 (compare Gunderson and Kopito, 1995). As NBD1 seems to stay nucleotide bound, this corroborates our recent conclusion, from analysis of the [MgATP] dependence of CFTR channel gating, that both NBDs must be occupied by nucleotide before a CFTR channel can open (Vergani et al., 2003). The higher apparent affinity for activation of CFTR channel opening by Mg8-N3ATP than by MgATP (Fig. 1 B) then suggests a higher affinity interaction of Mg8-N3ATP than of MgATP with NBD2.
The extremely slow release of nucleotide from NBD1, but rapid release from NBD2, satisfactorily explains the markedly asymmetric kinetics of inhibition by N-ethylmaleimide of CFTR channels with a single cysteine introduced into the phosphate-binding Walker A loop of NBD1 or NBD2 (Cotten and Welsh, 1998). Increasing concentrations of MgATP could protect against inhibition at either NBD, but, even with low [MgATP], inhibition at 35°C took many minutes when the cysteine was in NBD1 but only seconds when it was in NBD2, consistent with the different delays for nucleotide to leave the two binding sites predicted from our measurements at 30°C.
Our failure to observe photolabeling of NBD2 by [α32P]8-N3 nucleotide after extensive washing and postincubation with nucleotide-free solution at 30°C (Figs. 5 A and 6 B) agrees with Szabó et al. (1999) and Aleksandrov et al. (2001)(2002), who found that even a single ice-cold wash before UV irradiation abolished NBD2 labeling. Nor could we clearly discern a signal attributable to NBD2 after incubation with 5 or 50 μM [α32P]8-N3ATP at 0°C followed by UV irradiation without washing, although Aleksandrov et al. (2002) detected labeling of NBD2 under those conditions, at least at ≥25 μM [α32P]8-N3ATP. In our experiments, weak NBD2 signals could have been obscured by overlapping nonspecific signals that were also present in membranes from control oocytes not expressing CFTR (Fig. 3 A). The inability to clearly photolabel NBD2 cannot be attributed to a failure of nucleotide to bind to NBD2 in the split CFTR construct we used for distinguishing NBDs, for several reasons. First, the [MgATP] dependence of channel opening, interpreted above as reflecting effects at NBD2, showed an apparent affinity of this split channel, CFTR 3–835 + 837–1480 for MgATP ∼2-fold higher than that of WT CFTR (Csanády et al., 2000). Second, nucleotide occlusion (at NBD1) in this split channel was consistently augmented in the presence of Vi (Fig. 5 A), believed to temporarily trap ADP at NBD2 after hydrolysis of ATP there (discussed below). Finally, even a split CFTR channel lacking the entire R domain (CFTR 3–633 + 837–1480) showed markedly prolonged open bursts after mutation of its NBD2 Walker A lysine, K1250, suggesting nucleotide interaction at NBD2.
NBD1 in Intact CFTR Fails to Hydrolyze Mg8-N3ATP
Previous work suggested that 8-N3ATP might be more rapidly hydrolyzed at NBD2 than at NBD1 in CFTR (Aleksandrov et al., 2002), but the hydrolysis rate could not be estimated at either NBD, and kinase-mediated incorporation of 32P was not addressed. Here we show a strong UV-dependent component (i.e., not related to phosphorylation) of labeling by [γ32P]8-N3ATP of CFTR's NBD1 in both HEK293T-cell and oocyte membranes after washing and a 5-min postincubation at 30°C (Fig. 6). That more than half of the occluded nucleotide at NBD1 remained in the form of N3ATP, rather than N3ADP, after 5 min at 30°C implies a time constant of N3ATP loss >430 s, and hence a hydrolysis rate <0.002 s−1 under those conditions. This is at least two orders of magnitude lower than the rate of ATP hydrolysis by purified CFTR (Li et al., 1996), which may therefore essentially reflect hydrolysis at NBD2 alone.
This apparent failure of NBD1 in full-length CFTR to hydrolyze ATP seemed independent of phosphorylation status, as [γ32P]8-N3ATP was occluded (presumably at NBD1) both with and without PKA prephosphorylation. But the failure might reflect sequence asymmetry between CFTR's NBD1 and NBD2 that implies the presence of unorthodox residues at key positions in the NBD1 catalytic site. These include a serine (S573) instead of the conserved glutamate that normally follows the Walker B aspartate (D572 in NBD1), and another serine (S605) in place of the conserved histidine that in HisP interacts with the γ-phosphate and is believed to fulfill a switch function (Hung et al., 1998; Karpowich et al., 2001). Mutational analysis of the Walker B glutamate has shown that even the most conservative charge-removing substitution (glutamine) abolishes steady-state ATP hydrolysis by full-length P-gp (Urbatsch et al., 2000; Sauna et al., 2002), as well as by isolated bacterial NBDs (Moody et al., 2002). In addition, given recent compelling evidence for a head-to-tail dimeric organization of NBDs of Rad50 (Hopfner et al., 2000) and MutS (Junop et al., 2001), of bacterial ABC transporters (Fetsch and Davidson, 2002; Locher et al., 2002; Moody et al., 2002; Smith et al., 2002), and of human P-gp (Loo et al., 2002), the unorthodox signature sequence (LSHGH, instead of the ABC consensus, LSGGQ) from the NBD2 polypeptide is anticipated to complete the structure of the NBD1 catalytic site in intact CFTR (compare Jones and George, 1999). Interestingly, the NBDs of other ABC-C family members show asymmetric sequence abnormalities like those in CFTR, possibly accounting for the similar suggestions that in SUR1, SUR2A, SUR2B (Ueda et al., 1997; Matsuo et al., 1999, 2000), and in MRP1 (Hou et al., 2000), ATP hydrolysis occurs predominantly at NBD2 and far less (if at all) at NBD1. These findings contrast with the nearly symmetrical contributions to nucleotide binding and hydrolysis made by the two structurally similar NBDs of ABC-B family member P-gp (Senior et al., 1995; Urbatsch et al., 1995a,b; but cf. Hrycyna et al., 1999).
The conclusion that NBD1 in CFTR hydrolyzes ATP at a negligibly low rate is at odds with two kinds of earlier results. First, isolated NBD1 constructs, comprising the entire functional domain (at least residues 433–633; Chan et al., 2000) purified from E. coli as His-tagged (Duffieux et al., 2000) or tandem GST-NBD1-R domain (Howell et al., 2000) preparations, reportedly hydrolyzed MgATP at rates of 0.2 s−1 and 0.4 s−1 (at 30°C), with K0.5 values for [MgATP] of ∼250 and ∼60 μM, respectively, comparable to values (0.2 s−1, and 0.3–1 mM) found for intact CFTR (Li et al., 1996). Barring inadvertent contamination of the NBD1 preparations by copurifying ATPases, a possible explanation is that the presence of an orthodox LSGGQ signature sequence in both catalytic sites of an NBD1-NBD1 homodimer might permit them to efficiently hydrolyze MgATP despite the unorthodox NBD1 residues S573 and S605 at both sites. Second, purified NBD1 mutant, K464A, CFTR was reported to hydrolyze MgATP at a maximal rate 10–20-fold lower than that of wild-type CFTR, whereas the equivalent mutation in NBD2, K1250A, essentially abolished hydrolysis (Ramjeesingh et al., 1999). Unless the specific activity of that K464A CFTR was greatly underestimated (due to unrecognized nonfunctional K464A protein), if NBD1 is essentially catalytically inactive in intact CFTR as we propose, a possible explanation for the reduced hydrolysis by K464A CFTR is that this mutation somehow impairs MgATP hydrolysis at the NBD2 catalytic site. Gating measurements offer no support for this possibility, as the K464A mutation little affects normal CFTR-channel gating (Carson et al., 1995; Gunderson and Kopito, 1995; Powe et al., 2002; Vergani et al., 2003) other than reducing the apparent affinity for MgATP (Vergani et al., 2003). However, the mutation does substantially shorten prolonged (locked) open bursts ascribed to NBD2 catalytic-site occupancy by the equivalent of nonhydrolyzed nucleotide, due to the K1250A mutation or to exposure of wild-type CFTR to MgATP plus nonhydrolyzable ATP analogs (Powe et al., 2002; Vergani et al., 2003) or to MgATP plus Vi (Fig. 4 vs. Fig. 8, C and D). Although this destabilization could reflect loss of an allosteric influence of the wild-type NBD1 catalytic site on the NBD2 catalytic site, if transition to the open burst state involves formation of an NBD1/NBD2 dimer with interfacial catalytic sites (Vergani et al., 2003), then the K464A mutation could act locally within the interface to reduce stability of the NBD dimer.
Influence of Orthovanadate on Gating and Photolabeling of CFTR Channels
In several ATPases, upon ATP hydrolysis the analogue, Vi, can replace the dissociated phosphate and form a stable Mg–ADP–Vi complex that traps ADP in the catalytic site (Chabre, 1990). This property of Vi was exploited, together with 8-N3 nucleotide, to demonstrate obligate catalytic cooperativity between P-gp's two similar NBDs (Urbatsch et al., 1995a,b). In P-gp, no nucleotide trapping or photolabeling occurred without Vi (Urbatsch et al., 1995a). In contrast, even without Vi, CFTR tightly bound 8-N3 nucleotide at 30°C exclusively in NBD1 (Fig. 5 A), but the photolabeling was enhanced (on average by ∼30%) by the initial presence of Vi (Figs. 5, 7, and 8; cf. Szabó et al., 1999; Aleksandrov et al., 2001), although Vi little influenced dissociation of the occluded nucleotide from NBD1 (τ ∼15 min, Fig. 5 B). In addition, exposure of CFTR channels to Vi in either MgATP or Mg8-N3ATP resulted in marked prolongation of open bursts (compare Baukrowitz et al., 1994; Gunderson and Kopito, 1994), evident as a slow relaxation of macroscopic current after nucleotide withdrawal from patches with many channels (Fig. 4). But, the time constant of that slow current decay, a measure of the mean duration of Vi-locked open bursts, presumably reflecting slow loss from catalytic sites of Vi-trapped ADP (or 8-N3ADP) after hydrolysis, was only ∼30 s (Fig. 4 D). A consistent interpretation of these seemingly disparate time courses is made possible by the observation that Vi elicited comparable slow current relaxations whether it was applied before, or shortly after, exposure to MgATP alone (Fig. 4, C, F, and G). This allows us to conclude that Vi locked the channels open by acting at NBD2, because the ∼2-min interval between exposures in Fig. 4 F could permit dissociation of only ≤12% of tightly bound nucleotide from NBD1 (assuming dissociation τ ≥15 min at room temperature), whereas 46% of open channels were in locked-open bursts at the time of nucleotide withdrawal. Similarly, in MRP1, Vi-trapped ADP was found predominantly at NBD2 (Gao et al., 2000; Hou et al., 2000). The ∼30-s time constant we observed for current relaxation (Fig. 4) suggests a mean dwell time of ∼30 s for the Vi-trapped ADP at NBD2 of CFTR, readily accounting for the absence of photolabel at NBD2 (Fig. 5) after our extensive washing procedure, but its presence if the wash is omitted (Aleksandrov et al., 2001, 2002). In accord with this interpretation, NBD2 does appear to hydrolyze ATP (Aleksandrov et al., 2002; cf. Ramjeesingh et al., 1999) and is where the hydrolysis-abolishing mutation, K1250A, similarly (like Vi) prolongs bursts (Carson et al., 1995; Gunderson and Kopito, 1995; Ramjeesingh et al., 1999; Zeltwanger et al., 1999; Vergani et al., 2003).
Most of the nucleotide that dissociates slowly from NBD1 can bind tightly there even without Vi. If, as Fig. 5 B suggests, nucleotide occluded in the presence or absence of Vi dissociates at roughly the same rate, this would imply that in either case the nucleotide is released from the same CFTR conformation, one that can be reached without Vi. Presumably, therefore, Vi action at NBD2 somehow increases the steady-state fraction of CFTR channels in that tight nucleotide-binding conformation, resulting in increased photolabeling after the same period of incubation at 30°C. The alternative explanation, that the Vi-induced increment in occluded nucleotide reflects Vi-trapped ADP at NBD1, is less likely because the nucleotide occluded without Vi would have to dissociate as slowly as Vi-trapped nucleotide, the explanation would not account for Vi-locked open bursts of ∼30-s duration, and it would require some hydrolysis at NBD1.
Influence of Phosphorylation by PKA on Nucleotide Interactions with CFTR
Though CFTR channels require phosphorylation by PKA before they can be opened by nucleotides (for reviews see e.g., Gadsby and Nairn, 1999; Sheppard and Welsh, 1999), PKA had relatively little influence on either nucleotide binding at 0°C (compare Travis et al., 1993) or occlusion at 30°C (Fig. 8, A–C). But, at the low [8-N3ATP] used, both assays predominantly monitor NBD1, whereas the nucleotide events that control channel gating occur at NBD2 (above; Vergani et al., 2003). However, Aleksandrov et al. (2002) omitted washing to also observe photolabeling at NBD2 and noted that “none of the major features of the nucleotide interactions at each NBD…were strongly influenced by PKA phosphorylation”. So, it appears that PKA phosphorylation might not be required for nucleotide binding, occlusion, or hydrolysis in intact CFTR in cell membranes, as suggested by ATPase measurements on purified CFTR, in which phosphorylation merely increased apparent affinity for ATP ∼3-fold (Li et al., 1996).
Could exogenous PKA have had little effect in all the above studies because CFTR was already phosphorylated by endogenous kinases? Although some phosphorylation sites on CFTR might have remained phosphorylated (indeed, phosphatases did reduce ATPase activity at fixed [MgATP]; Li et al., 1996), it is unlikely that phosphorylation was sufficient to support channel gating because, for both CFTR purified from insect cells (Li et al., 1996) and CFTR in patches excised from insect cells (Bear et al., 1992), channels were not activated by MgATP until PKA was also added. Similarly, dephosphorylation by endogenous membrane-bound phosphatases of oocytes (e.g., Csanády et al., 2000), and of the BHK cells (Linsdell and Hanrahan, 1998) used by Aleksandrov et al. (2002), prevented activation of CFTR channels in excised patches until the channels were exposed to PKA. So these results suggest that CFTR channels can occlude and hydrolyze ATP under conditions in which channel gating does not occur. This conclusion does not contradict the proposed requirement for MgATP hydrolysis at the NBD2 catalytic site to normally terminate an open burst. But it does imply the existence of a mechanism, analogous to an automobile clutch, which links NBD function to the gating machinery and is modulated by phosphorylation.
The Rate-limiting Step for CFTR Channel Opening Follows Both Nucleotide Binding Steps
The rate at which CFTR channels leave the closed interburst state (of average duration, τib) and initiate open bursts, the opening rate (rCO, given by 1/ τib), is a saturating function of both MgATP and Mg8-N3ATP concentration (Fig. 1 B). At low concentrations of either nucleotide, opening is rate limited by infrequent binding. Because rCO does not increase linearly with [nucleotide], channel opening cannot be a single-step reaction, C→O, but must involve at least two closed states, and two distinct steps, e.g., C1→C2→O. If C1→C2 is the nucleotide-binding reaction, and at saturating [nucleotide] it occurs rapidly compared with the channel-opening step, C2→O, then rCO is expected to display Michaelis-Menten dependence on [nucleotide] as observed (Fig. 1 B), with maximal opening rate reflecting C2→O. However, in principle, the step that limits opening rate at saturating [nucleotide] could precede the binding step (e.g., it could reflect a slow isomerization among closed, nucleotide-free, states). This second possibility is ruled out, however, by our finding that the maximal opening rate is slowed ∼2-fold by the azido (N3) group substitution in the adenine ring of ATP (which does not preclude its hydrolysis by ABC proteins; e.g., Urbatsch et al., 1994), because no step that precedes nucleotide binding could respond to modification of the nucleotide structure.
We therefore conclude that, for both MgATP and Mg8-N3ATP, CFTR channel opening is rate limited at saturating [nucleotide] by a relatively slow step that follows nucleotide binding, that is strongly temperature dependent (Aleksandrov and Riordan, 1998; Mathews et al., 1998) and Mg2+ dependent (Dousmanis et al., 2002), and is sensitive to the detailed structure of the β and γ phosphate groups in the activating Mg2+–nucleoside triphosphate complex (Vergani et al., 2003). We have proposed (Vergani et al., 2003) that this slow step might be related to formation of an NBD1/NBD2 dimer stabilized by the two Mg2+–nucleotide complexes in the catalytic sites at the dimer interface (e.g., Hopfner et al., 2000; Smith et al., 2002) and, in particular, might correspond to formation of a prehydrolysis complex (Dousmanis et al., 2002), most likely at the NBD2 catalytic site.
Acknowledgments
We thank Pablo Artigas for help with the recordings in Fig. 4; László Csanády, Ellen Howard, and Muriel Lainé for helpful comments on the manuscript, and David Kopsco and Atsuko Horiuchi for excellent technical assistance.
This work was supported by the National Institutes of Health (DK51767) and the Cystic Fibrosis Association of Greater New York.
David Clapham served as guest editor.
Claudia Basso's present address is Instituto de Ciencias Biomedicas, Facultad de Medicina, Universidad de Chile, Independencia 1027, Santiago, Chile.
Abbreviation used in this paper: NBD, nucleotide-binding domain.
References
- Aleksandrov, A.A., and J.R. Riordan. 1998. Regulation of CFTR ion channel gating by MgATP. FEBS Lett. 431:97–101. [DOI] [PubMed] [Google Scholar]
- Aleksandrov, L., A. Mengos, X. Chang, A. Aleksandrov, and J.R. Riordan. 2001. Differential interactions of nucleotides at the two nucleotide binding domains of the cystic fibrosis transmembrane conductance regulador. J. Biol. Chem. 276:12918–12923. [DOI] [PubMed] [Google Scholar]
- Aleksandrov, L., A.A. Aleksandrov, X.B. Chang, and J.R. Riordan. 2002. The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J. Biol. Chem. 277:15419–15425. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., T.-C. Hwang, A.C. Nairn, and D.C. Gadsby. 1994. Coupling of CFTR Cl− channel gating to an ATP hydrolysis cycle. Neuron. 12:473–482. [DOI] [PubMed] [Google Scholar]
- Bear, C.E., C.H. Li, N. Kartner, R.J. Bridges, T.J. Jensen, M. Ramjeesingh, and J.R. Riordan. 1992. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell. 68:809–818. [DOI] [PubMed] [Google Scholar]
- Carson, M.R., S.M. Travis, and M.J. Welsh. 1995. The two nucleotide-binding domains of the cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J. Biol. Chem. 270:1711–1717. [DOI] [PubMed] [Google Scholar]
- Chabre, M. 1990. Aluminofluoride and beryllofluoride complexes: a new phosphate analogs in enzymology. Trends Biochem. Sci. 15:6–10. [DOI] [PubMed] [Google Scholar]
- Chan, K.W., L. Csanády, D. Seto-Young, A.C. Nairn, and D.C. Gadsby. 2000. Severed molecules functionally define the boundaries of the cystic fibrosis transmembrane conductance regulator's NH2-terminal nucleotide binding domain. J. Gen. Physiol. 116:163–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, X.B., Y.X. Hou, and J.R. Riordan. 1998. Stimulation of ATPase activity of purified multidrug resistance-associated protein by nucleoside diphosphates. J. Biol. Chem. 273:23844–23848. [DOI] [PubMed] [Google Scholar]
- Cotten, J.F., and M.J. Welsh. 1998. Covalent modification of the nucleotide binding domains of cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 273:31873–31879. [DOI] [PubMed] [Google Scholar]
- Csanády, L. 2000. Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophys. J. 78:785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády, L., K.W. Chan, D. Seto-Young, D.C. Kopsco, A.C. Nairn, and D.C. Gadsby. 2000. Severed channels probes regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J. Gen. Physiol. 116:477–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dousmanis, A.G., A.C. Nairn, and D.C. Gadsby. 2002. Distinct Mg2+-dependent steps rate limit opening and closing of a single CFTR Cl− channel. J. Gen. Physiol. 119:445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffieux, F., J.P. Annereau, J. Boucher, E. Miclet, O. Pamlard, M. Schneider, V. Stoven, and J.Y. Lallemand. 2000. Nucleotide-binding domain 1 of cystic fibrosis transmembrane conductance regulator production of a suitable protein for structural studies. Eur. J. Biochem. 267:5306–5312. [DOI] [PubMed] [Google Scholar]
- Fetsch, E.E., and A.L. Davidson. 2002. Vanadate-catalyzed photocleavage of the signature motif of an ATP-binding cassette (ABC) transporter. Proc. Natl. Acad. Sci. USA. 99:9685–9690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby, D.C., and A.C. Nairn. 1999. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol. Rev. 79:S77–S107. [DOI] [PubMed] [Google Scholar]
- Gao, M., H.R. Cui, D.W. Loe, C.E. Grant, K.C. Almquist, S.P. Cole, and R.G. Deeley. 2000. Comparison of the functional characteristics of the nucleotide binding domains of multidrug resistance protein 1. J. Biol. Chem. 275:13098–13108. [DOI] [PubMed] [Google Scholar]
- Gunderson, K.L., and R.R. Kopito. 1994. Effects of pyrophosphate and nucleotide analogs suggest a role for ATP hydrolysis in cystic fibrosis transmembrane regulator channel gating. J. Biol. Chem. 269:19349–19353. [PubMed] [Google Scholar]
- Gunderson, K.L., and R.R. Kopito. 1995. Conformational states of CFTR associated with channel gating: the role of ATP binding and hydrolysis. Cell. 82:231–239. [DOI] [PubMed] [Google Scholar]
- Haws, C., M.E. Krouse, Y. Xia, D.C. Gruenert, and J.J. Wine. 1992. CFTR channels in immortalized human airway cells. Am. J. Physiol. 263:L692–L707. [DOI] [PubMed] [Google Scholar]
- Hopfner, K.P., A. Karcher, D.S. Shin, L. Craig, L.M. Arthur, J.P. Carney, and J.A. Tainer. 2000. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 101:789–800. [DOI] [PubMed] [Google Scholar]
- Hou, Y., L. Cui, J.R. Riordan, and X. Chang. 2000. Allosteric interactions between the two non-equivalent nucleotide binding domains of multidrug resistance protein MRP1. J. Biol. Chem. 275:20280–20287. [DOI] [PubMed] [Google Scholar]
- Howard, M.E., M.D. Du Vall, D.C. Devor, J.Y. Dong, K. Henze, and R.A. Frizzell. 1995. Epitope tagging permits cell surface detection of functional CFTR. Am. J. Physiol. 269:C1565–C1576. [DOI] [PubMed] [Google Scholar]
- Howell, L.D., R. Borchardt, and J.A. Cohn. 2000. ATP hydrolysis by a CFTR domain: pharmacology and effects of G551D mutation. Biochem. Biophys. Res. Commun. 271:518–525. [DOI] [PubMed] [Google Scholar]
- Hung, L.-W., I.X. Wang, K. Nikaido, P.-Q. Liu, G. Ferro-Luzzi, and S.-H. Kim. 1998. Crystal structure of the ATP-binding subunit of an ABC transporter. Nature. 396:703–707. [DOI] [PubMed] [Google Scholar]
- Hrycyna, C.A., M. Ramachandra, U.A. Germann, P.W. Cheng, I. Pastan, and M.M. Gottesman. 1999. Both ATP sites of human P-glycoprotein are essential but not symmetric. Biochemistry. 38:13887–13899. [DOI] [PubMed] [Google Scholar]
- Jones, P.M., and A.M. George. 1999. Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol. Lett. 179:187–202. [DOI] [PubMed] [Google Scholar]
- Junop, M.S., G. Obmolova, K. Rausch, P. Hsieh, and W. Yang. 2001. Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol. Cell. 7:1–12. [DOI] [PubMed] [Google Scholar]
- Kaczmarek, L.K., K.R. Jennings, F. Strumwasser, A.C. Nairn, U. Walter, F.D. Wilson, and P. Greengard. 1980. Microinjection of catalytic subunit of cyclic AMP-dependent protein kinase enhances calcium action potentials of bag cell neurons in cell cultura. Proc. Natl. Acad. Sci. USA. 77:7487–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpowich, N., O. Martsinkevich, L. Millen, Y.R. Yuan, P.L. Dai, K. MacVey, P. Thomas, and J.F.J. Hunt. 2001. Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure (Camb). 9:571–586. [DOI] [PubMed] [Google Scholar]
- Li, C., M. Ramjeesingh, W. Wang, E. Garami, M. Hewryk, D. Lee, J. Rommens, K. Galley, and C.E. Bear. 1996. ATPase activity of the cystic fibrosis transmembrane regulator. J. Biol. Chem. 271:28463–28468. [DOI] [PubMed] [Google Scholar]
- Lindquist, R.N., J.L. Lynn Jr., and G.E. Lienhard. 1973. Possible Transition-state analogs for ribonuclease. The complexes of uridine with oxovanadium (IV)ion and vanadium (V) ion. J. Am. Chem. Soc. 95:8762–8768. [DOI] [PubMed] [Google Scholar]
- Linsdell, P., and J.W. Hanrahan. 1998. Adenosine triphosphate-dependent asymmetry of anion permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. J. Gen. Physiol. 111:601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher, K.P., A.T. Lee, and D.C. Rees. 2002. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 296:1091–1098. [DOI] [PubMed] [Google Scholar]
- Loo, T.W., M.C. Bartlett, and D.M. Clarke. 2002. The “LSGGQ” motif in each nucleotide-binding domain of human P-glycoprotein is adjacent to the opposing Walker A sequence. J. Biol. Chem. 277:41303–41306. [DOI] [PubMed] [Google Scholar]
- Marino, C.R., L.M. Matovcik, F.S. Gorelick, and J.A. Cohn. 1991. Localization of the cystic fibrosis transmembrane conductance regulator in pancreas. J. Clin. Invest. 88:712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, C.J., J.A. Tabcharani, and J.W. Hanrahan. 1998. The CFTR chloride channel: nucleotide interactions and temperature- dependent gating. J. Membr. Biol. 163:55–66. [DOI] [PubMed] [Google Scholar]
- Matsuo, M., N. Kioki, T. Amachi, and K. Ueda. 1999. ATP binding properties of the nucleotide-binding folds of SUR1. J. Biol. Chem. 274:37479–37482. [DOI] [PubMed] [Google Scholar]
- Matsuo, M., K. Tanabe, N. Kioka, T. Amachi, and K. Ueda. 2000. Different binding properties and affinities for ATP and ADP among sulfonylurea receptor subtypes, SUR1, SUR2A, and SUR2B. J. Biol. Chem. 275:28757–28763. [DOI] [PubMed] [Google Scholar]
- Moody, J.E., L. Millen, D. Binns, J.F. Hunt, and P.J. Thomas. 2002. Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J. Biol. Chem. 277:21111–21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata, K., M. Nishitani, M. Matsuo, N. Kioka, T. Amachi, and K. Ueda. 2000. Nonequivalent nucleotide trapping in the two nucleotide binding folds of the human multidrug resistance protein MRP1. J. Biol. Chem. 275:17626–17630. [DOI] [PubMed] [Google Scholar]
- Picciotto, M.R., J.A. Cohn, G. Bertuzzi, P. Greengard, and A.C. Nairn. 1992. Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 267:12742–12752. [PubMed] [Google Scholar]
- Powe, A.C.J., L. Al-Nakkash, M. Li, and T.C. Hwang. 2002. Mutation of Walker-A lysine 464 in cystic fibrosis transmembrane conductance regulator reveals functional interaction between its nucleotide-binding domains. J. Physiol. 539:333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramjeesingh, M., C. Li, E. Garami, L.-J. Huan, K. Galley, Y. Wang, and C.E. Bear. 1999. Walker mutations reveal loose relationship between catalytic and channel-gating activities of purified CFTR (Cystic Fibrosis Transmembrane Conductance Regulator). Biochemistry. 38:1463–1468. [DOI] [PubMed] [Google Scholar]
- Riordan, J.R., J.M. Rommens, B. Kerem, N. Alon, R. Rozmahel, Z. Grzelczak, J. Zielenski, S. Lok, N. Plavsic, J.L. Chou, et al. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 245:1066–1073. [DOI] [PubMed] [Google Scholar]
- Sauna, Z.E., M. Muller, X.H. Peng, and S.V. Ambudkar. 2002. Importance of the conserved Walker B glutamate residues, 556 and 1201, for the completion of the catalytic cycle of ATP hydrolysis by human P-glycoprotein (ABCB1). Biochemistry. 41:13989–14000. [DOI] [PubMed] [Google Scholar]
- Schultz, B.D., A. Takahashi, C. Liu, R.A. Frizzell, and M. Howard. 1997. FLAG epitope positioned in an external loop preserves normal biophysical properties of CFTR. Am. J. Physiol. 273:C2080–C2089. [DOI] [PubMed] [Google Scholar]
- Senior, A.E., M.K. al-Shawi, and I.L. Urbatsch. 1995. ATP hydrolysis by multidrug-resistance protein from Chinese hamster ovary cells. J. Bioenerg. Biomembr. 27:31–36. [DOI] [PubMed] [Google Scholar]
- Sheppard, D.N., and M.J. Welsh. 1999. Structure and function of the CFTR chloride channel. Physiol. Rev. 79:S23–S45. [DOI] [PubMed] [Google Scholar]
- Shyng, S.-L., and C.G. Nichols. 1997. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 110:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, P.C., N. Karpowich, L. Millen, J.E. Moody, J. Rosen, P.J. Thomas, and J.F. Hunt. 2002. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 10:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó, K., G. Szakács, T. Hegedus, and B. Sarkadi. 1999. Nucleotide occlusion in the human cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 274:12209–12212. [DOI] [PubMed] [Google Scholar]
- Travis, S.M., M.R. Carson, D.R. Ries, and M.J. Welsh. 1993. Interaction of nucleotides with membrane-associated cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 268:15336–15339. [PubMed] [Google Scholar]
- Ueda, K., N. Inagaki, and S. Seino. 1997. MgADP antagonism to Mg2+ independent ATP binding of the sulfonylurea receptor SUR1. J. Biol. Chem. 272:22983–22986. [DOI] [PubMed] [Google Scholar]
- Ueda, K., J. Komine, M. Matsuo, S. Seino, and T. Amachi. 1999. Cooperative binding of ATP and MgADP in the sulfonylurea receptor is modulated by glibenclamide. Proc. Natl. Acad. Sci. USA. 96:1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbatsch, I.L., M.K. al-Shawi, and A.E. Senior. 1994. Characterization of the ATPase activity of purified Chinese hamster P-glycoprotein. Biochemistry. 33:7069–7076. [DOI] [PubMed] [Google Scholar]
- Urbatsch, I.L., B. Sankaran, J. Weber, and A.E. Senior. 1995. a. P-glycoportein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 270:19383–19390. [DOI] [PubMed] [Google Scholar]
- Urbatsch, I.L., B. Sankaran, S. Bhagat, and A.E. Senior. 1995. b. Both P-glycoprotein nucleotide-binding sites are catalytically active. J. Biol. Chem. 270:26956–26961. [DOI] [PubMed] [Google Scholar]
- Urbatsch, I.L., L. Beaudet, I. Carrier, and P. Gros. 1998. Mutations in either nucleotide-binding site of P-glycoprotein (Mdr3) prevent vanadate trapping of nucleotide at both sites. Biochemistry. 37:4592–4602. [DOI] [PubMed] [Google Scholar]
- Urbatsch, I.L., K. Gimi, S. Wilke-Mounts, and A.E. Senior. 2000. Investigation of the role of glutamine-471 and glutamine-1114 in the two catalytic sites of P-glycoprotein. Biochemistry. 39:11921–11927. [DOI] [PubMed] [Google Scholar]
- Vergani, P., A.C. Nairn, and D.C. Gadsby. 2003. On the mechanism of Mg-ATP dependent gating of CFTR Cl− channels. J. Gen. Physiol. 121:17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter, M.C., D.N. Sheppard, M.R. Carson, and M.J. Welsh. 1994. Effect of ATP concentration on CFTR Cl channels: a kinetic analysis of channel regulation. Biophys. J. 66:1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltwanger, S., F. Wang, G.T. Wang, K.D. Gillis, and T.C. Hwang. 1999. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme. J. Gen. Physiol. 113:541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]