

Abstract
We tested the long-standing hypothesis that synaptotagmin 1 is the Ca2+ sensor for fast neurosecretion by analyzing the intracellular Ca2+ dependence of large dense-core vesicle exocytosis in a mouse strain carrying a mutated synaptotagmin C2A domain. The mutation (R233Q) causes a twofold increase in the KD of Ca2+-dependent phospholipid binding to the double C2A-C2B domain of synaptotagmin. Using photolysis of caged calcium and capacitance measurements we found that secretion from mutant cells had lower secretory rates, longer secretory delays, and a higher intracellular Ca2+-threshold for secretion due to a twofold increase in the apparent KD of the Ca2+ sensor for fast exocytosis. Single amperometric fusion events were unchanged. We conclude that Ca2+-dependent phospholipid binding to synaptotagmin 1 mirrors the intracellular Ca2+ dependence of exocytosis.
Keywords: chromaffin cell, membrane fusion, neurosecretion, capacitance measurement, amperometry
INTRODUCTION
Neurotransmitter release is characterized by a steep relationship between vesicular release rate constant and intracellular Ca2+ concentration ([Ca2+]i) (Dodge and Rahamimoff, 1967; Bollmann et al., 2000; Schneggenburger and Neher, 2000). This relationship has a slope of 3–5 in a double-logarithmic plot, indicating the minimal number of Ca2+-binding steps required to elicit vesicular fusion. Likewise, there is an inverse relationship between the secretory delay and [Ca2+]i, such that higher [Ca2+]i leads to a shorter delay. Keeping the delay short is critical for ensuring that secretion will take place during the duration of an evoked [Ca2+]i signal. For these reasons it is assumed that Ca2+-dependent triggering of exocytosis is a precisely regulated process, which is performed by a protein or a protein complex generally referred to as the calcium sensor for exocytosis (Augustine, 2001).
The best studied candidate for this Ca2+ sensor is the vesicular protein synaptotagmin 1, which contains two Ca2+- and phospholipid-binding C2 domains, C2A and C2B (Perin et al., 1990; Brose et al., 1992) and is anchored in the vesicle membrane via a transmembrane domain. Ca2+ binding was first demonstrated for the C2A domain (Davletov and Südhof, 1993; Chapman and Jahn, 1994), and for several years, the C2B domain was suggested to have other functions (Ca2+-dependent multimerization, Sugita et al., 1996; Chapman et al., 1996; inositol polyphosphate binding, Fukuda et al., 1995; Schiavo et al., 1996; binding to endocytic proteins, Zhang et al., 1994; von Poser et al., 2000; or Ca2+ channels, Sheng et al., 1997; Kim and Catterall, 1997). Recent studies have shown that the C2B domain also participates directly in Ca2+-dependent phospholipid binding (Fernandez et al., 2001), an observation which in itself does not rule out any of the other proposed functions of the C2B domain. Synaptotagmin 1 binds to the SNARE (soluble N-ethylmaleimide-sensitive fusion protein receptor) proteins syntaxin 1 (Bennett et al., 1992; Chapman et al., 1995; Li et al., 1995), and SNAP-25 (synaptosomal protein of 25 kD) (Gerona et al., 2000; Zhang et al., 2002), providing a link between the putative calcium sensor and the fusion machinery (the SNARE complex consisting of syntaxin 1, synaptobrevin 2, and SNAP-25).
In deletion studies it was found that synaptotagmin 1 is required for fast release of neurotransmitter in various organisms (Littleton et al., 1993; Nonet et al., 1993; Geppert et al., 1994) and it was suggested that synaptotagmin 1 may be the calcium sensor for fast release (Geppert et al., 1994). Recent experiments have indicated that Ca2+ binding to the C2B domain of synaptotagmin 1 is a prerequisite for fast release in neurons (Mackler et al., 2002; Yoshihara and Littleton, 2002; Shin et al., 2003). Furthermore, mutations that affected the Ca2+-binding affinities of the C2A domain without affecting binding to the double C2A-C2B domain were without effect in vivo (Fernández-Chacón et al., 2002). Whereas these data show that Ca2+ binding to synaptotagmin is of paramount importance for function, they do not show whether this Ca2+-binding step directly underlies the calcium dependence of exocytosis triggering. To make this point a mutation would be needed that would modify rather than eliminate calcium binding. The mutation R233Q in the C2A domain fulfills this criterion; it decreases the affinity of Ca2+-dependent phospholipid binding to the double C2A-C2B domain by a factor of two and causes a concomitant twofold shift in the dependence of EPSC (excitatory postsynaptic current) size on extracellular [Ca2+] in hippocampal autaptic cultures (Fernández-Chacón et al., 2001). This finding is widely accepted as proof of the role of synaptotagmin 1 as the Ca2+ sensor for release. However, two problems with recording in hippocampal autaptic neurons prevent a more detailed interpretation:
(1) Since the [Ca2+] at the release site probably deviates by 2–3 orders of magnitude (being in the order of 10–20 μM in the Calyx of Held; Schneggenburger and Neher, 2000) from the extracellular [Ca2+] (assayed in the range 1–12 mM; Fernández-Chacón et al., 2001) extrapolating a Ca2+ dependence between extracellular and local intracellular [Ca2+] can lead to apparent shifts along the [Ca2+]-axis for a number of reasons. For instance, it is well known that there is a nonlinear relationship between the extracellular [Ca2+] and the [Ca2+] at the release site which could cause distortion of the release-Ca2+ relationship.
(2) Synaptic vesicles in neurons (including hippocampal neurons) are known to have heterogeneous release probabilities (Rosenmund et al., 1993; Wu and Borst, 1999; Burrone and Lagnado, 2000; Sakaba and Neher, 2001). The observed effects could be explained by more or less stabilization or recruitment of the vesicles with the higher release probability depending on whether wild-type synaptotagmin 1 or the R233Q mutant is expressed. It is not known whether in neurons the heterogeneous release probability is a result of different intrinsic properties of the vesicles (e.g., due to differences in the maturation state), or different localization of the vesicles with respect to calcium channels. The observation that synaptotagmin 1 binds to the synprint site of calcium channels led to the suggestion that synaptotagmin promotes high release probability by positioning vesicles near Ca2+-channels (Sheng et al., 1997; Kim and Catterall, 1997).
To overcome these alternative explanations, a demonstration of Ca2+ sensor function should preferably involve measurement of intracellular [Ca2+] at the release site as a prerequisite for determining the relation between the biophysical properties of the calcium sensor for exocytosis and the biochemical properties of synaptotagmin 1. Particularly, it would be important to demonstrate that a change in the response to Ca2+ caused by a mutation in synaptotagmin 1 is not caused by an overall increase or decrease in release, but a change in Ca2+ affinity of the release apparatus.
Chromaffin cells from adrenal medulla allow exactly this type of experiment. Even though primed vesicles in chromaffin cells have a heterogeneous release probability, this heterogeneity is well-defined and can be accurately assayed. Furthermore, a detailed biophysical model of calcium triggered exocytosis has been derived (Voets, 2000). The response to a step elevation of [Ca2+]i (caused by flash photolysis of caged-Ca2+) typically consists of a triphasic capacitance increase. The two faster components (collectively referred to as the exocytotic burst) represent the heterogeneity of vesicular release probability in the sense that they comprise release of two types of vesicles that were in a Ca2+-triggerable state at the time of the flash (Voets, 2000). The fast exocytotic burst phase represents fusion of vesicles residing in the readily releasable pool (RRP, high release probability), whereas the slow exocytotic burst phase represents fusion of vesicles from a pool of slowly releasable vesicles (SRP, low release-probability). The slowest phase of the flash response (τ of seconds) is called the sustained phase and is thought to represent priming of new vesicles (i.e., refilling of SRP and RRP), followed by fusion as long as the [Ca2+]i remains high (Voets, 2000). In chromaffin cells from synaptotagmin knock-out mice, the fastest exocytotic burst phase—and therefore the RRP—was missing, but the slow burst phase and the sustained phase were still present (Voets et al., 2001). The conclusion from that study was therefore that synaptotagmin 1 is required for the formation or stabilization of the RRP, or for triggering fusion from the RRP. These two possibilities, however, could not be distinguished.
Here we examine secretion from chromaffin cells from mutant mice carrying the R233Q point mutation in the C2A-domain of synaptotagmin 1 (Fernández-Chacón et al., 2001). It should be noted that the R233Q point mutation, although localized to the C2A-domain, affects the overall Ca2+ affinity of the double C2A-C2B domain fragment in which both the C2A and the C2B domain bind Ca2+ (Fernández-Chacón et al., 2001). We wanted to test the hypothesis that Ca2+-dependent lipid binding to synaptotagmin 1 is the event that triggers exocytosis. If this is the case a concomitant shift in the intracellular Ca2+ dependence of secretion should be apparent in mutant cells. Alternatively, if synaptotagmin stabilized an intrinsically fast and highly Ca2+-sensitive state of the release apparatus one would expect a smaller relative amplitude of the readily releasable component (RRP) for the R233Q mutant, since its stabilizing action should be weaker.
MATERIALS AND METHODS
Preparation of Chromaffin Cells
For preparation of cells we took advantage of a newly developed method for enzymatic (papain-based) isolation of chromaffin cells from the adrenals of a single mouse (Sørensen et al., 2003). The method was developed for single mouse embryos, but was used in the current work on young adult mouse adrenals. The only modifications required for adult mice were to rip open the adrenals by tweezers before the papain treatment, and increase the duration of this treatment from 40 min to 45–75 min. All analysis were performed on cell preparations prepared in parallel from homozygous wild-type (WT) and R233Q littermates derived from matings between heterozygotes of WT/R233Q knock-in (Fernández-Chacón et al., 2001).
Capacitance and Calcium Measurements
Experimental procedures related to whole-cell patch-clamp and capacitance measurements were performed essentially as described (Voets, 2000). Flashes of UV light were generated by a flash lamp (Dr. Rapp Optoelektronik, Hamburg, Germany) and coupled to the epifluorescence port of an inverted Zeiss 135 TV microscope equipped with a 40× FLUAR oil immersion objective (NA 1.3) (ZEISS). Fluorescence excitation light was generated by a monochromator (Polychrome II; TILL Photonics) and a two-port epifluorescence condensor was used for coupling 85% of the flash light and 15% of the monochromator light into the microscope (TILL Photonics). NP-EGTA was supplied by G. Ellis-Davies (MCP Hahnemann University, Philadelphia). Cells were bathed in extracellular solution (in mM: 150 NaCl, 2.8 KCl, 2 CaCl2, 1 MgCl2, 10 Na-HEPES, 2 mg/ml glucose, pH 7.2, Osmolarity ∼310 mOsm) and patched using a pipette solution containing the photolysable calcium chelator NP-EGTA (in mM: 109 Cs-glutamate, 35 Cs-HEPES, 2 Mg-ATP, 0.3 Na-GTP, 5 NP-EGTA, 1 CaCl2, 400 μM Fura-4F (Molecular Probes), 400 μM Furaptra (Molecular Probes), pH 7.2, Osmolarity ∼300 mOsm). Calcium measurements were performed by dual-wavelength ratiometric fluorimetry as described (Voets, 2000; Sørensen et al., 2002) using a 1:1 mixture of the calcium-sensitive dyes Fura-4F (KD = 1 μM) and Furaptra (KD = 40 μM). The signal (ratio of flurescence with excitation at 350 and 380 nm) was calibrated intracellularly by dialyzing the cells in whole-cell configuration with eight different pipette solutions with known [Ca2+]i. Calcium ramps were elicited by the fluorescence excitation light, alternating between 350 and 380 nm, so that photolysis of NP-EGTA could be combined with simultaneous measurement of [Ca2+]i. All measurements were done at room temperature.
For kinetic analysis individual capacitance traces were fitted with a triple exponential function:
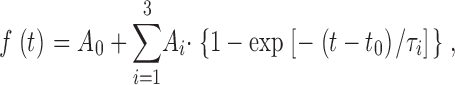 |
(1) |
for t > t 0, where A0 is the capacitance of the cell before the flash, t 0 is the time of the flash. The amplitudes (Ai) and time constants (τi) of the two faster exponentials define the size and release kinetics of the fast and the slow exocytotic burst, respectively. The secretory delay (time from the flash until the beginning of the capacitance increase) was found by extrapolating the fit function backward in time until it crossed the baseline value (ΔCm= 0, see Fig. 1 C). The slowest exponential was included in order to correct for the sustained component of release during the fit, but it was not used to estimate any parameters, since the time constant of the sustained component is too slow to be measured accurately in this way. The sustained component was in some cases fitted better by a straight line, in other cases an exponential with a time constant in the several-second range seemed more appropriate. By the fitting of Eq. 1 the linear case was in practice fitted with a third-exponential with a very long time constant (so that the exponential approaches a straight line). To obtain the rate of sustained release (Fig. 2 D, right) we subtracted the amplitudes of the fast and slow burst (A1 and A2) from the total amount of secretion up to 5 s after the flash and calculated the linear rate.
Figure 1.

R233Q mutant cells display robust capacitance responses to flash photolysis of caged Ca2+. (A) After whole-cell dialysis of an example WT cell with a pipette solution containing a photolysable Ca2+-cage, the [Ca2+]i was increased by a brief flash of UV light at 0.5 s (top). The simultaneous capacitance measurement with high time resolution shows a fast secretory response (bottom). (B) Similar experiment as in A shown for an example R233Q cell where the post-flash [Ca2+]i reached comparable levels. Note that the difference in the sustained rate of secretion seen between the two examples was not a general feature (compare Fig. 2 D, right). (C) The first 200 ms after the flash. By plotting the two example responses on the same time scale it was found that the R233Q secretion lacked behind WT secretion. Fitting of a sum of three exponential components (gray lines) to the capacitance increase was possible in both cases. Fast burst secretion from the R233Q cell displayed a longer time delay (13.5 ms instead of 4.3 ms) and a slower fast time constant (32.5 ms instead of 22.9 ms) than the WT cell.
Figure 2.

Slow-down of secretion in R233Q cells is caused by a decrease in fast secretory rate constant and a increase in secretory delay in a Ca2+-dependent manner. (A) Rate constant (=1/time constant) of the fast burst component as a function of post-flash [Ca2+]i for WT (open circles) and R233Q (closed squares) cells (left). The mean (±SEM) rates in R233Q cells (filled bar) were significantly lower than WT cells (open bar) when considering cells with post-flash [Ca2+]i > 10 μM (right). (B) Secretory delay as a function of post-flash [Ca2+]i (left). The secretory delays in R233Q cells (filled bar) were significantly longer than WT cells (open bar) when considering cells with post-flash [Ca2+]i > 10 μM (right). (A and B) The black lines are simulation of a 4-Ca2+ binding site model with parameters obtained by fitting to calcium ramp data (full line, WT; dashed line, R233Q. For parameters see legend to Fig. 3 and text). Gray lines are simulation of a 3-Ca2+ binding site model, for comparison. (C) Rate constant of the slow burst component as a function of post-flash [Ca2+]i (left) and mean ± SEM rates (right). Line is the simulation of a 3-Ca2+ binding site model according to Voets (2000). (D). Mean ± SEM of the amplitudes of the fast and slow burst component (left), and rate of sustained release (right) for WT (open bars) and R233Q (closed bars) cells. *, P < 0.05; **, P < 0.01 (Mann-Whitney test).
Mathematical Model
The model (Fig. 3 D) contains 11 free parameters and is underdetermined by the secretion detected during a calcium ramp. However, some of these parameters are specified by flash photolysis data and using this information it is possible to determine some other parameters or parameter combinations. For instance, the sustained component (steady secretion at high [Ca2+]i after release of the pools) will be given by k2. The size of the SRP before the ramp (assuming equilibrium) will be proportional to the ratio k2/k−2, and the proportion that the RRP makes up of the entire releasable pool (SRP+RRP) will be given by k1/(k−1 + k1), even though no information can be gained on each of the parameters, k1 and k−1. The model was fitted to calcium ramp data by first solving the steady-state condition in order to identify pool sizes (which depends on the ratios k2/k−2 and k1/(k−1 + k1)). We varied k2, k−1, k−2, and α during the fit while keeping k1 equal to the previously measured value (0.12 s−1; Voets, 2000). The model was then integrated forward in time using a fourth order Runge-Kutta routine with automatic step size control (Press et al., 1997) and with the calcium ramp providing the input. Given the order of magnitude of α- and β-values, as determined by flash experiments, the timing of the secretion of the pools will be governed by β/α (which is equal to KD, the equilibrium constant for calcium binding), as shown in Fig. 4 B and discussed in the text. Parameter optimization was obtained by performing repetitive simulations in a backward simplex scheme to minimize the sum of squared deviations between data and model (Press et al., 1997).
Figure 3.

A higher Ca2+ threshold for secretion in R233Q cells can be explained by an increase in the KD of the Ca2+-sensor for secretion. (A and B) Example calcium ramps (top) and simultaneously measured capacitance changes (bottom) for a WT (A) and a R233Q (B) cell. Superimposed is shown a regression line (straight dashed line) used for deriving the Ca2+-threshold for secretion (C), and the fit of a model of secretion (D) to the ramp data (gray curve) together with the assumed exocytosis timecourse from the RRP and SRP (gray dashed curves). (C) Mean ± SEM of the Ca2+-threshold for secretion from WT (open bar, N = 22 cells) and R233Q (closed bar, N = 25) cells. (D). Model for secretion from the SRP and RRP pools. Fixed parameters during the fit were: βs = 4 s−1, γs = 20 s−1,βf = 56 s−1, γf = 1450 s−1, k1 = 0.12 s−1 (Voets, 2000). (E) Mean ± SEM of the fitted KD (=β/α) for the fast and slow calcium sensor for WT (open bar, N = 22 cells) and R233Q (closed bar, N = 25) ***, P < 0.001 (Mann-Whitney test).
Figure 4.

Analysis of the model for secretion presented in Fig. 3 D. (A) Simulation of fusion of the RRP after flash stimulation to 23 μM calcium (black curve). Illustrated is the fitting of a single exponential component to the fusion curve (red curve) and the determination of the delay. (B) Simulation of fusion of both RRP and SRP (including a sustained component) according to the model in Fig. 3 D during ramp stimulation. The capacitance units were normalized to the sum of the SRP and RRP pools (which had the same size). (Top) Typical calcium ramp. Parameters used for the thick black curve are those found for WT cell in the present paper. Color coded is shown the effect of decreasing α (by a factor of 2), increasing β (by a factor of 2.4), decreasing γ (by a factor of 4), or increasing b (from 0.55 to 1). (C and D) Delay (C) and rate (D) versus post-flash calcium (determined as shown in A) for RRP fusion using the parameters found for WT cells (thick black line), or following the changes of α (decrease by a factor of 2), β (decrease by a factor of 2.4), γ (increase by a factor of 4), or b (from 0.55 to 1). The model simulation show that only a decrease in α cause simultaneous decrease of rates at low calcium and increase of delays.
For analyzing flash-data a simplified model encompassing only one vesicle pool (the SRP or the RRP) with a Ca2+ sensor was used for deriving rate constant-versus-calcium and delay-versus-calcium fits (lines in Fig. 2, A–C, and Fig. 4). For these computations the calcium-input was taken to be a step-function, jumping from zero to some set value. The model was now integrated forward in time to simulate the fusion of a single pool of vesicles following the step (see Fig. 4 A). After the simulation a single exponential component was fitted to the fusion curve, and extrapolation to zero was used to determine the delay (Fig. 4 A) in the same way as for the recorded capacitance data. This procedure was repeated at different calcium concentrations between 2 and 100 μM and the rate constant versus calcium and delay versus calcium relationships were drawn (curves in Fig. 2, A–C, and Fig. 4, C–D). Computations were performed by macros written for Igor Pro version 3.16 PPC (Wavemetrics).
Amperometry
Carbon fiber electrodes were prepared from 10-μm diameter fibers (Amoco Performance Products) as described previously (Schulte and Chow, 1996). Secretion was elicited by dialyzing the cells in the whole-cell configuration with a pipette solution containing 1 μM calcium (88 mM Cs-glutamate, 28 mM Cs-HEPES, 2 mM Mg-ATP, 0.3 mM Na-GTP, 10 mM BAPTA, 8–10 mM CaCl2, 400 μM Fura-4F, 400 μM Furaptra, pH 7.2). The carbon fiber was held at 700 mV over the bath Ag/AgCl electrode by an EPC-7 and pressed against the cell membrane. The [Ca2+]i varied from 500 nM to several μM due to differences in leak current and Ca2+ extrusion capacity of the cells. Cells which did not show secretion because of too low [Ca2+]i were depolarized in order to induce Ca2+ influx. The carbon fiber electrodes were cut by a new scalpella blade against a clean glass surface between each measurement. The electrodes themselves were used only a few times (1–4 times and when possible symmetrical between the two cell types) in order to avoid introducing a significant between-electrode variation component into the dataset. Currents were filtered at 3-kHz bandwidth and sweeps of 10-s measurements recorded by an EPC-9 controlled by Pulse software v. 8.31 (HEKA Elektronik). Upon loading into Igor Pro ver. 3.16 PPC (Wavemetrics) data were digitally filtered at 1 kHz by a custom-written Gaussian filter. The rms noise of the background current was 0.4–0.7 pA. Data analysis was performed as described in the text by a custom-written macro running under Igor Pro. Further details of this macro are described elsewhere (Sørensen et al., 2003). Analysis was restricted to amperometric peaks with amplitude >10 pA. Statistical analysis was performed by JMP 5 (SAS Institute).
RESULTS
Flash photorelease of caged calcium typically increased [Ca2+]i to >10 μM and resulted in robust secretion from both WT and R233Q cells as assayed by capacitance increase (Fig. 1, A and B) . When fitting a sum of exponential functions to the capacitance traces (see materials and methods) the inclusion of a rapid component (τ = ∼20 ms) was necessary for both R233Q and WT cells. Therefore, in contrast to synaptotagmin knockout cells, in the R233Q mutants the fast exocytotic burst component, and therefore the RRP, persisted.
Closer inspection of the time course of WT and R233Q secretion revealed a difference in kinetics, with the R233Q secretion being slower than WT secretion for UV flashes resulting in comparable post-flash [Ca2+]i (Fig. 1 C). This difference was caused by both a longer time constant of fast burst secretion in the R233Q cells and by a longer secretory delay (Fig. 1 C). Plotting the rate constant (=1/time constant) for fast burst secretion and the secretory delay against the post-flash [Ca2+]i (Fig. 2, A and B) showed that increasing [Ca2+]i resulted in increasing rate constants for release and shorter secretory delays, as described previously (Voets, 2000). The relationships for the R233Q mutant cells were displaced toward higher [Ca2+]i (Fig. 2, A and B), indicating a difference in the Ca2+ dependence of fusion. Considering only those points where post-flash [Ca2+]i > 10 μM revealed significantly lower rate constants for the R233Q cells (Fig. 2 A, right), and longer delays (Fig. 2 B, right). In this subset of data average post-flash [Ca2+]i was similar for wild-type cells (26 ± 2 μM) and R233Q cells (25 ± 2 μM). A similar plot of the rate constant for the slow exocytotic burst against post-flash [Ca2+]i revealed no striking difference, even though rates did seem slower at low [Ca2+]i in R233Q cells (Fig. 2 C). Finally, we considered the pool sizes (the amplitudes in the exponential fits). We found significantly larger pool sizes, both for the RRP (amplitude of fast burst component) and the SRP (amplitude of slow burst component) in the R233Q mutant cells (Fig. 2 D). The rate of the sustained component of release was also on average higher in R233Q mutant cells, but this was not significant.
These data show that a mutation in the C2A domain that changes Ca2+-dependent phospholipid binding, alters the calcium dependence of the fast fusion rate constant and the secretory delay. The effect on the pool sizes was not expected and points to the possibility that the C2A domain of synaptotagmin may also act during priming (the process filling the two releasable vesicle pools, the SRP and RRP). Another possibility is that due to defective secretion in these cells the pool sizes are up-regulated either passively (by the accumulation of unfused granules) or actively by a regulatory mechanism. It should be noted that the relative proportion of the releasable vesicles pools that was made up of the RRP (e.g., the quantity RRP/(RRP+SRP), where SRP and RRP denotes pool sizes) was not changed in the R233Q mutant. Therefore, the R233Q mutation and (by inference) Ca2+-dependent phospholipid binding of synaptotagmin 1 does not influence the equilibrium between the SRP and RRP. This alternative explanation for the role of synaptotagmin 1 (Voets et al., 2001) can therefore be ruled out.
If the effect of the R233Q mutation is to decrease the calcium-affinity (increase the KD) of the calcium sensor for exocytosis, as would be expected from the biochemical data, then this should be even more noticeable at lower [Ca2+]i, where the calcium sensor would be far from saturation. We therefore stimulated the cells using a calcium-ramp protocol, where calcium was liberated from the calcium cage by light from the fluorescence light source alternating between 350 and 380 nm, combining slow Ca2+ release with measurement of [Ca2+]i (Sørensen et al., 2002). Simultaneous capacitance measurements showed sigmoid-shaped capacitance increases during the calcium-ramp protocol (Fig. 3, A and B) . Fig. 3, A and B, show responses from WT and R233Q cells chosen for display because the shape of the calcium ramp was almost identical between the two cells, allowing for easy comparison. Note that these cells had uncharacteristic large responses. Inspecting the ramp responses showed that R233Q cells started secretion at a significantly higher Ca2+-threshold than WT cells (Fig. 3, A–C).
To obtain a more quantitative description we next considered a biophysical model of the calcium sensor for exocytosis (Fig. 3 D), which in its essential features was previously shown to describe exocytosis from chromaffin cells (Voets et al., 1999; Voets, 2000). The model assumes that a number of Ca2+ ions bind to Ca2+-binding sites on the calcium sensor with association rate constant (“on-rate”) α, and dissociation rate constant (“off-rate”) β. Once all binding sites are occupied the vesicle can undergo exocytosis in an irreversible (and Ca2+-independent) process with rate constant γ. In the model in Fig. 3 D we have included separate calcium sensors (with separate α, β, and γs) for the RRP-associated calcium sensor (right part of the scheme), and the SRP-associated calcium sensor (left part of the scheme). The previous model (Voets, 2000) assumed three Ca2+-binding sites for both the RRP-associated and the SRP-associated calcium sensor. For reasons that will become apparent below we assumed four Ca2+-binding sites for the RRP-associated calcium sensor, and we also included a cooperativity factor (b) that increases the affinity of subsequent Ca2+-binding steps dependent on how many Ca2+ ions that have already bound. We further allowed for priming and unpriming through the rate k2, and the rate constant k−2, respectively, and for movement of vesicles between the RRP and SRP (but for simplicity only in the Ca2+-unbound state) through rate constants k1 and k−1. The values of k−2, k1, and k−1 were measured previously (Voets, 2000), and k2 was found to be calcium dependent, modeled as k2 = (rmax [Ca2+])/([Ca2+] + KD), where KD = 2.3 μM and rmax = 55 fF/s (Voets, 2000).
The model (Fig. 3 D) contains 11 free parameters and is thus quite complicated. We therefore first examined the model itself to see what one can conclude from flash photolysis and Ca2+-ramp experiments, respectively. During flash photolysis the [Ca2+]i changes in a step-like manner from a relatively low to a high value. This causes rapid fusion of vesicles in the RRP, and also the vesicles in the SRP will fuse, albeit somewhat slower. In this case the calcium binding and fusion reactions (governed by α, β, and γ) are much faster than the movement of vesicles between pools (governed by k1 and k−1; k−2 and k2). For purposes of flash photolysis experiments, therefore, one can perceive each of the releasable vesicle pools (SRP and RRP) as fusing in isolation. Thus, k1, k−1, k−2, and k2 set the initial pool sizes and can be ignored subsequently. Therefore, we constructed a mathematical model that encompasses a single vesicle pool (for both the SRP and the RRP), each fusing according to its α, β, γ, and b after a step elevation of [Ca2+]i (Voets, 2000). Fig. 4 A shows that under these conditions the fusion of the RRP follows a near-exponential time course, except for some delay. To derive fusion rate constants and delays, we fitted these simulations with a single exponential component and used extrapolation to zero to derive the exocytotic delay (Fig. 4 A), exactly as we had done when analyzing experimental capacitance data (Fig. 1 C). This was repeated with calcium steps to several values between 2 and 100 μM and the rate constant versus calcium and the delay versus calcium relationships were displayed in Fig. 4, C and D (thick black line is simulation using parameters for WT cells). The assumption that each pool can be seen in isolation may break down at the lower levels of [Ca2+] seen during calcium ramps. For these experiments we therefore simulated the entire model (Fig. 3 D) using the measured calcium ramp as an input. Fig. 4 B shows a typical calcium ramp together with the simulation of the model (thick black line is a simulation using the parameters found for WT cells). We next asked which parameters of the fast calcium sensor can be distinguished using flash photolysis and ramp measurements. We therefore repeated simulations while changing one of the parameters of the fast calcium sensor at a time in the direction that would tend to cause slower secretion (for α this would be a reduction, for β an increase, for γ a decrease, and for b an increase, see color coded traces in Fig. 4, B–D). The result from flash simulation showed the changes that one would expect; for instance if γ was decreased by a factor of four the rates were indistinguishable at lower [Ca2+]i and only at higher concentrations were a slightly lower rate and a longer delay observed. Increasing β (dissociation rate constant for Ca2+) caused markedly lower rates at low [Ca2+]i, whereas the delays were similar. Notably, only if α (association rate constant for Ca2+) was decreased were lower rates and longer delays found at lower [Ca2+]i. When comparing these simulations to the kinetic data obtained with the R233Q mutation and WT cells (Fig. 2, A and B), it is clear that the observed shift in calcium-dependence over the assayed range of [Ca2+]i can be explained within the framework of a sequential Ca2+-binding model only by a decrease of α in the R233Q mutant.
From the ramp experiments it is not possible to gain information about whether α is decreased or β increased, since in both cases is the capacitance increase displaced toward later times or higher calcium concentrations (Fig. 4 B). The reason for this is that the Ca2+ ramp is so slow that the sensor is in a “quasi steady-state” during the ramp, i.e., it will be almost in equilibrium with [Ca2+]i, and therefore information about on- and off-rates cannot be obtained. However, the ramp shape will depend critically on the ratio between β and α, i.e., the KD (=β/α) for calcium binding. Also a change in cooperativity (b) will change secretion since it will modify the mean affinity of the calcium-binding step. Finally, secretion is going to depend only very weakly on γf (as long as γf is relatively large) since the calcium concentrations during the ramp will not lead to saturation of the sensor (green trace, Fig. 4 B). The increased Ca2+-threshold for secretion in R233Q cells (Fig. 3) would therefore be qualitatively in agreement with the decrease in α observed in flash experiments. To check for quantitative agreement between the two stimulation regimes we fitted the model to ramp data by varying α for both calcium sensors (SRP and RRP) between conditions (WT or R233Q cells), while keeping β and γ constant at the previously estimated values (βf = 56 s−1, γf = 1450 s−1, Voets, 2000). The resulting differential equation model was first solved in the steady-state at resting [Ca2+]i in order to find the initial pool sizes and then integrated forward in time using the calcium ramp measurements to drive the model (materials and methods). It was found that the model could fit ramp capacitance increases quite satisfactorily (Fig. 3, A and B). One of the advantages of ramp stimulation is that each cell is assayed over a range of calcium concentrations, rather than at only one concentration (as by flash photolysis stimulation). Therefore, by fitting the model an estimate of α and hence of the KD for calcium binding to the calcium sensor is obtained for each cell. Thus, an accurate estimate of the mean KD can be obtained by measuring in several cells. By fitting several ramp responses we estimated the KD for the fast Ca2+ sensor to 11.4 ± 0.9 μM in WT cells (corresponding to αf = 4.9 μM−1s−1) whereas in R233Q cells it was 23.3 ± 1.6 μM (P < 0.001, corresponding to αf = 2.4 μM−1s−1) (Fig. 3 E). We also found that the KD for the slow Ca2+ sensor was increased (from 5.0 ± 0.7 μM to 9.1 ± 0.7 μM, P < 0.001) (Fig. 3 E); however, we are less confident about this result because fusion of the SRP is temporally “sandwiched” between the fusion of the RRP and the sustained component. We therefore cannot exclude that the apparent difference in KD for the slow Ca2+ sensor is due to a difference in kinetics of the sustained component. However, the increase in KD for the SRP sensor would be in agreement with the difference in kinetics at lower [Ca2+] seen in Fig. 2 C. The result for the RRP Ca2+ sensor is more solid, because the initiation of fusion from the RRP is well resolved in both flash and ramp data. After the fitting to ramp data we simulated flash experiments using the fitted values of α and the (fixed) values of β and γ without further fitting or parameter modification. The simulations agreed well with flash data from WT and R233Q cells (lines in Fig. 2, A and B), indicating that the two stimulation regimes (flash and calcium ramps) are in agreement.
The model for the Ca2+ sensor for exocytosis in previous work on chromaffin cells assumed three noncooperative (i.e., b = 1) Ca2+-binding steps for both the fast and the slow calcium sensor (Voets, 2000). We also fitted ramp data and flash data with this model. This did not change the conclusion of a twofold shift in the KD of the fast calcium sensor (unpublished data), and the fit to rate-calcium data were essential unchanged (gray lines in Fig. 2 A). However, in the present study we found that with that model the long delays after flashes found in the R233Q mutant, especially at low [Ca2+]i, could not be readily accounted for (gray lines in Fig. 2 B). Similar observations led to the assumption of cooperative Ca2+-binding in secretion models for retinal bipolar cells (Heidelberger et al., 1994), the Calyx of Held synapse (Schneggenburger and Neher, 2000), and cochlear inner hair cells (Beutner et al., 2001). In our work, the cooperativity coefficient was found to be ∼0.55 (this value was used through-out, also when fitting ramp data), which means that the first Ca2+ ion binds with a KD of β/α = 11 μM for WT cells, whereas the last Ca2+ ion bind with a KD of b3 × β/α = 1.9 μM. The displayed KDs (Fig. 3 E) therefore apply to the first Ca2+-binding event. It must be stressed that there is not a substantial or principal difference between the three or four calcium binding site models, and because of the considerable scatter in this kind of data it is possible that differences in recording conditions can lead to the preference of different models.
It has been reported that overexpression of synaptotagmin 1 in PC12 cells increased the duration of the amperometric “foot” signal that is thought to reflect the formation of a fusion pore preceding full vesicle fusion (Wang et al., 2001). We therefore investigated whether the synaptotagmin 1 mutation R233Q changed the characteristics of single fusion events. In these experiments, the cells were patched in the whole-cell configuration and dialyzed with a relatively low [Ca2+]i (∼1 μM), where the calcium sensor would be far from saturation and the effect, if any, of the R233Q mutation therefore noticeable. Both WT and R233Q cells displayed robust amperometric spikes (Fig. 5 A). Detailed analysis of spike kinetics was performed by a semiautomatic software routine which identified the spikes and ∼50–90% risetime, peak amplitude, halfwidth (duration at 50% peak amplitude), and charge (Fig. 5 B). To inspect the statistical structure of the data we displayed the cumulative distribution of each parameter for each cell investigated. Fig. 5 C shows the distributions of halfwidths from 10 randomly selected WT cells and 10 randomly selected R233Q cells. It is clear that substantial cell-to-cell variability was present. This was confirmed by a hierarchic (nested) analysis of variance (ANOVA), which showed highly significant among-cell (subgroup) variability regardless of the parameter tested (P < 0.0001, peak amplitude, risetime, halfwidth, and foot duration were tested after logarithmic transformation, cubic-root of the charge was tested without transformation). This result means that the spikes are not statistical independent but their characteristics depend strongly on the cell from which the spike was recorded, as has previously been described (Colliver et al., 2000). The consequence for the analysis of two different populations of cells is that pooling all spikes for a two-sample test assuming independence is not a valid test for the null hypothesis of no difference between the two cell populations. Instead, one can use the spikes to calculate one statistic for each cell, then compare this statistic between populations of cells (Colliver et al., 2000). We therefore calculated the cell median of each parameter. Furthermore, we averaged the cumulative distributions of each parameter between cells and displayed the mean distribution (in these distributions each cell has the same weight, regardless of the number of spikes obtained from that cell) of the cubic-root of the charge (Fig. 5 D, the cubic-root of the charge is expected to be proportional to the diameter of the fused vesicle), the 50–90% risetime (Fig. 5 E), the foot duration (Fig. 5 F), the halfwidth (Fig. 5 G), and the peak amplitude (Fig. 5 H) for WT and R233Q cells, respectively. The resulting mean distributions showed no striking differences between WT and R233Q spikes (Fig. 5, D–H). The cell medians for each parameters were averaged across cells and the mean (±SEM) for each cell population displayed as the insets in Fig. 5, D–H (for the foot duration the 80% fractile was chosen, because close to 40% of the cells did not have any detectable foot signal). Mann-Whitney tests between medians confirmed that there was no statistical significant differences (P > 0.1) between WT and R233Q cells in any of the parameters investigated. The above-mentioned hierarchic ANOVA also leads to a test of the hypothesis of no difference between WT and R233Q cell populations (groups) based on moments. This test was also insignificant for all parameters investigated (P > 0.1). We conclude that the mutation R233Q in our hands do not cause detectable changes in single-spike characteristics.
Figure 5.

Single event characteristics are unchanged in R233Q cells. (A) Amperometric current recordings from a WT and a R233Q cell. (B) Extraction of single event characteristics: A semiautomatic routine detected the spikes, performed baseline subtraction, identified the halfwidth (thick line with two crosses across the peak), the risetime, peak amplitude, duration of the foot signal (line with two crosses connects the beginning and end of the foot signal), and integrated the spike (gray area) to derive the charge. (C) Cell-distributions of halfwidths for 10 randomly selected WT cells (full lines) and 10 randomly selected R233Q cells (dashed lines). Note the large cell-to-cell variations in the position of the distribution. (D) Averaged cumulative distribution of cubic-root of the charge for WT cells (full lines), and R233Q cells (dashed lines). (Inset) Mean ± SEM of median Q1/3 for WT and R233Q cells. (E) Averaged cumulative distribution of 50–90% risetime. (Inset) Median risetime. (F) Averaged cumulative distribution of foot duration. (Inset) 80% fractile foot duration. (G) Averaged cumulative distribution of halfwidth. (Inset) Median halfwidth. (H) Averaged cumulative distribution of peak amplitude. (Inset) Median peak amplitude. A total of 2,436 spikes from 22 WT cells (41–236 spikes pr cell) and 2,086 spikes from 22 R233Q cells (19–165 spikes per cell) were analyzed.
DISCUSSION
Our data indicate that synaptotagmin 1 acts as a Ca2+-trigger during vesicle fusion. In the introduction we hypothesized that if Ca2+-dependent phospholipid binding to the C2 domains of synaptotagmin 1 was the parameter that determines the rate of release, then changing the KD for this binding reaction should shift the intracellular Ca2+ dependence of the vesicle release rate along the [Ca2+]i-axis. This prediction was born out by experiments on chromaffin cells from the R233Q mutant, which showed that mutant cells had lower secretory rates (Fig. 2 A), and longer secretory delays (Fig. 2 B) for a given [Ca2+]i, and a higher Ca2+ threshold of release (Fig. 3, A–C). In a mathematical model of the calcium sensor, which in its essential features was previously shown to describe secretion from chromaffin cells (Voets, 2000), all of these changes were consistent with a change in the KD of the fast calcium-sensor for exocytosis by a factor of two (Figs. 3 E and 4). The alternative interpretations set out in the introduction, which would predict a smaller amplitude of the fast RRP could clearly be rejected. Our assay system bypassed calcium influx through calcium channels and instead stimulated cells by photorelease of caged calcium. Thus, a function of synaptotagmin in colocalization of vesicles with calcium channels cannot be the basis for the observed difference in exocytosis triggering. This, of course, does not rule out that synaptotagmin may influence the spatial relationship between calcium channels and release sites and, thereby, lead to additional effects on release during depolarizations. Our amperometric measurements ruled out another alternative hypothesis: The synaptotagmin mutation did not alter the rate of fusion pore expansion. Together, our results show that Ca2+ binding of synaptotagmin 1 is unlikely to regulate events immediately upstream of fusion triggering (transitions between vesicles pools, colocalization with calcium channels), and also does not seem to be involved in events immediately following triggering (regulating fusion pore expansion). In biochemical measurements the R233Q mutations induced a twofold lower affinity for Ca2+-dependent lipid binding, whereas the binding to syntaxin was unchanged (Fernández-Chacón et al., 2001). This supports the view that Ca2+-dependent lipid binding, rather than Ca2+-dependent binding to components of the SNARE complex, is the event that triggers exocytosis (see also Shin et al., 2003).
It should be noted, though, that a shift in the Ca2+ dependence of exocytosis could also come about without changing properties of the Ca2+ sensor itself. This could be the case if the Ca2+-sensor coupled allosterically to exocytosis by shifting the equilibrium between two conformational states (“relaxed” and “tense”, which may correspond to conformations of the SNARE complex), of which only one (tense) supports exocytosis. Ca2+ would bind more tightly to the Ca2+ sensor in the tense state. In such a scheme a change of the energy level of any of the two states could lead to a change in the apparent Ca2+ sensitivity of exocytosis, even though the Ca2+ sensor per se was left unaffected. Examples for such effects can be found in the literature on Ca2+-dependent K+ channels (e.g., Cui et al., 1997). However, in the current work we can correlate changes in apparent affinity of the Ca2+ sensor with observed biochemical changes of Ca2+ binding, suggesting that the effect we see is on the Ca2+ sensor itself.
Recently, data obtained with the mutation of an aspartate residue in the C2A-domain of synaptotagmin 1 in Drosophila were proposed to suggest that the C2A domain of synaptotagmin 1 may not be involved in Ca2+ triggering (Robinson et al., 2002). We have found that similar mutations have a severe effect on Ca2+-dependent phospholipid binding only on the isolated C2A domain, but not on the double C2-domain fragment (Fernández-Chacón et al., 2002). In vivo, these mutations show no major changes in synaptic transmission consistent with the finding that the two C2-domains cooperate and may be partially redundant in Ca2+-dependent phospholipid binding (Fernández-Chacón et al., 2002). This is in contrast to the R233Q mutation studied here which, although the site is localized on the C2A-domain too, alters the apparent Ca2+-affinity of the entire C2A/B-domain fragment (Fernández-Chacón et al., 2001). Recent mutagenesis data on the C2A and C2B domain may indeed suggest that Ca2+-binding to the C2B domain is the prevailing event leading to phospholipid binding (Mackler et al., 2002; Shin et al., 2003). These and other results (Fernández-Chacón et al., 2002) suggest that the C2A domain contributes to the apparent Ca2+ affinity of the C2B domain, but that actual Ca2+ binding to the C2A domain may not be essential for triggering release. Whether this is correct or not is of no consequence for the interpretation of the data presented here.
It has been argued that synaptotagmin 1 acts not only to promote fusion at higher [Ca2+]i, but also to prevent release at lower [Ca2+]i, and that these functions may be mechanistically distinct (Yoshihara and Littleton, 2002). This idea was based on the observations that a synaptotagmin-null mutant displayed increased secretion during sustained Ca2+ increases (Yoshihara and Littleton, 2002), and that mutated synaptotagmins defective in triggering had an even more severe phenotype than the complete absence of synaptotagmin (Mackler et al., 2002). We note that in a simple sequential Ca2+-binding model of the calcium sensor the steep dependence of the fusion rate on the [Ca2+]i can be described simultaneously as promotion of fusion at high-[Ca2+]i and inhibition of secretion at low [Ca2+]i. Within this framework, we suggest that the increased secretion in synaptotagmin-null mutants may result from functional substitution with another calcium sensor with a higher affinity for calcium and lower maximal triggering rates. This calcium sensor could be one of several other synaptotagmin isoforms, for instance synaptotagmins 3–7 or 9 which are expressed in neuroendocrine cells and neurons (Mizuta et al., 1994; Li et al., 1995; Sugita et al., 2001; Fukuda et al., 2002; Saegusa et al., 2002). Of these, synaptotagmins 3, 5, and 7 have a higher calcium-affinity than synaptotagmin 1 (Sugita et al., 2001, 2002), whereas calcium binding and affinities of synaptotagmins 4, 6, and 9 are at present unknown. Thus, the action of synaptotagmin as a “fusion clamp” would result simply from its ability to displace the high-affinity sensor. This interpretation removes the potentially confusing discussion of whether the action of the R233Q mutant should be described as an increase in fusion clamping activity at low [Ca2+]i (for instance in ramp experiments), or whether the function is to impair fusogenic function at higher [Ca2+]i (flash data). In a sequential Ca2+-binding model of the Ca2+ sensor these two functions cannot be separated.
Overexpression of synaptotagmin 1 in PC12 cells was found to increase the duration of the fusion pore, whereas overexpression of synaptotagmin 4 decreased fusion pore duration (Wang et al., 2001). We did not find differences in any amperometric parameter with the R233Q mutation, even though vesicles were brought to fuse at relatively low [Ca2+]i, where a difference in synaptotagmin function due to the mutation should have been noticed. The most likely explanation for this is that the R233Q mutation regulates only the (calcium dependent) probability that a vesicle will fuse; however, once the initial fusion pore is formed the function of synaptotagmin 1 in triggering release is terminated. The interpretation of the overexpression approach is quite different since in this case the amount of synaptotagmin 1 in the vesicle membrane will increase over wild-type levels. This may cause fusion with a different stochiometry of calcium sensors and lead to interference with fusion pore dilation at a time after exocytosis triggering.
Although our data provide direct evidence that synaptotagmin 1 functions in the Ca2+-triggering step as opposed to immediately preceding or following steps, the data also raise many questions. We currently have no explanation for the increase in the total number of vesicles in the releasable pools in the R233Q mutant. Furthermore, our data do not provide insight into the nature of the Ca2+ sensors that are responsible for release from the SRP. Critical to addressing these questions will be the analysis of additional synaptotagmin 1 mutants, as well as mutants in other candidate Ca2+-sensor proteins.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft Grant SFB 523 (E. Neher).
Angus C. Nairn served as editor.
Abbreviations used in this paper: NP-EGTA, nitrophenyl-EGTA; RRP, readily releasable pool; SNARE, soluble N-ethylmaleimide–sensitive fusion protein receptor; SRP, slowly releasable pool; WT, wild-type.
References
- Augustine, G.J. 2001. How does calcium trigger neurotransmitter release? Curr. Opin. Neurobiol. 11:320–326. [DOI] [PubMed] [Google Scholar]
- Bennett, M.K., N. Calakos, and R.H. Scheller. 1992. Syntaxin: A synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 257:255–259. [DOI] [PubMed] [Google Scholar]
- Beutner, D., T. Voets, E. Neher, and T. Moser. 2001. Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron. 29:681–690. [DOI] [PubMed] [Google Scholar]
- Bollmann, J.H., B. Sakmann, and J.G.G. Borst. 2000. Calcium sensitivity of glutamate release in a calyx-type terminal. Science. 289:953–957. [DOI] [PubMed] [Google Scholar]
- Brose, N., A.G. Petrenko, T.C. Südhof, and R. Jahn. 1992. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 256:1021–1025. [DOI] [PubMed] [Google Scholar]
- Burrone, J., and L. Lagnado. 2000. Synaptic depression and the kinetics of exocytosis in retinal bipolar cells. J. Neurosci. 20:568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, E.R., S. An, J.M. Edwardson, and R. Jahn. 1996. A novel function of the second C2 domain of synaptotagmin. Ca2+-triggered dimerization. J. Biol. Chem. 271:5844–5849. [DOI] [PubMed] [Google Scholar]
- Chapman, E.R., P.I. Hanson, S. An, and R. Jahn. 1995. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J. Biol. Chem. 270:23667–23671. [DOI] [PubMed] [Google Scholar]
- Chapman, E.R., and R. Jahn. 1994. Calcium-dependent interaction of the cytoplasmic region of synaptotagmin with membranes. Autonomouse function of a single C2-homologous domain. J. Biol. Chem. 269:5735–5741. [PubMed] [Google Scholar]
- Colliver, T.L., E.J. Hess, E.N. Pothos, D. Sulzer, and A.G. Ewing. 2000. Quantitative and statistical analysis of the shape of amperometric spikes recorded from two populations of cells. J. Neurochem. 74:1086–1097. [DOI] [PubMed] [Google Scholar]
- Cui, J., D.H. Cox, and R.W. Aldrich. 1997. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca-activated K+ channels. J. Gen. Physiol. 109:647–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davletov, B.A., and T.C. Südhof. 1993. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J. Biol. Chem. 268:26386–26390. [PubMed] [Google Scholar]
- Dodge, F.A., and R. Rahamimoff. 1967. Co-operative action of calcium ions in transmitter release at the neuromuscular junction. J. Physiol. 193:419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez, I., D. Arac, J. Ubach, S.H. Gerber, O. Shin, Y. Gao, R.G.W. Anderson, T.C. Südhof, and J. Rizo. 2001. Three-dimensional structure of the synaptotagmin 1 C2B-domain: Synaptotagmin 1 as a phospholipid binding machine. Neuron. 32:1057–1069. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón, R., A. Königstorfer, S.H. Gerber, J. Garcia, M.F. Matos, C.F. Stevens, N. Brose, J. Rizo, C. Rosenmund, and T.C. Südhof. 2001. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 410:41–49. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón, R., O. Shin, A. Königstorfer, M.F. Matos, A.C. Meyer, J. Garcia, S.H. Gerber, J. Rizo, T.C. Südhof, and C. Rosenmund. 2002. Structure/function analysis of Ca2+-binding to the C2A-domain of Synaptotagmin I. J. Neurosci. 22:8438–8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, M., T. Kojima, J. Aruga, M. Niinobe, and K. Mikoshiba. 1995. Functional diversity of C2 domains of synaptotagmin family. J. Biol. Chem. 270:26523–26527. [DOI] [PubMed] [Google Scholar]
- Fukuda, M., J.A. Kowalchyk, X. Zhang, T.F.J. Martin, and K. Mikoshiba. 2002. Synaptotagmin IX regulates Ca2+-dependent secretion in PC12 cells. J. Biol. Chem. 277:4601–4604. [DOI] [PubMed] [Google Scholar]
- Geppert, M., Y. Goda, R.E. Hammer, C. Li, T.W. Rosahl, C.F. Stevens, and T.C. Südhof. 1994. Synaptotagmin I: A major Ca2+ sensor for transmitter release at a central synapse. Cell. 79:717–727. [DOI] [PubMed] [Google Scholar]
- Gerona, R.R.L., E.C. Larsen, J.A. Kowalchyk, and T.F.J. Martin. 2000. The C terminus of SNAP25 is essential for Ca2+-dependent binding of synaptotagmin to SNARE complexes. J. Biol. Chem. 275:6328–6336. [DOI] [PubMed] [Google Scholar]
- Heidelberger, R., R. Heinemann, E. Neher, and G. Matthews. 1994. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 371:513–515. [DOI] [PubMed] [Google Scholar]
- Kim, D.K., and W.A. Catterall. 1997. Ca2+-dependent and –independent interactions of the isoforms of the α1A subunit of brain Ca2+ channels with presynaptic SNARE proteins. Proc. Natl. Acad. Sci. USA. 94:14782–14786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C., B. Ullrich, J.Z. Zhang, R.G.W. Anderson, N. Brose, and T.C. Südhof. 1995. Ca2+-dependent and –independent activities of neural and non-neural synaptotagmins. Nature. 375:594–599. [DOI] [PubMed] [Google Scholar]
- Littleton, J.T., M. Stern, K. Schulze, M. Perin, and H.J. Bellen. 1993. Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca2+-activated neurotransmitter release. Cell. 74:1125–1134. [DOI] [PubMed] [Google Scholar]
- Mackler, J.M., J.A. Drummond, C.A. Loewen, I.M. Robinson, and N.E. Reist. 2002. The C2B Ca2+-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature. 418:340–344. [DOI] [PubMed] [Google Scholar]
- Mizuta, M., N. Inagaki, Y. Nemoto, S. Matsukura, M. Takahashi, and S. Seino. 1994. Synaptotagmin III is a novel isoform of rat synaptotagmin expressed in endocrine and neuronal cells. J. Biol. Chem. 269:11675–11678. [PubMed] [Google Scholar]
- Nonet, M.L., K. Grundahl, B.J. Meyer, and J.B. Rand. 1993. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 73:1291–1305. [DOI] [PubMed] [Google Scholar]
- Perin, M.S., V.A. Fried, G.A. Mignery, R. Jahn, and T.C. Südhof. 1990. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 345:260–263. [DOI] [PubMed] [Google Scholar]
- Press, W.H., S.A. Teukolsky, W.T. Vetterling, and B.P. Flannery. 1997. Numerical recipes in C. The art of scientific computing, 2nd edition. Cambridge University Press.
- Robinson, I.M., R. Ranjan, and T.L. Schwarz. 2002. Synaptotagmins I and IV promote transmitter release independently of Ca2+ binding in the C2A domain. Nature. 418:336–340. [DOI] [PubMed] [Google Scholar]
- Rosenmund, C., J.D. Clements, and G.L. Westbrook. 1993. Nonuniform probability of glutamate release at hippocampal synapse. Science. 262:754–757. [DOI] [PubMed] [Google Scholar]
- Saegusa, C., F. Mitsunori, and K. Mikoshiba. 2002. Synaptotagmin V is targeted to dense-core vesicles that undergo calcium-dependent exocytosis in PC12 cells. J. Biol. Chem. 277:24499–24505. [DOI] [PubMed] [Google Scholar]
- Sakaba, T., and E. Neher. 2001. Quantitative relationship between transmitter release and calcium current at the Calyx of Held synapse. J. Neurosci. 21:462–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo, G., Q.-M. Gu, G.D. Prestwich, T. Söllner, and J.E. Rothman. 1996. Calcium-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc. Natl. Acad. Sci. USA. 93:13327–13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger, R., and E. Neher. 2000. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 406:889–893. [DOI] [PubMed] [Google Scholar]
- Schulte, A., and R.H. Chow. 1996. A simple method for insulating carbon-fiber microelectrodes using anodic electrophoretic deposition of paint. Anal. Chem. 68:3054–3058. [DOI] [PubMed] [Google Scholar]
- Sheng, Z.-H., C.T. Yokoyama, and W.A. Catterall. 1997. Interaction of the synprint site of N-type Ca2+ channels with the C2B domain of synaptotagmin I. Proc. Natl. Acad. Sci. USA. 94:5405–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, O., J.-S. Rhee, J. Tang, S. Sugita, C. Rosenmund, and T.C. Südhof. 2003. Sr2+ binding to the Ca2+ binding site of the synaptotagmin 1 C2B domain triggers fast exocytosis without stimulating SNARE interactions. Neuron. 37:99–108. [DOI] [PubMed] [Google Scholar]
- Sugita, S., W. Han, S. Butz, X. Lu, R. Fernandéz-Chacon, Y. Lao, and T.C. Südhof. 2001. Synaptotagmin VII as a plasma membrane Ca2+ sensor in exocytosis. Neuron. 30:459–473. [DOI] [PubMed] [Google Scholar]
- Sugita, S., Y. Hata, and T.C. Südhof. 1996. Distinct Ca2+-dependent properties of the first and second C2-domains of synaptotagmin I. J. Biol. Chem. 271:1262–1265. [DOI] [PubMed] [Google Scholar]
- Sugita, S., O.H. Shin, W. Han, Y. Lao, and T.C. Sudhof. 2002. Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J. 21:270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen, J.B., U. Matti, S. Wei, R.B. Nehring, T. Voets, U. Ashery, T. Binz, E. Neher, and J. Rettig. 2002. The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc. Natl. Acad. Sci. USA. 99:1627–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen, J.B., G. Nagy, F. Varoqueaux, R.B. Nehring, N. Brose, M.C. Wilson, and E. Neher. 2003. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 114:75–86. [DOI] [PubMed] [Google Scholar]
- Voets, T. 2000. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 28:537–545. [DOI] [PubMed] [Google Scholar]
- Voets, T., E. Neher, and T. Moser. 1999. Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron. 23:607–615. [DOI] [PubMed] [Google Scholar]
- Voets, T., T. Moser, P. Lund, R.H. Chow, M. Geppert, T.C. Südhof, and E. Neher. 2001. Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc. Natl. Acad. Sci. USA. 98:11680–11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Poser, C., J.Z. Zhang, C. Mineo, W. Ding, Y. Ying, T.C. Südhof, and R.G.W. Anderson. 2000. Synaptotagmin regulation of coated pit assembly. J. Biol. Chem. 275:30916–30924. [DOI] [PubMed] [Google Scholar]
- Wang, C.T., R. Grishanin, C.A. Earles, P.Y. Chang, T.F.J. Martin, E.R. Chapman, and M.B. Jackson. 2001. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science. 294:1111–1115. [DOI] [PubMed] [Google Scholar]
- Wu, L.-G., and J.G.G. Borst. 1999. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 23:821–832. [DOI] [PubMed] [Google Scholar]
- Yoshihara, M., and J.T. Littleton. 2002. Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron. 36:897–908. [DOI] [PubMed] [Google Scholar]
- Zhang, J.Z., B.A. Davletov, T.C. Sudhof, and R.G. Anderson. 1994. Synaptotagmin I is a high affinity receptor for clathrin AP-2: implications for membrane recycling. Cell. 78:751–760. [DOI] [PubMed] [Google Scholar]
- Zhang, W., M.J. Kim-Miller, M. Fukuda, J.A. Kowalchyk, and T.F.J. Martin. 2002. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron. 34:599–611. [DOI] [PubMed] [Google Scholar]