

Abstract
Both intracellular calcium and transmembrane voltage cause inactivation, or spontaneous closure, of L-type (CaV1.2) calcium channels. Here we show that long-lasting elevations of intracellular calcium to the concentrations that are expected to be near an open channel (≥100 μM) completely and reversibly blocked calcium current through L-type channels. Although charge movements associated with the opening (ON) motion of the channel's voltage sensor were not altered by high calcium, the closing (OFF) transition was impeded. In two-pulse experiments, the blockade of calcium current and the reduction of gating charge movements available for the second pulse developed in parallel during calcium load. The effect depended steeply on voltage and occurred only after a third of the total gating charge had moved. Based on that, we conclude that the calcium binding site is located either in the channel's central cavity behind the voltage-dependent gate, or it is formed de novo during depolarization through voltage-dependent rearrangements just preceding the opening of the gate. The reduction of the OFF charge was due to the negative shift in the voltage dependence of charge movement, as previously observed for voltage-dependent inactivation. Elevation of intracellular calcium concentration from ∼0.1 to 100–300 μM sped up the conversion of the gating charge into the negatively distributed mode 10–100-fold. Since the “IQ-AA” mutant with disabled calcium/calmodulin regulation of inactivation was affected by intracellular calcium similarly to the wild-type, calcium/calmodulin binding to the “IQ” motif apparently is not involved in the observed changes of voltage-dependent gating. Although calcium influx through the wild-type open channels does not cause a detectable negative shift in the voltage dependence of their charge movement, the shift was readily observable in the Δ1733 carboxyl terminus deletion mutant, which produces fewer nonconducting channels. We propose that the opening movement of the voltage sensor exposes a novel calcium binding site that mediates inactivation.
Keywords: calcium channels, gating currents, calcium signaling
INTRODUCTION
Inactivation of L-type channels has traditionally been attributed to two independent mechanisms initiated by either intracellular calcium or membrane depolarization (for reviews see Eckert and Chad, 1984; Hering et al., 2000; Budde et al., 2002). Local calcium concentrations on the intracellular side of a functioning calcium channel rapidly change from 0.1 μM at rest to >100 μM when the channel is open. High intracellular calcium affects the gating of calcium channels, limiting their ability to reopen after a period of activity. A 25-yr search for the intracellular calcium-binding site responsible for inactivation (Brehm and Eckert, 1978) has culminated in the current view that calcium controls inactivation by associating with a calmodulin molecule tethered to the channel (Lee et al., 1999; Peterson et al., 1999; Qin et al., 1999; Zuhlke et al., 1999). In L-type channels, calcium binding to the carboxyl-terminal lobe of the tethered calmodulin was proposed to initiate inactivating transitions that may be linked to a complex translocation of the calmodulin along the carboxyl-terminal portion of the α1 subunit (Peterson et al., 1999). In cardiac L-type channels, the exclusive selectivity of calcium-dependent inactivation for the calcium ion (Lee et al., 1985) is a logical corollary of the calmodulin-based theory.
When calcium channels pass ions other than calcium, or when they are blocked, they are thought to inactivate by a process that depends only on voltage. However, a current-dependent component of inactivation of barium currents has also been suggested (Ferreira et al., 1997). Decay of barium currents through inactivating calcium channels has multiexponential kinetics, suggesting that voltage-dependent inactivation comprises multiple, possibly interrelated (Hering et al., 2000) processes. Studies of channel inactivation based on ionic current decay are complemented by analysis at the level of gating currents.
Gating currents, which are generated by charged moieties of voltage-gated channels, reflect directly the transitions in closed and/or inactivated channels. When recorded in blocked channels, gating currents are not distorted by the variable ionic environment near open channels. At the level of gating currents, inactivation of virtually all types of voltage-gated channels can be described by two mechanisms with different kinetics. A fast mechanism involves blocking by cytoplasmic portions of the channel protein itself and characteristically “immobilizes” the gating charge (“ball-and-chain” mechanism) (Armstrong and Bezanilla, 1977; Hoshi et al., 1990; Bezanilla et al., 1991; Eaholtz et al., 1994). A slower (C-type) inactivation mechanism appears to involve multiple conformational changes. During slow inactivation, the intramembrane charge is not immobilized but converts to a different modality (charge 2; Brum and Rios, 1987), which moves only at voltages much more negative than those typical of the noninactivated channels (charge 1). Charge 2 remains mobile at negative voltage until the channel recovers via slow transition(s) not limited by the charge movement itself. Gating currents in blocked cardiac L-type calcium channels have been shown to inactivate by the charge interconversion mechanism (Shirokov et al., 1992), because the return movement of the voltage sensors during recovery at very negative voltages occurs more rapidly than the recovery of ion currents. Furthermore, the onset of the reduction of charge mobile at positive voltages is slower than the inactivation decay of calcium, or barium, currents (Ferreira et al., 1997). Recent work of Ferreira et al. (2003) implied that in addition to the charge 1/charge 2 interconversion, cardiac L-type channels also have a rapid component of voltage-dependent inactivation, which may be seen as immobilization of the gating charge. It remains unclear, however, how the two kinetic components of the changes in gating currents during inactivation (immobilization vs. interconversion of charge) relate to the inactivation mechanisms of unblocked calcium channels.
Gating charge movements are not always well correlated with channel activity. For example, in virtually every experimental system, calcium currents through L-type channels experience run-down during whole-cell patch-clamp recording. The process apparently involves multiple causes, including an influence of intracellular calcium (Belles et al., 1988; Costantin et al., 1999; Kepplinger et al., 2000; Rueckschloss and Isenberg, 2001). At the same time, gating currents in L-type channels are much more stable (Kostyuk et al., 1981; Hadley and Lederer, 1991b). In other words, voltage sensors remain mobile in the channels that do not pass calcium currents. As a second example, in native cardiac cells (Bean and Rios, 1989) and apparently in various expression systems, a substantial fraction of L-type channels contribute to gating charge movement but do not open to pass calcium currents. This may reflect a negative regulatory control of channel opening by the distal parts of the carboxyl-terminal segment of the α1 subunit (Gao et al., 2001), since certain carboxyl-terminal deletion mutants show normal ionic current amplitudes but much smaller gating charge movements than wild-type channels (Wei et al., 1994). In the present study, we use one such mutant (Δ1733) to investigate the relation between calcium ions permeating the channel and alterations in gating charge movements.
Previous attempts to demonstrate possible effects of intracellular, or permeating, calcium ions on gating currents in L-type calcium channels have yielded negative or indirect results. Alterations in gating currents were not detected during calcium flux through L-type calcium channels (Shirokov et al., 1993), or when flash photolysis was employed to manipulate intracellular calcium (Hadley and Lederer, 1991a). More recent experiments, however, have revealed that gating currents may be profoundly affected by calcium in the patch-clamp pipette solution (Ferreira et al., 1998), by calcium incoming through the channels (for N-type channels) (Shirokov, 1999), or by caffeine-induced calcium release from the sarcoplasmic reticulum (Leroy et al., 2002).
In this report, we show that stationary elevation of intracellular calcium to ∼100 μM blocks L-type calcium channel activity and converts the voltage dependence of charge mobility to more negative values than in noninactivated channels. Increasing concentrations of calcium progressively accelerate the transition of the voltage sensor into these inactivated state(s). Using a carboxyl terminus deletion mutant of the α1 subunit (Δ1733), we show that these inactivating transitions are also induced by calcium permeating the open channels; in this respect, the behavior of Δ1733 mutant is similar to that previously reported for N-type calcium channels (Shirokov, 1999). Detailed analysis of the effects of intracellular calcium on the intramembrane charge movements supports the view that the interaction site between calcium and the voltage sensor is isolated from the intracellular space in resting channels and becomes accessible after a cooperative movement of several voltage sensors. We observed similar effects of elevated cytosolic calcium in the “IQ-AA” mutation within the “IQ-motif”, where calmodulin binding was proposed to cause inactivation. This mutation has been shown to eliminate the control of inactivation by calmodulin (Zuhlke et al., 2000), tethered to the channel molecule (Pitt et al., 2001). Taken together, our results suggest that in addition to the calmodulin, calcium ions interact during inactivation with a site that lies within the channel's permeation pathway, or is formed after the opening movement of the voltage sensors.
MATERIALS AND METHODS
The following proteins were transiently expressed in tsA-201 cells: CaV1.2 channel, as a combination of α1C (Entrez protein database accession no. CAA33546), β2a (AAK14821), α2aδ (AAA81562) subunits, and the NCX1.1 (AAA30509) sodium–calcium exchanger (Aceto et al., 1992). The cDNAs were in pcDNA3.1 mammalian expression vector (Invitrogen). Mutations were done by PCR overlap extension, or by the QuickChange site-directed mutagenesis kit (Stratagene) and verified by sequencing. In the “IQ-AA” mutant, I1654A and Q1655A amino acid mutations were combined with amino-terminal deletion Δ134 and carboxyl-terminal deletion Δ1733.
Cells were transfected by a modified calcium-phosphate–based transfection procedure using 20 μg of each DNA per 60-mm culture dish, as described previously (Shirokov et al., 1998). Selection of the expressing cells was aided by EYFP fluorescence (1–3 μg per dish, pEYFP-N1 vector; CLONTECH Laboratories, Inc.). Expression of NCX was assessed by the presence of a characteristic outward steady-state current (at least 2 pA/pF at the holding potential of −90 mV) observed when NCX worked in the reversed mode (Fig. S1). The exchanger current had little effect on the measurement of asymmetric ionic and gating currents (Fig. S2). Online supplemental figures available at http://www.jgp.org/cgi/content/full/jgp.200308876/DC1.
Composition of solutions is given at Table I . We used chloride as the main anion as it has minimal Ca2+ buffering capacity. Free Ca2+ in the pipette solution was adjusted by adding CaCl2 and measured by a Ca2+-selective electrode (WPI, Inc.). All solutions were adjusted to 320 mosmol·kg−1 and pH 7.3. Experiments were performed at room temperature. Except for the experiment illustrated in Fig. 1, recordings started 5–8 min after establishing the whole-cell configuration. The delay was sufficient for calcium currents to stabilize after the initial run-up.
TABLE I.
Composition of Solutions
| Chemical | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Ca2+ | NaCl | CsCl | TEA-Cl | Tris-Cl | HEPES | EGTA | Mg-ATP | GdCl3 |
| mM | mM | mM | mM | mM | mM | mM | mM | mM | |
| Pipette | |||||||||
| 1 EGTA | — | — | 155 | — | — | 10 | 1 | 5 | — |
| 0.01 Ca | 0.01a | — | 155 | — | — | 10 | — | 5 | — |
| 0.1 Ca | 0.1a | — | 155 | — | — | 10 | — | 5 | — |
| 0.3 Ca | 0.3a | — | 155 | — | — | 10 | — | 5 | — |
| 0 Ca, 20 Na | — | 20 | 130 | — | — | 10 | 10 | 5 | — |
| Bath | |||||||||
| 0 Na | 10b | — | — | 150 | 10 | — | — | — | — |
| 150 Na | 10b | 150 | — | — | 10 | — | — | — | — |
| 150 Na, Gd | 10b | 150 | — | — | 10 | — | — | — | 0.015 |
| Gd | 10b | — | — | 150 | 10 | — | — | — | 0.015 |
CaCl2 was added for the indicated concentration of free Ca2+, as measured by Ca-selective electrode.
CaCl2 was added for the indicated total concentration of Ca2+.
Figure 1.

Ca2+ load through the patch pipette blocked Ca2+ current through L-type Ca2+ channels and reduced their OFF gating current. Current traces were elicited by voltage pulses from the holding potential from −90 to 20 mV. Pipette solution had 0.3 mM of free Ca2+ (0.3 Ca, Table I), and bath solution was 0 Na.
Symmetric capacitive and leak currents were subtracted electronically by capacitance compensation circuitry of the patch-clamp amplifier (Axopatch 200B; Axon Instruments, Inc.). We chose small cells and used relatively large pipettes (0.8–1.2 MΩ). Cell capacitance (11 ± 5 pF, n = 126), series resistance (Rs = 6.4 ± 3.1 MΩ) and leak resistance were set during a control pulse applied from the holding potential of −90 to −100 mV. No additional subtraction of symmetric currents were required to record gating currents. Previously (Shirokov et al., 1998), we demonstrated the validity of this approach, which works well for the small cells we used.
To calculate charge transfer, the current transient was integrated after subtraction of the steady-state component. The steady-state component was determined as a 10 ms average, 50 ms after the beginning of the voltage pulse. Curve fitting was done by a nonlinear least-squares routine of SigmaPlot (SPSS, Inc.). Data are presented as means ± SEM.
Online Supplemental Material
The additional material (available at http://www.jgp.org/cgi/content/full/jgp.200308876/DC1) describes the following: technical aspects of the experiments in cells expressing NCX and Ca channels (Figs. S1 and S2), estimation of the magnitude of gating charge during voltage pulses to the reversal potential (Fig. S3), comparison of activation/deactivation kinetics of whole-cell Ca2+ currents in the Δ1733 mutant and in the wild-type channel (Figs. S4 and S5), and a consideration of a three-particle allosteric model explaining why Ca2+ binding to a low-affinity site might preferentially affect the onset, but not the recovery, kinetics (Fig. S6).
RESULTS
High Cytosolic Calcium Blocks Ionic Current and Affects Channel Gating
We examined the effects of directly perfusing high calcium concentrations through the whole-cell patch pipette on ionic and gating currents in L-type calcium channels. As illustrated in Fig. 1 , when the pipette contained 0.3 mM calcium, calcium currents initially increased during the first several minutes of perfusion (trace 2), but were completely blocked by 18 min (trace 3). After ionic currents became very small, gating currents could be recorded directly, i.e., without any ionic current blocker. The outward ON transients of the intramembrane charge movement appeared to be unaffected by the calcium load. However, the OFF transients were dramatically reduced. The asymmetric effect of calcium on gating currents suggests that calcium interacts with the channel only after the opening movements of the voltage sensors (see below).
The reduction of the OFF transient could be clearly observed only when most of the channels were blocked by intracellular calcium so that ionic tail currents were small. Thus, it was impossible to correlate directly the reduction of calcium currents with the reduction in the OFF gating current transient throughout the range of results. To overcome this difficulty, we designed a two-pulse experiment, in which we compared the effects of calcium load on the magnitude of ionic currents during the first pulse and on the amount of intracellular charge movement of charge 1 type available for the second pulse. The reduction in OFF currents would be manifest as a loss of charge 1 availability, which in turn would be reflected in a reduction in the ON gating current transient during the second pulse.
The pulse protocol is illustrated in Fig. 2 A (top). The first pulse to +20 mV activated nearly maximal calcium currents. Then, the voltage went to −50 mV for 20 ms. Although the tail currents decayed to zero by the end of the interpulse, indicating that the channels had closed, the interpulse was too brief to allow recovery from the inactivation elicited by the first depolarization. To minimize ionic currents, the voltage applied during the second pulse was very close to the reversal potential for the particular cell under study. The reversal potential varied from 50 to 70 mV in different cells, and it generally drifted by 3–5 mV during the time of experiment. Once the voltage of the second pulse was chosen, it remained the same during the experiment. For the cells illustrated in Fig. 2, A and B, the second pulse went to 53 and 68 mV, respectively. Since ionic currents were relatively small and slow during the second pulse, the ON gating current transient at the second pulse was clearly resolvable. When the cell was recorded with 0.1 Ca pipette solution (Fig. 2 A), calcium currents during the first pulse and the ON gating currents during the second pulse each became progressively smaller with the time of recording. As expected, it took much longer for calcium currents to run down when the pipette contained 1 EGTA solution (Fig. 2 B). Importantly, however, the gating current transients at the second pulse were much more stable and did not decay despite the eventual loss of ionic current. The results provide qualitative evidence that calcium loading exerts similar effects on ionic and gating currents.
Figure 2.

Comparison of Ca2+ current magnitude and availability of the intramembrane charge movement measured in a double-pulse experiment. (A) Currents elicited by the pulse protocol shown at the top were recorded in a cell loaded with Ca2+ through the patch pipette. Pipette solution had 0.1 mM of free Ca2+ (0.1 Ca), and bath solution was 0 Na. The time after establishing the whole-cell recording configuration is indicated near traces. Ca2+ current at the first pulse to +20 mV and gating currents elicited by the second pulse to the reversal potential of ionic currents were decreasing in parallel during the course of experiment. (B) Currents obtained similarly to those shown in A, but in a cell recorded with 1 EGTA pipette solution. While Ca2+ currents at the first pulse to +20 mV slowly run-down, gating currents at the second pulse to the reversal potential did not change. (C) Correlation between the magnitude of Ca2+ current and the gating charge during the second pulse for the cell shown in A. The gating charge was obtained by integrating the ON transient during the second pulse, as described in the text. Linear regression is shown by the thin line, the dashed lines show 99% prediction interval. (D) Correlation between the magnitude of Ca2+ current and the gating charge during the second pulse for the cell shown in B. Lines are as in C.
A quantitative analysis of these results requires a means of quantifying the ON charge movement under the conditions of these experiments. In the absence of ionic current blocker, however, it is impossible to eliminate the contribution of residual ionic currents to the charge movement during the second pulse. Therefore, we estimated the bulk value of the gating charge by integrating the ON gating current transient after subtracting the 2-ms current average near the minimal value reached 4–5 ms after the beginning of the second pulse. Most of the gating charge had moved at this time and the slowly developing outward current did not significantly obstruct the transient. This procedure is illustrated in Fig. S3, available at http://www.jgp.org/cgi/content/full/jgp.200308876/DC1.
Fig. 2, C and D, displays the correlations between ionic currents and the estimated ON charge movement during the period in which the cells were perfused with or without 0.1 mM calcium. As shown in Fig. 2 C for the cell loaded with 0.1 mM calcium, the gating charge at the second pulse (from −50 mV to the reversal potential) went almost to zero in parallel with the reduction of calcium current at the first pulse. In four cells tested with 0.1 Ca pipette solution, the linear regression, Q(Vrev) = A·Ipeak(20 mV) + B, had the r 2 value of at least 0.89, the slope A was −0.47 ± 0.07 fC/pA, and the intercept B was 25 ± 62 fC, a value not significantly different than zero. Fig. 2 D illustrates comparable results for the cells with 1 EGTA pipette solution. In this case, there was practically no loss in gating charge even after ionic currents had run down to near zero. For the five cells perfused with 1 EGTA, the linear regressions were also of a good quality (the r 2 value was at least 0.92), but the slope was only −0.07 ± 0.03 fC/pA, and the origin of coordinates was never within the 99% interval for prediction. These results agree with previous findings that calcium currents recorded in cells with EGTA in the pipette run down to zero without a significant change in gating currents (Kostyuk et al., 1981; Hadley and Lederer, 1991b). Overall, the results demonstrate that during calcium loading, the calcium current during the first pulse and the gating charge available to the second pulse were reduced in parallel (Fig. 2 C). We infer that calcium affected both ionic and gating currents by a common mechanism.
Alteration of Cytosolic Calcium by Sodium–Calcium Exchange
Inhibition of channel activity during direct perfusion with calcium required long perfusion times (13 ± 6 min with 0.3 Ca pipette solution, n = 14). This probably reflects the influence of calcium buffering and organellar sequestration on calcium diffusion into the cell, since 90% equilibration of calcium between the pipette and a 20-pF cell would occur within only 2 min if diffusion of calcium were free (Oliva et al., 1988; Pusch and Neher, 1988). The long perfusion times could also be compatible with elaboration of a second messenger, or the activation of proteases or other degradative enzymes. Thus, it would be desirable to alter the calcium concentration in the vicinity of the channel by a process that was both rapid and readily reversible. For this purpose, we transfected cells with the cardiac sodium–calcium exchanger NCX1.1 (Aceto et al., 1992), so that submembrane calcium concentrations could be rapidly and reversibly altered by changing the sodium gradient across the membrane.
The effects of exchange activity on channel function are illustrated in the experiment in Fig. 3 A. Here, calcium (0.3 mM) was loaded through the pipette into cells expressing both L-type channels and NCX. At first, neither the pipette nor the bathing solution contained sodium and under these conditions the exchanger was not active. After 3 min of loading, calcium currents were partially inhibited (trace 1). When the bathing solution was changed to that with 150 mM sodium, calcium current rapidly increased (trace 2). In four similar experiments, current increased by an average of 112 ± 16% within 20 s after the switch to 150 Na. When external sodium was removed for a longer period of time, ionic and gating currents became similar to those observed previously in calcium-loaded cells (compare trace 3 in Fig. 1 and Fig. 3 A). Finally, when NCX was again activated by 150 mM sodium, the ionic currents (trace 4) were partially restored. Extracellular sodium had little or no effect on channel currents in control experiments in cells without NCX (−4 ± 3%; n = 5). Therefore, the sodium-dependent increase in calcium currents reflected the efflux of intracellular calcium by sodium–calcium exchange. The rapidity of the restoration of calcium currents, in the face of continued cellular perfusion with 0.3 mM calcium, suggests that the exchanger's effects are local, i.e., exchange activity rapidly alters the calcium concentration in a submembrane space sensed by the calcium channel.
Figure 3.

Effects of manipulations of intracellular Ca2+ by sodium–calcium exchanger (NCX) on L-type Ca2+ channels. Voltage pulses were from −90 to 20 mV. (A) Activation of NCX reverses the inhibition of Ca2+ current by intracellular Ca2+. Pipette solution was 0.3 Ca. When bathing solution was changed from 0 Na (indicated by open boxes) to 150 Na (indicated by gray boxes), NCX reduced local [Ca2+]. (B) NCX functioning in the reverse mode reversibly blocks Ca2+ currents. Pipette solution: 0 Ca, 20 Na. When bathing solution was changed from 150 Na to 0 Na, NCX increased local [Ca2+].
NCX was also used to load calcium into the cell (Fig. 3 B). Here, the pipette solution had 20 mM sodium and 10 mM EGTA and the bath solution had 150 mM sodium. Under these conditions, calcium currents were large (trace 1). Removing sodium from the bath solution induced a biphasic decline in channel currents as calcium entered the cell in exchange for intracellular sodium. In this experiment, the calcium currents initially declined by about one-half within the time between two consecutive pulses (20 s, trace 2) and then decreased more slowly to nearly zero during the ensuing 5 min (trace 3). At this time, the OFF gating current was greatly reduced (trace 3), indicating that the exchanger had increased intracellular calcium sufficiently to reproduce the effects seen during direct perfusion with high calcium pipette solutions. Readdition of sodium to the bath caused a substantial increase of calcium current (trace 4). Removing extracellular sodium a second time again made calcium currents small and the gating currents asymmetric (trace 5). Similar results were obtained in seven other cells selected for measurable NCX currents (see materials and methods).
The mechanisms underlying the biphasic time course of calcium current decline are unclear. One possibility is the presence of two calcium-dependent blocking mechanisms with different calcium affinities. Alternatively, diffusion of calcium away from the membrane and the interaction with cytosolic EGTA may account for the peculiar time course. Yet another possibility is that calcium influx through NCX decreased (“inactivated”) with time. Regardless of the precise reasons for the kinetics of this response, one point is clear: both ionic currents and the OFF transients were affected by calcium influx through NCX in the same manner as by calcium load through the pipette. The reversible and more rapid action of calcium delivered by NCX, rather than by the pipette, speaks for a direct, diffusion-limited mechanism.
The reversibility of calcium's effects on gating currents is documented in the experiment shown in Fig. 4 . Calcium was loaded into the cell by the reverse mode of the NCX under conditions where the pipette had 20 mM sodium and 10 mM EGTA. When sodium in the bath solution was replaced with TEA, calcium currents were reduced to zero and gating currents became asymmetric (compare traces 1 and 2). When sodium was restored to the bath medium, calcium currents partially recovered (trace 3). At this point, we blocked the restored calcium currents with a brief application of 15 μM gadolinium (trace 4). The remaining gating current transients were symmetric, as expected when gating currents are recorded at low intracellular calcium (e.g., with 1 EGTA pipette solution). After partial recovery in gadolinium-free solution (trace 5), extracellular sodium was again replaced with TEA. The resulting influx of calcium through the exchanger again greatly reduced calcium currents through calcium channels (trace 6). Blockade of these currents with gadolinium revealed an asymmetric reduction in the OFF gating current transient (trace 7). Addition of sodium to the bath again restored both calcium currents (traces 8 and 10) and the symmetry of the gating current transients (trace 9).
Figure 4.

Intracellular Ca2+ loaded through NCX reversibly blocked Ca2+ currents and reduced the OFF gating current transients. Voltage pulses were from −90 to 20 mV. Pipette solution was 0 Ca, 20 Na. (A) Protocol of application of bath solutions (boxes) and amplitudes of Ca2+ currents (dots). When bathing solution was changed from 150 Na (gray boxes) to 0 Na (open boxes), NCX functioning in the reversed mode increased local [Ca2+]. When bathing solution was 150 Na, Gd, Ca2+ currents were blocked. (B) Current traces recorded at times indicated by corresponding numbers in A.
The effects of calcium loading through reverse exchange activity are summarized as follows. Calcium currents were reduced to 10% of their original value within 6 ± 1 min, n = 12, of calcium load through NCX. This was significantly faster than current run-down in the absence of intracellular calcium: With 0 Ca 20 Na solution in the pipette and 150 Na solution in the bath, it took 39 ± 12 min, n = 18, for calcium currents to run down to 50% of their maximal magnitude, if intracellular calcium was not manipulated. In five cells tested for the effect of calcium load through NCX on gating currents, determined in the presence of gadolinium as illustrated in Fig. 4, elevation of intracellular calcium reduced the ratio of the OFF charge to the ON charge (QOFF/QON) to 0.21 ± 0.06. Readdition of sodium to the bath restored only 48 ± 9% of calcium current magnitude before the second removal of extracellular sodium (compare traces 1 and 4 in Fig. 3 A, or traces 1 and 3 in Fig. 4). However, QOFF/QON was equal to 0.97 ± 0.06, n = 5, when the restored calcium currents were blocked by gadolinium. Given that elevation of intracellular calcium by perfusion through the pipette, or by NCX, rapidly and reversibly affected both ionic and OFF gating currents, it is likely that both the availability of ionic currents and the voltage-dependent gating are controlled by the same calcium-dependent mechanism.
Cytosolic Calcium Shifts the Voltage Dependence of QOFF
An analysis of the voltage dependence of calcium's effects on intramembrane charge movement provided further insights into the mechanism and location of the interaction between calcium and voltage sensors. For the experiments illustrated in Figs. 5–7, we blocked ionic currents with 15 μM gadolinium and studied the changes in voltage distribution of the intramembrane charge movement that accompany combined application of high intracellular calcium and depolarization. Fig. 5 A shows representative traces for experiments conducted with either 1 EGTA or 0.1 Ca solutions in the perfusion pipette, and Fig. 5 B compares voltage dependencies for the ON charge (circles) in pipette solutions with different calcium concentrations. Before averaging, the individual ON charge transfer functions were normalized in each cell by the maximal charge (Qmax), which was determined by fitting the Boltzmann distribution Q = Qmax/(1 + exp(−(V − V1/2)/K)) to the values of the ON charge. The solid lines in panel B are the best fits to the averaged values. Loading of 0.3 mM calcium negatively shifted the voltage distribution of the ON charge movement by ∼20 mV. This was probably due to the screening of the surface charge. The symbols in panel C represent the averaged OFF charges under several different perfusion conditions. Before averaging, the OFF charge values were normalized in each cell by the Qmax, which was determined from the ON transients as described above. The OFF transients recorded during repolarizations to −90 mV were dramatically reduced in high calcium if the test pulse went more positive than −10 mV. The dashed lines in panel C are the fits to the ON charge transfer functions in panel B. In calcium-loaded cells, they deviated significantly from the OFF charge transfer functions at potentials above 0 mV. The difference between the ON and the OFF charges is plotted in panel D. The effect of calcium, as judged by the apparent loss of the mobile charge in the OFF transient, had a more positively distributed dependence on voltage when compared with that of the ON charge and a significantly steeper slope (K = 11 vs. 25 mV).
Figure 5.

OFF gating currents elicited by repolarizations to −90 mV were reduced by intracellular Ca2+. Bathing solution: Gd. Pipette solutions are indicated in C. (A) Representative tracings for pulses to −40, 0, 40, and 80 mV. (B) Average ON charge transfer functions obtained with different pipette solutions, as illustrated in A. The averaging procedure is described in the text. Different symbol sizes correspond to different [Ca2+], as listed in C. Only the maximal SEM value is shown for clarity. The curves are single Boltzmann fits. V1/2 was: 15 ± 3 mV (1 EGTA), 13 ± 4 mV (0.01 Ca), 7 ± 3 mV (0.1 Ca), and −5 ± 5 mV (0.3 Ca); K was 25 ± 3 mV. Each group had n = 6 cells. (C) Average OFF charge transfer functions. The dashed lines are the fits to the ON charge transfer from B. (D) The difference between ON and OFF charge transfer functions. The solid lines are single Boltzmann fits. V1/2 was: 31 ± 12 mV, and K was 11 ± 3 mV for all fits.
Figure 7.

Intracellular Ca2+ accelerates inactivation of gating currents. Bathing solution: Gd. Pipette solutions are indicated. (A) Representative OFF gating current traces elicited by repolarizations to −60 mV are spaced according to the time of conditioning at 20 mV. (B) Average charge transferred during the OFF transients plotted versus logarithm of conditioning duration. Different symbols correspond to different [Ca2+] in the pipette. Charge transfers are normalized to the maximal value in each cell and averaged, n = 4 for 0.3 Ca, n = 7 for other concentrations. The maximal values were achieved at 10 ms with 1 EGTA and 0.01 Ca, at 5 ms with 0.1 Ca, and at 2.5 ms with 0.3 Ca pipette solutions. Only the maximal SEM value is shown for clarity. Curves are single-exponential fits. The time constants were: 670 ± 47 ms in 1 EGTA, 280 ± 35 ms in 0.01 Ca, 77 ± 12 ms in 0.1 Ca, and 8 ± 2 ms in 0.3 Ca.
When the pipette solution contained 0.3 mM calcium, the OFF transients could be recovered if the cell was repolarized to very negative voltages. This is illustrated in Fig. 6 A, where the OFF transient carried about half of the charge in the ON transient when the membrane was repolarized to −120 mV. During repolarization to −200 mV, almost all the OFF charge could be recovered (Fig. 6 A, bottom trace). The voltage distributions of the OFF charge (QOFF) were derived as follows. The QOFF charge transfer function in each cell was fit by: Q = Q−∞ + Qmax /(1 + exp(−(V − V1/2)/K)). Then, the (QOFF − Q−∞)/Qmax values were averaged. As shown in Fig. 6 B, the voltage distribution of the OFF charge movement recorded after a 20-ms depolarization to 20 mV was shifted by about −90 mV. The reduction of the OFF charge measured in the experiments shown in Fig. 5 is a manifestation of the same effect. A similar negative shift in the voltage dependence of charge movement was observed with various voltage-gated channels in the absence of calcium, i.e., during voltage-dependent inactivation (Bezanilla et al., 1982; Brum and Rios, 1987; Shirokov et al., 1992, 1998; Olcese et al., 1997). However, in the absence of calcium, the shift in charge movement required much longer depolarizations. For example, in cardiac calcium channels recorded with a low-calcium intracellular solution, the full conversion of the charge movement into the negatively distributed mode required several seconds of depolarization to voltages above 0 mV (Shirokov et al., 1998). In contrast, in our experiments with high intracellular calcium, two-thirds of the charge moved more negatively than −80 mV after only 20 ms of depolarization to 20 mV (Fig. 6 B).
Figure 6.

Combination of high intracellular [Ca2+] and a brief depolarization caused a large negative shift of the voltage distribution of the intramembrane charge movement in L-type Ca2+ channels. (A) Representative tracings of the OFF charge for repolarizations to 40, −40, −120, and −200 mV applied after a 20-ms prepulse to 20 mV. Bathing solution: Gd. Pipette solutions are indicated. (B) Average OFF charge distributions obtained with 1 EGTA (open circles), or 0.3 Ca (filled circles) pipette solutions, as illustrated in A. The curves are single-Boltzmann fits. In 1 EGTA solution: V1/2 = −27 ± 2 mV, K = 27 ± 3 mV, n = 6. With 0.3 Ca solution: V1/2 = −116 ± 6 mV, K = 29 ± 5 mV, n = 4.
Additional information on time dependence of the calcium's effects on charge movement is presented in Fig. 7 , where we measured reduction in OFF charge movement after depolarizations of different duration. Because the return voltage was −60 mV, most of the recorded charge was of the positively distributed mode. The OFF transients shown in Fig. 7 A are spaced according to the duration of the preceding conditioning at 20 mV. The transients recorded after a depolarization >5 ms were progressively smaller for increasing conditioning duration. With the 1 EGTA pipette solution, the reduction was slow and had the time constant of 0.7 s, somewhat less than reported previously for 10 mM EGTA (2.2 s; Shirokov et al., 1998). The reduction of the mobile charge was ∼10 times faster when cells where loaded with 0.1 mM calcium (77 ms) and ∼100 times faster (8 ms) with 0.3 mM calcium in the pipette. The more rapid reduction of the OFF transients at −60 mV in high intracellular calcium was not accompanied by a detectable slowing of their kinetics. Also, there was no slow component significantly deviating from the baseline over the 50 ms recording interval at −60 mV. Since charge immobilization, or rapid inactivation (Ferreira et al., 2003), would be characterized by a measurable slow component of charge movement cotemporal with recovery from inactivation, we conclude that this indicates that there was no detectable immobilization of charge by calcium under our experimental conditions. Additional support to this view comes from the analysis of the kinetics of the OFF transients at very negative voltages (like those shown in Fig. 6 for −200 mV). In contrast to control traces, the decay of the OFF transients at −200 mV in cells recorded with 0.1 mM calcium in the pipette was biexponential with a significant slow component (τ = 7.4 ± 1.1 ms, n = 4). This time constant is similar to that of the charge 2 movement in cardiac cells (Shirokov et al., 1992). Recovery of calcium currents takes much longer at −200 mV (τ = 46 ± 8 ms, n = 4, after 100 ms conditioning to 20 mV; unpublished data). Thus, the bulk of the intramembrane charge (Fig. 6) moves at −200 mV before the channels become able to open. Taken together, the data in Figs. 5–7 indicate that intracellular calcium acts on the channel by accelerating the same transitions that occur during voltage-dependent inactivation (charge 1/charge 2 interconversion).
The Effects of Calcium Are Independent of Calmodulin
To test whether calmodulin was involved in the observations described above, we used the “IQ-AA” mutant. In this mutant, the key amino acids I1654 and Q1655 of the so-called “IQ” motif, where calcium/calmodulin is thought to bind and cause inactivation, were mutated to alanines. As expected from the work of Zuhlke et al. (2000), calcium currents through the mutant channel inactivated slowly, with kinetics similar to those of barium currents. As shown in Fig. 8 , when intracellular calcium was increased by reverse exchange activity in cells expressing the “IQ-AA” mutant (n = 5), both ionic and the OFF gating currents were reduced in the same manner as in the wild-type channel (Figs. 3 and 4). Calcium loading by direct perfusion also blocked ionic and the OFF gating currents in the “IQ-AA” mutant (unpublished data; n = 4). Thus, calcium/calmodulin binding to the “IQ” motif does not promote the changes in voltage-dependent gating seen at high calcium.
Figure 8.

The “IQ-AA” mutant, in which Ca2+/calmodulin does not accelerate inactivation of Ca2+ current, was affected in the same manner as the wild-type channel. Both ionic and OFF gating currents were reduced by the Ca2+ load. Experimental protocol was as in Fig. 3 B.
Calcium Permeating the Channel Affects Gating Currents in the Δ1733 Mutant
It is possible that the effects of intracellular calcium load on the intramembrane charge movement are exerted only when calcium increases are long lasting relative to the channel-gating kinetics and that the calcium ions actually passing through the channel may not act the same way. Our previous attempts to demonstrate that calcium currents through L-type channels affected their gating currents were unsuccessful (Shirokov et al., 1993). While the negative observations were in accord with the widely held view that calcium- and voltage-dependent components of inactivation have distinct molecular mechanisms, we also proposed a trivial explanation. Both native cardiac cells and expression systems usually produce a large number of calcium channel molecules that contribute to the gating current but do not open to pass calcium into the cell (Bean and Rios, 1989). Because gating currents are recorded from all channels, while only a fraction of channels open, the local effect of incoming calcium could go undetected.
The dissociation between ionic conductance and voltage-dependent gating in calcium channels was originally thought to reflect the properties of the mechanism by which calcium channels run down (Kostyuk et al., 1981; Hadley and Lederer, 1991b). However, in view of more recent studies of the negative control of L-type channels by the distal parts of the carboxyl terminus of their pore-forming α1 subunit (Wei et al., 1994; Gao et al., 2001), we propose that the excess of the voltage sensors is not due to ionic current run-down, but is brought about by the interactions involving the carboxyl terminus. Carboxyl terminus deletions, which do not remove the sequence above position 1700 and leave calmodulin binding domains untouched, produce channels that pass calcium currents of similar or greater magnitude than the wild-type, but their gating currents are much smaller. Based on the relationship: I = N · PO · i s.sh., where I is the whole-cell current, N is the number of channels available to be open, PO is the probability of a channel being open, and i s.sh. is the current through an open channel, Wei et al.(1994) proposed that carboxyl terminus deletion mutants have higher PO, since i s.sh. was not changed and the maximal gating charge (hence, N) was less. However, an increase in PO must be accompanied by changes in the kinetics of channel opening and/or closing. Both the published works of others (Wei et al., 1994; Gao et al., 2001) and our experiments with the carboxyl terminus deletion mutant Δ1733 (see Figs. S4 and S5) demonstrate that the activation/deactivation kinetics of the macroscopic calcium currents through the carboxyl terminus deletion mutants are virtually indistinguishable from those of the wild-type. Since the opening/closing kinetics (hence, PO) are not altered significantly in the deletion mutant, the mutation increases the number of channels available for opening, rather than PO. This also means that in the mutant, unlike in the wild-type, gating currents are generated mostly by the channels that pass calcium.
Encouraged by our previous finding that calcium current through N-type channels does in fact accelerate the appearance of the negative shift in the voltage dependence of the intramembrane charge movements (Shirokov, 1999), we did similar experiments on the Δ1733 channels, which, as discussed above, have higher availability for opening. The pulse protocol shown on the top of Fig. 9 A allowed us to monitor ionic currents and the intramembrane charge movement in inactivated channels. First, the conditioning pulse activated calcium current. Then, the membrane was briefly repolarized to −150 mV, and a test pulse to 20 or −50 mV was applied. The magnitude of calcium current during the test pulse to 20 mV (trace 1) reflected the number of noninactivated channels at the moment when the test pulse was applied. If the test pulse went to −50 mV (trace 2), it moved little charge in noninactivated channels, because most of the charge in noninactivated channels is mobile at voltages more positive than −50 mV (Fig. 5). However, because inactivation is accompanied by a negative shift in the voltage distribution of the intramembrane charge movement (see Fig. 6; also Shirokov et al., 1998), an increase of the charge movement elicited by the pulse from −150 to −50 mV would provide evidence that inactivation during the conditioning pulse had affected the voltage sensor and made it mobile at negative voltages.
Figure 9.

Ca2+ current promotes inactivated states of the voltage sensor in the Δ1733 deletion mutant channels. (A) Pulse protocols used to monitor availability of ionic currents (current trace 1, test pulse from −150 to 20 mV) and the intramembrane charge movement in inactivated channels (current trace 2, test pulse from −150 to −50 mV). Conditioning pulses were from −90 mV to Vcond. Pipette solution: 1 EGTA. (B) Test currents recorded as shown in A. Conditioning voltages are indicated by the figures above the traces. Traces 3 (test voltage 20 mV) and 4 (test voltage −150 mV) were recorded with 0 Na bath solution. Traces 5 (test voltage −150 mV) were recorded in the same cell after the blockade of ionic currents by Gd bath solution.
Fig. 9 B displays currents associated with test depolarizations from −150 to 20 mV (traces 3) or to −50 mV (traces 4) after conditioning pulses (Vcond) to various voltages. For test depolarizations to 20 mV (traces 3), calcium currents progressively declined with increasing Vcond and attained a minimum at Vcond ∼40 mV. At more positive Vcond, calcium currents during the conditioning pulse were smaller and inactivated less (not shown); the reduced inactivation was evident in the increasing current magnitudes for test pulses at Vcond > 40 mV. An opposite pattern was seen for charge movements recorded after test pulses to −50 mV (traces 4). At test voltages of −50 mV, ionic currents were very small and gating charge movements were easily resolvable. Here, charge movements increased with Vcond to a maximum at 20–40 mV, and then declined at more positive values. Importantly, blockade of the calcium currents by gadolinium eliminated the effect of conditioning (traces 5). The results provide a clear demonstration that calcium current passing through the channel shifts the distribution of gating current transients to negative potentials.
The pooled results from five similar experiments with the Δ1733 mutant are shown in Fig. 10 A. There was a bell-shaped voltage dependence for current inactivation during the conditioning pulse (circles), with maximal inactivation occurring at 20–40 mV. The increase in gating charge movement following depolarization from −150 to −50 mV (filled bars) showed a generally similar dependence on Vcond, with a maximal value at Vconc = 20 mV. When calcium currents were blocked with gadolinium, intramembrane charge movements were small and did not vary with Vcond (gray bars). The correlation between ionic currents and charge movement, and its dependence upon channel activity, strongly support our conclusion that calcium influx through the Δ1733 channels during the conditioning pulse promoted inactivating transitions that both reduced calcium currents and shifted the voltage dependence of intramembrane charge movements to negative voltages. In all probability, these inactivating transitions are equivalent to those induced by increasing the cytosolic calcium concentration (Fig. 6).
Figure 10.

Comparison of the reduction of calcium current and the availability of charge movement from −150 to −50 mV after conditioning at different voltages, measured as in Fig. 9. (A) Current inactivation (circles) and charge 2 (filled bars) in the Δ1733 mutant. Current inactivation was measured as the reduction of the magnitude of test calcium current relative to the magnitude without conditioning (Vcond = −90 mV), charge 2 was measured as the difference between charge transfers at −50 mV obtained with and without (Vcond = −90 mV) conditioning. Before averaging, the increase of the charge movement between −150 and −50 mV, was related to the maximal charge transfer in each cell. The maximal charge transfer was obtained in the Gd bath solution by pulsing from −150 to 60 mV. In the Δ1733 mutant, the charge movement recorded in 0 Na solution (filled bars) was significantly increased by conditioning pulses more positive than −20 mV (P < 0.05, unpaired Student's t test). The charge movement in Gd solution (gray bars) was significantly increased only after 100 mV conditioning. (B) Current inactivation (circles) and charge 2 (bars) in wild-type channels. Current inactivation was similar to that in the Δ1733 mutant, but there was no significant change of charge movement between −150 and −50 mV (0 Na bath solution, open bars).
Fig. 10 B displays the results of similar experiments obtained with the wild-type channel. The bell-shaped dependence of current inactivation (circles) on Vcond was essentially identical to that seen with the Δ1733 mutant. In contrast, however, there was no dependence of charge movement on Vcond in the wild-type channels (bars), indicating that were was no correlation between intramembrane charge movement and channel inactivation. The results are consistent with previous negative observations for the native channels. For the reasons described above, the negative observation does not necessarily imply a lack of the interaction between calcium influx and the voltage sensors in wild-type calcium channels. Indeed, the presence of the effect in the Δ1733 deletion suggests that the interaction does exist in native L-type channels, but that the presence of the carboxyl terminus obscures its detection, perhaps by enhancing a subpopulation of channels that gate but do not open.
DISCUSSION
Inactivation of L-type channels has traditionally been subdivided into voltage-dependent and calcium-dependent components. Here we propose that the voltage- and calcium-dependent components proceed by a common mechanism, and that calcium governs the rate at which inactivation occurs through its interaction with the channel in the permeation pathway. The following lines of evidence and comparisons with previous work support this hypothesis.
Calcium Affects Voltage-dependent Gating
Direct perfusion of calcium into the cell through the patch pipette, at concentrations ≥100 μM, produced complete blockade of ionic current, and this was associated with a large negative shift in the voltage dependence of charge movement in the OFF gating transition. The shift has been previously associated with voltage-dependent inactivation. The effects of intracellular calcium were reversed by NCX activity operating in a calcium efflux mode (Fig. 3 A), and were mimicked by NCX operating in the calcium influx mode (Figs. 3 B and 4). Thus, channel inactivation involves the reversible interaction with high concentrations of intracellular calcium (∼100 μM); these concentrations are probably similar to the local calcium concentrations the channels experience during activity.
Both direct and reversed modes of NCX altered ionic and gating currents very rapidly in comparison to the much slower response during direct perfusion with calcium. Even more surprising was the inability of 10 mM EGTA in the pipette solution to prevent the inhibition of calcium current during reversed NCX activity. This implies that NCX is very efficient in affecting calcium concentrations local to the channel in this expression system. Reverse NCX activity has previously been shown to load transfected CHO cells with as much as 6 mmole calcium per liter cell water within 5 min (Reeves et al., 1996). In the present experiments, the effects of NCX activity on the global cellular calcium concentration are not known. Most likely standing concentration gradients of calcium are generated, reflecting the balance between NCX activity on the one hand and cellular perfusion on the other, with the steepest change in calcium occurring in the region beneath the plasma membrane, a region that would be directly sensed by the calcium channels. We have tried various combinations of different buffers, including BAPTA, to prevent the inhibition of calcium channels by reverse NCX activity; the most effective system was the use of the pipette solutions containing large (100 mM) quantities of glutamate or citrate, which are small, rapidly diffusing low-affinity calcium buffers.
Calcium Accelerates the Changes of Gating Currents That Were Previously Associated with Voltage-dependent Inactivation
The negative shift in the voltage dependence of the intramembrane charge movement is characteristic of calcium channels that have undergone voltage-dependent inactivation (Brum and Rios, 1987; Shirokov et al., 1998; Ferreira et al., 2003). In our experiments, increasing concentrations of intracellular calcium progressively increased the rate at which the OFF charge at −90 mV was reduced (Fig. 6). At 0.3 mM calcium in the pipette solution, the OFF charge declined nearly 100 times faster (8 ms) than with 1 mM EGTA (0.7 s). The simplest interpretation of these results is that the rate of the inactivating transition induced by depolarization increases with the calcium concentration, i.e., that there is no mechanistic distinction between “voltage-dependent” and “calcium-dependent” inactivation under our experimental conditions.
Recent work of Ferreira et al. (2003) suggests that a rapid component of voltage-dependent inactivation of L-type calcium channels may reflect the charge immobilization mechanism. Within the limitations of our ability to define slow components of the relaxation of the charge movement transients, it appears that intracellular calcium did not promote charge immobilization. We did not detect a measurable slow component of relaxation of the OFF transient, as it would be expected for remobilization of the voltage sensor (e.g., Fig. 7). This is also reflected in the double pulse experiments (Fig. 2 A), where complete decay of the OFF transient occurred during the interpulse interval, and yet the subsequent ON transient during the second pulse was dramatically reduced.
It is hypothetically possible that a shift in gating mode, due to voltage- or calcium-dependent facilitation, could alter the voltage distribution of charge movements. Voltage-dependent facilitation, however, would be minimized in our experiments through our use of the β2a splice variant (Cens et al., 1996; Qin et al., 1998). Moreover, calcium-dependent facilitation typically occurs at much lower cytosolic calcium concentrations (0.4 μM; Hirano and Hiraoka, 1994) than those required in our experiments to shift the voltage-distribution of the gating currents. Thus, we conclude that in our experimental conditions the effect of cytosolic calcium on channel-gating currents was to accelerate the inactivating transitions conventionally associated with voltage-dependent inactivation.
A minimal four-state diagram describes the charge 1/charge 2 interconversion mechanism as an allosteric interaction between two gates/processes (diagram 1; Brum and Rios, 1987). Voltage-dependent activating transition rest↔active and voltage-independent inactivating transition prime↔inactive are coupled according to microscopic reversibility: KAP→AI · exp(V1/K) = KRP→RI · exp(V2/K), where K is the slope factor, V1 and V2 are half-distribution voltages for charge 1 and charge 2, respectively, and KAP→AI and KRP→RI are equilibrium constants. Results of our current work allow one to propose that the prime↔inactive transition of the scheme depends on intracellular calcium (diagram 2). The coupling equation then becomes: (1/KDA) · exp(V1/K) = (1/KDR) · exp(V2/K), where KDR and KDA are dissociation constants for calcium in rest and active states. Although the oversimplified diagram 2 explains well why intracellular calcium affects the voltage sensor only after it moved into the active state: KDR = KDA · exp((V1 − V2)/K) ≈ 500 · KDA, it clearly is not sufficient, since inactivation of the voltage sensor occurs even at very low (0.1 μM) intracellular calcium, yet high concentrations of calcium (>10 μM) are required to speed it up. This problem is resolved by the eight-state model for three coupled processes illustrated in Fig. S6, available at http://www.jgp.org/cgi/content/full/jgp.200308876/DC1.
Results of our work on the expressed cardiac L-type channels are in accord with the findings of Leroy et al. (2002) on native cardiac L-type calcium channels. In guinea-pig ventricular heart cells, applications of caffeine promoted a negative shift of the voltage dependence of the intramembrane charge movement (Leroy et al., 2002). Since pretreatment with ryanodine and/or thapsigargin reduced the effect of caffeine on the charge movement, the authors proposed that the caffeine-induced interconversion between charge 1 and 2 was mediated by calcium released from the sarcoplasmic reticulum. Other studies of inactivation of L-type calcium channels by calcium released from the sarcoplasmic reticulum (Sham et al., 1995; Adachi-Akahane et al., 1996) provided strong evidence for functional coupling between L-type calcium channels and ryanodine receptors in microdomains with restricted diffusion of calcium (for review see Rios and Stern, 1997). Our results indicate that the concentration of intracellular calcium has to be in the order of 10–100 μM to affect kinetics of inactivation, therefore providing independent, albeit indirect, support for the existence of calcium microdomains between the sarcolemma and the sarcoplasmic reticulum.
Calcium Interacts with Channels That Have Moved Their Voltage-dependent Gate
The simplest interpretation of how intracellular calcium affects gating currents is that calcium interacts with the channels after they have undergone voltage-dependent gating. The OFF transients, but not the ON transients, were reduced, suggesting that the opening transition of voltage sensors was required for calcium to affect gating. Calcium had no effect on charge movement at potentials more negative than −10 mV (Fig. 5), a voltage at which about one-third of the charge movement associated with channel opening has occurred. At more positive potentials, the reduction of gating currents had a steep voltage dependence (Fig. 5 D), indicating that the cooperative movement of voltage sensors is required for calcium to interact with the channel. In agreement with this interpretation, the OFF gating currents were reduced by calcium only when the depolarizing pulse was long enough (∼5 ms) for most of the gating current transient to be complete (see Fig. 7).
Taken together, these findings indicate that the calcium binding site responsible for inactivation is not available in the resting channel, and that calcium interacts with the channel only after it underwent a substantial voltage-driven transition leading to the opening. Such a transition may be the movement of the voltage-dependent gate. The site of interaction with calcium might therefore lie within the water-filled cavity between the selectivity filter and the voltage-dependent gate (Doyle et al., 1998; Roux and MacKinnon, 1999). Alternatively, the site might be generated at a cytosolically disposed location by a voltage-dependent conformational change of the S6 transmembrane segments that precedes the final opening transition. Consistent with either scenario, the S6 transmembrane segments have been shown to be important determinants of inactivation of various types of calcium channels (Zhang et al., 1994; Hockerman et al., 1995, 1997; Hering et al., 1996, 1997, 1998; Berjukow et al., 1999, 2000; Sokolov et al., 2000; Stotz et al., 2000; Stotz and Zamponi, 2001). Zong et al. (1994) previously proposed that Ca-dependent inactivation is closely linked with ion selectivity, implying an interaction of calcium with the channel's permeation pathway
Calcium-calmodulin Binding to the Cytoplasmic Amino and/or Carboxyl Termini Is Not Required for Calcium-dependent Inactivation
Inactivation of L-type channels depends on calcium/calmodulin binding to the amino terminus (Ivanina et al., 2000), and to the carboxyl terminus (Lee et al., 1999; Peterson et al., 1999; Qin et al., 1999; Zuhlke et al., 1999). Because intracellular calcium blocked calcium currents and reduced OFF gating current transients in the “IQ-AA” mutant (Fig. 8), in which neither mechanism would be applicable, we conclude that calcium/calmodulin-dependent regulation of inactivation is not necessary for calcium-dependent channel closure and the acceleration of inactivation at the level of gating currents. How can this conclusion be reconciled with the by now well-established role of calcium/calmodulin in the calcium-dependent inactivation of ionic currents? Hypothetically, it is possible that the calmodulin-dependent mechanism, in contrast to the direct effects of calcium, has no significant effect on intramembrane charge movements. This seems unlikely, however, in view of the parallel reduction in ionic currents and charge movements documented in Figs. 2 and 10.
A more plausible, although speculative, possibility is that calmodulin is required to maintain the inactivating effects of calcium during its permeation through the open channel. Calcium ions passing through an open channel are thought to affect the channel gating by promoting occupancy of a closed state from which the channel can reopen with a lower probability (Rose et al., 1992). The probability of reopening is reduced rapidly, after the first opening in calcium, as compared with barium (Yue et al., 1990). This strongly suggests that calcium acts inside or near the permeation pathway and induces an inactivated state from which reopening is blocked. With low cytosolic calcium concentrations, local calcium gradients in the vicinity of an individual channel would dissipate rapidly once channel closure occurs. Assuming that inactivation requires the continued interaction of calcium with its inactivation site, one can envision that modest conformational changes in the inactivated channel protein could lead to dissociation of the bound calcium and hence to the loss of the inactivated state. Calcium/calmodulin-mediated voltage-dependent interactions with the cytoplasmic tails of the α1 subunit (Kobrinsky et al., 2003), might act as a “latch” to prevent such conformational changes and lock in the inactivated state of the channel. According to this view, calcium/calmodulin would not be required for inactivation induced by high cytosolic calcium concentrations, as in our experiments, because the inactivation site would be immediately reoccupied by calcium if dissociation occurred.
Calcium Influx Through the Open Channels Accelerates the Inactivating Transitions of the Voltage Sensors
In Δ1733 channels, calcium influx during conditioning depolarizations induced additional inactivation and a proportional shift in the voltage distribution of intramembrane gating charge (Fig. 9). A similar observation was made in the case of expressed N-type channels (Shirokov, 1999), but not for native L-type cardiac channels (Shirokov et al., 1993). As discussed in results, it is possible that calcium influx shifts the voltage dependence of charge movement in wild-type channels, but the effect is undetectable because most wild-type channels gate but do not open. At the same time, high cytosolic calcium affects movement of the voltage sensor in most if not all wild-type channels. This implies that the interaction does not require channel opening. This conclusion seems at odds with our finding, discussed above, that calcium interacts with the channels only after the ON intramembrane charge movements have been largely completed. Movement of the voltage-dependent gate, however, might not be sufficient to open the channel. The final closed-to-open transitions are likely to have little intrinsic voltage dependence (Zheng and Sigworth, 1997, 1998; Sukhareva et al., 2003), and they may be blocked or inhibitied in the nonconducting wild-type channels. Therefore, we propose that both calcium incoming through the open channels and elevated bulk calcium promote the same molecular events by interacting with the inactivation site, access to which is controlled by the voltage sensor.
Overview
Here we summarize our findings in the form of a working hypothesis that provides a reasonable account for all the results presented here. We realize that there may be alternate explanations for many of the individual elements discussed below and that charge immobilization may contribute to inactivation in some circumstances. Nevertheless, the hypothesis provides a plausible viewpoint for interpreting the data presented here and for designing experiments in future studies. Channel opening is thought to be a sequential process involving initial movements of the voltage sensors that are linked to rearrangements of the S6 transmembrane segments, followed by additional transitions that result in ionic conduction. These voltage-independent transitions, but not the preceding movements of the voltage sensors, appear to be blocked by interactions involving the carboxyl terminal segments of the α1 subunit in most wild-type channels; hence, most channels gate but do not conduct. The initial movements of the S6 segments generate a calcium binding site which, when occupied with calcium, blocks the subsequent voltage-independent transitions; hence, high calcium concentrations block ionic current. The change in the chemical environment of the voltage sensor, as reflected in the negative shift of the voltage dependence of its movement, is preferentially associated with the channel configuration stabilized by the binding of calcium to the inactivation site, accounting for the acceleration of charge interconversion by calcium. In conducting channels, the high local calcium concentration generated as a result of channel activity promotes channel closure and the entry of the channel protein into the same inactivated state described above—i.e., reopening of the channel is prevented by calcium's effects on the voltage-independent transitions of the S6 residues. Nonconducting wild-type channels obviously cannot generate the local calcium gradients that would lead to charge interconversion. Because most wild-type channels are nonconducting, little if any change of the intramembrane charge movement can be detected after inactivation of ionic currents. Upon channel closure, the local calcium concentration gradient dissipates very rapidly and this would lead to dissociation of calcium from the inactivation site and channel reopening. The interactions involving calcium/calmodulin at the IQ motif and other sites stabilize this configuration of the channel and block or inhibit calcium dissociation from the inactivation site; hence, calcium/calmodulin accelerates inactivation of calcium currents, but it is not necessary when cytosolic calcium is directly elevated (by perfusion through the pipette or by reverse NCX activity).
Further structure-function analysis is required in order to identify the inactivation site and to understand the influence of bound calcium on voltage-dependent gating of these channels.
Supplemental Material
Acknowledgments
The authors would like to thank Molecular Resource Facility of NJMS, Drs. J. Berlin, A. Harris, M. Nowycky, and N. Shirokova for comments, and Dr. M. Condrescu for the NCX 1.1 clone.
The work was supported by NIH R01MH62838 to R. Shirokov.
Olaf S. Andersen served as editor.
The online version of this article contains supplemental material.
Dr. D. Isaev's permanent address is Department of General Physiology, Nervous System, Bogomoletz Institute of Physiology, Ukrainian Academy of Science, 4 Bogomoletz St., 01024 Kyiv, Ukraine.
References
- Aceto, J.F., M. Condrescu, C. Kroupis, H. Nelson, N. Nelson, D. Nicoll, K.D. Philipson, and J.P. Reeves. 1992. Cloning and expression of the bovine cardiac sodium-calcium exchanger. Arch. Biochem. Biophys. 298:553–560. [DOI] [PubMed] [Google Scholar]
- Adachi-Akahane, S., L. Cleemann, and M. Morad. 1996. Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. J. Gen. Physiol. 108:435–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M., and F. Bezanilla. 1977. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70:567–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, B.P., and E. Rios. 1989. Nonlinear charge movement in mammalian cardiac ventricular cells. Components from Na and Ca channel gating. J. Gen. Physiol. 94:65–93. (published erratum appears in J. Gen. Physiol. 1989. 94:401) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belles, B., C.O. Malecot, J. Hescheler, and W. Trautwein. 1988. Run-down” of the Ca current during long whole-cell recordings in guinea pig heart cells: role of phosphorylation and intracellular calcium. Pflugers Arch. 411:353–360. [DOI] [PubMed] [Google Scholar]
- Berjukow, S., F. Gapp, S. Aczel, M.K. Sinnegger, J. Mitterdorfer, H. Glossmann, and S. Hering. 1999. Sequence differences between α1C and α1S Ca2+ channel subunits reveal structural determinants of a guarded and modulated benzothiazepine receptor. J. Biol. Chem. 274:6154–6160. [DOI] [PubMed] [Google Scholar]
- Berjukow, S., R. Marksteiner, F. Gapp, M.J. Sinnegger, and S. Hering. 2000. Molecular mechanism of calcium channel block by isradipine. Role of a drug-induced inactivated channel conformation. J. Biol. Chem. 275:22114–22120. [DOI] [PubMed] [Google Scholar]
- Bezanilla, F., E. Perozo, D.M. Papazian, and E. Stefani. 1991. Molecular basis of gating charge immobilization in Shaker potassium channels. Science. 254:679–683. [DOI] [PubMed] [Google Scholar]
- Bezanilla, F., R.E. Taylor, and J.M. Fernandez. 1982. Distribution and kinetics of membrane dielectric polarization. 1. Long-term inactivation of gating currents. J. Gen. Physiol. 79:21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm, P., and R. Eckert. 1978. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 202:1203–1206. [DOI] [PubMed] [Google Scholar]
- Brum, G., and E. Rios. 1987. Intramembrane charge movement in frog skeletal muscle fibres. Properties of charge 2. J. Physiol. 387:489–517. (published erratum appears in J. Physiol. 1988. 396:581) [DOI] [PMC free article] [PubMed]
- Budde, T., S. Meuth, and H.C. Pape. 2002. Calcium-dependent inactivation of neuronal calcium channels. Nat. Rev. Neurosci. 3:873–883. [DOI] [PubMed] [Google Scholar]
- Cens, T., M. Mangoni, S. Richard, J. Nargeot, and P. Charnet. 1996. Coexpression of the β2 subunit does not induce voltage-dependent facilitation of the class C L-type Ca channel. Pflugers Arch. 431:771–774. [PubMed] [Google Scholar]
- Costantin, J.L., N. Qin, M.N. Waxham, L. Birnbaumer, and E. Stefani. 1999. Complete reversal of run-down in rabbit cardiac Ca2+ channels by patch-cramming in Xenopus oocytes; partial reversal by protein kinase A. Pflugers Arch. 437:888–894. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J.M. Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: Molecular basis of K conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Eaholtz, G., T. Scheuer, and W.A. Catterall. 1994. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 12:1041–1048. [DOI] [PubMed] [Google Scholar]
- Eckert, R., and J.E. Chad. 1984. Inactivation of Ca channels. Prog. Biophys. Mol. Biol. 44:215–267. [DOI] [PubMed] [Google Scholar]
- Ferreira, G., J. Yi, E. Rios, and R. Shirokov. 1997. Ion-dependent inactivation of barium current through L-type calcium channels. J. Gen. Physiol. 109:449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, G., H. Takeshima, E. Rios, and A. Gonzalez. 1998. High intracellular calcium affects L-type calcium channel gating. Biophys. J. 74:A101. [Google Scholar]
- Ferreira, G., E. Rios, and N. Reyes. 2003. Two components of voltage-dependent inactivation in Ca(v)1.2 channels revealed by its gating currents. Biophys. J. 84:3662–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, T., A.E. Cuadra, H. Ma, M. Bunemann, B.L. Gerhardstein, T. Cheng, R. Ten Eick, and M.M. Hosey. 2001. C-terminal fragments of the α1C (CaV1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated α1C subunits. J. Biol. Chem. 276:21089–21097. [DOI] [PubMed] [Google Scholar]
- Hadley, R.W., and W.J. Lederer. 1991. a. Ca2+ and voltage inactivate Ca2+ channels in guinea-pig ventricular myocytes through independent mechanisms. J. Physiol. 444:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley, R.W., and W.J. Lederer. 1991. b. Properties of L-type calcium channel gating current in isolated guinea pig ventricular myocytes. J. Gen. Physiol. 98:265–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering, S., S. Aczel, M. Grabner, F. Doring, S. Berjukow, J. Mitterdorfer, M.J. Sinnegger, J. Striessnig, V.E. Degtiar, Z. Wang, and H. Glossmann. 1996. Transfer of high sensitivity for benzothiazepines from L-type to class A (BI) calcium channels. J. Biol. Chem. 271:24471–24475. [DOI] [PubMed] [Google Scholar]
- Hering, S., S. Aczel, R.L. Kraus, S. Berjukow, J. Striessnig, and E.N. Timin. 1997. Molecular mechanism of use-dependent calcium channel block by phenylalkylamines: role of inactivation. Proc. Natl. Acad. Sci. USA. 94:13323–13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering, S., S. Berjukow, S. Aczel, and E.N. Timin. 1998. Ca2+ channel block and inactivation: common molecular determinants. Trends Pharmacol. Sci. 19:439–443. [DOI] [PubMed] [Google Scholar]
- Hering, S., S. Berjukow, S. Sokolov, R. Marksteiner, R.G. Weiss, R. Kraus, and E.N. Timin. 2000. Molecular determinants of inactivation in voltage-gated Ca2+ channels. J. Physiol. 528:237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, Y., and M. Hiraoka. 1994. Dual modulation of unitary L-type Ca2+ channel currents by [Ca2+]i in fura-2-loaded guinea-pig ventricular myocytes. J. Physiol. 480:449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockerman, G.H., B.D. Johnson, T. Scheuer, and W.A. Catterall. 1995. Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels. J. Biol. Chem. 270:22119–22122. [DOI] [PubMed] [Google Scholar]
- Hockerman, G.H., B.D. Johnson, M.R. Abbott, T. Scheuer, and W.A. Catterall. 1997. Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels in transmembrane segment IIIS6 and the pore region of the alpha1 subunit. J. Biol. Chem. 272:18759–18765. [DOI] [PubMed] [Google Scholar]
- Hoshi, T., W.N. Zagotta, and R.W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. [DOI] [PubMed] [Google Scholar]
- Ivanina, T., Y. Blumenstein, E. Shistik, R. Barzilai, and N. Dascal. 2000. Modulation of L-type Ca2+ channels by Gβγ and calmodulin via interactions with N and C termini of α1C. J. Biol. Chem. 275:39846–39854. [DOI] [PubMed] [Google Scholar]
- Kepplinger, K.J., G. Forstner, H. Kahr, K. Leitner, P. Pammer, K. Groschner, N.M. Soldatov, and C. Romanin. 2000. Molecular determinant for run-down of L-type Ca2+ channels localized in the carboxyl terminus of the α1C subunit. J. Physiol. 529:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyuk, P.G., O.A. Krishtal, and V.I. Pidoplichko. 1981. Calcium inward current and related charge movements in the membrane of snail neurones. J. Physiol. 310:403–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobrinsky, E., E. Schwartz, D.R. Abernethy, and N.M. Soldatov. 2003. Voltage-gated mobility of the Ca2+ channel cytoplasmic tails and its regulatory role. J. Biol. Chem. 278:5021–5028. [DOI] [PubMed] [Google Scholar]
- Leroy, J., J.M. Lignon, F. Gannier, J.A. Argibay, and C.O. Malecot. 2002. Caffeine-induced immobilization of gating charges in isolated guinea-pig ventricular heart cells. Br. J. Pharmacol. 135:721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K.S., E. Marban, and R.W. Tsien. 1985. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. J. Physiol. 364:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, A., S.T. Wong, D. Gallagher, B. Li, D.R. Storm, T. Scheuer, and W.A. Catterall. 1999. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 399:155–159. [DOI] [PubMed] [Google Scholar]
- Olcese, R., R. Latorre, L. Toro, F. Bezanilla, and E. Stefani. 1997. Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J. Gen. Physiol. 110:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva, C., I.S. Cohen, and R.T. Mathias. 1988. Calculation of time constants for intracellular diffusion in whole cell patch clamp configuration. Biophys. J. 54:791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, B.Z., C.D. DeMaria, J.P. Adelman, and D.T. Yue. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 22:549–558. [DOI] [PubMed] [Google Scholar]
- Pitt, G.S., R.D. Zuhlke, A. Hudmon, H. Schulman, H. Reuter, and R.W. Tsien. 2001. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J. Biol. Chem. 276:30794–30802. [DOI] [PubMed] [Google Scholar]
- Pusch, M., and E. Neher. 1988. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflugers Arch. 411:204–211. [DOI] [PubMed] [Google Scholar]
- Qin, N., D. Platano, R. Olcese, J.L. Constantin, E. Stefani, and L. Birnbaumer. 1998. Unique regulatory properties of the type 2a Ca channel β subunit caused by palmitoylation. Proc. Natl. Acad. Sci. USA. 95:4690–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, N., R. Olcese, M. Bransby, and L. Birnbaumer. 1999. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc. Natl. Acad. Sci. USA. 96:2435–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, J.P., G. Chernaya, and M. Condrescu. 1996. Sodium-calcium exchange and calcium homeostasis in transfected Chinese hamster ovary cells. Ann. NY Acad. Sci. 779:73–85. [DOI] [PubMed] [Google Scholar]
- Rios, E., and M.D. Stern. 1997. Calcium in close quarters: microdomain feedback in excitation-contraction coupling and other cell biological phenomena. Annu. Rev. Biophys. Biomol. Struct. 26:47–82. [DOI] [PubMed] [Google Scholar]
- Rose, W.C., C.W. Balke, W.G. Wier, and E. Marban. 1992. Macroscopic and unitary properties of physiological ion flux through L-type Ca2+ channels in guinea-pig heart cells. J. Physiol. 456:267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, B., and R. MacKinnon. 1999. The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science. 285:100–102. [DOI] [PubMed] [Google Scholar]
- Rueckschloss, U., and G. Isenberg. 2001. Cytochalasin D reduces Ca2+ currents via cofilin-activated depolymerization of F-actin in guinea-pig cardiomyocytes. J. Physiol. 537:363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham, J.S., L. Cleemann, and M. Morad. 1995. Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes. Proc. Natl. Acad. Sci. USA. 92:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov, R. 1999. Interaction between permeant ions and voltage sensor during inactivation of N-type Ca2+ channels. J. Physiol. 518:697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov, R., G. Ferreira, J. Yi, and E. Rios. 1998. Inactivation of gating currents of L-type calcium channels. Specific role of the α2δ subunit. J. Gen. Physiol. 111:807–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov, R., R. Levis, N. Shirokova, and E. Rios. 1992. Two classes of gating current from L-type Ca channels in guinea pig ventricular myocytes. J. Gen. Physiol. 99:863–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov, R., R. Levis, N. Shirokova, and E. Rios. 1993. Ca2+-dependent inactivation of cardiac L-type Ca2+ channels does not affect their voltage sensor. J. Gen. Physiol. 102:1005–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov, S., R.G. Weiss, E.N. Timin, and S. Hering. 2000. Modulation of slow inactivation in class A Ca2+ channels by β-subunits. J. Physiol. 527:445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz, S.C., J. Hamid, R.L. Spaetgens, S.E. Jarvis, and G.W. Zamponi. 2000. Fast inactivation of voltage-dependent calcium channels. A hinged-lid mechanism? J. Biol. Chem. 275:24575–24582. [DOI] [PubMed] [Google Scholar]
- Stotz, S.C., and G.W. Zamponi. 2001. Identification of inactivation determinants in the domain IIS6 region of high voltage-activated calcium channels. J. Biol. Chem. 276:33001–33010. [DOI] [PubMed] [Google Scholar]
- Sukhareva, M., D.H. Hackos, and K.J. Swartz. 2003. Constitutive activation of the shaker Kv channel. J. Gen. Physiol. 122:541–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, X., A. Neely, A.E. Lacerda, R. Olcese, E. Stefani, E. Perez-Reyes, and L. Birnbaumer. 1994. Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac alpha 1 subunit. J. Biol. Chem. 269:1635–1640. [PubMed] [Google Scholar]
- Yue, D.T., P.H. Backx, and J.P. Imredy. 1990. Calcium-sensitive inactivation in the gating of single calcium channels. Science. 250:1735–1738. [DOI] [PubMed] [Google Scholar]
- Zhang, J.F., P.T. Ellinor, R.W. Aldrich, R.W. Tsien. 1994. Molecular determinants of voltage-dependent inactivation in calcium channels. Nature. 372:97–100. [DOI] [PubMed] [Google Scholar]
- Zheng, J., and F.J. Sigworth. 1997. Selectivity changes during activation of mutant Shaker potassium channels. J. Gen. Physiol. 110:101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J., and F.J. Sigworth. 1998. Intermediate conductances during deactivation of heteromultimeric Shaker potassium channels. J. Gen. Physiol. 112:457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhlke, R.D., G.S. Pitt, K. Deisseroth, R.W. Tsien, and H. Reuter. 1999. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 399:159–162. [DOI] [PubMed] [Google Scholar]
- Zuhlke, R.D., G.S. Pitt, R.W. Tsien, and H. Reuter. 2000. Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the α1C subunit. J. Biol. Chem. 275:21121–21129. [DOI] [PubMed] [Google Scholar]
- Zong, S., J. Zhou, and T. Tanabe. 1994. Molecular determinants of calcium-dependent inactivation in cardiac L-type calcium channels. Biochem. Biophys. Res. Commun. 201:1117–1123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
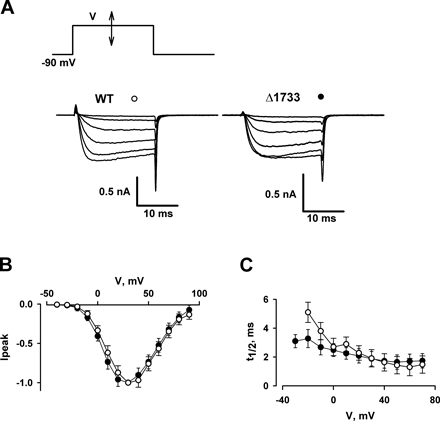{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}