

Abstract
Rod vision begins when 11-cis-retinal absorbs a photon and isomerizes to all-trans-retinal (ATR) within the photopigment, rhodopsin. Photoactivated rhodopsin triggers an enzyme cascade that lowers the concentration of cGMP, thereby closing cyclic nucleotide–gated (CNG) ion channels. After isomerization, ATR dissociates from rhodopsin, and after a bright light, this release is expected to produce a large surge of ATR near the CNG channels. Using excised patches from Xenopus oocytes, we recently showed that ATR shuts down cloned rod CNG channels, and that this inhibition occurs in the nanomolar range (aqueous concentration) at near-physiological concentrations of cGMP. Here we further characterize the ATR effect and present mechanistic information. ATR was found to decrease the apparent cGMP affinity, as well as the maximum current at saturating cGMP. When ATR was applied to outside-out patches, inhibition was much slower and less effective than when it was applied to inside-out patches, suggesting that ATR requires access to the intracellular surface of the channel or membrane. The apparent ATR affinity and maximal inhibition of heteromeric (CNGA1/CNGB1) channels was similar to that of homomeric (CNGA1) channels. Single-channel and multichannel data suggest that channel inhibition by ATR is reversible. Inhibition by ATR was not voltage dependent, and the form of its dose–response relation suggested multiple ATR molecules interacting per channel. Modeling of the data obtained with cAMP and cGMP suggests that ATR acts by interfering with the allosteric opening transition of the channel and that it prefers closed, unliganded channels. It remains to be determined whether ATR acts directly on the channel protein or instead alters channel–bilayer interactions.
Keywords: retinoids, phototransduction, photoreceptors, cGMP, CNG channels
INTRODUCTION
Cyclic nucleotide–gated (CNG) channels are members of the voltage-gated channel family and are the subject of many reviews (Finn et al., 1996; Pugh, 1996; Zagotta and Siegelbaum, 1996; Karpen, 1997; Li et al., 1997; Molday and Molday, 1998; Biel et al., 1999; Broillet and Firestein, 1999; Frings, 1999; Richards and Gordon, 2000; Bradley et al., 2001; Flynn et al., 2001; Kaupp and Seifert, 2002; Warren and Molday, 2002; Zimmerman, 2002; Matulef and Zagotta, 2003). CNG channels that mediate the light response in retinal rods are nonselective cation channels that are weakly voltage dependent and are thought to be tetramers consisting of one β- and three α-subunits. They can be modulated by a number of factors, including phosphorylation enzymes, Ca2+/calmodulin, and divalent cations. We recently found that rod CNG channels can also be modulated by all-trans-retinal (ATR) and related compounds (Dean et al., 2002).
ATR is a member of a class of compounds called retinoids that includes vitamin A and its derivatives (Nau and Blaner, 1999). Retinoids regulate a wide variety of physiological processes, including gene transcription, immune responses, and visual transduction. In rod visual transduction, the only direct action of light is the photoisomerization of the retinoid chromophore 11-cis-retinal to ATR within opsin, the protein portion of the photopigment rhodopsin, in the rod disk membrane. This isomerization induces a conformational change in rhodopsin, which leads to the activation of a GTP-binding protein, transducin. Transducin then activates a phosphodiesterase, which hydrolyzes cGMP to 5′-GMP. The resulting decrease in cGMP concentration causes closure of CNG ion channels in the rod outer segment plasma membrane, hyperpolarizing the cell, and reducing the release of glutamate onto retinal bipolar cells (Rodieck, 1998; Pugh and Lamb, 2000; Roof and Makino, 2000; Burns and Baylor, 2001; Fain et al., 2001; Zimmerman, 2001).
After light activates the transduction cascade, ATR is released from its binding pocket in rhodopsin into the rod disk interior, where it apparently binds an ABCR transporter that shuttles it across the disk membrane into the cytosol to be converted by retinol dehydrogenase to ATR (Sun et al., 1999; Weng et al., 1999; Ahn et al., 2000). Next, all-trans-retinol is transferred from the rod outer segment to the extracellular space, where it may be complexed with interphotoreceptor retinoid binding protein (IRBP; Pepperberg et al., 1993), which appears to protect it from degradation. All-trans-retinol then enters the surrounding retinal pigment epithelial cells, where it is converted back into 11-cis-retinal, which is eventually returned to the rod to be reinserted into opsin (Crouch et al., 1996; Saari, 1999). Each of these processes is occurring in the tens of minutes required to reset rhodopsin during dark adaptation after exposure to bright light. At the same time, other components in the transduction cascade are also being reset. This includes production of cGMP by guanylate cyclase, so that CNG channels can reopen.
Approximately 3 mM ATR is expected to be released from rhodopsin in bright light, if the rod outer segment is considered as a single compartment (Saari, 1999). Since retinoids have been shown to modulate several kinds of ion channels (Sidell and Schlichter, 1986; Bosma and Sidell, 1988; Fukuhara et al., 1997; Vellani et al., 2000; Zhang and McMahon, 2000, 2001), it seemed natural to ask whether they might also modulate rod CNG channels. We recently showed that ATR can induce long shut states in rod CNG channels, and that it acts in the nanomolar range at near physiological cGMP concentrations (Dean et al., 2002). Here we further characterize the inhibition of rod CNG channels by ATR, and suggest that ATR is a closed-state inhibitor that prefers unliganded channels and has profound effects on the channel's cGMP sensitivity. Our findings raise the question of whether ATR might contribute to the slow time course of dark adaptation by making it more difficult for channels to reopen despite the increase in cGMP concentration after cessation of light.
MATERIALS AND METHODS
Expression of Channels in Xenopus Oocytes
CNG channel clones for the bovine rod α (CNGA1; accession no. NM-174278) and rod β (CNGB1; EMBL/GenBank/DDBJ accession no. X89626) were provided by William N. Zagotta (University of Washington, Seattle, WA) (in the pGEMHE plasmid) and Robert S. Molday (University of British Columbia, Vancouver, Canada), respectively. The CNGB1 subunit was subcloned into the pGEMHE plasmid in our lab. The pGEMHE plasmid contained the untranslated sequence of the Xenopus β-globin gene to promote high protein expression in oocytes (Liman et al., 1992). Channel cRNA was made by in vitro transcription using Ambion's mMessage mMachineTM kit.
Partial ovariectomies were performed on anesthetized Xenopus laevis frogs, and individual oocytes were isolated by treatment with 1 mg/ml collagenase type 1A (Worthington) in a low-calcium solution (82.5 mM NaCl, 2.5 mM KCl, 5 mM HEPES, 1 mM MgCl2, at pH 7.6). Channel cRNA was injected into oocytes using a Drummond “NANOJECT” injector. Typically, ∼50 nl of 1 μg/μl cRNA was injected into each egg. For coexpression of α and β subunits, cRNA for CNGA1 and CNGB1 were mixed in a ratio of 1:4 before injection. For current measurements from multichannel patches, injected oocytes were incubated at 16°C for 1–12 d before patch clamp experiments. For single-channel studies, the same amount of cRNA was injected, but the oocytes were kept at 16°C overnight and then incubated at 4°C (to slow expression) for 1–12 d before patch-clamp experiments. Oocytes were stored in a solution containing: 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 5 mM HEPES, 1 mM MgCl2, 2.5 mM pyruvic acid, 100 U/ml penicillin, and 100 μg/ml streptomycin, at pH 7.6. The vitelline membrane was removed by mechanical dissection after treatment with a hypertonic solution containing: 100 mM N-methyl-D-glucamine, 2 mM KCl, 10 mM EGTA, 10 mM HEPES, and 1 mM MgCl2, at pH 7.4.
Electrophysiological Solutions and their Application
The cell chamber for patch-clamp experiments was a glass Petri dish. Water-soluble solutions were applied using a 36-solution patch perfusion system, RSC-100 rapid solution changer (Molecular Kinetics). Both sides of the patches were bathed in a low-divalent sodium solution consisting of: 130 mM NaCl, 200 μM EDTA, and 2 mM HEPES, at pH 7.2. The solution bathing the intracellular surface of the patch contained various concentrations of cGMP (Sigma-Aldrich) dissolved in the low-divalent solution. Niflumic acid (500 μM; Sigma-Aldrich) was added to the extracellular solution to block Ca2+-activated Cl− channels endogenous to Xenopus oocytes.
ATR stocks were made in 100% ethanol and kept in amber glass vials covered in aluminum foil and stored at −80°C or −20°C until use. The purity and stability of the ATR stock was checked by measuring the absorption spectrum (200–800 nm) with a Beckman DU640 spectrophotometer. ATR was applied to the intracellular or extracellular surface of patches by removing 50% of the bath volume, vigorously mixing the retinoid stock into this solution using a glass Pasteur pipette in a glass beaker, and then pouring this solution back into the remaining bath and mixing again. We found that the greatest concentration of ethanol (0.1%) applied to any patch had no effect on cGMP-activated current or on the seal resistance. Petri dishes and agar bridges were replaced after each ATR experiment. ATR (Sigma-Aldrich) stocks were applied to patches under dim room light conditions. Spectroscopic measurements showed no degradation of ATR under these conditions; however, degradation was apparent in brighter room light.
Electrophysiological Recordings and Analysis
Standard patch clamp methods were used to record currents from excised, inside-out, and outside-out oocyte patches. Pipette openings were typically 0.5–5 μm in diameter with resistances of 1.0–15 MΩ after fire polishing. All recordings were obtained at room temperature. Macroscopic currents were recorded at voltages ranging from −100 to +100 mV in 50-mV steps from a holding potential of 0 mV. Leak currents were measured in the low divalent solution without cGMP for inside-out patches, and in 5 mM MgCl2 to block the CNG channels for outside-out patches. These leak currents were subtracted from each record. All macroscopic currents from multichannel patches were measured in the steady-state after completion of voltage-dependent gating (Karpen et al., 1988) and before significant ion depletion effects (Zimmerman et al., 1988). For heteromeric channels, the presence of the β (CNGB1) subunit was confirmed by exposing the patch to l-cis-diltiazem, which sensitively blocks heteromeric channels but has little effect on homomeric channels (Chen et al., 1993). Single-channel records were obtained as sequential sweeps lasting 8 s each, while membrane voltage was held continuously at +80 mV. Pipettes were coated with dental wax before polishing to reduce the noise from electrode capacitance.
ATR was added to patches only after allowing for completion of the spontaneous increases in apparent cGMP affinity of the rod channel due to dephosphorylation by endogenous patch-associated phosphatases (Gordon et al., 1992; Molokanova et al., 1997). These increases in apparent cGMP affinity took tens of minutes and were monitored in inside-out patches by sampling the current periodically at cGMP concentrations (typically 10 μM) that were below the K1/2, while incubating the patch the rest of the time in saturating cGMP (2 mM) to accelerate the process (Molokanova et al., 1999). Since changing the cGMP concentration was not practical for outside-out patches, we simply incubated these patches in 2 mM cGMP for 40–60 min before applying ATR.
For ATR dose–response curves on inside-out patches, the bath contained either a saturating concentration of cGMP (2 mM) or a low concentration of cGMP that activated ∼7–8% of the maximal current at 2 mM cGMP. The current was monitored for about an hour after each addition of ATR to ensure that steady-state had been reached. Typically, one or two ATR concentrations were tested per patch. Experiments addressing the voltage dependence of ATR inhibition were performed at a constant holding potential of either +50 or −50 mV, with frequent sampling of the current at that holding potential until steady-state had been reached. Then the holding potential was jumped to the opposite voltage and again monitored until steady-state was reached. For most patches, the leak was rechecked at the end of the experiment by applying the low divalent solution to the patch through a glass capillary tube anchored in the bath and attached to a syringe.
Patch currents were recorded using an Axopatch 1B or 200 patch-clamp amplifier (Axon Instruments, Inc.) with analogue-to-digital converters connected to a Macintosh Quadra or G4 computer running Pulse software (Instrutech). The data were low-pass filtered at 2 kHz and sampled at 10 or 25 kHz. Data analysis was performed using the IgorPro software package (WaveMetrics) or QuB Suite (www.qub.buffalo.edu).
The model depicted in Fig. 8 B and used to fit the data in Figs. 3 and 8 A has the following definitions and equations:
Figure 8.

ATR appears to prefer closed, unliganded channels, and to interfere with channel opening. ATR dose–response relations for homomeric (CNGA1) channels activated by cGMP or cAMP. (A) Data were measured as described in Fig. 2 for ATR dose–response curves in saturating (2 mM) cGMP (blue filled circles), saturating (20 mM) cAMP (red squares; giving only 5% of the activation obtained with 2 mM cGMP), and low (∼10–15 μM) cGMP (green open circles; giving ∼7.4% of the activation obtained with 2 mM cGMP). Error bars are SEM values. Averaged data points from 2–7 patches were normalized to Io and fit with the model (B) described above (see materials and methods and results), where K1 = 9.0 × 1010 M−n, K2 = 4.0 × 1010 M−n, K3 = 2.8 × 1010 M−n, K4 = 2.0 × 109 M−n, and n = 1.43. For saturating and low cGMP, L = 19.8 and KcN = 7,200 M−1; for cAMP, L = 0.055 and KcN = 1,144 M−1. The red dashed line represents the prediction of the model for cAMP if ATR binding depends only on the open probability and not on ligand occupancy (i.e., K1 = K2 = K3 = 9 × 1010 M−n).
Figure 3.

cGMP dose–response relation for homomeric (CNGA1) rod channels with and without 400 nM ATR. Steady-state, cGMP-evoked currents were measured at +100 mV from several patches with and without 400 nM ATR. Averaged data were normalized to the maximum current obtained with 2 mM cGMP and no ATR, and fit with an allosteric model (smooth curves; see materials and methods and Fig. 8 for details). Data points are averaged from 2–7 patches, plotted with SEM (error bars). Circles: cGMP dose–response relation without ATR. Model parameters: L = 19.8, KcN = 7,200 M−1. Triangles: cGMP dose–response relation with 400 nM ATR. Model parameters: L = 19.8, KcN = 7,200 M−1, K1 = 9.0 × 1010 M−n, K2 = 4.0 × 1010 M−n, K3 = 2.8 × 1010 M−n, K4 = 2.0 × 109 M−n, R = 400 nM, and n = 1.43.
O = open, ion-conducting channel.
C1 − C4 = closed, nonconducting channels.
R = ATR.
I = cyclic nucleotide–activated patch current.
Imax = maximum possible current when all channels are open.
I0 = maximum possible current at a fixed cGMP or cAMP concentration with [R] = 0.
I/Imax = current normalized to Imax.
I/I0 = current normalized to I0.
KcN = association constant for cyclic nucleotide binding.
L = equilibrium constant for the allosteric opening transition of the fully liganded channel.
K1 − K4 = association constants for ATR binding.
n = cooperativity constant for ATR binding.
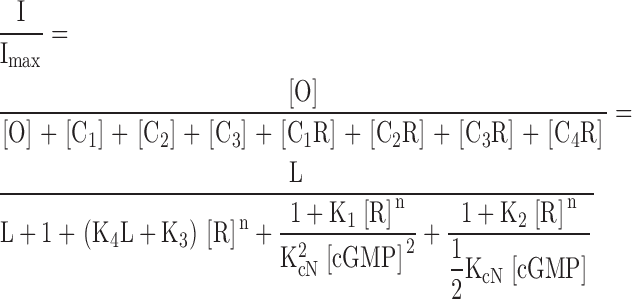 |
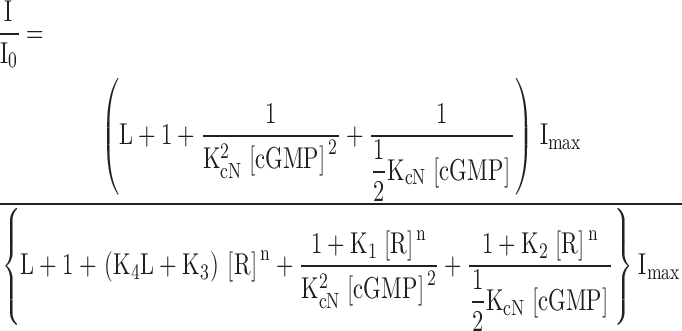 |
 |
Values of KcN and L for activation of the channel by cGMP and cAMP were derived from fits of cyclic nucleotide dose–response curves obtained in the absence of ATR (for cGMP: Fig. 3, circles; and for cAMP, Fig. 2 of Crary et al., 2000). These values of KcN and L are comparable to those in the literature (e.g., Fodor et al., 1997; Rosenbaum et al., 2003) and predict an open probability of 0.95 in 2 mM cGMP, which is similar to that measured in our single-channel recordings (Dean et al., 2002). An open probability of 0.05 in saturating cAMP is consistent with the ratio of maximum currents obtained for cAMP and cGMP after the spontaneous dephosphorylation process that shifts the cyclic nucleotide dose–response curves of these channels in excised patches (see materials and methods; Gordon et al., 1992; Molokanova et al., 1997).
Figure 2.

The ATR dose–response relations for homomeric (CNGA1) and heteromeric (CNGA1 + CNGB1) rod CNG channels. Cyclic GMP–activated currents were measured at +100 mV before and after ATR addition. Currents were monitored for ∼1 h after ATR addition to ensure that the ATR inhibition had reached steady-state. Averaged data, plotted with SEM (error bars) were fit with the Hill equation (solid or dashed curves): I/I0 = IC50 n/(IC50 n + [ATR]n), where I is current remaining at a given [ATR], I0 is the current before ATR addition, IC50 is the concentration of ATR required to achieve half maximal inhibition, and n is the Hill (cooperativity) coefficient. Filled circles, homomeric (CNGA1 only) rod channel dose–response relation for inhibition by ATR in saturating (2 mM) cGMP. Data points are averaged from 19 patches; IC50 = 220 nM and n = 1.4. Open triangles, heteromeric (CNGA1 and CNGB1) rod channel dose–response relation for ATR inhibition in saturating (2 mM) cGMP. Data points are from five patches; IC50 = 185 nM and n = 1.8. Open circles, homomeric (CNGA1) rod channel dose–response relation for ATR inhibition at cGMP concentrations evoking ∼7.4% activation. Data are from five patches; IC50 = 20 nM and n = 1.4.
RESULTS
Rod CNG channels were markedly inhibited by ATR, especially at low cGMP concentrations. This is illustrated in Fig. 1 , which presents current families from two multi-channel patches in response to a series of voltage pulses in the presence of saturating (left) or low (right) cGMP, with or without ATR. Greater inhibition was seen with only 30 nM ATR in low cGMP than with ten times as much ATR in high cGMP. Fig. 2 shows average ATR dose–response curves for patches exposed to low and saturating cGMP. For these data, “low” cGMP was that required to give ∼7.4% of the current obtained for each patch with saturating cGMP. Because there is some patch-to-patch (i.e., channel-to-channel) variability in apparent cGMP affinity, the exact concentration of cGMP needed to obtain 7.4% Imax also varied, but was ∼10–15 μM. The ATR dose–response curves were fit with Hill relations with cooperativity constants of 1.4–1.8, suggesting that more than one ATR molecule may interact with each channel.
Figure 1.

ATR inhibits homomeric (CNGA1) rod channels more potently at low than at saturating cGMP concentrations. Currents were measured from multichannel, inside-out patches of homomeric (CNGA1) rod channels. The raw traces represent families of cGMP-activated currents in response to voltage steps ranging from −100 to +100 mV in 50-mV increments, from a holding potential of 0 mV. Currents measured in the absence of cGMP were subtracted from all traces. (Left) Current families depicting control and inhibition at saturating cGMP (2 mM cGMP) by 300 nM ATR (50.6% inhibition). (Right) Current families showing control and inhibition by 30 nM ATR (67.6% inhibition) at a low cGMP concentration that elicits ∼7.4% of maximal cGMP induced current.
The native rod channel appears to be a heteromer containing one β subunit (CNGB1) and three α subunits (CNGA1; Weitz et al., 2002; Zheng et al., 2002; Zhong et al., 2002; for review see Zimmerman, 2002). As shown in Fig. 2, ATR was approximately equally effective at inhibiting heteromeric (CNGA1/CNGB1; open triangles) and homomeric (CNGA1; filled circles) rod channels expressed in the oocytes. Thus, in contrast to channel inhibition by pseudechetoxin (Brown et al., 1999), calmodulin (Chen et al., 1994), and l-cis-diltiazem (Chen et al., 1993), inhibition by ATR appears not to be substantially affected by the presence of the β subunit (CNGB1). This figure also gives quantitative evidence for the high apparent affinity of CNGA1 for ATR at low cGMP concentrations (open circles), as introduced in Fig. 1.
Fig. 3 shows that the effect of ATR inhibition was to reduce both the apparent affinity of the channel for cGMP and the maximal current at saturating cGMP. The saturating form of the lower curve suggests that ATR is not a competitive inhibitor of cGMP binding, since raising cGMP over a wide range does not relieve ATR inhibition. The downward shift of the curve with ATR suggests that ATR either inhibits opening of fully liganded channels and/or partially blocks the open channels, reducing the single-channel conductance. Previous single-channel analysis (Dean et al., 2002) suggests that a partial reduction in single-channel conductance is unlikely, and that ATR may induce long-lived closed or inactivated states. Although a complete block of the pore was not ruled out by those studies, open-pore block is unlikely because ATR inhibits more effectively at low than at high cGMP, i.e., at low, rather than high, open probability. Furthermore, the data shown in Fig. 3 are well-described by a model (solid curves) which assumes that ATR interferes with the allosteric opening transition of the channel and does not alter its single-channel conductance (see materials and methods and Fig. 8 for details of the model).
Inhibition of rod CNG channels (CNGA1) by ATR was slow and often demonstrated two components. Fig. 4 A shows a typical time course of inhibition by 400 nM ATR applied to the intracellular surface of an excised, inside-out, multichannel patch. This time course was described by two exponentials, with time constants of 119 and 1,667 s; for 11 patches showing two components, the mean time constants (±SEM) were 110 ± 18.1 s and 2,081 ± 666 s. Many patches demonstrated only a single exponential time course with a time constant of hundreds of seconds (854 ± 105 s, 12 patches). Because of the variability in the time course, we were unable to find any correlation between the relative magnitude or time constant of each component and the ATR concentration. We wondered whether access of the lipophilic ATR to its interaction site(s) depended on patch geometry, the lipid or protein composition of the membrane, or other variable patch properties (Karpen et al., 1988; Zimmerman et al., 1988; Ruknudin et al., 1991). When the amount of inhibition was changed by jumping voltage (see Fig. 7 below) or cGMP concentration (see Fig. 6 below) after loading the patch with ATR, changes in inhibition were more rapid and were always described by a single exponential. This supports the notion that the slow, variable, sometimes complex time course observed with ATR addition may reflect access problems relating to patch structure.
Figure 4.

Time course of inhibition by 400 nM ATR in 2 mM cGMP for inside-out and outside-out, multichannel patches containing homomeric (CNGA1) rod channels. (A) Time course of inhibition for this inside-out patch is best fit with a double exponential (τfast = 119 s and τslow = 1,667 s), although patches were often fit with a single exponential. (B) The time course for outside-out patches was so much slower than for inside-out patches that very little inhibition was measured during the lifetime of the patch; for this patch, a single-exponential fit gave τ = 6,748 s, although clearly the current had not reached steady-state by the end of the record.
Figure 7.

Inhibition of homomeric (CNGA1) rod channels by 200 nM ATR in saturating (2 mM) cGMP showed no significant voltage dependence other than that expected with voltage-dependent changes in open probability. For the top panel, an inside-out, multichannel patch was maintained at a holding potential of +50 mV in 200 nM ATR and 2 mM cGMP and monitored to steady-state. A double exponential provided the best fit to the time course of the inhibition, with τfast = 206 s and τslow = 3,779 s. After steady-state was achieved, the holding potential was changed to −50 mV. The time course of the resulting decrease in current (increase in inhibition) was best fit with a single exponential, with τ = 44 s. For the lower panel, the same experiment was performed on another patch, except that the initial membrane potential was −50 mV, and it was then switched to +50 mV. A double exponential provided the best fit to the initial time course of inhibition, with τfast = 77 s and τslow = 769 s. The time course of the increase in current (decrease in inhibition) after switching the voltage to +50 mV was best fit with a single exponential, with τ = 64 s. These changes in ATR inhibition with voltage are consistent with the expected voltage-dependent changes in open probability and the greater ATR inhibition of closed versus open channels.
Figure 6.

Current recovered with high cGMP after inhibition by ATR in low cGMP. After applying 100 nM ATR to a bath solution containing low cGMP (activating ∼7.5% of maximal current), the patch current was monitored until the inhibition reached a steady-state (99.5% inhibition). The cGMP concentration in the bath was then raised to 5 mM, with the continued presence of 100 nM ATR in the bath. The increase in [cGMP] opens some channels that were previously unliganded, and also presumably relieves some of the inhibition by ATR, since ATR is a less effective inhibitor in high than in low cGMP (Figs. 1 and 2). After a gap representing the time when cGMP was mixed into the chamber, the current recovery was fit by a single-exponential rise with a time constant of 82 s.
If inhibition by ATR is slow because it involves restricted access of ATR to a crevice or binding pocket of the channel that is accessible primarily from the intracellular surface, then inhibition might be slower or weaker when ATR is applied to the outside surface of the membrane. To test for this possibility, we measured the time course of inhibition when ATR was applied to outside-out patches. A typical time course for application of 400 nM ATR to an outside-out patch is shown in Fig. 4 B. These data demonstrate a single exponential with a time constant of 6,748 s (112 min). Similar results were obtained with five other patches. The slower, weaker action of ATR from the outside of the membrane would be consistent with a mechanism in which ATR exerts its effects via the intracellular surface of the membrane or channel (see discussion). As reported previously (Dean et al., 2002), reversal of ATR inhibition by washout from the chamber was difficult, presumably because of the preference of this lipophilic inhibitor for the membrane or hydrophobic binding sites over water. However, single-channel and multichannel records provide evidence for reversibility of inhibition by ATR. The one-channel patch shown in Fig. 5 shows channel opening after a long shut period characteristic of inhibition by ATR (Dean et al., 2002). This may reflect dissociation of ATR from its interaction site(s), and therefore reversibility, but we cannot rule out the appearance of a new channel in the patch (however, simultaneous openings of two channels were never seen in several minutes of recording from this patch).
Figure 5.

Single-channel recordings in the presence of ATR suggest that the channel can reopen after long-lived closed or inactivated states. Raw current traces in A–C were recorded at a holding potential of +80 mV from an inside-out patch containing one homomeric (CNGA1) channel. Data were filtered at 2 kHz, and the sampling rate was 25 kHz. The lower solid line represents the zero-current level when the channel was closed. The patch was bathed in saturating (2 mM) cGMP (A), and then 200 nM ATR was added to the bath (B and C). (A) A representative portion of a control trace depicting normal channel activity in saturating cGMP; the channel spends the majority of its time open. (B) After adding ATR, the channel activity remained relatively normal for a time. (C) Sometime before the start of this sweep, the channel entered a long-lived closed state, but reopening of the channel (with ATR still in the bath) occurred near the end of the sweep. Of the 3.2 s shown, the channel was in a closed state for 2.5 s; another portion of the record for this patch showed a 6-s closure. These long-lived closed states with ATR have been documented previously (Dean et al., 2002).
Better evidence for reversibility of ATR inhibition was provided by multichannel patches exposed to jumps in either cGMP or voltage. At the start of the experiment in Fig. 6 , the addition of 100 nM ATR to a patch in low cGMP gave >99% inhibition. This ATR concentration normally inhibits much less at saturating cGMP (see Fig. 2). When the cGMP concentration was raised to 5 mM in the continued presence of 100 nM ATR, there was an increase in current thought to reflect two processes: (a) the opening of channels that were initially associated with little or no cGMP and no ATR; and (b) the opening of channels that were initially associated with ATR, but lost ATR upon binding cGMP. The latter process is thought to account for most of the current increase, since most of the channels are inhibited by ATR at low cGMP, as seen in the data in Fig. 6 and supported by calculations of the model from Fig. 8.
If ATR is a closed-state inhibitor, it should work better at negative than at positive voltages, since the channel is slightly voltage dependent, with depolarization favoring opening. Since ATR has no net charge, no additional voltage dependence of inhibition is expected unless the ATR interaction site moves in the electric field or is obscured by the movement of another region of the channel. Fig. 7 illustrates a weak voltage dependence consistent with that of channel gating. Thus, in the left panel, applying ATR with a holding potential of +50 mV reduced the current as channels became inhibited, but switching to −50 mV reduced the current even more, as more channels closed and therefore became more susceptible to ATR inhibition. Reversing the order of the applied voltages (right panel) gave the reverse response. This slight voltage dependence was compared quantitatively with the voltage dependence of gating in the absence of ATR and was found to be similar. Thus, ATR did not induce any significant voltage dependence beyond the existing weak voltage dependence of channel gating. Interestingly, in both cases, the time course of the voltage-induced jump in current is much faster (τ = 44 s for −50 mV and 64 s for +50 mV) than that of the initial change in current during application of ATR and may reflect the time required for preequilibrated ATR to interact with the channel. The initial slow decreases in current shown in Fig. 7 are consistent with that depicted in Fig. 4 A and appear to reflect complex, patch-dependent parameters, as discussed above.
The finding that ATR is a much more effective inhibitor at low than at high cGMP (Figs. 1 and 2) could reflect a preference of ATR either for closed channels or for channels without bound cyclic nucleotide (unliganded channels). One way to distinguish between these two possibilities is with the partial agonist, cAMP (Gordon and Zagotta, 1995). For rod CNG channels, a saturating concentration of cAMP (20 mM) gives only ∼5% of the maximal activation obtained in saturating cGMP (2 mM). Fig. 8 A compares ATR dose–response relations for low cGMP, saturating cAMP and saturating cGMP on homomeric rod CNG channels. The model described in Fig. 8 B (equations in materials and methods) approximates all three relations, and assumes that ATR prefers closed over open channels, but also unliganded over liganded channels (K1 > K2 > K3 >> K4). We were not able to fit the data by assuming that all ATR equilibrium constants were equal, i.e., that inhibition depended only on whether the channel was closed, and not on the number of bound ligand molecules. This is illustrated by the red dashed curve in Fig. 8 A, which is a prediction for the cAMP experiment if ligand occupancy does not affect ATR inhibition (i.e., K1 = K2 = K3 >> K4). The model in Fig. 8 B further assumes that when the fully liganded, open rod channel binds ATR, it closes and cannot reopen with ATR bound. The data could not be fit with a model in which the rod channel could open with ATR bound. Thus, the data and model in Fig. 8 suggest that ATR prefers unliganded, closed channels over channels that are liganded and/or open.
Finally, the model depicted in Fig. 8 B also gave a reasonable description of the cGMP dose–response curves with and without ATR (Fig. 3). For these fits, we used the same constants as for the curves in Fig. 8 A, changing only the concentrations of cGMP and ATR to match the experimental conditions. Thus, the reduction in current by ATR at saturating concentrations of cGMP can be explained simply by an inhibition of channel opening by ATR, and it is not necessary to invoke a reduction in single-channel conductance or competition between ATR binding and cGMP binding.
DISCUSSION
Results presented here suggest that ATR is a potent, reversible, closed-state inhibitor of both homomeric (CNGA1) and heteromeric (CNGA1/CNGB1) rod CNG channels. ATR acts best from the intracellular side of the membrane to decrease the apparent agonist affinity, but it does not seem to compete with cyclic nucleotides at their binding domain. The inhibition is not appreciably voltage dependent, and may involve multiple ATR molecules per channel. Descriptions of the data with an allosteric model suggest that ATR prefers unliganded channels, but is also very effective on closed, liganded channels. The model assumes that ATR interacts more weakly with open channels and drives them to close as well.
It is not clear whether ATR has its effects by interacting directly with the channel protein or some intermediary protein, or by altering bilayer–channel interactions. If ATR alters bilayer–protein interactions, it may disrupt lipid rafts in the membrane and thereby alter the behavior of channels embedded in the rafts (Edidin, 1997; Brown and London, 1998; Jacobson and Dietrich, 1999; Martens et al., 2000; Brady et al., 2004), it may form retinoid-rich microdomains (Boeck and Zidovetzki, 1988) that affect channel function, or it may alter physical properties of the bilayer, such as curvature, at the lipid–protein interface (Lundbaek et al., 1996; Cantor, 1999; Lundbaek and Andersen, 1999). Changes in bilayer curvature, for example, have been shown to have dramatic effects on the dimerization of gramicidin channels (Lundbaek and Andersen, 1994). Evidence for lipid rafts has been found in rod outer segment membranes (Seno et al., 2001) and in oocytes (Luria et al., 2002), and a recent report (Brady et al., 2004) shows that disruption of lipid rafts drastically reduces the apparent cAMP affinity of olfactory CNG (CNGA2) channels. Finally, it should be mentioned that Schiff base reactions can occur between ATR and lipids; this offers another potential action of ATR via the bilayer. However, this mechanism seems unlikely here in light of our previous finding (Dean et al., 2002) that the rod CNG channel is inhibited quite well by all-trans-retinol, which cannot form a Schiff base.
On the other hand, retinoids, including ATR, are known to interact directly with proteins, such as rhodopsin, retinoid binding proteins, and transcription factors (Newcomer et al., 1998; Nau and Blaner, 1999; Noy, 2000; Kefalov et al., 2001). These interactions may involve covalent bonds. A putative ATR binding pocket within the channel protein might include residues at locations that are distant from each other in the primary sequence, but near each other in the three-dimensional structure of the channel; for example, in rhodopsin, the binding pocket is made up of residues that are contributed by adjacent transmembrane α helices. Direct interactions with the channel could involve hydrophobic crevices or domains, either among the transmembrane segments or within hydrophobic areas in the cytoplasmic amino- and carboxy-terminal domains. It is also possible that ATR acts by way of another protein associated with the channel, rather than directly on the channel; however, we have found similar inhibition of the channel in the native rod outer segment membrane (unpublished data), where protein (and lipid) components are different.
Our results suggest that ATR is a closed-state inhibitor, as are some other lipophilic agents, including tetracaine (Fodor et al., 1997) and dequalinium (Rosenbaum et al., 2004). However, unlike these inhibitors, ATR appears to discriminate not only between closed and open channels, but also between liganded and unliganded channels. This raises the question of whether ATR may interact at sites within or near the cyclic nucleotide binding domain. Although ATR does not appear to be a competitive inhibitor of cyclic nucleotide binding, it may interact with a region whose conformation changes in response to cyclic nucleotide binding (Matulef and Zagotta, 1999).
A comparison of channel inhibition by extracellularly and intracellularly applied ATR may give clues as to the mechanism of its action. Retinoids insert into bilayers, and flip-flop across them, very rapidly—on a millisecond time scale. However, they take at least seconds to leave a bilayer into an aqueous solution, and retinoid interactions with proteins take seconds to minutes (Noy, 1999). We found that channel inhibition was orders of magnitude slower and less effective when ATR was applied to the extracellular surface of the membrane than when it was applied to the intracellular surface. One possible explanation for this result is that the ATR interaction site(s) was accessible only from the intracellular surface of the channel. Thus, when ATR was applied extracellularly, the channel only sensed the small fraction of the applied ATR that had passed through the bilayer and remained near the intracellular surface long enough to find the interaction site(s).
The physiological relevance of ATR inhibition of rod CNG channels remains to be determined. A bright light sufficient to bleach all rhodopsin molecules in a rod is expected to produce 3 mM ATR in the rod outer segment (Saari, 1999). Although most of this ATR is expected to associate with the disk membranes, some will most likely be in or near the plasma membrane and therefore near the CNG channels. To date, the only retinoid binding protein that has been described in rods is IRBP, which is produced by the rods and secreted into the extracellular matrix. It is possible that IRBP may buffer intracellular, as well as extracellular, ATR. Furthermore, retinol dehydrogenase, which converts ATR to all-trans-retinol after light absorption, would also reduce the amount of ATR available to inhibit the CNG channels. However, the IC50 for ATR inhibition of the channel is 25 nM with ∼10–15 μM cGMP, and should be much lower when the free cGMP concentration is in the range expected in vivo: ∼6 μM in the dark and even lower in the light (Nakatani and Yau, 1988). Thus, it is reasonable to assume that some inhibition is possible in a rod exposed to bright, prolonged light. One potential caveat to this proposal is the fact that rod CNG channels are modulated by phosphorylation enzymes, Ca2+-binding proteins, and perhaps other agents; thus, their sensitivity to ATR in vivo may not be the same as that in our excised patches. If ATR does indeed inhibit the channels in the intact rod, what role might this inhibition play in visual transduction? One possibility is that it could contribute to the slow time course of dark adaptation after an exposure to bright and/or prolonged light, since the reduced apparent cGMP affinity of the channels would tend to make them remain closed even when the cGMP levels are recovering. Finally, inhibition of the channels by ATR may occur in certain disease conditions, such as in Stargardt's disease, in which there is an abnormal accumulation of ATR.
Acknowledgments
We thank Drs. Peter Calvert, Carter Cornwall, M.F. Crary, Rosalie Crouch, Clint Makino, Jeff Martens, Koji Nakanishi, David Ong, and Jack Saari for helpful discussions, and Elizabeth Seed for technical assistance.
This work was supported by a grant from the National Eye Institute, National Institutes of Health (EY07774).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: ATR, all-trans-retinal; CNG, cyclic nucleotide–gated; IRBP, interphotoreceptor retinoid binding protein.
References
- Ahn, J., J.T. Wong, and R.S. Molday. 2000. The effect of lipid environment and retinoids on the ATPase activity of ABCR, the photoreceptor ABC transporter responsible for Stargardt macular dystrophy. J. Biol. Chem. 275:20399–20405. [DOI] [PubMed] [Google Scholar]
- Biel, M., X. Zong, and F. Hofmann. 1999. Cyclic nucleotide gated channels. Adv. Second Messenger Phosphoprotein Res. 33:231–250. [DOI] [PubMed] [Google Scholar]
- Boeck, H.D., and R. Zidovetzki. 1988. NMR study of the interaction of retinoids with phospholipid bilayers. Biochim. Biophys. Acta. 946:244–252. [DOI] [PubMed] [Google Scholar]
- Bosma, M., and N. Sidell. 1988. Retinoic acid inhibits Ca2+ currents and cell proliferation in a β-lymphocyte cell line. J. Cell. Physiol. 135:317–323. [DOI] [PubMed] [Google Scholar]
- Bradley, J., S. Frings, K.-W. Yau, and R. Reed. 2001. Nomenclature for ion channel subunits. Science. 294:2095–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady, J.D., T.C. Rich, X. Le, K. Stafford, C.J. Fowler, L. Lynch, J.W. Karpen, R.L. Brown, and J.R. Martens. 2004. Functional role of lipid raft microdomains in cyclic nucleotide-gated channel activation. Mol. Pharmacol. 65:503–511. [DOI] [PubMed] [Google Scholar]
- Broillet, M.C., and S. Firestein. 1999. Cyclic nucleotide-gated channels. Molecular mechanisms of activation. Ann. NY Acad. Sci. 868:730–740. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- Brown, R.L., T.L. Haley, K.A. West, and J.W. Crabb. 1999. Pseudechetoxin: A peptide blocker of cyclic nucleotide-gated ion channels. Proc. Natl. Acad. Sci. USA. 96:754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns, M.E., and D.A. Baylor. 2001. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu. Rev. Neurosci. 24:779–805. [DOI] [PubMed] [Google Scholar]
- Cantor, R. 1999. Lipid composition and lateral pressure profile in bilayers. Biophys. J. 76:2625–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T.-Y., Y.-W. Peng, R.S. Dhallan, B. Ahamed, R.R. Reed, and K.-W. Yau. 1993. A new subunit of the cyclic nucleotide-gated cation channel in retinal rods. Nature. 362:764–767. [DOI] [PubMed] [Google Scholar]
- Chen, T.-Y., M. Illing, L.L. Molday, Y.T. Hsu, K.-W. Yau, and R.S. Molday. 1994. Subunit 2 (β) of retinal rod cGMP-gated cation channel is a component of the 240-kDa channel-associated protein and mediates Ca2+-calmodulin modulation. Proc. Natl. Acad. Sci. USA. 91:11757–11761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary, J.I., D.M. Dean, F. Maroof, and A.L. Zimmerman. 2000. Mutation of a single residue in the S2-S3 loop of CNG channels alters the gating properties and sensitivity to inhibitors. J. Gen. Physiol. 116:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch, R.K., G.J. Chader, B. Wiggert, and D.R. Pepperberg. 1996. Retinoids and the visual process. Photochem. Photobiol. 64:613–621. [DOI] [PubMed] [Google Scholar]
- Dean, D.M., W. Nguitragool, A. Miri, S.L. McCabe, and A.L. Zimmerman. 2002. All-trans-retinal shuts down rod cyclic nucleotide-gated ion channels: a novel role for photoreceptor retinoids in the response to bright light? Proc. Natl. Acad. Sci. USA. 99:8372–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edidin, M. 1997. Lipid micromains in cell surface membranes. Curr. Opin. Struct. Biol. 7:528–532. [DOI] [PubMed] [Google Scholar]
- Fain, G., H. Matthews, M.C. Cornwall, and Y. Koutalos. 2001. Adaptation in vertebrate photoreceptors. Physiol. Rev. 81:117–151. [DOI] [PubMed] [Google Scholar]
- Finn, J.T., M.E. Grunwald, and K.-W. Yau. 1996. Cyclic nucleotide-gated ion channels: an extended family with diverse functions. Annu. Rev. Physiol. 58:395–426. [DOI] [PubMed] [Google Scholar]
- Flynn, G.E., J.P.J. Johnson, and W.N. Zagotta. 2001. Cyclic nucleotide-gated channels: shedding light on the opening of a channel pore. Nat. Rev. Neurosci. 2:643–652. [DOI] [PubMed] [Google Scholar]
- Fodor, A.A., S.E. Gordon, and W.N. Zagotta. 1997. Mechanism of tetracaine block of cyclic nucleotide-gated channels. J. Gen. Physiol. 109:3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frings, S. 1999. Tuning Ca2+ permeation in cyclic nucleotide-gated channels. J. Gen. Physiol. 113:795–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara, S., H. Mukai, and E. Munekata. 1997. Activin A and all-trans-retinoic acid cooperatively enhanced the functional activity of L-type Ca2+ channels in the neuroblastoma C1300 cell line. Biochem. Biophys. Res. Commun. 241:363–368. [DOI] [PubMed] [Google Scholar]
- Gordon, S.E., and W.N. Zagotta. 1995. A histidine residue associated with the gate of the cyclic nucleotide-activated channels in rod photoreceptors. Neuron. 14:177–183. [DOI] [PubMed] [Google Scholar]
- Gordon, S.E., D.L. Brautigan, and A.L. Zimmerman. 1992. Protein phosphatases modulate the apparent agonist affinity of the light-regulated ion channel in retinal rods. Neuron. 9:739–748. [DOI] [PubMed] [Google Scholar]
- Jacobson, K., and C. Dietrich. 1999. Looking at lipid rafts? Trends Cell Biol. 9:87–91. [DOI] [PubMed] [Google Scholar]
- Karpen, J.W. 1997. Why do cyclic nucleotide-gated channels have the jitters? Biophys. J. 72:986–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpen, J.W., A.L. Zimmerman, L. Stryer, and D.A. Baylor. 1988. Gating kinetics of the cyclic-GMP-activated channel of retinal rods: flash photolysis and voltage-jump studies. Proc. Natl. Acad. Sci. USA. 85:1287–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp, U.B., and R. Seifert. 2002. Cyclic nucleotide-gated ion channels. Physiol. Rev. 82:769–824. [DOI] [PubMed] [Google Scholar]
- Kefalov, V., R. Crouch, and M. Cornwall. 2001. Role of noncovalent binding of 11-cis-retinal to opsin in dark adaptation of rod and cone photoreceptors. Neuron. 29:749–755. [DOI] [PubMed] [Google Scholar]
- Li, J., W.N. Zagotta, and H.A. Lester. 1997. Cyclic nucleotide-gated channels: structural basis of ligand efficacy and allosteric modulation. Q. Rev. Biophys. 30:177–193. [DOI] [PubMed] [Google Scholar]
- Liman, E.R., J. Tytgat, and P. Hess. 1992. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 9:861–871. [DOI] [PubMed] [Google Scholar]
- Lundbaek, J.A., and O.S. Andersen. 1994. Lysophospholipids modulate channel function by altering the mechanical properties of lipid bilayers. J. Gen. Physiol. 104:645–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbaek, J.A., and O.S. Andersen. 1999. Spring constraints for gramicidin-induced lipid bilayer deformations estimates using gramicidin channels. Biophys. J. 76:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbaek, J.A., P. Birn, J. Girshman, A.J. Hansen, and O.S. Andersen. 1996. Membrane stiffness and channel function. Biochemistry. 35:3825–3830. [DOI] [PubMed] [Google Scholar]
- Luria, A., V. Vegelyte-Avery, B. Stith, N.M. Tsvetkova, W.F. Wolkers, J.H. Crowe, F. Tablin, and R. Nuccitelli. 2002. Detergent-free domain isolation from Xenopus egg plasma membrane with properties similar to those of detergent-resistant membranes. Biochemistry. 41:13189–13197. [DOI] [PubMed] [Google Scholar]
- Martens, J., R. Navarro-Polanco, E. Coppock, A. Nishiyama, L. Parshley, T. Grobaski, and M. Tamkun. 2000. Differential targeting of Shaker-like potassium channels to lipid rafts. J. Biol. Chem. 275:7443–7446. [DOI] [PubMed] [Google Scholar]
- Matulef, K., and W.N. Zagotta. 1999. Molecular rearrangements in the ligand-binding domain of cyclic nucleotide-gated channels. Neuron. 24:443–452. [DOI] [PubMed] [Google Scholar]
- Matulef, K., and W.N. Zagotta. 2003. Cyclic nucleotide-gated ion channels. Annu. Rev. Cell Dev. Biol. 19:23–44. [DOI] [PubMed] [Google Scholar]
- Molday, R.S., and L.L. Molday. 1998. Molecular properties of cGMP-gated channel of rod photoreceptors. Vision Res. 38:1315–1323. [DOI] [PubMed] [Google Scholar]
- Molokanova, E., B. Trivedi, A. Savchenko, and R.H. Kram. 1997. Modulation of rod photoreceptor cyclic nucleotide-gated channels by tyrosine phosphorylation. J. Neurosci. 17:9068–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molokanova, E., F. Maddox, C.W. Luetje, and R.H. Kram. 1999. Activity-dependent modulation of rod photoreceptor cyclic nucleotide-gated channels mediated by phosphorylation of a specific tyrosine residue. J. Neurosci. 19:4786–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani, K., and K.-W. Yau. 1988. Guanosine 3′,5′-cyclic monophosphate-activated conductance studied in a truncated rod outer segment of the toad. J. Physiol. 395:731–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau, H., and W.S. Blaner. 1999. Retinoids: The Biochemical and Molecular Basis of Vitamin A and Retinoid Action. Springer, New York. 619 pp.
- Newcomer, M.E., R.S. Jamison, and D.E. Ong. 1998. Structure and function of retinoid-binding proteins. Subcell. Biochem. 30:53–80. [DOI] [PubMed] [Google Scholar]
- Noy, N. 1999. Physical-chemical properties and action of retinoids. Retinoids: The Biochemical and Molecular Basis of Vitamin A and Retinoid Action. H. Nau and W.S. Blaner, editors. Springer-Verlag, NY. 3–29.
- Noy, N. 2000. Retinoid-binding proteins: mediators of retinoid action. Biochem. J. 348:481–495. [PMC free article] [PubMed] [Google Scholar]
- Pepperberg, D.R., T.L. Okajima, B. Wiggert, H. Ripps, R.K. Crouch, and G.J. Chader. 1993. Interphotoreceptor retinoid-binding protein (IRBP): molecular biology and physiological role in the visual cycle of rhodopsin. Mol. Neurobiol. 7:61–85. [DOI] [PubMed] [Google Scholar]
- Pugh, E.N.J. 1996. Cooperativity in cyclic nucleotide-gated ion channels. J. Gen. Physiol. 107:165–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh, E.N.J., and T.D. Lamb. 2000. Phototransduction in Vertebrate Rods and Cones: Molecular Mechanisms of Amplification, Recovery and Light Adaptation. Elsevier Science, Amsterdam, Netherlands. 596 pp.
- Richards, M.J., and S.E. Gordon. 2000. Cooperativity and cooperation in cyclic nucleotide-gated ion channels. Biochemistry. 39:14003–14011. [DOI] [PubMed] [Google Scholar]
- Rodieck, R.W. 1998. The First Steps in Seeing. Sinauer Associates, MA. 562 pp.
- Roof, D.J., and C.L. Makino. 2000. The structure and function of retinal photoreceptors. The Principals and Practice of Ophthalmology, 2nd edition. D.M. Alberts and F.A. Jakobiec, editors. W.B. Saunders, Philadelphia, PA. 1624–1673.
- Rosenbaum, T., L.D. Islas, A.E. Carlson, and S.E. Gordon. 2003. Dequalinium: a novel, high-affinity blocker of CNGA1 channels. J. Gen. Physiol. 121:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum, T., A. Gordon-Shaag, L.D. Islas, J. Cooper, M. Munari, and S.E. Gordon. 2004. State-dependent block of CNG channels by dequalinium. J. Gen. Physiol. 123:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruknudin, A., M.J. Song, and F. Sachs. 1991. The ultrastructure of patch-clamped membranes: a study using high voltage electron microscopy. J. Cell Biol. 112:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saari, J.C. 1999. Retinoids in mammalian vision. Retinoids: The Biochemical and Molecular Basis of Vitamin A and Retinoid Action. H. Nau and W.S. Blaner, editors. Springer-Verlag, NY. 563–588.
- Seno, K., M. Kishimoto, M. Abe, Y. Higuchi, M. Mieda, Y. Owada, W. Yoshiyama, H. Liu, and F. Hayashi. 2001. Light- and guanosine 5′,3-O′ (thio) triphosphate-sensitive localization of a G protein and its effector on detergent-resistant membrane rafts in rod photoreceptor outer segments. J. Biol. Chem. 276:20813–20816. [DOI] [PubMed] [Google Scholar]
- Sidell, N., and L. Schlichter. 1986. Retinoic acid blocks potassium channels in human lymphocytes. Biochem. Biophys. Res. Commun. 138:560–567. [DOI] [PubMed] [Google Scholar]
- Sun, H., R.S. Molday, and J. Nathans. 1999. Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J. Biol. Chem. 274:8269–8281. [DOI] [PubMed] [Google Scholar]
- Vellani, V., A.M. Reynolds, and P.A. McNaughton. 2000. Modulation of the synaptic Ca2+ current in salamander photoreceptors by polyunsaturated fatty acids and retinoids. J. Physiol. 529:333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, R., and R.S. Molday. 2002. Regulation of the rod photoreceptor cyclic nucleotide-gated channel. Adv. Exp. Med. Biol. 514:205–223. [DOI] [PubMed] [Google Scholar]
- Weitz, D., N. Ficek, E. Kremmer, P.J. Bauer, and U.B. Kaupp. 2002. Subunit stoichiometry of the CNG channel of rod photoreceptors. Neuron. 36:881–889. [DOI] [PubMed] [Google Scholar]
- Weng, J., N.L. Mata, S.M. Azarian, R.T. Tzekov, D.G. Birch, and G.H. Travis. 1999. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from phenotype in abcr knockout mice. Cell. 98:13–23. [DOI] [PubMed] [Google Scholar]
- Zagotta, W.N., and S.A. Siegelbaum. 1996. Structure and function of cyclic nucleotide-gated channels. Annu. Rev. Neurosci. 19:235–263. [DOI] [PubMed] [Google Scholar]
- Zhang, D.-Q., and D.G. McMahon. 2000. Direct gating by retinoic acid of retinal electrical synapses. Proc. Natl. Acad. Sci. USA. 97:14754–14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D.-Q., and D.G. McMahon. 2001. Gating of retinal horizontal cell hemi gap junction channels by voltage, Ca2+, and retinoic acid. Mol. Vis. 7:247–252. [PubMed] [Google Scholar]
- Zheng, J., M.C. Trudeau, and W.N. Zagotta. 2002. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron. 36:891–896. [DOI] [PubMed] [Google Scholar]
- Zhong, H., L.L. Molday, R.S. Molday, and K.-W. Yau. 2002. The heteromeric cyclic nucleotide-gated channel adopts a 3A:1B stoichiometry. Nature. 420:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, A.L. 2001. Visual transduction. Cell Physiology Sourcebook: A Molecular Approach, 3rd edition. N. Sperelakis, editor. Academic Press, Boston, MA. 807–814.
- Zimmerman, A.L. 2002. Two B or not two B? Questioning the rotational symmetry of tetrameric ion channels. Neuron. 36:997–999. [DOI] [PubMed] [Google Scholar]
- Zimmerman, A.L., J.W. Karpen, and D.A. Baylor. 1988. Hindered diffusion in excised patches from retinal rod outer segments. Biophys. J. 54:351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]