

INTRODUCTION
Since antiquity, motion has been looked upon as the index of life. The organ of motion is muscle. Our present understanding of the mechanism of contraction is based on three fundamental discoveries, all arising from studies on striated muscle. The modern era began with the demonstration that contraction is the result of the interaction of two proteins, actin and myosin with ATP, and that contraction can be reproduced in vitro with purified proteins. The second fundamental advance was the sliding filament theory, which established that shortening and power production are the result of interactions between actin and myosin filaments, each containing several hundreds of molecules and that this interaction proceeds by sliding without any change in filament lengths. Third, the atomic structures arising from the crystallization of actin and myosin now allow one to search for the changes in molecular structure that account for force production.
Mostly I will discuss how biochemical studies from 1941 to 1972 contributed to our understanding of contraction. I shall particularly focus on two aspects of the history: the work of the Szeged school, since the papers in the Studies from the Institute of Medical Chemistry University Szeged were and are not readily available;1 and the history of the proteolytic fragments of myosin HMM and S1 that allowed studies of in vitro contraction in solution. In a few cases more recent information will be quoted for clarity.
The Cold Spring Harbor Meeting in 1972 was perhaps a watershed in muscle research where the outlines of contraction and its regulation were enunciated. Indeed, the general atmosphere at this time was most optimistic and it was thought that the full solution to the problem was imminent. In fact, it took another 30 yr of intensive study to begin to understand the conformational changes undergone by myosin during the contractile cycle.2
The Beginning
A viscous protein was extracted from muscle with concentrated salt solution by Kühne (1864), who called it “myosin” and considered it responsible for the rigor state of muscle. Muralt and Edsall (1930) showed that the “myosin” in solution had a strong flow birefringence with indications that the particles were uniform in size and shape. Occasionally a preparation was obtained that lacked flow birefringence, which was discarded. In 1935, Weber (1935) developed a new technique for the in vitro study of contraction. He squirted “myosin” dissolved in high salt into water where it formed threads that became strongly birefringent upon drying. Engelhardt and Lyubimova (1939) reported in a careful study that myosin had ATPase activity. The importance of the finding was underlined by the earlier findings of Lohmann (1934) that ATP was likely to be the energy source for contraction. Nevertheless, the idea that myosin was an ATPase was not universally accepted because enzymes were considered to be small globular proteins, which myosin clearly was not; however, efforts by Polis and Meyerhof (1947) to separate an ATPase from myosin failed. Engelhardt et al. (1941) also checked the effect of ATP on the “myosin” fibers of H.H. Weber and found that the fibers became more extensible.
Engelhardt and Lyubimova's experiments represented the opening salvo in the revolution of muscle biochemistry. Albert Szent-Györgyi and colleagues then established that the “myosin” used by previous investigators consisted of two proteins. These were purified and shown to be necessary for the contraction elicited by ATP. This work took place during the war years in complete scientific isolation, without access even to Nature and Science. The results were published in three volumes of Studies of the Institute of Medical Chemistry University of Szeged during the years 1941–1943.1 The most important scientific instruments available were a simple Ostwald viscometer and polarizing filters to detect double refraction of flow. Banga and Szent-Györgyi (1942) observed that exposure of ground muscle to high salt concentrations for 20 min extracted a protein of low viscosity (myosin A), whereas overnight exposure solubilized a protein with high viscosity (myosin B). The viscosity of myosin B was reduced by adding ATP while the viscosity of myosin A remained essentially unaffected. The effect of ATP on Kühne's “myosin” was independently discovered by Needham et al. (1942). On account of the war these two groups were never able to communicate. Needham et al. (1942) found that ATP reduced the viscosity and flow birefringence of “myosin”. These changes were reversed upon exhaustion of ATP. They proposed that ATP caused a reversible change in the asymmetry of the “myosin” molecule possibly due to the shortening of the molecule, or changes in the interaction between micellae formed by myosin molecules. They thought it was likely that the change in birefringence resulted from enzyme-substrate combination.
Szent-Györgyi (1942a) discovered that the threads prepared from myosin B using H.H. Weber's method shortened on addition of boiled muscle juice (Fig. 1), but when fibers of myosin A were tested these remained unchanged. The shortening was apparently due to exclusion of water. The active material in the boiled extract was identified as ATP. In his autobiography, Szent-Györgyi (1963) describes that “to see them (the threads) contract for the first time, was perhaps the most thrilling moment of my life.”
Figure 1.

Contraction of actomyosin threads (“myosin B”) on addition of ATP. Shown are the same thread A before and B after addition of boiled muscle juice (a source of ATP) (Szent-Györgyi, 1942a).
Straub joined Szent-Györgyi about this time and it became clear that the difference between myosin B and A was due to the presence of another protein that they called “actin”, which, when combined with myosin, was responsible for the high viscosity and for contractility. Myosin A was purified as paracrystals by Szent-Györgyi (1943a) and retained the name myosin. In a very elegant series of experiments actin was purified by Straub (1942). Myosin B was renamed actomyosin (Szent-Györgyi, 1942b). Straub (1943) showed that the newly discovered protein existed in two forms: globular actin (G-actin) that was stable in the absence of salt, and in the presence of ions it polymerized to form fibrous actin (F-actin). The steady-state ATPase activity of actomyosin, but not of myosin alone, was activated by magnesium (Banga, 1942). The effect of ATP on viscosity and birefringence was imitated only by one other trinucleotide ITP (Needham et al., 1941; Dainty et al., 1944). Contraction was not elicited by ADP (Szent-Györgyi, 1943b). The formation of a rapidly sedimenting coarse precipitate of suspended actomyosin formed by adding ATP and called “superprecipitation” was also taken as a measure of contraction. Furthermore, Szent-Györgyi demonstrated that ATP had a dual function that depended on ionic strength. At low ionic strength ATP induced contraction, at high ionic strength it dissociated actin from myosin (Guba, 1943). It was realized that the rigor state was due to the formation of actomyosin in the absence of ATP. In fact, rigor mortis was the result of the depletion of ATP (Erdös, 1943). It was further shown that the steady-state ATPase activity was increased during the contraction of actomyosin or of minced and washed muscle (Biró and Szent-Györgyi, 1949). The claim that the in vitro contraction of actomyosin threads and superprecipitation (mostly due to dehydration) were equivalent to contraction of living muscle was not universally accepted. Astbury (1947), in his Croonian lecture, proposed that the cross-β pattern (a structure produced by stretching and releasing hair) represented contracted myosin. Both he and Meyerhof believed that superprecipitation was an artifact. (In spite of their scientific differences Astbury and Szent-Györgyi remained close friends and Astbury spent the summer of 1953 in Szent-Györgyi's cottage in Woods Hole.) However, the development and the behavior of the glycerol extracted psoas muscle preparation by Szent-Györgyi (1949) brought conclusive evidence that the interaction of ATP with actomyosin was the basic contractile event. The glycerol extracted psoas muscle preparation consists of a chemically skinned muscle fiber bundle that is permeable to ions. On addition of Mg2+ ATP the preparation develops a tension that is comparable to the tension development of living muscle. Moreover, the preparation behaves somewhat like actomyosin. The glycerinated psoas muscle preparation, with some modification, is still used today for structural studies.
The demonstration that contraction can be reproduced in vitro by two proteins, actin and myosin, opened up the modern phase of muscle biochemistry. It made possible the interpretation of structural features of striated muscle that formed the basis of the sliding filament theory. It simplified the study of contraction, allowed one to focus on the way the ATP energy is used and facilitated the beginning of the discussion that relates structural changes with biochemical events.
The Myosin Molecule
About 400 myosin molecules assemble to form a filament, which interacts with actin filaments containing about the same number of actin monomers (Hanson and Lowy, 1963; Huxley, 1963). Similar filaments form readily in vitro by self-assembly except that they display variable filament lengths (Huxley, 1963).
Myosin therefore has multiple functions: filament formation, ATPase activity, and reversible combination with actin. The use of proteolytic enzymes revealed which regions of the myosin molecule were responsible for each of these different functions. This approach was initiated independently by Gergely (1950) and by Perry (1951), who wanted to see if the ATPase activity might be separated from myosin. Both of these authors observed that the ATPase activity was solubilized by a short tryptic digestion. However, they concluded that the soluble fraction did not combine with actin since the rise of viscosity on addition of actin was relatively small (Gergely, 1953). Further investigations by Mihályi and Szent-Györgyi (1953a) confirmed that the ATPase was solubilized, but ultracentrifuge evidence indicated that the soluble fraction also bound to actin. Tryptic digestion of myosin resulted in the formation of two well-defined components that sedimented differently at high ionic strength (0.6 M KCl). The two new peaks formed in an all or none fashion, at intermediate digestion times only the two new peaks and that of intact myosin were observed. In the presence of actin the faster peak sedimented very rapidly, indicating that it bound to actin (Mihályi and Szent-Györgyi, 1953a). The splitting of the native myosin into two main components during the first rapid phase of tryptic digestion did not decrease ATPase activity. The viscosity of the digestion products was considerably reduced; nevertheless the amount of actin needed to saturate the increase in viscosity was not altered by the rapid phase of tryptic digestion. The two principle components were separated using differential centrifugation in the presence of actin, which was removed by a second centrifugation in the presence of ATP. Both ATPase activity and the ability to combine with actin was retained by the rapidly sedimenting component (Mihályi and Szent-Györgyi, 1953b). Further investigations used a simple method for the separation of the two components of tryptic digestion (Szent-Györgyi, 1953). The slow component was named light meromyosin (LMM), and the faster sedimenting component heavy meromyosin (HMM). The separation was based on differences of solubility at low concentrations of monovalent cations and also in fractionation by ammonium sulfate. LMM had a very similar solubility to intact myosin in both of these reagents; it precipitated below 0.2 M KCl and at 30% ammonium sulfate saturation. HMM remained fully soluble at low ionic strengths while LMM formed paracrystals.
The region connecting LMM with HMM was also available to some other proteolytic enzymes, such as chymotrypsin, producing fragments very similar to the meromyosins (Gergely et al., 1955). Mihályi and Harrington (1959) reported that trypsin rapidly attack a 64-residues long region between LMM and HMM, suggesting the possible absence of the coiled coil structure at the LMM-HMM junction. The paracrystals of LMM showed a 420 ± 25 Å periodicity even without staining or shadowing (Philpott and Szent-Györgyi, 1954). This periodicity agreed with the fundamental fiber period obtained by Huxley (1953a), indicating a structural role for LMM in filament formation and interactions between myosin molecules. LMM was further purified by ethanol precipitation (Szent-Györgyi et al., 1960), probably removing peptide material attached to myosin or formed during digestion (LMM fraction 1). It showed the characteristic periodic structure in electron microscopy. Fibers prepared from purified LMM fraction 1 showed 10 orders of 428 A repeat with strong meridional reflections at 143 and 70 Å. Optical rotatory dispersion indicated that LMM is a fully coiled α-helix (Cohen and Szent-Györgyi, 1957). Sedimentation data suggested that LMM and HMM were linearly attached in myosin (Lauffer and Szent-Györgyi, 1955)
The division of roles between LMM and HMM then became clear. LMM is responsible for filament formation, whereas HMM contains the sites responsible for ATPase activity and also the sites for interacting with actin. The working out of how the meromyosins form a myosin molecule was made difficult by three factors: the low estimates of the molecular weights of the meromyosins—uncertainties of the molecular weight of myosin ranging from 400,000 to 1,000,000; uncertainty in the number of parallel peptide chains; and uncertainties in the estimation of lengths both of myosins and meromyosins. Clarification came from two directions: electron microscopy and analysis of the products of prolonged tryptic digestion.
Mueller and Perry (1962) showed that exposure of HMM to trypsin for an extended time converted it to a single major component sedimenting more slowly, called subfragment 1 (S1), but also yielded a more heterogeneous component known as subfragment 2 (S2). Kominz et al. (1965), using papain, were able to obtain a rather homogeneous S1 component, which very likely was responsible for the thickening seen at the ends of the 1,600 Å long myosin molecules (Huxley, 1963). The length of LMM was estimated to be ∼900 Å from gap-overlap structures of LMM paracrystals (Huxley, 1963). The important electron microscope studies of Slayter and Lowey (1967) established that a myosin molecule ended in two globules (heads). This proved that myosin was made up of two parallel peptide chains, in contrast to previous studies proposing a three-chain structure (Woods et al., 1963). HMM is therefore a two-headed molecule connected to LMM via S2; LMM plus S2 forms the rod portion of the molecule, which for most of its length is a coiled-coil α-helix (Fig. 2). Lowey et al. (1969) showed that, at low ionic strengths, papain or chymotrypsin splits myosin into S1 and rod. The S1 combined with actin and was a fully active ATPase. In addition, a homogeneous helical S2 fragment was obtained (Lowey et al., 1967). Later a longer S2 was isolated (Sutoh et al., 1978), which was flexible. A portion of it had a low melting point. It appears therefore that the LMM is responsible for the core of the filaments and the flexible S2 allows the myosin heads to reach out to the actin filaments. Thus, HMM consists of two cross-bridges connected by a short rod. Moreover S1, containing the active site and the site interacting with actin, is essentially an isolated cross-bridge and is a very suitable preparation for the study of most aspects of the cross-bridge cycle (Huxley, 1963).
Figure 2.

The myosin molecule (adapted from Alberts et al., 2002).
The Light Chains
The presence of small subunits of myosin was first observed by Tsao (1953), who found that prolonged urea treatment produced low molecular weight components in addition to the larger subunits. Various denaturing agents including alkali treatment (Kominz et al., 1959) also dissociated small molecular weight components from purified myosin preparations. To decide if these were true subunits, their stoichiometry, their effects on ATPase activity, and their location within the myosin molecule had to be demonstrated. Two classes of light chains have been reported. One class could be removed with LiCl with the concomitant loss of ATPase activity (Stracher 1969; Dreizen and Gershman, 1970). Partial recovery of ATPase was reported by recombination of the isolated light and heavy chains after removal of LiCl. This class of light chains was named the “essential light chains”. Another class of light chains could be reversibly removed by 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) treatment without significant loss of ATPase activity (Weeds, 1969; Gazith et al., 1970). This class of light chains was named the “regulatory light chains” because they are directly involved in regulation of ATPase activity and contraction in molluscan muscles (see Szent-Györgyi, 1975). There are two moles of regulatory and two moles of essential light chains in a mole of myosin (Lowey and Risby, 1971; Weeds and Lowey, 1971). In myosin from fast rabbit muscle there are two types of essential light chains, A1 and A2. A1 has an extra 41 amino acid extension possibly indicating the presence of two populations of myosin in adult rabbit (Weeds and Lowey, 1971). The light chains bind to S1 (Trotta et al., 1968; Weeds and Lowey, 1971). S1 obtained by digestion with chymotrypsin at low ionic strengths contains only essential light chains (Weeds and Lowey, 1971), However, significant amounts of the regulatory light chains are preserved in S1 that is prepared by digestion with papain at low ionic strengths. Sequences of both light chains indicate the presence of 4 EF hands; but only one of these contain the ligands necessary for divalent cation binding (Collins, 1991). The light chains play a crucial role in regulating contraction in molluscan muscles and phosphorylation of the light chains is necessary for smooth muscle activity.
Actin
Actin was discovered by Straub (1942). Together with myosin and ATP it constitutes the contractile system. In the absence of salt, actin molecules are stable as monomers (G-actin); in the presence of salt, especially divalent cations, actin polymerizes. The high asymmetry of the polymerized actin (F-actin) is indicated by its high viscosity, thyxotropy and strong double refraction (Straub, 1942). The preparation of actin necessitated overcoming a number of problems. Actin in muscle is present as F-actin that usually is extracted together with myosin after ATP is completely hydrolyzed. The strategy therefore was to remove myosin from fresh muscle still containing ATP. The remainder of the myosin was denatured by acetone. Actin then was depolymerized and extracted in a mildly alkaline salt-free solution. Use of salt or acidity below pH 6.0 had to be avoided to prevent repolymerization (Straub, 1943). Straub and Feuer (1950) found that ATP was a functional group of G-actin, its removal from G-actin by dialysis resulted in loss of polymerizability. Asakura (1961), using sonication, demonstrated that polymerization is associated with ATP hydrolysis. However, hydrolysis of nucleotides is not essential for F-actin formation. ADP alone can support a slow polymerization (Hayashi and Rosenbluth, 1960). Nonhydrolizable ATP analogs are also effective (Cooke and Murdoch, 1973). Polymerization begins very slowly, because nucleation is slow, but introduction of nuclei in the form of small amounts of sonicated fragments leads to an explosive reaction (Higashi and Oosawa, 1965). It was proposed that F-actin formation is a condensation process and that a nucleus consists of about four monomers (Asakura et al., 1963).
Actin polymerizes in the presence of salts to form a long-pitch two-stranded helix with a periodicity of ∼360 Å (Hanson and Lowy, 1963). In negatively stained actin filaments ∼13 actin subunits can be visualized within the two strands that form the helical repeat of 360 Å.
The Sliding Filament
Cross-striated muscle is organized in sarcomeres, repeating units ∼2–3-μm long (Fig. 3). Huxley, in his Ph.D. thesis in 1952 (see Huxley, 1953a), observed that the basic meridional periodicities of muscle remain constant at various muscle lengths. Equatorial reflections indicated the presence of two filamentous structures. Moreover, electron microscopy revealed the presence of two types of filaments: 1.6-μm long thick filaments located in the A-band and 1-μm long thin filaments stretching from the Z-band to the H-zone (Huxley, 1953b, 1957a). Hanson and Huxley (1953) observed that high concentration of salts in the presence of ATP removed the A-band protein, which was therefore identified as myosin. Hasselbach (1953) also observed independently the removal of myosin from the A-band with pyrophosphate solution. Myosin added to the ghost fibers bound to the thin filaments demonstrating that these contained actin. Light microscopic observations distinguished a zone of high refractive index, the A (anisotropic) band from the I (isotropic) band.
Figure 3.

Cross-striated muscle is organized in sarcomeres that extend from one Z-line to the next. The distance between Z-lines is 2–3 μm. The thin filaments contain actin and the thick filaments contain myosin. The thick filaments have bipolar symmetry with a central bare zone in which there are no cross-bridges. The actin fiber symmetry reverses in the Z-line. The area not penetrated by the thin filaments is variable, and is known as the H-zone.
The sliding filament theory was based on the observations of constancy of the length of the A-band and the shortening of the I band during a contraction. As pointed out by A.F. Huxley, this observation was made by applying interference microscopy to the most differentiated motile system available, namely intact frog muscle fibers (Huxley and Niedergerke, 1954). A very similar observation was made on glycerol-extracted myofibrils using phase contract microscopy (Huxley and Hanson, 1954). These authors were also able to associate actin with the thin filaments and myosin with the thick filaments. The sliding filament hypothesis was proposed to explain these observations (Fig. 4). The association of thin filaments with actin and thick filaments with myosin was later verified by electron microscopy (Huxley, 1957a; Hanson and Lowy, 1963). Previous theories took it for granted that contraction was the result of a length change in long polymer-like molecules. Until the epoch-making papers in 1954 the idea that movement might result from a process other than the shortening of molecular structures had just not been considered. However, the studies cited above clearly showed that the filaments did not change their lengths during contraction and thus heralded a new era.
Figure 4.
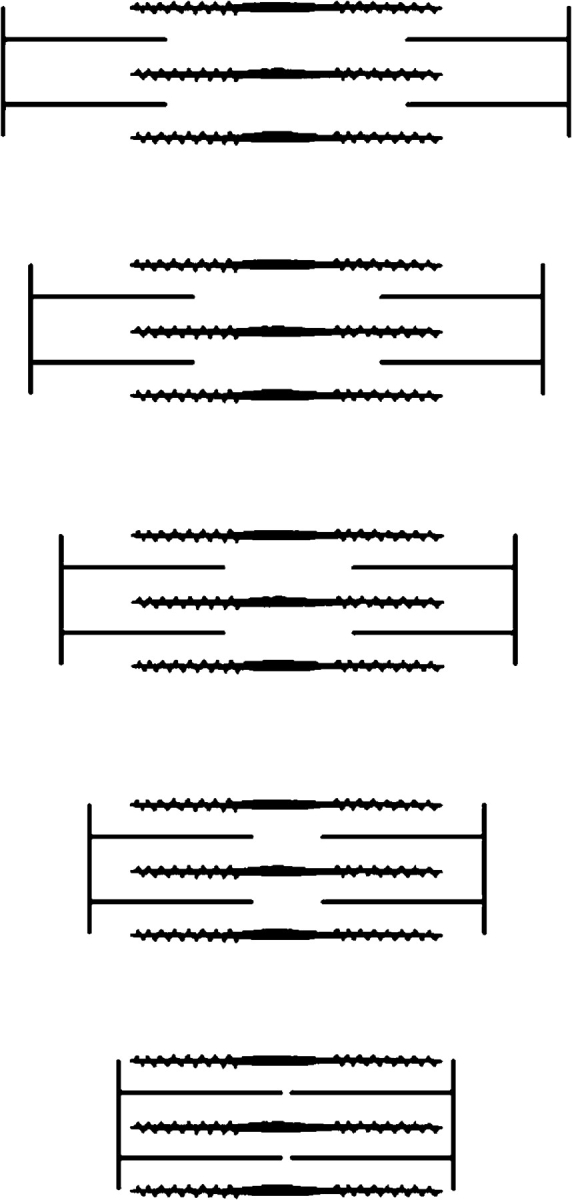
2-μm long myosin containing thick filaments with cross-bridges and 1-μm long actin containing thin filaments are shown. As the sarcomere shortens, the myosin cross-bridges react with actin and propel the thin filaments toward the center of the sarcomere. Both filament types remain at constant lengths during contraction. The sliding of the filaments explains the constancy of the A-band and the changes of the I-band and the H-zone.
During a contraction actin filaments move toward the center from both halves of the sarcomere. This necessitates a change in direction (orientation) of the actin filaments every half sarcomere. The directionality is built into the way actin and myosin assembled into filaments. Thick filaments are bipolar structures. Their assembly begins with the tail-to-tail association of the LMM fractions so that the heads come out pointing in opposite directions. Then filaments grow by addition of myosin molecules onto these bipolar nuclei. The overall result is a smooth central region 0.2-μm long that is free of myosin heads, while the molecules in the two halves of the filaments face in opposite direction (Huxley, 1963). The polarity of the actin filaments was established from the asymmetric structures seen by electron microscopy when complexed with S1 or HMM (“decorated actin”). The filaments showed a pointed and a barbed end. The barbed ends attached to both sides of the Z-line so that the actin molecules faced toward the center of the sarcomere (Huxley, 1963). This symmetric structure of the filaments attached to the Z-line assures that actin containing filaments slide toward the center of the sarcomere.
Contraction is driven by cross-bridge activity. Cross-bridges were clearly visualized by Huxley (1957a) by electron microscopy of ultra-thin sections. Shortly afterwards, Huxley (1958) proposed a mechanical cross-bridge cycle that is similar to present-day models. A.F. Huxley (1957b) investigated the idea that the entropy of cross-bridge attachment may be used to drive the cross-bridge cycle, which is still a relevant idea. However, the first direct evidence for a change in cross-bridge shape that might provide the basis for movement was obtained by Reedy et al. (1965), who discovered that the angle of the myosin cross-bridge in insect muscles depended on the state of the muscle. Electron microscopy combined with X-ray diffraction showed that at rest the cross-bridges extended at right angle from the thick filament (90°), whereas in rigor (no ATP present) the cross-bridges protruded at an acute angle (45°). Therefore, when Huxley (1969) put forward a swinging cross-bridge model, proposing that the myosin head attached to actin changes its angle during the contraction cycle, the idea was widely supported. Nevertheless, in point of fact it took many years to produce direct evidence in support of the swinging cross-bridge model.
The Cross-bridge Cycle
Filament sliding is generated by interactions of the cross-bridges with actin. This interaction can be studied with the soluble fragments of myosin, S1, and HMM. The use of transient kinetics to explore the steps of the cross-bridge cycle was introduced by Tonomura and colleagues, who showed that there is an initial rapid liberation of phosphate by myosin (Kanazawa and Tonomura, 1965). The kinetic analysis with S1 indicated the existence of several ATP states and several ADP states (Bagshaw and Trentham, 1974). Kinetic analysis also demonstrated that the bound ATP was in equilibrium with the bound ADP and inorganic phosphate. An equilibrium constant of ∼7 indicated the reversibility between the states of the bound ATP and bound ADP and Pi. Therefore, hydrolysis of the ATP does not dissipate its energy while the nucleotide is bound. The cross-bridge cycle was proposed by Lymn and Taylor (1971) based on two fundamental findings: they provided evidence that hydrolysis of ATP occurs in the detached state when myosin is not bound to actin; they also showed that the addition of ATP to myosin results in a burst of ATP hydrolysis that was nearly stoichiometric with the myosin heads. The burst occurred when the active site was unoccupied, so this finding indicated that the dissociation of ADP was the limiting reaction of the cycle. They proposed a four-state model (Fig. 5). In the detached state the myosin undergoes a structural change from the state reached at the end of contraction to the state formed after ATP hydrolysis. When attached to actin the conformation of the cross-bridge is altered, which propels actin forward by a step. Force production is associated with products release whereupon the cross-bridge assumes the rigor configuration. The release of ADP enables the rebinding of ATP. The affinity of myosin to actin is greatly reduced by ATP binding and myosin detaches from actin. This is a simplified model omitting a number of intermediates, nevertheless it describes the essential steps of the cycle and continues to be used. The ATP and the product-bound states are weak-binding states; the transition to the strong-binding state and concomitant release of products is required for force production. In striated muscle there is a strong coupling between ATP binding to myosin and actin binding to myosin. This is reflected in a large mutual reduction in affinities.
Figure 5.
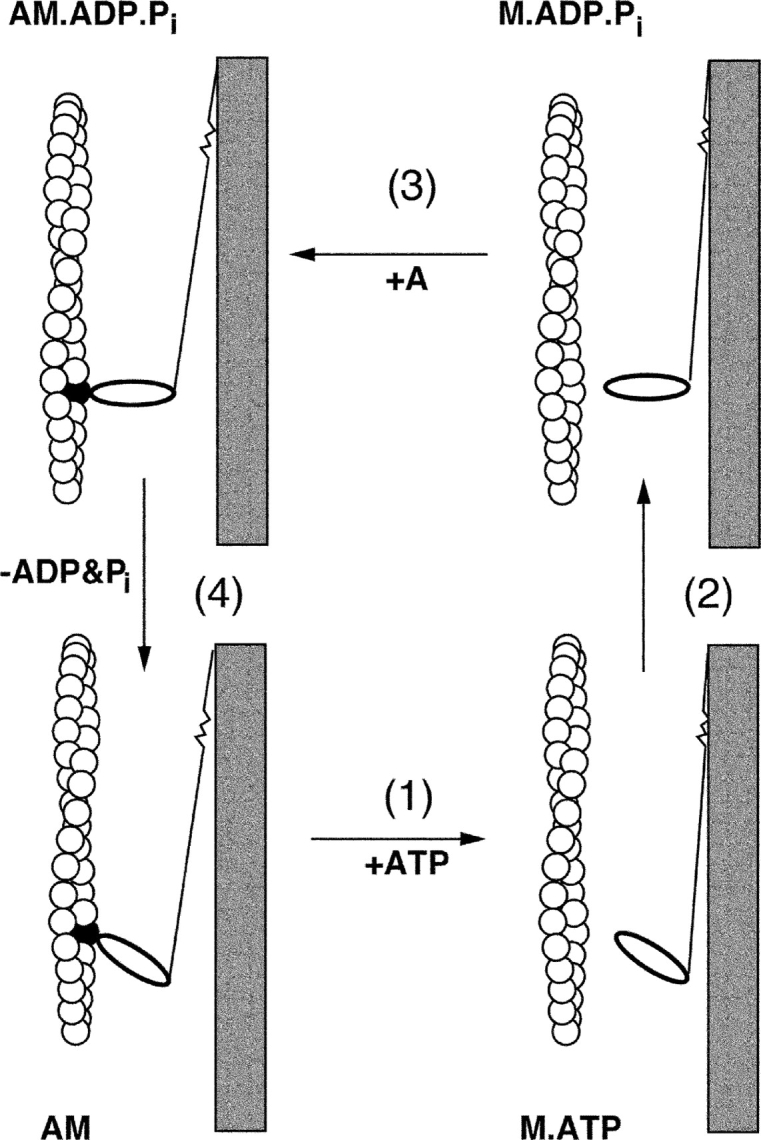
The cross-bridge cycle. Note that ATP hydrolysis takes place in the detached state. In the actin-bound state contraction is associated with the dissociation of the hydrolysis products; recovery of the resting state structure follows dissociation of myosin from actin by ATP (see Taylor, 2001).
A mechanistic relationship between possible cross-bridge movements and the mechanical properties of muscle first was proposed by Huxley (1957a). However, it turned out that an understanding of the structural changes that the myosin cross-bridge undergoes during a cycle necessitated the determination of the structures of actin and the myosin cross-bridge at atomic resolution. This took another 30 yr! In the meantime, several important techniques were developed and yielded new understanding at the molecular level. Application of electron spin resonance and fluorescence energy transfer for the study of muscle proteins were introduced by M.F. Morales (Dos Remedios et al., 1972). New techniques were developed to deal with the physiology of single molecules—the mechanical equivalent of patch-clamps. The first was a direct demonstration of the in vitro sliding of actin filaments over lawn of myosin molecules attached to a cover-slip (Kron and Spudich, 1986). Later came measurements of the step size and tension induced by single myosin molecules acting on an actin filament attached to a very thin glass needle (Kishino and Yanagida, 1988). This method was refined by holding the actin filament between beads in a laser trap (Finer et al., 1994). Other very important developments were the expression of myosin (De Lozanne and Spudich, 1987), the production of mutants in Dictyostelium discoideum (Patterson and Spudich, 1995), and the expression of smooth muscle myosin in insect cells using baculo virus as a vector (Trybus, 1994).
Tropomyosin
Tropomyosin was discovered and isolated by Bailey (1946)(1948). The molecule has a very high α-helix content. (Cohen and Szent-Györgyi, 1957). The presence of nonpolar side chains in every three or four residues in the amino acid sequences indicate that it is a two-stranded coiled coil (Hodges and Smiley, 1972). Tropomyosin is located in the thin filaments, lying on a flat surface formed by the two strands of actin. In studies using electron microscopy and small angle X-ray diffraction (Cohen and Longley, 1966) magnesium salts of tropomyosin form paracrystals that have a repeat period of 396 Å, indicating an elongated structure with an end-to-end overlap. Tropomyosin can be removed from actin at low temperatures (Drabikowski and Gergely, 1962). It also combines with troponin, the complex responsible for thin filament regulation by blocking the actin sites necessary for binding myosin in a calcium-dependent manner.
Regulation of Contraction
By the time of the Cold Spring Harbor Symposium in 1972, the basic principles of regulation were understood. A low Ca2+ concentration in the sarcoplasm activated the muscle; removal of Ca2+ resulted in relaxation. Marsh (1951), working in Bailey's laboratory, followed contraction by measuring the ATP-induced loss in centrifuged volume of homogenized muscle fibrils. He observed that soluble muscle extract prevented the volume change, i.e., prevented contraction. Boiling or acid treatment of the extract inactivated the factor, which was also nondialyzable, indicating that it was a protein. The effect of the factor was inhibited by 2 mM Ca2+. Bendall (1952) tested the muscle extract on glycerinated fibers and found that it relaxed the fibers. Initially the “relaxing factor”, or Marsh-Bendall factor, was considered to be an ATP-regenerating enzyme such as creatine kinase or myokinase. However, it was not really soluble and could be collected by high-speed centrifugation. Moreover, Kumagai et al. (1955) found it to be identical with the Kielley-Meyerhof granular ATPase. The factor was later identified by Hasselbach and Makinose (1961)(1963) and by Ebashi and Lipmann (1962) as fragmented sarcoplasmic reticulum that acted as Ca2+-pump. The triggered release of sequestered Ca2+ to ∼10 μM caused muscle to contract.
Actin-linked Regulation
Perry and Grey (1956) reported that EDTA inhibited only a crude actomyosin preparation but not a synthetic preparation, a first indication of the involvement of a protein modulating the activity of actomyosin. Weber and Winicur (1961) observed that the Ca2+ sensitivity of different actomyosin preparations varied and that the variation was due to differences in the way the actin was prepared. Ebashi and colleagues discovered that for the relaxing effect tropomyosin was required. However, only “native” tropomyosin was effective. This was due to an additional protein, named troponin (Ebashi, 1963; Ebashi and Ebashi, 1964). In the thin filaments tropomyosin lies on the flat surface formed between the two strands of actin. Tropomyosin's length somewhat exceeds the pitch of the long actin helix so that the tropomyosin molecules overlap when binding to actin. The presence of tropomyosin and the overlap between the tropomyosin molecules confers cooperativity to the regulatory system (Bremel and Weber, 1972). Troponin is arranged periodically, each tropomyosin binds one troponin molecule (Ohtsuki et al., 1967). Greaser and Gergely (1971) showed that troponin consists of three different subunits. TroponinC (TnC) binds Ca2+ and is related to calmodulin; troponinI (TnI) is an inhibitory subunit that binds to TnC and to actin and troponinT (TnT) in a Ca2+-dependent manner, and TnT binds to tropomyosin. It is thought that in the absence of Ca2+ the affinity between TnC and TnI is strong so that tropomyosin is held over the myosin-binding site of actin. In the presence of Ca2+ the binding between TnI and TnC weakens, tropomyosin is allowed to roll azimuthally around actin to open up the binding-site for myosin. Combination with S1 evidently leads to its further movement. This steric hindrance model of regulation was based on observations of the low angle X-ray diffraction patterns from muscle fibers. The second-order reflection of the actin period changes dramatically on activation (Haselgrove, 1973; Huxley, 1973; Parry and Squire, 1973). This result was later supported by electron microscopy (Vibert et al., 1997).
Myosin-linked Regulation
Molluscan myosins are regulated molecules. In contrast to skeletal muscle myosins, these myosins have a specific high affinity Ca2+ binding-site. Binding of Ca2+ to these sites is necessary for activity (Kendrick-Jones et al., 1970). A detailed study of scallop myosin has shown that the light chains are regulatory subunits. Depletion of the regulatory light chains by EDTA results in the loss of regulation and of the ability to bind Ca2+. Rebinding of the regulatory light chains restores regulation. Only two-headed molecules, myosin and HMM, are regulated. While retaining the ability to bind Ca2+, S1 is fully active in its absence, although it can still be regulated by the troponin-tropomyosin system (Szent-Györgyi et al., 1973). Ca2+ binding requires that both regulatory and essential light chains are complexed with the heavy chain. Isolated light chains are unable to bind Ca2+. Later studies have shown why this is so. Szentkiralyi (1984) isolated the light chain-binding region, called the regulatory domain, consisting of both light chains complexed with the associated heavy chain fragment. The atomic structure of the regulatory domain located the bound Ca2+ on domain 1 of the essential light chain. The Ca2+-binding loop is stabilized by the regulatory light chain, mainly by interactions between Gly23 of the essential light chain and Gly117 of the regulatory light chain (Xie et al., 1994). The mutation of Gly117 to alanine abolished Ca2+ binding. Furthermore, conversion of cysteine into glycine of the inactive skeletal regulatory light chain resulted in a “gain in function” mutation (Jancsó and Szent-Györgyi, 1994).
Epilogue
The years of research between 1941 and 1972 were exciting. The results were often quite surprising. However, peu a peu, a solid foundation for our understanding of muscle function at the molecular level was established. The realization that movement and force production require interaction between two proteins, the discovery of actin, the sliding filament mechanism, the way contraction is controlled, and an understanding of the manner in which the energy of ATP may be used all opened up new vistas. The field also served as an example of how the combination of structural and biochemical approaches can lead to a detailed understanding of a cellular function. The success of the analysis was beyond most expectations. The road was not easy and errors abounded. Nevertheless, a solid base for the next 30 yr of research has been established which, no doubt, will often lead to results no one foresaw. The importance of a detailed knowledge of motility in cell biology is manifest. Moreover, discoveries remain surprising and repeatedly find unexpected applications in numerous cellular functions. In spite of the expectations of 1972, the work is still far from complete. One may quote Albert Szent-Györgyi:
“A discovery is said to be an accident meeting a prepared mind.”
We should stay prepared.
Acknowledgments
I thank Kenneth C. Holmes for twisting my arm at the 2004 Biophysics Meeting to write this review, for suggestions, and for translating some of my sentences into the Queen's English.
Kenneth C. Holmes served as editor.
Footnotes
The experiments in Szeged were published in three volumes as “Studies from the Institute of Medical Chemistry University of Szeged”. While the publisher is given as S. Karger Basel, New York, it is not clear whether the publisher ever proceeded with the publication. Volume I (1941–1942) entitled “Myosin and Muscular Contraction” by I. Banga, T. Erdös, M. Gerendás, W.F.H.M. Mommaerts, F.B. Straub, and A. Szent-Györgyi, edited by A. Szent-Györgyi was sent to press on July 6, 1942, and printed by R. Gergely Budapest. (At this time Hungary was at war with the Allies.) Volume II (1942), no title, edited by A. Szent-Györgyi was sent to press on December 22, 1942, and printed by the Town Printer and Book Publisher KFT of Szeged. Volume III (1943) entitled “Muscular Contraction, Blood Coagulation”, edited by A. Szent-Györgyi was sent to press on December 13, 1943, and printed by the Town Printer and Book Publisher KFT of Szeged. Albert Szent-Györgyi originally planned to publish a fourth volume of the studies. This became impossible because he had to go into hiding from the GESTAPO. He doubted that he would survive the war and while in hiding he wrote up the work he and his collaborators had carried out in Szeged, including his speculations and theories on the biochemistry of muscle contraction. The manuscript entitled “Muscle” was sent to his friend, Hugo Theorell on the August 1, 1944 to be published in Acta Physiologica Scandinavica. Szent-Györgyi had been given Swedish citizenship to protect him from the GESTAPO, The citizenship also qualified him to publish in the Acta. Hugo Theorell, not knowing how to contact Szent-Györgyi, sent a message to the Swedish Legation that the manuscript had been received, having no idea that Szent-Györgyi was sheltering there. He had to be smuggled out from the Legation to escape the rabble organized by the Arrow Cross Party, the Hungarian Nazi party, which broke into the Legation the next day and looted it while looking for him. The long paper was published in 1945 (Szent-Györgyi, 1945).
A detailed compendium of data on all aspects of muscle function available from antiquity until 1970 can be found in the monumental book by Needham (1971), which contains some 2,400 references. The structural studies leading to the sliding filament concept are described by Huxley (1958)(1969). The early history of regulation is discussed by Ebashi and Endo (1968). A more up to date short review on myosin-linked regulation is also available (Szent-Györgyi, 1996).
References
- Alberts, B., A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter. 2002. Molecular Biology of the Cell. Fourth edition. Garland Publishing. 950 pp.
- Asakura, S. 1961. F-actin adenosine triphosphatase activated under sonic vibration. Biochim. Biophys. Acta. 52:65–75. [DOI] [PubMed] [Google Scholar]
- Asakura, S., M. Taniguchi, and F. Oosawa. 1963. Mechano-chemical behaviour of F-actin. J. Mol. Biol. 7:55–69. [Google Scholar]
- Astbury, W.T. 1947. Croonian lecture. On the structure of biological fibers and the problem of muscle. Proc. R. Soc. Lond. B. Biol. Sci. 134:303–328. [DOI] [PubMed] [Google Scholar]
- Bagshaw, C.R. and D.R Trentham. 1974. The characterization of myosin-product complexes and of product-release steps during the magnesium ion-dependent adenosine triphosphatase reaction. Biochem. J. 141:331–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, K. 1946. Tropomyosin: A new asymmetric protein component of muscle. Nature. 157:368–369. [DOI] [PubMed] [Google Scholar]
- Bailey, K. 1948. Tropomyosin: A new asymmetric component of the muscle fibril. Biochem. J. 43:271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banga, I. 1942. The phosphatase activity of myosin. Stud. Inst. Med. Chem. Univ. Szeged. I:27–36. [Google Scholar]
- Banga, I., and A. Szent-Györgyi. 1942. Preparation and properties of myosin A and B. Stud. Inst. Med. Chem. Univ. Szeged. I:5-15. [Google Scholar]
- Bendall, J.R. 1952. Effect of the “Marsh factor” on the shortening of muscle fibre models in the presence of adenosine triphosphate. Nature. 170:1058–1060. [DOI] [PubMed] [Google Scholar]
- Biró, N.A., and A.G. Szent-Györgyi. 1949. The effect of actin and physicochemical changes on the myosin ATPase system, and on washed muscle. Hung. Acta Physiol. II:1–14. [PubMed] [Google Scholar]
- Bremel, R.D., and A. Weber. 1972. Cooperation within actin filament in vertebrate skeletal muscle. Nature New Biol. 238:97–101. [DOI] [PubMed] [Google Scholar]
- Cohen, C., and W. Longley. 1966. Tropomyosin paracrystals formed by divalent cations. Science. 152:794–796. [DOI] [PubMed] [Google Scholar]
- Cohen, C., and A.G. Szent-Györgyi. 1957. Optical rotation and helical polypeptide chain configuration in α-proteins. J. Am. Chem. Soc. 79:248. [Google Scholar]
- Collins, J.H. 1991. Myosin light chains and troponin C: structural and evolutionary relationships revealed by amino acid sequence comparisons. J. Muscle Res. Cell Biol. 12:3–25. [DOI] [PubMed] [Google Scholar]
- Cooke, R., and L. Murdoch. 1973. Interaction of actin with analogs of adenosine triphosphate. Biochemistry. 12:3927–3932. [DOI] [PubMed] [Google Scholar]
- Dainty, N., A. Kleinzeller, A.S.C. Lawrence, M. Miall, J. Needham, D.M. Needham, and S.-C. Shen. 1944. Studies on the anomalous viscosity and flow birefringence of protein solutions. III. Changes in these properties of myosin solutions in relation to adenosinetriphosphate and muscular contraction. J. Gen. Physiol. 27:355–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lozanne, A. and J.A. Spudich. 1987. Disruption of the Dictyostelium myosin heavy chain gene by homologous recombination. Science. 236:1086–1091. [DOI] [PubMed] [Google Scholar]
- Dos Remedios, C.G., R.G. Millikan, and M.F. Morales. 1972. Polarization of tryptophan fluorescence from single striated muscle fibers. A molecular probe of contractile state. J. Gen. Physiol. 59:103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabikowski, W., and J. Gergely. 1962. The effect of the temperature of extraction on the tropomyosin content in actin. J. Biol. Chem. 237:3412–3417. [Google Scholar]
- Dreizen, P., and L.C. Gershman. 1970. Relationship of structure to function in myosin. II. Salt denaturation and recombination experiments. Biochemistry. 9:1688–1693. [DOI] [PubMed] [Google Scholar]
- Ebashi, S. 1963. Third component participating in the superprecipitation of “natural actomyosin”. Nature. 200:1010. [DOI] [PubMed] [Google Scholar]
- Ebashi, S., and F. Ebashi. 1964. A new protein component participating in the superprecipitation of myosin B. J. Biochem. 55:604–613. [DOI] [PubMed] [Google Scholar]
- Ebashi, S., and M. Endo. 1968. Calcium ion and muscle contraction. Progr. Biophys. Mol. Biol. 125–183. [DOI] [PubMed]
- Ebashi, S., and F. Lipmann. 1962. Adenosine triphosphate-linked concentration of calcium ions in a particulate fraction of rabbit muscle. J. Cell Biol. 14:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt, V.A., and M.N. Lyubimova. 1939. Myosin and adenosinetriphosphatase. Nature. 144:668–669. [Google Scholar]
- Engelhardt, V.A., M.N. Ljubimova and R.A. Meitina. 1941. Chemistry and mechanics of the muscle studied on myosin threads. Compt. Rend. De l'acad Sci. de l'USSR. 30:644. [Google Scholar]
- Erdös, T. 1943. Rigor, contracture and ATP. Stud. Inst. Med. Chem. Univ. Szeged. III:51–56. [Google Scholar]
- Finer, J.T., R.M. Simmons, and J.A. Spudich. 1994. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 368:113–119. [DOI] [PubMed] [Google Scholar]
- Gazith, J., S. Himmelfarb, and W.J. Harrington. 1970. Studies on the subunit structure of myosin. J. Biol. Chem. 245:15–22. [PubMed] [Google Scholar]
- Gergely, J. 1950. On the relationship between myosin and ATPase. Fed. Proc. 9:176. [Google Scholar]
- Gergely, J. 1953. Studies on myosin-adenosinetriphosphatase. J. Biol. Chem. 200:543–550. [PubMed] [Google Scholar]
- Gergely, J., M.A. Gouvea, and D. Karibian. 1955. Fragmentation of myosin by chymotrypsin. J. Biol. Chem. 212:165–177. [PubMed] [Google Scholar]
- Greaser, M.L., and J. Gergely. 1971. Reconstitution of troponin activity from three protein components. J. Biol. Chem. 246:4226–4233. [PubMed] [Google Scholar]
- Guba, F. 1943. Observations on myosin and actomyosin. Stud. Inst. Med. Chem. Univ. Szeged. III:40–45. [Google Scholar]
- Hanson, J., and H.E. Huxley. 1953. Structural basis of cross-striations in muscle. Nature. 172:530–532. [DOI] [PubMed] [Google Scholar]
- Hanson, J., and J. Lowy. 1963. The structure of F-actin and of actin filaments isolated from muscle. J. Mol. Biol. 6:46–60. [Google Scholar]
- Haselgrove, J.C. 1973. X-ray evidence for a conformational change in actin-containing filaments of vertebrate striated muscle. Cold Spring Harb. Symp. Quant. Biol. 37:225–234. [Google Scholar]
- Hasselbach, W. 1953. Elektronmikroskopische untersuchungen an muskelfibrillen bei totaler und partieller extraktion des L-myosins. Z. Naturforsch. 8b:449–454. [Google Scholar]
- Hasselbach, W., and M. Makinose. 1961. Die calciumpumpe der “erschlaffungsgrana” des muskels und ihre abhangigkeit von der ATP-spaltung. Biochem. Z. 333:518–526. 13712164 [Google Scholar]
- Hasselbach, W., and N. Makinose. 1963. Über der mechanismus des calciumtransportes durch die membranen des sarcoplasmatischen reticulums. Biochem. Z. 339:94–111. [PubMed] [Google Scholar]
- Hayashi, T., and R. Rosenbluth. 1960. Studies on actin. II. Polymerization and the bound nucleotide. Biol. Bull. 119:290. [Google Scholar]
- Higashi, S., and F. Oosawa. 1965. Conformational changes associated with polymerisation and nucleotide binding in actin molecules. J. Mol. Biol. 12:843–865. [DOI] [PubMed] [Google Scholar]
- Hodges, R.S., and L.B. Smiley. 1972. Cysteine sequences of rabbit skeletal tropomyosin. Can. J. Biochem. 50:330. [DOI] [PubMed] [Google Scholar]
- Huxley, A.F. 1957. b. Muscle structure and theories of contraction. Progr. Biophys. Biophys. Chem. 7:255–318. [PubMed] [Google Scholar]
- Huxley, A.F., and R. Niedergerke. 1954. Structural changes in muscle during contraction. Interference microscopy of living muscle fibers. Nature. 173:971–978. [DOI] [PubMed] [Google Scholar]
- Huxley, H.E. 1953. a. X-ray analysis and the problem of muscle. Proc. R. Soc. Lond. B. Biol. Sci. 141:59–61. [DOI] [PubMed] [Google Scholar]
- Huxley, H.E. 1953. b. Electron microscope studies of the organisation of the filaments in striated muscle. Biochim. Biophys. Acta. 12:387–394. [DOI] [PubMed] [Google Scholar]
- Huxley, H.E. 1957. a. The double array of filaments in cross-striated muscle. J. Biophys. Biochem. Cyt. 3:631–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley, H.E. 1958. The contraction of muscle. Sci. Am. 199:66–82. [PubMed] [Google Scholar]
- Huxley, H.E. 1963. Electron microscope studies on the structure of natural and synthetic protein filaments from striated muscle. J. Mol. Biol. 7:281–308. [DOI] [PubMed] [Google Scholar]
- Huxley, H.E. 1969. The mechanism of muscle contraction. Science. 164:1356–1366. [PubMed] [Google Scholar]
- Huxley, H.E. 1973. Structural changes in actin- and myosin-containing filaments during contraction. Cold Spring Harb. Symp. Quant. Biol. 37:371–376. [Google Scholar]
- Huxley, H.E., and J. Hanson. 1954. Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. Nature. 173:979–988. [DOI] [PubMed] [Google Scholar]
- Jancsó, A., and A.G. Szent-Györgyi. 1994. Regulation of scallop myosin by the regulatory light chain depends on a single glycine residue. Proc. Natl. Acad. Sci. USA. 91:8762–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa, T., and Y. Tonomura. 1965. The pre-steady state of the myosin-adenosine triphosphatase system. I. Initial rapid liberation of inorganic phosphate. J. Biochem. 57:604–615. [PubMed] [Google Scholar]
- Kendrick-Jones, J., W. Lehman, and A.G. Szent-Györgyi. 1970. Regulation in molluscan muscles. J. Mol. Biol. 54:313–326. [DOI] [PubMed] [Google Scholar]
- Kishino, A., and T. Yanagida. 1988. Force measurements by micromanipulation of a single actin filament by glass needles. Nature. 334:74–76. [DOI] [PubMed] [Google Scholar]
- Kominz, D.R., E.R. Mitchell, T. Nihei, and C.M. Kay. 1965. The papain digestion of skeletal myosin A. Biochemistry. 4:2373–2382. [Google Scholar]
- Kominz, D.R., W.R. Caroll, E.N. Smith, and E.R. Mitchell. 1959. A subunit of myosin. Arch. Biochem. Biophys. 79:191–199. [Google Scholar]
- Kron, S.J., and J.A. Spudich. 1986. Fluorescent actin filaments move on myosin fixed to a glass surface. Proc. Natl. Acad. Sci. USA. 83:6272–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühne, W. 1864. Untersuchungen über das Protoplasma und die Contractilitat. W. Engelmann, Leipzig.
- Kumagai, H., S. Ebashi, and F. Takeda. 1955. Essential relaxing factor in muscle other than myokinase or creatine phosphokinase. Nature. 176:166. [DOI] [PubMed] [Google Scholar]
- Lauffer, M.A., and A.G. Szent-Györgyi. 1955. Comments of the structure of myosin. Arch. Biochem. Biophys. 56:542–548. [DOI] [PubMed] [Google Scholar]
- Lohmann, K. 1934. Über die enzymatische ausspaltung der kreatinephosphorsaure; zugleich ein beitrag zum chemismus der muskelkontraktion. Biochem. Z. 271:264–277. [Google Scholar]
- Lowey, S., L. Goldstein, C. Cohen, and S.M. Luck. 1967. Proteolytic degradation of myosin and the meromyosins by a water-insoluble polyanionic derivative of trypsin. J. Mol. Biol. 23:287–304. [DOI] [PubMed] [Google Scholar]
- Lowey, S., and D. Risby. 1971. Light chains from fast and slow muscle myosins. Nature. 234:81–85. [DOI] [PubMed] [Google Scholar]
- Lowey, S., H.S. Slayter, A.G. Weeds, and H. Baker. 1969. Substructure of the myosin molecule. I. Subfragments of myosin by enzymic degradation. J. Mol. Biol. 42:1–29. [DOI] [PubMed] [Google Scholar]
- Lymn, R.W., and E.W. Taylor. 1971. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry. 10:4617–4624. [DOI] [PubMed] [Google Scholar]
- Marsh, B.B. 1951. A factor modifying muscle fiber syneresis. Nature. 167:1065–1066. [DOI] [PubMed] [Google Scholar]
- Mihályi, E., and W.F. Harrington. 1959. Studies on the tryptic digestion of myosin. Biochim. Biophys. Acta. 36:447–466. [Google Scholar]
- Mihályi, E., and A.G. Szent-Györgyi. 1953. a. Trypsin digestion of muscle proteins. I. Ultracentrifugal analysis of the process. J. Biol. Chem. 201:189–196. [PubMed] [Google Scholar]
- Mihályi, E., and A.G. Szent-Györgyi. 1953. b. Trypsin digestion of muscle proteins. J. Biol. Chem. III:211–219. [PubMed] [Google Scholar]
- Mueller, H., and S.V. Perry. 1962. The degradation of heavy meromyosin by trypsin. Biochem. J. 85:431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralt, A. and J.T. Edsall. 1930. Studies in the physical chemistry of muscle globulin. IV. The anisotropy of myosin and double refraction of flow. J. Biol. Chem. 89:351–386. [Google Scholar]
- Needham, D.M. 1971. Machina carnis, the Biochemistry of Muscular Contraction in its Historical Development. Cambridge University Press. 782 pp.
- Needham, J., A. Kleinzeller, M. Miall, M. Dainty, D.M. Needham, and A.S.C. Lawrence. 1942. Is muscle contraction essentially an enzyme-substrate combination? Nature. 150:46–49. [Google Scholar]
- Needham, J., S-C. Shen., D.M. Needham, and A.S.C. Lawrence. 1941. Myosin birefringence and adenylpyrophosphate. Nature. 147:766–768. [Google Scholar]
- Ohtsuki, I.T., T. Masaki, T. Nonomura, and S. Ebashi. 1967. Periodic distribution of troponin along the thin filament. J. Biochem. 61:817–819. [DOI] [PubMed] [Google Scholar]
- Patterson, B., and J.A. Spudich. 1995. A novel positive selection for identifying cold-sensitive mutants in Dictyostelium. Genetics. 140:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry, D., and J.M. Squire. 1973. Structural role of tropomyosin in muscle regulation: analysis of the X-ray patterns from relaxed and contracting muscles. J. Mol. Biol. 75:33–55. [DOI] [PubMed] [Google Scholar]
- Perry, S.V. 1951. The adenosinetriphophatase activity of myofibrils isolated from skeletal muscle. Biochem. J. 48:257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, S.V., and T.C. Grey. 1956. Ethylenediaminetetra-acetate and the adenosinetriphosphatase activity of actomyosin systems. Biochem. J. 64:5P. [Google Scholar]
- Philpott, D., and A.G. Szent-Györgyi. 1954. The structure of light-meromyosin: an electron microscopic study. Biochim. Biophys. Acta. 15:165–173. [DOI] [PubMed] [Google Scholar]
- Polis, B.D., and O. Meyerhof. 1947. Studies on adenosinetriphosphatase in muscle. I. Concentration of the enzyme on myosin. J. Biol. Chem. 169:389–401. [PubMed] [Google Scholar]
- Reedy, M.K., K.C. Holmes, and R.T. Tregear. 1965. Induced changes in orientation of the cross-bridges of glycerinated insect flight muscle. Nature. 207:1276–1280. [DOI] [PubMed] [Google Scholar]
- Slayter, H.S., and S. Lowey. 1967. Substructure of the myosin molecule as visualized by electron microscopy. Proc. Natl. Acad. Sci. USA. 58:1611–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutoh, K., K. Sutoh, T. Karr, and W.F. Harrington. 1978. Isolation and physico-chemical properties of a high molecular weight subfragment-2 of myosin. J. Mol. Biol. 126:1–22. [DOI] [PubMed] [Google Scholar]
- Stracher, A. 1969. Evidence for the involvement of light chains in the biological functioning of myosin. Biochem. Biophys. Res. Commun. 35:519–525. [DOI] [PubMed] [Google Scholar]
- Straub, F.B. 1942. Actin. Stud. Inst. Med. Chem. Univ. Szeged. II:3–15. [Google Scholar]
- Straub, F.B. 1943. Actin, II. Stud. Inst. Med. Chem. Univ. Szeged. III:23–37. [Google Scholar]
- Straub, F.B., and G. Feuer. 1950. Adenosinetriphosphate the functional group of actin. Biochim. Biophys. Acta. 4:455–470. [PubMed] [Google Scholar]
- Szent-Györgyi, A. 1942. a. The contraction of myosin threads. Stud. Inst. Med.Chem. Univ. Szeged. I:17–26. [Google Scholar]
- Szent-Györgyi, A. 1942. b. Discussion. Stud. Inst. Med. Chem. Univ. Szeged. I:67–71. [Google Scholar]
- Szent-Györgyi, A. 1943. a. The crystallization of myosin and some of its properties and reactions. Stud. Inst. Med. Chem. Univ. Szeged. III:76–85. [Google Scholar]
- Szent-Györgyi, A. 1943. b. Observations on actomyosin. Stud. Inst. Med. Chem. Univ. Szeged. III:86–92. [Google Scholar]
- Szent-Györgyi, A. 1945. Studies on muscle. Acta Physiol. Scand. 9:25. [DOI] [PubMed] [Google Scholar]
- Szent-Györgyi, A. 1949. Free energy relations and contraction of actomyosin. Biol. Bull. 96:140–161. [PubMed] [Google Scholar]
- Szent-Györgyi, A. 1963. Lost in the twentieth century. Annu. Rev. Biochem. 32:1–13. [DOI] [PubMed] [Google Scholar]
- Szent-Györgyi, A.G. 1953. Meromyosins, the subunits of myosin. Arch. Biochem. Biophys. 42:305–320. [DOI] [PubMed] [Google Scholar]
- Szent-Györgyi, A.G. 1975. Calcium regulation of muscle contraction. Biophys. J. 15:707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Györgyi, A.G. 1996. Regulation of contraction by calcium binding myosins. Biophys. Chem. 59:357–363. [DOI] [PubMed] [Google Scholar]
- Szent-Györgyi, A.G., C. Cohen, and D. Phipott. 1960. Light meromyosin fraction I: A helical molecule from myosin. J. Mol. Biol. 2:133–142. [Google Scholar]
- Szent-Györgyi, A.G., E.M. Szentkiralyi, and J. Kendrick-Jones. 1973. The light chains of scallop myosin as regulatory subunits. J. Mol. Biol. 74:179–203. [DOI] [PubMed] [Google Scholar]
- Szentkiralyi, E.M. 1984. Tryptic digestion of scallop S1: evidence for a complex between the two light chains and a heavy chain peptide. J. Muscle Res. Cell Motil. 5:147–164. [DOI] [PubMed] [Google Scholar]
- Taylor, E.W. 2001. E.B. Wilson lecture: the cell as molecular machine. Mol. Biol. Cell. 12:251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trybus, K. 1994. Regulation of expressed truncated smooth muscle myosins; role of the essential light chain and tail length. J. Biol. Chem. 269:20819–20822. [PubMed] [Google Scholar]
- Trotta, P.P., P. Dreizen, and A. Stracher. 1968. Studies on subfragment-I, a biologically active fragment of myosin. Proc. Natl. Acad. Sci. USA. 61:659–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao, T.C. 1953. Fragmentation of the myosin molecule. Biochim. Biophys. Acta. 11:368–382. [DOI] [PubMed] [Google Scholar]
- Vibert, P., R. Craig, and W. Lehman. 1997. Steric-model for activation of muscle thin filaments. J. Mol. Biol. 266:8–14. [DOI] [PubMed] [Google Scholar]
- Weber, A., and S. Winicur. 1961. The role of calcium in the superprecipitation of actomyosin. J. Biol. Chem. 236:3198–3202. [PubMed] [Google Scholar]
- Weber, H.H. 1935. Der feinbau und die mechanischen eigenschaften des myosin-fadens. Arch. Physiol. 235:205–233. [Google Scholar]
- Weeds, A.G. 1969. Light chains of myosin. Nature. 223:1362–1364. [DOI] [PubMed] [Google Scholar]
- Weeds, A.G., and S. Lowey. 1971. The substructure of the myosin molecule II. The light chains of myosin. J. Mol. Biol. 61:701–725. [DOI] [PubMed] [Google Scholar]
- Woods, E.F., S. Himmelfarb, and W.F. Harrington. 1963. Studies of the structure of myosin in solution. J. Biol. Chem. 238:2374–2385. [PubMed] [Google Scholar]
- Xie, X., D.H. Harrison, I. Schlichting, R.M. Sweet, V.N. Kalabokis, A.G. Szent-Györgyi and C. Cohen. 1994. Structure of the regulatory domain of scallop myosin at 2.8 Å resolution. Nature. 368:306–312. [DOI] [PubMed] [Google Scholar]