

The Picture in 1972
This review will describe the investigation of the mechanism of muscle contraction and cell motility from 1972 to the present. The preceding article in this issue by Andrew Szent-Gyorgyi covers the period up to 1972. In 1972 the field of actomyosin interactions was summarized in a conference at Cold Spring Harbor, published in the Cold Spring Harbor Symposia on Quantitative Biology XXXVII, 1973. After this meeting many participants thought that the problem of muscle contraction was solved “in principle”. In many ways this attitude was correct. In the mid-1950s it had been established that during muscle contraction two sets of filaments of constant length slide past each other. Prior to the sliding filament model, the most popular theories held that contraction was produced by the shortening of some large, rubber-like polymers. Since 1954, the motor that produced filament sliding, the myosin head, had been observed both by electron microscopy and X-ray diffraction. Kinetics studies had shown that ATP dissociated actin from myosin and that ATP was hydrolyzed when myosin was detached from actin leading to a four-state model of the kinetics of the actin myosin interaction. The synthesis of these results led to a model of myosin action in which a rigid myosin head attached to actin and changed its orientation producing the power stroke before being detached. This simple elegant model was able to explain much of the existing data on the interaction of myosin with actin in muscle and in solution.
Although the model proposed in 1972 was in essence correct, there was in fact a long road ahead leading to the present level of understanding of the motor proteins. Major milestones on this road include solving the three-dimensional structures of the actin monomer and the myosin head in a number of their different nucleotide states. The structural information was synthesized with, unthinkable in 1972, measurements of the mechanics of single myosin molecules. This development was facilitated by the demonstration that the muscle myosin, studied so intensely in 1972, is in fact only one member of a large superfamily of myosin molecules, some of which were more amenable to study by biophysical techniques. Genetically engineered proteins provided novel preparations for enzymatic and structural studies. In addition an entirely new super family of microtubule motors was discovered. This review will describe these studies leading to our current models of force production by motor proteins in eukaryotic cells. Due to the limitations of length, the review will not be comprehensive, but will concentrate on some of the key experiments leading to our understanding of force production by the two motor proteins, myosin and kinesin. An excellent book covers much of this material (Howard, 2001).
The Period from 1972 to 1986
In 1986 I wrote an extensive review which described the experimental studies leading to the first major modification of the model of myosin action proposed in 1972 (Cooke, 1986). Several lines of experimental evidence suggested that myosin did not act as a rigid oar, but that there was one region of myosin attached rigidly to actin during the power stroke while another region of myosin changed its orientation. Low angle X-ray diffraction patterns had shown that, although a sizable fraction of myosin heads were attached to actin, only a small fraction of the mass of these heads contributed to the actin layer lines (Holmes and Goody, 1984; Huxley and Kress, 1985). In addition, probes on two sites on the myosin head, attached to a reactive sulfhydryl or placed at the nucleotide site, gave no indication that these regions of the protein changed orientation during force production or that their orientation was altered by a application of force to the muscle (Cooke et al., 1982; Yanagida, 1985). A lower resolution structure of the myosin head seen by electron microscopy in a crystal showed that it resembled a tadpole, with a large globular domain attached to a more slender cylindrical region. Together these data suggested that the large globular domain of myosin is attached to actin in a rigid fashion with the cylindrical region, which came to be known as the neck, acting as the lever producing the power stroke. Once again this model has proven to be essentially correct, but again a long road lay ahead to reach our current level of understanding.
In 1972 a simple model for the kinetics of the actomyosin interaction in solution had been determined. Between 1972 and 1986 many details of this interaction in solution were determined. Taylor and coworkers showed that a large decrease in free energy occurred upon the binding of myosin to actin following the release of phosphate (Taylor, 1979). The binding of ATP to myosin was a second reaction involving a large change in free energy, with an extraordinarily strong association constant of 1010 M−1 (Mannherz et al., 1974). In contrast the hydrolysis step was found to be freely reversible (Trentham et al., 1976). A significant step forward was the development of caged compounds, which could be rapidly released by photolysis and in particular caged ATP. This allowed the kinetic investigations, all of which had been performed in solution, to be extended to muscle fibers. It would be expected that the kinetics observed in solution will be modified by the steric constraints that exist in the myofibril. The studies of the mechanics and biochemistry after rapid release of caged compounds inside muscle fibers showed that the kinetics scheme derived from the solution studies was generally applicable to muscle fibers, with rapid and strong binding of ATP dissociating the myosin head from actin, followed by hydrolysis while the myosin was either detached from actin or weakly associated with it (Goldman et al., 1982).
Myosin Structures
The determination of the high-resolution structures of actin and myosin was a crucial step in understanding the functions of these proteins. Both structures were particularly difficult to obtain, myosin because of its size and actin because of its tendency to polymerize. After a 10-yr struggle by several laboratories, the structure of G-actin was solved by Kabsch and Holmes in 1990 in a complex with a small molecule which prevented polymerization (Kabsch et al., 1990). Actin was found to be composed of four globular subdomains with a nucleotide bound in a prominent cleft between them. A number of structures of the actin monomer have now been obtained, all in a complex with molecules that prevent polymerization. The two outer subdomains have been shown to alter their orientation relative to the inner two subdomains, thereby producing an opening and closing of the cleft that contains the bound nucleotide. Although conformational changes in actin undoubtedly occur during force generation, the nature of these changes and their role in force generation remain unknown. Actin is now known to undergo reversible polymerization cycles that are involved in force production in nonmuscle cells, which will be discussed in a later section. The polymerization is controlled by binding and hydrolysis of the bound nucleotide, which probably is mediated by the domain movements described above; but the connection between structure and kinetics remains undeciphered.
Obtaining a high-resolution structure of the myosin head was a heroic effort. Problems included the size of the molecule and heterogeneity in the protein preparation due both to myosin isoforms and to the fact that it was a product of proteolysis. These problems were finally overcome by Rayment, Winkelman, and colleagues, who determined the structure of the entire head of a chemically modified form of chicken myosin including both light chains (Rayment et al., 1993b). The NH2-terminal portion of the myosin heavy chain (called S1), ∼770 amino acids, formed a large globular domain, which contained the sites for binding actin and nucleotide, now known as the catalytic domain (Fig. 1). A prominent cleft, known as the actin binding cleft, originated close to the nucleotide site and extended down to the region where actin was bound. The myosin heavy chain then extended in a single α-helix for ∼70 amino acids around which were wrapped the two myosin light chains. A surprising result was that the region in the vicinity of the binding site for the nucleotide had structural homology with the G-proteins. A number of structures of the G-proteins have been solved for different nucleotide states and several structural elements designated as switch regions had been shown to change conformation in response to whether the bound nucleotide was a di- or triphosphate. Changes in the conformation of these regions influence the affinity of the G-protein for other proteins, and, in turn, the binding of these proteins affects the hydrolysis rate and the affinity of the nucleotide. These homologies immediately suggested mechanisms for the function of myosin as well.
Figure 1.

The structure of the head region of myosin, determined by Rayment et al. (1993b). The catalytic domain is shown on the left. The upper 50K, lower 50K, nucleotide binding, and converter domains are colored red, white, green, and blue respectively. The relay helix is colored blue with its distal end connected to the actin binding side in the lower 50K domain by a blue strand, shown at the top of he molecule.
The high-resolution structures of the actin monomer and of the myosin head could be combined with lower resolution structures obtained by X-ray diffraction of actin fibers or three-dimensional reconstructions obtained from electron micrographs of actomyosin complexes to produce models of the actin filament and of the actin filament decorated with myosin in the rigor, e.g., nucleotide-free conformation. This showed that the structure of the myosin head, which was obtained in the absence of nucleotides, could fit reasonably well into the electron density of the myosin head in the rigor conformation, a conformation that is very likely to represent the end of the power stroke (Rayment et al., 1993a). However, it also was noted that the fit of the globular catalytic domain into the structure could be improved if the actin binding cleft was more closed, suggesting that although the light chain domain observed in the crystal resembled that of the rigor complex, changes probably occurred in the catalytic domain.
The structure of the myosin head, along with the fit to the actomyosin complex, represented an immense breakthrough in the field, which now can be subdivided into pre- and poststructural periods. The structures made sense of many previous results, and allowed more rational design of new experiments. The interpretation of previous data in terms of the structures immediately strengthened the hypothesis that the power stroke consisted of a rotation of the light-chain domain relative to the catalytic domain, which was bound and rigidly oriented on actin.
The crystallization of the full myosin head remained difficult, and structural studies switched to a truncated form of myosin from the slime mold Dictyostelium, which involve only the catalytic domain. This construct was crystallized in complexes with ADP and analogs of ATP or ADP-Pi. These structures fell into two classes and provided the first view of the major conformational changes that occur in the catalytic domain in response to the bound nucleotide—conformational changes that are now thought to produce force. One class resembled the original structure of chicken S1, which was obtained without nucleotides. This class included the catalytic domain complexed with analogs thought to resemble ATP, including ADP•BeF3 or ATPγS at the active site (for reviews see Holmes, 1996, 1997; Houdusse et al., 2000). However, Holmes and colleagues found the structure of S1•ADP•BeF3 resembled the second class, discussed below, showing that structures are not tightly coupled to the type of nucleotide bound. Although the binding of ATPγS or ADP•BeF3 causes a major decrease in the affinity of myosin for actin the structures obtained with these ligands resembled those found in the absence of nucleotides. Either the affinity can be altered by subtle changes in structure, or more dramatic changes occur that have not been trapped in these crystal forms.
A second class of structures showed significant shifts in selected secondary elements (relative to the first class, described above) and includes the structures obtained with ADP•AlF4 and ADP•VO4 at the active site (Fisher et al., 1995). The geometry of the these groups suggested that these structures may represent transition states of the hydrolysis step. The presence of these phosphate analogs at the active site induced significant shifts in the positions of several secondary elements. There was a partial closure of the actin binding cleft that bisects the catalytic domain and significant shifts in the region adjacent to the light chains. The shift in the structure around the nucleotide is stabilized by formation of a hydrogen bond between the γ phosphate and a conserved glycine that is part of switch 2, a region known to mediate nucleotide conformational changes in the G-proteins. This conformational change is transmitted to the distal COOH-terminal region via a conserved helix, known as the relay helix (Fig. 1). The COOH-terminal region, which has been termed the “converter region”, has translated by more that 23 Å and rotated by ∼70°. Although the light chains were not a part of these constructs, they are attached to the converter domain and could be expected to also change their orientation (Holmes, 1996). This swing was later visualized with a smooth myosin construct that contained the essential light chain (Dominguez et al., 1998). The current hypothesis is that the binding of myosin to actin and release of phosphate reverses this swing, producing the power stroke shown in Fig. 2.
Figure 2.

The lever arm movement of the light chain domain. Reconstructions of the actin–myosin complex at the beginning and end of the power stroke. (A) The “beginning” of the power stroke, based on the truncated S1–ADP.vanadate coordinates (PDB-1VOM). The missing lever arm has been restored using the chicken S1 coordinates (PDB-MYS) with an appropriate rotation. The break in the chain at the beginning of the lever arm marks the extent of the fragment of S1 used in the crystal structure analysis. (B) The “end” state, or rigor complex. Note that the end of the lever arm moves ∼12 nm between the two states. Regions of myosin are colored as in Fig. 1, with the exception that the colors of the light chains have been switched. Diagram prepared with GRASP (Nicholls et al., 1991). Reproduced with permission from Holmes (1997).
The binding of actin to myosin promotes the release of first phosphate, then ADP, and recent structures have suggested that this is caused by an opening of the nucleotide cleft, and in particular to a movement of the switch 1 region away from the bound nucleotide. Two crystal structures have been solved with an open nucleotide pocket, and fitting the myosin head into models of the acto–S1 complex, derived from electron micrographs shows that the pocket must open when myosin is bound to actin (Coureux et al., 2003; Holmes et al., 2003; Reubold et al., 2003). The switch 1 region is known to open in the G-proteins, in response to the binding of proteins that promote nucleotide exchange. In the kinesin motors, described below, crystal structures show an open conformation of switch 1, in contrast to the closed conformation seen in most myosin structures. Spectroscopic studies have shown that the kinesin nucleotide pocket closes upon binding to microtubules (Naber et al., 2003). Thus, the binding of the motors to their polymeric partners produces conformational changes at the nucleotide sites, which appear to be complementary in the two motor families. This will be discussed in more detail in a section below.
In summary, the crystal structures have shown that the relative orientation between the catalytic domain and the light chain domain is extremely variable, with the two orientations described above and yet another orientation seen for scallop myosin with bound ADP (Houdusse et al., 2000). Although the orientation of the light chain domain is influenced by the bound nucleotide via switch 2, it is not tightly coupled, and the same nucleotide can produce different structures. The interactions between the nucleotide and actin binding are probably mediated in part by changes in the actin cleft, and also involve changes in switch 1. Much of the flexibility within the myosin head is the result of the relative motions of several relatively rigid domains, which include the converter domain, the nucleotide binding domain and the upper and lower 50K domains (on either side of the actin cleft) (Houdusse et al., 2000). Thus, in the absence of actin the conformation of myosin appears to be very flexible, with little free energy change between conformations. These conformational changes will be synthesized with data on mechanics, and fit into the contractile cycle in a section below.
In Vitro Assays of Force and Displacement
A second major breakthrough in the field of motility was the development of in vitro measurements of the force and displacement produced by single myosin molecules. Measuring the force produced by actomyosin in a reconstituted system dated back to the very early days of work in this field, when Albert Szent-Gyorgyi showed that slender threads of actomyosin could shorten, an early demonstration that these proteins were in fact the ones which produce the shortening of active muscle (described in this issue, Szent-Gyorgyi, 2004). Although this system was used to demonstrate that a single-headed myosin produced the same force as two-headed myosin, the system remained poorly defined and was not extensively used. The next innovation was made by Sheetz and Spudich, who showed that beads coated with myosin could move rapidly along bundles of actin filaments found in a plant cell (Sheetz and Spudich, 1983). A more defined system was then developed in which myosin or myosin subfragments attached to a glass substrate translated actin filaments, attaining velocities approaching those seen in vivo (Kron and Spudich, 1986). The system was used to answer a long standing question, by showing that the myosin head is sufficient to produce motility, thus localizing the force generating elements to the myosin head (Toyoshima et al., 1987). Small beads can be manipulated by forces exerted by tightly focused laser beams. Spudich and colleagues applied this technology to the actomyosin system measuring the displacement and force generated by single myosin molecules, obtaining ∼10 nm and 3–4 pN, respectively (Finer et al., 1994). Thus opened a whole new field involving the use of optical tweezers in increasingly complex systems to measure the mechanics produced by single myosin motors. However, these measurements proved to be difficult, with thermal fluctuations playing major roles. Yanagida and his group measured very large displacements, which were incompatible with a swinging lever arm of myosin. Eventually these investigations converged on the conclusion that muscle myosin has a stroke length of between 5 and 10 nanometers, and produces forces of 10 pN or greater (for review see Knight et al., 2001). Multiple steps have been found for some myosin isoforms, and the length of the displacement produced has been shown to depend on the number of light chains incorporated into the neck region. Both will be discussed below in more detail.
The Expanding Myosin Superfamily
In the Cold Spring Harbor meeting in 1972, Pollard and Korn reported the purification of a novel single-headed myosin from Acanthamoeba, which they named Myosin I (Pollard and Korn, 1972). Although there was skepticism at the time, with some proposing that this protein may be a proteolytic fragment of a more conventional myosin, this was in fact the first demonstration that myosin has different isoforms with widely varying structures and properties. Myosin I has now been shown to be ubiquitous in eukaryotic cells. To date at least 17 classes of myosin isoforms have been identified in the myosin superfamily. All of these share a highly conserved catalytic domain, followed by a neck region that binds a variable number of light chains. The light chains are either calmodulin or homologous to calmodulin and they bind to the consensus sequence known as an IQ motif (IQxxxRGxxxR). As in muscle myosin the neck region consists of a central α-helix with a variable number of IQ motifs (from zero to six). The neck region is followed by a tail region, which can be very diverse and which is involved in dimerization, if the molecule is dimeric, and in attachment of the myosin to its cargo. An alignment of the various myosin isoforms can be found at http://www.mrc-lmb.cam.ac.uk/myosin/myosin.html.
An extensive review of the isoforms of the myosin superfamily is beyond the scope of this review, and I will mention only two members because of the role they have played in understanding the mechanism of force generation. The first is myosin V, which is a processive motor, i.e., it walks along an actin filament making many steps without letting go. Myosin V has a long neck region with six light chains and it takes a step of 36 nm, which is equal to a periodicity of the actin filament allowing it to move down one side of the filament (Mehta et al., 1999). The long neck region allows this myosin to bridge across the 36 nm distance between successive actin sites. The second isoform is myosin VI, which moves in the opposite direction to all other myosins. Although myosin VI has only two light chains it can nonetheless also take long steps of ∼30 nm (Rock et al., 2001). The mechanism of action of these two myosin isoforms will be discussed in a later section on motor mechanisms.
The Mechanism of Force Production by Myosin
As described in the 1986 review, discussed above, evidence from spectroscopic probes and X-ray diffraction had led to the conclusion that the catalytic domain attached in a rigid fashion to the actin filament, with the neck region acting as a lever arm during the power stroke (Cooke, 1986). However, in 1986 there was no direct evidence showing movement of the light chain domain. In the intervening years, this hypothesis has been proven conclusively.
The first direct observation of the movement of the neck region came from electron micrographs of actin decorated with a myosin head from smooth muscle, which showed that the release of ADP generates a substantial movement of the neck region, with little motion of the catalytic domain (Whittaker et al., 1995). It has now been shown that a variety of myosin molecules, but not all, undergo a rotation upon release of ADP. This rotation is accompanied by a closure of the actin binding cleft (Volkmann et al., 2000). ADP-produced rotations have been seen in electron micrographs, and a step length of several nanometers associated with ADP release has been measured using optical traps (Veigel et al., 1999).
Whereas the release of ADP produces a small step in some myosins, the major displacement occurs before ADP release. Several studies of myosin interacting with actin in vitro have now shown that this larger displacement is also produced by a swing of the light chain lever arm. Myosin molecules with necks of increasing lengths, generated by either different numbers of light chains or by constructing completely artificial lever arms, produce greater velocities and also greater displacements (Anson et al., 1996; Guilford et al., 1996; Uyeda et al., 1996). Thus, mechanically, the neck region appears to operate as a lever arm whose length is its most important property. Large conformational changes within the light-chain domain are not playing a significant active role in generating displacement. Finally, the definitive evidence that the light-chain region undergoes a large change in orientation producing a power stroke came from studies of myosin V. Because this myosin is processive it is more amenable to some biophysical techniques than is muscle myosin. Electron micrographs directly showed the light-chain domain undergoing a change in orientation, from one that resembled the prepower stroke orientation seen in crystal structures to one similar to the post-power stroke (Walker et al., 2000). Measurements using an optical trap showed that the movement consisted of an initial power stroke of 25 nm produced by rotation of the neck, followed by a diffusive search in which the unattached head completed the step using Brownian fluctuations to reach the full 36 nm to the next equivalent actin site (Veigel et al., 2002). In addition the polarization of fluorescent probes attached to the light chains were observed to undergo large changes in orientation on single myosin V molecules as they translated along an actin filament (Forkey et al., 2003). To complete the picture, the position of fluorescently tagged light chains was measured with nanometer precision, showing the expected step length if the myosin V moved hand over hand along the filament (Yildiz et al., 2003). Thus, the question of the mechanism of myosin action, which began in muscle many decades ago, was finally answered most definitively by applying single-molecule techniques to a myosin isoform, and neither the techniques nor the isoform were even imagined at the time the question was first posed. As discussed above, this motion of the LC domain is hypothesized to be driven by changes in the positions and orientations of a few secondary elements that have homologues in both the kinesin superfamily and the G proteins (for review see Vale and Milligan, 2000)).
The evidence obtained in vitro discussed above shows definitively that the light-chain region acts as a lever arm. The studies of similar conformational changes in fibers are more complex because the heads are distributed throughout the cycle. The rotation of the neck region during the power stroke has been supported directly by the observation that fluorescent probes change their angle during force transients of both rigor and active muscle fibers (Corrie et al., 1999). The direction of the angular change was different for rigor and active fibers, indicating that the average angle of the neck region was different in these two states. The observed magnitude of the change was small, however, but this could be indicative of a larger change if only a few heads are attached. Two distinct orientations of a paramagnetic probe attached to the LC domain have been observed in scallop muscle (Baker et al., 1998). One orientation was found in rigor and two in relaxed fibers, with a shift in the populations of these two observed in active fibers, suggesting that the LC domain undergoes a substantial rotation during the power stroke. The interference pattern between the two sets of myosin heads on either side of the thick filament splits the myosin 145 meridional reflection into two peaks whose ratio provides a very accurate measurement of the movements of the heads. Changes in these peaks shows that the heads move inwards, supporting the rotating cross-bridge theory (Piazzesi et al., 2002). Changes in the intensity show heads move toward the vertical then past it. Electron micrographs of active insect flight muscle show a large swing of the light chain domain, and also suggest a change in the angle of the catalytic domain (Taylor et al., 1999). Although the rotation of the LC domain during the power stroke in muscle is now established, the exact angular distribution and changes that occur in this distribution are still not known in skeletal muscle. The interpretation of the data is difficult because we still do not know the exact fraction of the myosin heads producing force, or whether all myosin heads attached to actin are in force-producing states.
As discussed above, the structural changes that produce myosin motility largely have been determined. What are the energetic changes that drive these structural events? Ultimately the energy comes from the hydrolysis of ATP, but change hydrolysis does not occur during the power stroke, and free energy appears to be “shuttled” among different forms during the cycle (for reviews see Geeves, 1991; Cooke, 1997). The two processes that involve large changes in free energy are the formation of tight bonds between actin and myosin, and between myosin and ATP. A similar process appears to occur with the microtubule motors, with a tight bond between motor and polymer being modulated by nucleotides. The hydrolysis step involves little change in free energy. The formation of the actomyosin interface would bury an extensive hydrophobic area (∼750–1,000 Å2 on each protein), liberating a considerable amount of free energy. Experimentally, the formation of the rigor actomyosin interface is observed to release ∼30 kJ/mole at 2°C and physiological salt concentration (for review see Smith et al., 1984). This change in free energy is similar to the work performed in an actomyosin interaction, ∼30–40 kJ/mole (for review see Cooke, 1997). The release of so much free energy from the formation of the actomyosin interface simultaneously with the production of mechanical work suggests strongly that the free energy driving the power stroke comes directly from the formation of a strong bond between actin and myosin. Due to the high mechanical efficiency of muscle, much of this free energy must be used to produce mechanical work. Formation of the interface has been shown to be a two step process (Geeves, 1991). An initial complex, the A-state, has a lower affinity and is largely electrostatic in nature. An isomerization to a stronger complex, the R-state, is associated with the release of phosphate and with the formation of an extensive hydrophobic interface.
Thus, a combination of high-resolution structures, spectroscopic studies, and enzymology has suggested a reasonably detailed picture of the kinetic cycle, shown in Figs. 2 and 3. At the end of the power stroke myosin is bound tightly to actin, State 4 in Fig. 3. The bond with actin has closed the actin binding cleft, and has opened switch 1 releasing ADP. In the transition from State 4 to 1, myosin trades one ligand, actin, for another ligand, ATP, for which it has even greater affinity. Hydrolysis of the ATP occurs when myosin is either detached from actin or bound weakly to it, leading to State 2. The transition from State 1 to 2 involves first closing of switch 2, with a closed conformation required for hydrolysis. In addition, the movement of switch 2 alters the conformation of the helix running from the nucleotide to the converter domain. This helix is known as the “relay helix” because it forms the major pathway of communication between the nucleotide and the converter domain, shown in blue in Fig. 1. The altered helix conformation changes the orientation of the converter domain to the “up” position, primed for the next power stroke, State 2. Although the structure of the myosin is not tightly coupled to hydrolysis, hydrolysis tends to promote the closed conformation. In the transition from State 2 to 3, myosin binds to actin, forming a weak bond. This state is probably the A-state identified in solution studies (Geeves, 1991), and it has been shown in fibers to be a transient state that is in rapid equilibrium with the detached states. In State 3 phosphate remains bound to the myosin active site. Is force generated before phosphate release? This question remains controversial. However, the weak bond becomes stronger with increasing temperature, and the energy released in its formation would be considerable under physiological conditions. The high efficiency of the actomyosin interaction during muscle contraction suggests that this energy be used, and kinetic studies have suggested that it indeed is (Dantzig et al., 1992). The release of phosphate leads to a stronger bond and to force generation. The release of phosphate may be associated with the opening of switch 1 and/or switch 2, both of which open at some point between State 3 and the end of the power stroke that is reached following a displacement in State 4. After phosphate release the lever arm swing generates force and displacement leading back to State 4 at the end of the power stroke. As argued in this review, the energy released in the formation of the actomyosin interface is used to generate force. How could the formation of the interface be translated into motion of the light chain domain? The direct connection between the interface in the lower 50K domain and the converter domain, shown at the top of the molecule in Fig. 1, could be involved in this process. However, the myosin structure in the absence of actin appears to be flexible, with little change in free energy between conformations, and the effect of the bond with actin may be to provide a more rigid structure with the rigor myosin conformation now being favored over all others.
Figure 3.
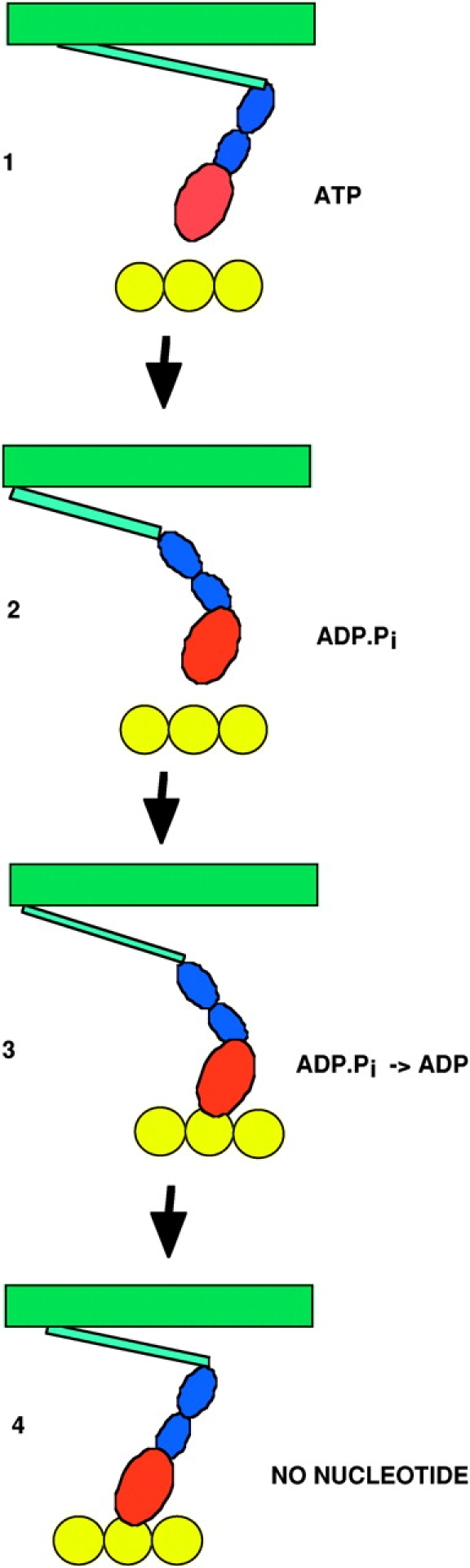
A schematic of the actomyosin cycle. In state 1 the myosin has just been released from actin and is in the post power stroke conformation. In the transition from state 1 to 2, nucleotide hydrolysis promotes the transition to the prepower stroke conformation, although the coupling between nucleotide state and conformation is not tight, with rapid interconversion between conformations and nucleotide states occurring between States 2 and 3. Myosin then binds weakly to actin in State 3. Subsequent release of phosphate leads to a stronger bond in State 4. State 3 probably has multiple substates, some force generating and some not. In State 4 the myosin is strongly bound to actin, with a rigid conformation that represents the end of the power stroke. An important aspect of the cycle that is omitted in the cartoon, is that the molecules are in constant motion, undergoing energetic Brownian motions in times of micro- to milliseconds.
Distant Cousins, the Kinesin Superfamily
The motor protein kinesin, which carries cargoes along microtubules, was discovered in squid axons by Vale, Reese, and Sheetz in 1985 (Vale et al., 1985). The kinesins are now known to form a superfamily of motors with widely different properties. Some motors move processively while others do not, and different isoforms move toward different ends of the microtubule (for review see Goldstein, 2001). During the separation of sister chromosomes in cell division, kinesin family proteins generate a relative sliding of two sets of microtubules. The motion of flagella is produced by a member of another family of microtubule motors, dynein. Dyneins bound to one set of microtubules interact with an adjacent set of microtubules, causing the two sets to slide past each other. Thus, the sliding filament model expands to include the microtubule motors in addition to the myosin motors.
The best studied kinesin motor, known as conventional kinesin, has an architecture similar to myosin, with two globular heads connected via a coiled-coil stalk to a region that binds to a cargo. The structures of the head domains of two members of the family, kinesin and ncd, (ncd moves in the opposite direction to kinesin) were determined by Fletterick and coworkers in 1996 (Kull et al., 1996; Sablin et al., 1996). The surprising result was that for both motors the structure of the region in the vicinity of the bound nucleotide resembled that of myosin (and of the G-proteins). Both switch 1 and 2 were present in all three families. Thus, the myosin, kinesin, and G-protein superfamilies all derive from a common ancestor. Topologically, the binding site for microtubules, although smaller than the actin binding site of myosin, occurred at an equivalent location. In contrast, the region homologous to the light chain domain on myosin consisted of a short 15 amino acid peptide, which was disordered in the original structure. Subsequent crystal structures showed that this same peptide could be “docked” in an extended β sheet–like conformation along one side of the motor domain (Kozielski et al., 1997). Thus, although kinesin takes eight nanometer steps it does not contain a lever arm that could produce such a step. A number of studies suggested that the 15 amino acid polypeptide, known as the neck linker, was involved in producing motility (Case et al., 1997; Endow and Waligora, 1998). Spectroscopic studies, described in a section below, suggested a mechanism in which cyclic docking of the neck linker could produce motility.
Force Production by Kinesin Motors
Structural studies performed by Vale and collaborators led to a proposal for the mechanism of motility of kinesin (Rice et al., 1999). This model, shown in Fig. 4, involves the alternate docking of this neck linker to the core of the motor domain with the undocked form leading to an entropic spring generated by its random conformation. Spectroscopic probes attached to the neck linker provided information on its mobility, while visualization of gold nanoparticles in electron micrographs provided information on its location. The neck linker was in a mobile conformation when the kinesin was not bound to microtubules or when it was bound in the presence of ADP. The neck linker became immobilized when kinesin bound to microtubules in the presence of ATP or ADP.Pi analogs. The position of the gold particles suggested that the immobilized spectral component was associated with a conformation in which the neck linker was docked to the motor domain in a β-sheet conformation as it is seen in the structure of rat kinesin (Kozielski et al., 1997). Docking of the neck linker would position the unattached kinesin head toward the plus end of the microtubule, State 2 in Fig. 4 where it could bind to the next site along the microtubule. Several other lines of evidence now support this model. Rosenfeld and coworkers observed the fluorescence of probes placed on the neck linker, showing that the transition involved two steps, and providing further evidence for this model (Rosenfeld et al., 2001, 2002). Mechanical studies showed that the kinesin cycle contained one ATP dependent, and one ATP independent, force generating step as suggested by the model (Visscher et al., 1999).
Figure 4.
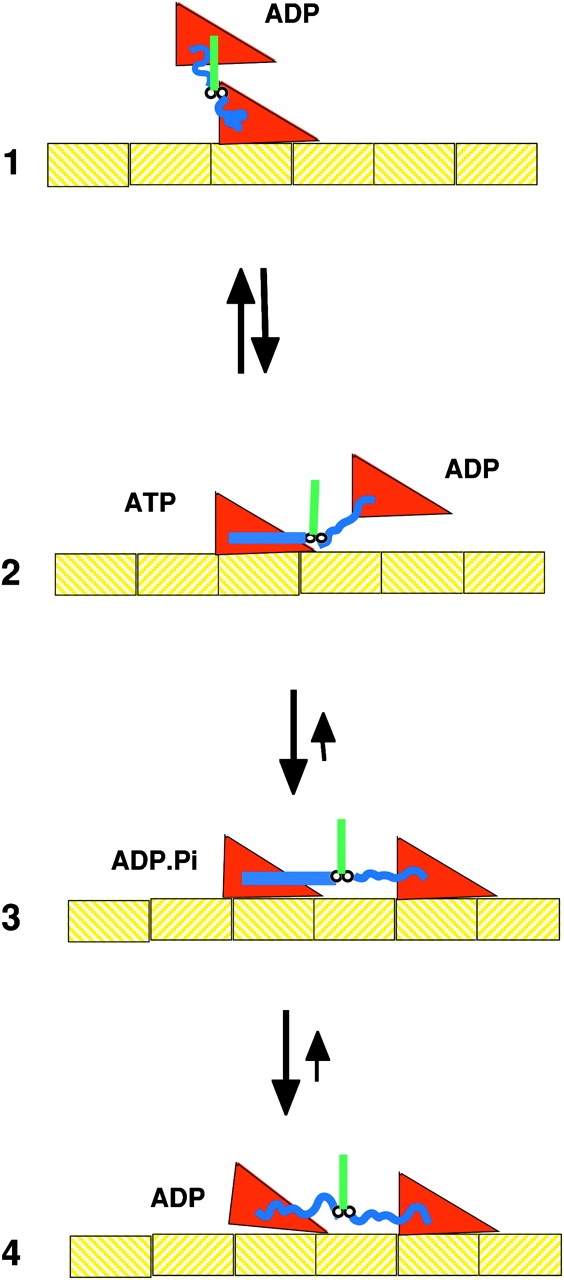
A schematic of the kinesin microtubule interaction. In State 1 the microtubule bound head has no bound nucleotide, and the unattached head has just been released from the trailing site on the microtubule in the transition from State 4 to 1. The binding of ATP to the bound head in State 2 promotes docking of the neck linker along one side of the motor domain, as shown by the straight blue line. This positions the unbound head toward the plus end of the microtubule, toward which the kinesin is walking. However, the free energy favoring docking is small and docked and undocked conformations are in rapid equilibrium. The binding of the kinesin head to the microtubule in State 3 leads to a stable configuration, and produces an effective step of ∼4 nm. The release of phosphate from the trailing head leads to undocking of the neck linker, whose random coil configuration produces a large force between the heads, releasing the trailing head. State 4 then transitions back to State 1, having move 8 nm to the right. When viewing this figure the reader should remember that the molecules are undergoing constant Brownian motions.
Further spectroscopic studies, probed the energetics of the transition from docked to undocked conformations. For kinesin bound to microtubules in the presence of AMPPNP, ΔG was small and favorable, ΔH was large and favorable, and TΔS was large and unfavorable (Rice et al., 2003). The magnitude of the ΔH and TΔS terms were appropriate for a transition from a denatured random peptide to a folded protein. Thus, the undocked state is likely to be highly disordered, and can be approximated by a random coil. The properties of a random coil neck linker will play an important role in the production of force. Random polymers act like rubber with an end-to-end distance much shorter than that of the extended conformation, and there is a lot of free energy, in the form of entropy, that can drive this shortening. As described below the elastic nature of the undocked neck linkers will generate a force between the two heads, detaching the trailing head. Thus, the shortening of a rubber-like polymer can contribute to force production. Long ago, rubber-like shortening was thought to produce muscle contraction, a theory displaced by the sliding filament model. Now, many decades later this mechanism does appear to play a role on a nanometer scale in a motor protein, although not the one that powers muscle.
As with myosin, a combination of structural and enzymatic studies have led to a model of the mechanism of kinesin motility, shown in Fig. 4. State 1 shows kinesin bound to a microtubule by one head with both neck linkers undocked. In order for the unbound head to bind to a microtubule site to either the left or the right, both neck linkers must be stretched into an extended conformation, which is energetically costly due to the decrease in entropy. Docking of the neck linker on the bound head, transition from state 1 to state 2, promotes binding of the unattached head to the right, because the entropic cost of straightening out the neck linker is counterbalanced by the enthalpy released by docking. Docking is more favorable when bound to microtubules and with ATP or ADP.Pi at the active site. Although docking of the neck linker is reversible, the transition from State 1 to 3 is strongly favored by the formation of a strong bond between the now leading head and the microtubule. The nucleotide controls the docking of the neck linker by a mechanism that is similar to that described above for myosin (Vale and Milligan, 2000; Kikkawa et al., 2001). Binding of triphosphate or ADP.Pi analogs is thought to alter the position of switch 2 via a hydrogen bond with a glycine that is conserved across both superfamilies. This movement is translated to the helix homologous to the relay helix of myosin, which in turn allows the docking to occur. The mechanism by which the bond with the microtubule affects docking is not known, however, just as in myosin there is a direct connection between the switch 2 helix and the site where microtubules bind. As described above binding of the motor domain to the polymer closes the nucleotide pocket, most probably by closing the switch 1 to cover the phosphates as seen in myosin (Naber et al., 2003). This would promote nucleotide hydrolysis in State 2. The release of phosphate then opens switch 2, which moves the switch 2 helix into a position where it sterically blocks the docking site for the neck linker. Undocking of the neck linker of the trailing head in State 4 would generate a force between the heads that could pull the trailing head off the microtubule. Kinetic studies of a mutant kinesin show that phosphate release precedes detachment (Klumpp et al., 2004). The trailing head is probably released preferentially because it binds more weakly to the microtubule (due to bound ADP) and because heads are pulled off more easily in the plus direction (Uemura et al., 2002). This transition also would be facilitated if the neck linker of the unattached head was also docked, but in the backward direction. Such a docking is seen in the crystal structure of Eg5, but further studies are needed in order to determine whether this conformation also plays a role in the kinesin cycle (Turner et al., 2001).
While the conformational changes proposed for kinesin, described above, probably operate for a number of other members of the kinesin family of motors, there is evidence that one member of that family, ncd, operates by a different mechanism. In this motor the neck linker of kinesin is replaced by a section of coiled coil α-helix that is continuous with the rod. This coiled coil interacts with many of the same residues of the motor domain that also interact with the neck linker in kinesin. The velocity generated in in vitro assays is correlated with the length of the coiled coil rod (Stewart et al., 1993). Crystal structures of ncd have shown that the coiled coil region can exist in two different orientations relative to the motor domain (Kozielski et al., 1999). Together, these results have led to the hypothesis, as yet unproven, that the coiled coil rod operates as a lever in a manner analogous to the mechanism of myosin.
Force Production by Actin Polymerization
The thin filaments of a muscle cell are stable structures, and in 1972 actin was viewed as a mostly passive track, along which myosin moved. Although actin had been shown to exist in nonmuscle cells, as discussed at the meeting in Cold Spring Harbor by Bray, its roles had not yet been determined (Bray, 1972). In the intervening period, the field of the cytoskeleton and of actin's role in the dynamics of the cytoskeleton has exploded. Actin filaments are very dynamic, polymerizing in some regions, while depolymerizing in others. The dynamic nature of the actin cytoskeleton is orchestrated by a host of other proteins that nucleate new filaments, sever existing filaments, cap them on either end, form branches, and promote nucleotide exchange on actin monomers (for review see Pollard et al., 2000). The first demonstration that actin polymerization could produce a force came from the extension of the acrosomal process of the sperm. This very rapid extension was produced by a bundle of actin filaments that polymerized at their distal ends. An even more dramatic example of actin-based motility comes from the bacterium Lysteria (for review see Pantaloni et al., 2001). Lysteria “rocket” around inside living cells, propelled by a tail that consists of rapidly polymerizing bundles of actin filaments. Myosin has been shown to play no role in this motility. A branched meshwork of actin filaments has been shown to produce the force that generates the protrusion of the cytoplasm at the leading edge of motile cells (for review see Pollard et al., 2000).
How does actin exert a force through polymerization? In the cytoplasm the high concentration of actin monomers provides a strong driving force for polymerization. Thus, the free energy released by the addition of a monomer to the growing end of a filament can be harnessed to perform mechanical work. It is now thought that this process involves a thermal ratchet mechanism (Mogilner and Oster, 2003). Fluctuations between the position of the growing polymer end and the load against which it is pushing are generated by either Brownian movement of the load or by flexibility in the filaments (transition from States 1 to 2 in Fig. 5). When a space opens between the filament and the load, a monomer can insert onto the filament end (transition from States 2 to 3). Once the monomer is in place the fluctuating filament pushes on the load, which has now moved to a new equilibrium position.
Figure 5.
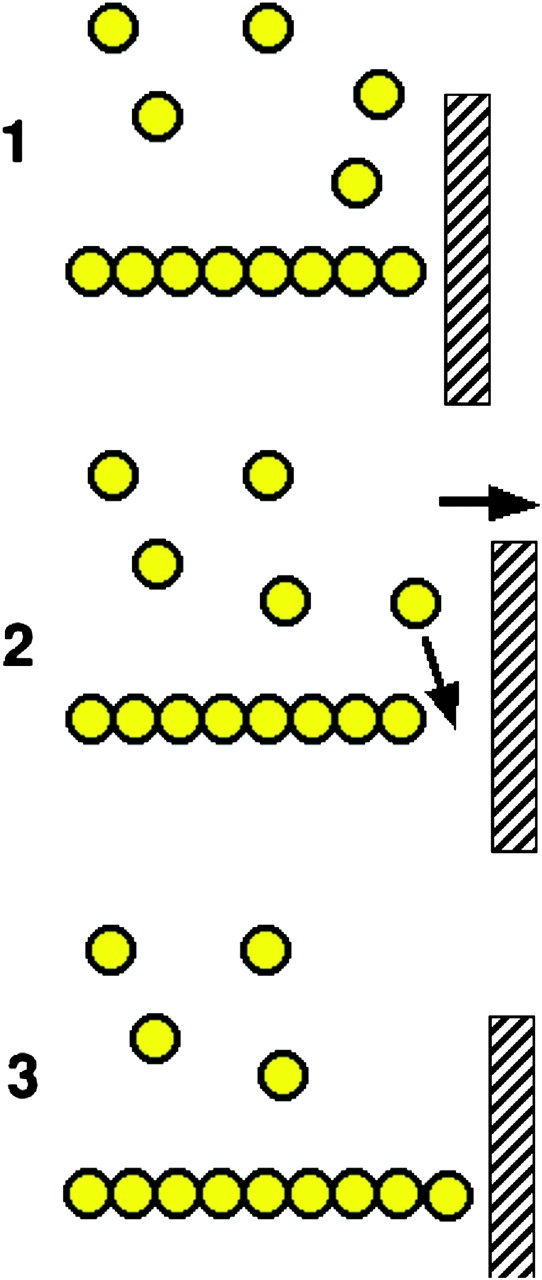
A schematic of force production by actin polymerization. A single actin polymer is shown adjacent to a load, against which it is pushing. In the transition from State 1 to 2 the gap between the end of the filament and the load is increased by either a fluctuation in the position of the load, or in the position of the actin filament. This allows the insertion of an additional actin monomer, thus extending the filament, and leading to a new equilibrium position that has moved 2.7 nm to the right in State 3.
The reversible polymerization of actin is controlled by hydrolysis of its tightly bound nucleotide. In vivo, the prevailing bound nucleotide on the monomer is ATP. G-actin-ATP has a high affinity for the end of a polymer (Pollard et al., 2000). The affinity and the kinetics of association are both greater at one end of the polymer than at the other, and both are greater if ATP is still bound to the monomers at the end of the polymer. Once the monomer has incorporated into a growing filament end, the nucleotide is slowly hydrolyzed and phosphate is released, leaving ADP tightly bound in the central cleft of the actin. G-actin-ADP has a lower affinity for a polymer end, meaning that G-actin-ATP can rapidly incorporate into a growing end of a newly forming polymer, while actin-ADP monomers can be dissociating from the end of an adjacent older filament in which the nucleotides have been hydrolyzed. Thus, the binding of actin monomers to the polymer is orchestrated by nucleotides bound to the actin, a process that has some similarities to the binding of the motor proteins to their polymers.
Thus, although the sliding filament model proposed in the 1950s has proven to be applicable to a wide range of systems, including muscles of all types and much of the cell motility produced by myosin and the microtubule motors, finally we have an example of motility that does not involve sliding filaments, but filament extension.
Some Common Features of the Diverse Mechanisms for Producing Force
What features of force generation are common to all of the force-generating mechanisms discussed above? All of them involve tight binding to a polymer with an affinity that is modulated by the binding of nucleotides. In turn, the binding and hydrolysis of the nucleotides is modulated by the binding of the polymers. The mechanisms of myosin and kinesin motors both involve communication between polymer binding and the nucleotide via switch 1. The conformation of the myosin neck, or the kinesin neck-linker, is controlled by both the nucleotide and by binding to the polymer. The nucleotide operates via switch 2, and the polymer may exert control through structural connections discussed below. In these respects both motors resemble the G-proteins.
The free energy involved in the interactions of the motors with the polymers is large, and it must be converted into work during the cycle, if the motors are to work efficiently. Formation of the actomyosin interface is endothermic, it is driven by a favorable increase in entropy, not by release of enthalpy (Smith et al., 1984). In this respect it is like most protein–protein interactions, and the kinesin–microtubule interaction is likely to be the same. Studies in muscle have shown that force is generated via an endothermic interaction. All of the above argue that the energy released in the formation of the actomyosin bond is used to generate force and produce mechanical work. Formation of the bond could stabilize the rigor conformation of the light-chain domain. In fact there is a direct connection between the relay helix and the actin-binding domain that could provide a pathway of communication between them. If formation of the actomyosin bond pulled on this structure, it would stabilize the relay helix, and thus the converter domain and the light chain domain, in the post power stroke rigor conformation. Alternatively, the bond with actin could provide a more rigid myosin structure with the rigor conformation being the most favorable. Exploring these hypotheses must await high-resolution structures of the actin–myosin complex, one of the major unrealized goals of this field. A similar argument applies to the interaction between the docking of the neck linker and microtubule binding in kinesin. In the case of the kinesin motor the involvement of the kinesin-microtubule bond in producing work is more evident, with the bond stabilizing the position of the kinesin head at a new site that was reached via Brownian fluctuations in position. These fluctuations are rectified to produce unidirectional motion by the docking of the neck linker. Likewise, the work performed by actin involves the formation of a protein–protein bond, in this case between an actin monomer and an actin filament.
The first law of thermodynamics requires that the production of mechanical work, which is enthalpic in nature, must be paid for by a corresponding change in the enthalpy of the system. How does an entropically driven process, such as formation of a protein–protein interface perform work? When entropy is used to perform work, the required enthalpy is acquired by capture of a thermal fluctuation. In fact, the first model of a motor protein, proposed by Sir Andrew Huxley in 1957, involved a classic thermal ratchet (Huxley, 1957) (Fig. 6). In this model, fluctuations in the position of myosin are generated by thermal energy, and a favorable fluctuation is captured by formation of the bond with actin. The relaxation of the spring then provides the enthalpy to perform the mechanical work of the power stroke. At the end of the power stroke the myosin is bound tightly to the actin, and this bond is broken by the binding of ATP to the myosin. Thus, a cycle of binding of the motor to a polymer, orchestrated by the binding of nucleotides, can rectify random thermal fluctuations into unidirectional mechanical work. The evidence discussed in this review argues that the generation of mechanical work by myosin, kinesin, and actin all operate by such a mechanism.
Figure 6.

The first model of a motor protein, proposed by Sir Andrew Huxley in 1957, before the myosin cross-bridge had been visualized. The myosin is shown connected to the filament backbone by two springs. As shown, a thermal fluctuation has moved the myosin part of the way toward the actin site, which is entering from the right. A further fluctuation can carry it the rest of the distance, where binding to actin traps the springs in distorted positions. Relaxation of the springs then drags the actin to the left. When the actin myosin positions reach the center, where both springs are in their relaxed positions, rapid binding of ATP releases the myosin from actin. A similar cyclic interaction between motors and polymers, orchestrated by the binding of nucleotides is thought to produce the motility of both actin-myosin and kinesin-microtubule motor systems.
The time required to capture a thermal fluctuation depends on the energy involved in the fluctuation: the more energy, the longer it takes. Capture of a fluctuation equivalent to the hydrolysis of ATP requires several seconds, while capture of half that amount requires a few milliseconds. This led Huxley and Simmons to propose a multistep power stroke for myosin in 1971 (Huxley and Simmons, 1971). More recently direct measurements have shown multiple steps in the working strokes of many myosins, and if work is performed before the release of phosphate, all myosins may make multiple steps. The compliant elements in the cross-bridge are very stiff, stretching by only ∼1.4 nm under full load, so they would have to be stretched out several times to produce even a 5-nm power stroke, again suggesting several steps (Ford et al., 1977). Kinesin takes an 8-nm power stroke in at least two steps, one involving the release of the trailing head, and one involving its subsequent binding to the next site on the microtubule. Thus, the motor proteins appear to have evolved cycles consisting of several mechanical steps that would allow the energy of ATP to be rapidly converted to mechanical energy using a thermal ratchet mechanism.
Summary
The sliding filament model, proposed to explain muscle contraction in 1954, has proven to be very robust. Muscle contraction as well as much of the motility of nonmuscle cells has now been shown to be produced by the relative motion of actin filaments and myosin filaments or myosins attached to cargoes. In addition the mechanism of the microtubule motors kinesin and dynein, a system unknown in 1954, has been shown to work in the same fashion. Although there is considerable homology in the sequences of the catalytic domains of the myosins and kinesins, they appear to have evolved diverse mechanisms for translating changes in the catalytic domain into motion. Most myosins appear to operate by a lever arm motion of the light-chain domain. However, myosin VI takes a long step with a short lever arm, suggesting a mechanism where some region of the protein melts, possibly the coiled-coil rod (Rock et al., 2001). This protein may thus have a mechanism with similarities to that proposed for conventional kinesin. Kinesin appears to operate by alternate docking of its neck linker; however, ncd may operate by a lever arm motion. The only area of motility that has not followed the sliding filament model is the force produced by actin polymerization.
The molecular mechanisms of the motor proteins that produce motility are now reasonably well understood, although gaps in our knowledge remain, particularly in the structures of the motor–polymer complexes. These mechanisms were elucidated by innovations and technologies that largely were unimaginable in 1954. We should expect a similar rate of progress in the next 50 yr.
Acknowledgments
Kenneth C. Holmes served as editor.
References
- Anson, M., M.A. Geeves, S.E. Kurzawa, and D.J. Manstein. 1996. Myosin motors with artificial lever arms. EMBO J. 15:6069–6074. [PMC free article] [PubMed] [Google Scholar]
- Baker, J.E., I. Brust-Mascher, S. Ramachandran, L.E. LaConte, and D.D. Thomas. 1998. A large and distinct rotation of the myosin light chain domain occurs upon muscle contraction. Proc. Natl. Acad. Sci. USA. 95:2944–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, D. 1972. Cytoplasmic Actin: A Comparative Study. Cold Spring Harbor Symposia on Quantitative Biology. The Cold Spring Laboratory, Cold Spring Harbor. XXXVII:567–571.
- Case, R., D. Pierce, N. HomBooher, C. Hart, and R. Vale. 1997. The directional preference of kinesin motors is specified by an element outside of the motor catalytic domain. Cell. 90:959–966. [DOI] [PubMed] [Google Scholar]
- Cooke, R. 1986. The mechanism of muscle contraction. CRC Crit. Rev. Biochem. 21:53–118. [DOI] [PubMed] [Google Scholar]
- Cooke, R. 1997. Actomyosin interaction in striated muscle. Physiol. Rev. 77:671–697. [DOI] [PubMed] [Google Scholar]
- Cooke, R., M.S. Crowder, and D. Thomas. 1982. Orientation of spin labels attached to cross-bridges in contracting muscle fibres. Nature. 300:776–778. [DOI] [PubMed] [Google Scholar]
- Corrie, J.E.T., B.D. Brandmeier, R.E. Ferguson, D.R. Trentham, I. Kendrick-Jones, S.C. Hopkins, U.A. van der Heide, Y.E. Goldman, C. Sabido-David, R.E. Dale, et al. 1999. Dynamic measurement of myosin light-chain-domain tilt and twist in muscle contraction. Nature. 400:425–430. [DOI] [PubMed] [Google Scholar]
- Coureux, P.D., A.L. Wells, J. Menetrey, C.M. Yengo, C.A. Morris, H.L. Sweeney, and A. Houdusse. 2003. A structural state of the myosin V motor without bound nucleotide. Nature. 425:419–423. [DOI] [PubMed] [Google Scholar]
- Dantzig, J.A., Y.E. Goldman, J. Lacktis, N.C. Millar, and E. Homsher. 1992. Reversal of the cross-bridge force generating transition by photogeneration of phosphate in rabbit psoas muscle fibres. J. Physiol. 451:247–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez, R., Y. Freyson, K.M. Trybus, and C. Cohen. 1998. Crystal structure of a verterbrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-powerstroke state. Cell. 94:559–571. [DOI] [PubMed] [Google Scholar]
- Endow, S.A., and K.W. Waligora. 1998. Determinants of kinesin motor polarity. Science. 281:1200–1202. [DOI] [PubMed] [Google Scholar]
- Finer, J.T., R.M. Simmons, and J.A. Spudich. 1994. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 368:113–119. [DOI] [PubMed] [Google Scholar]
- Fisher, A.J., C.A. Smith, J. Thoden, R. Smith, K. Sutoh, H.M. Holden, and I. Rayment. 1995. X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP•BeFx and MgADP•AlF4-. Biochemistry. 34:8960–8972. [DOI] [PubMed] [Google Scholar]
- Ford, L.E., A.F. Huxley, and R.M. Simmons. 1977. Tension responses to sudden length change in stimulated frog muscle fibres near slack length. J. Physiol. 269:441–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forkey, J.N., M.E. Quinlan, M.A. Shaw, J.E. Corrie, and Y.E. Goldman. 2003. Three-dimensional structural dynamics of myosin V by single-molecule fluorescence polarization. Nature. 422:399–404. [DOI] [PubMed] [Google Scholar]
- Geeves, M.A. 1991. The dynamics of actin and myosin association and the crossbridge model of muscle contraction. Biochem. J. 274:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, Y.E., M.G. Hibberd, J.A. McCray, and D.R. Trentham. 1982. Relaxation of muscle fibres by photolysis of caged ATP. Nature. 300:701–705. [DOI] [PubMed] [Google Scholar]
- Goldstein, L.S. 2001. Kinesin molecular motors: transport pathways, receptors, and human disease. Proc. Natl. Acad. Sci. USA. 98:6999–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilford, W.H., M.J. Tyska, Y. Freyzon, and D.M. Warshaw. 1996. Smooth muscle myosin with an elongated neck region produces greater unitary displacement in vitro. Biophys. J. 70:A127. [Google Scholar]
- Holmes, K.C. 1996. Muscle proteins - their actions and interactions. Curr. Opin. Struct. Biol. 6:781–789. [DOI] [PubMed] [Google Scholar]
- Holmes, K.C. 1997. The swinging lever-arm hypothesis of muscle contraction. Curr. Biol. 7:R112–R118. [DOI] [PubMed] [Google Scholar]
- Holmes, K.C., I. Angert, F.J. Kull, W. Jahn, and R.R. Schroder. 2003. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 425:423–427. [DOI] [PubMed] [Google Scholar]
- Holmes, K.C., and R.S. Goody. 1984. The nature of the actin cross-bridge interaction. Adv. Exp. Med. Biol. 170:373–384. [DOI] [PubMed] [Google Scholar]
- Houdusse, A., A.G. Szent-Gyorgyi, and C. Cohen. 2000. Three conformational states of scallop myosin S1. Proc. Natl. Acad. Sci. USA. 97:11238–11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard, J. 2001. Mechanics of Motor Proteins and the Cytoskeleton. Sinauer, Sunderland, MA.
- Huxley, A.F. 1957. Muscle structure and theories of contraction. Prog. Biophys. Biophys. Chem. 7:255–318. [PubMed] [Google Scholar]
- Huxley, A.F., and R.M. Simmons. 1971. Proposed mechanism of force generation in striated muscle. Nature. 233:533–538. [DOI] [PubMed] [Google Scholar]
- Huxley, H.E., and M. Kress. 1985. Crossbridge behaviour during muscle contraction. J. Muscle Res. Cell Motil. 6:153–161. [DOI] [PubMed] [Google Scholar]
- Kabsch, W., H.G. Mannherz, D. Suck, E.F. Pai, and K.C. Holmes. 1990. Atomic structure of the actin:DNase I complex. Nature. 347:37–44. [DOI] [PubMed] [Google Scholar]
- Kikkawa, M., E.P. Sablin, Y. Okada, H. Yajima, R.J. Fletterick, and N. Hirokawa. 2001. Switch-based mechanism of kinesin motors. Nature. 411:439–445. [DOI] [PubMed] [Google Scholar]
- Klumpp, L.M., A. Hoenger, and S.P. Gilbert. 2004. Kinesin's second step. Proc. Natl. Acad. Sci. USA. 101:3444–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight, A.E., C. Veigel, C. Chambers, and J.E. Molloy. 2001. Analysis of single-molecule mechanical recordings: application to acto-myosin interactions. Prog. Biophys. Mol. Biol. 77:45–72. [DOI] [PubMed] [Google Scholar]
- Kozielski, F., S. De Bonis, W.P. Burmeister, C. Cohen-Addad, and R.H. Wade. 1999. The crystal structure of the minus-end-directed microtubule motor protein ncd reveals variable dimer conformations. Structure Fold. Des. 7:1407–1416. [DOI] [PubMed] [Google Scholar]
- Kozielski, F., S. Sack, A. Marx, M. Thormahlen, E. Schonbrunn, V. Biou, A. Thompson, E.M. Mandelkow, and E. Mandelkow. 1997. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 91:985–994. [DOI] [PubMed] [Google Scholar]
- Kron, S.J., and J.A. Spudich. 1986. Fluorescent actin filaments move on myosin fixed to a glass surface. Proc. Natl. Acad. Sci. USA. 83:6272–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kull, F.J., E.P. Sablin, R. Lau, R.J. Fletterick, and R.D. Vale. 1996. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 380:550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannherz, H.G., H. Schenck, and R.S. Goody. 1974. Synthesis of ATP from ADP and inorganic phosphate at the myosin-subfragment 1 active site. Eur. J. Biochem. 48:287–295. [DOI] [PubMed] [Google Scholar]
- Mehta, A.D., R.S. Rock, M. Rief, J.A. Spudich, M.S. Mooseker, and R.E. Cheney. 1999. Myosin-V is a processive actin-based motor. Nature. 400:590–593. [DOI] [PubMed] [Google Scholar]
- Mogilner, A., and G. Oster. 2003. Force generation by actin polymerization II: the elastic ratchet and tethered filaments. Biophys. J. 84:1591–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naber, N., T.J. Minehardt, S. Rice, X. Chen, J. Grammer, M. Matuska, R.D. Vale, P.A. Kollman, R. Car, R.G. Yount, et al. 2003. Closing of the nucleotide pocket of kinesin-family motors upon binding to microtubules. Science. 300:798–801. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., K.A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 11:281–296. [DOI] [PubMed] [Google Scholar]
- Pantaloni, D., C. Le Clainche, and M.F. Carlier. 2001. Mechanism of actin-based motility. Science. 292:1502–1506. [DOI] [PubMed] [Google Scholar]
- Piazzesi, G., M. Reconditi, M. Linari, L. Lucii, Y.B. Sun, T. Narayanan, P. Boesecke, V. Lombardi, and M. Irving. 2002. Mechanism of force generation by myosin heads in skeletal muscle. Nature. 415:659–662. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., L. Blanchoin, and R.D. Mullins. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29:545–576. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., and E.D. Korn. 1972. The “Contractile” Proteins of Acanthamoeba castellanii. Cold Spring Harbor Symposia on Quantitative Biology. The Cold Spring Laboratory, Cold Spring Harbor, NY. XXXVII:573–583.
- Rayment, I., H.M. Holden, M. Whittaker, C.B. Yohn, M. Lorenz, K.C. Holmes, and R.A. Milligan. 1993. a. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 261:58–65. [DOI] [PubMed] [Google Scholar]
- Rayment, I., W.R. Rypniewski, K. Schmidt-Base, R. Smith, D.R. Tomchick, M.M. Benning, D.A. Winkelmann, G. Wesenberg, and H.M. Holden. 1993. b. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science. 261:50–57. [DOI] [PubMed] [Google Scholar]
- Reubold, T.F., S. Eschenburg, A. Becker, F.J. Kull, and D.J. Manstein. 2003. A structural model for actin-induced nucleotide release in myosin. Nat. Struct. Biol. 10:826–830. [DOI] [PubMed] [Google Scholar]
- Rice, S., Y. Cui, C. Sindelar, N. Naber, M. Matuska, R. Vale, and R. Cooke. 2003. Thermodynamic properties of the Kinesin neck-region docking to the catalytic core. Biophys. J. 84:1844–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, S., A.W. Lin, D. Safer, C.L. Hart, N. Naber, B.O. Carragher, S.M. Cain, E. Pechatnikova, E.M. Wilson-Kubalek, M. Whittaker, et al. 1999. A structural change in the kinesin motor protein that drives motility. Nature. 402:778–784. [DOI] [PubMed] [Google Scholar]
- Rock, R.S., S.E. Rice, A.L. Wells, T.J. Purcell, J.A. Spudich, and H.L. Sweeney. 2001. Myosin VI is a processive motor with a large step size. Proc. Natl. Acad. Sci. USA. 98:13655–13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld, S.S., G.M. Jefferson, and P.H. King. 2001. ATP reorients the neck linker of kinesin in two sequential steps. J. Biol. Chem. 276:40167–40174. [DOI] [PubMed] [Google Scholar]
- Rosenfeld, S.S., J. Xing, G.M. Jefferson, H.C. Cheung, and P.H. King. 2002. Measuring kinesin's first step. J. Biol. Chem. 277:36731–36739. [DOI] [PubMed] [Google Scholar]
- Sablin, E.P., F.J. Kull, R. Cooke, R.D. Vale, and R.J. Fletterick. 1996. Crystal structure of the motor domain of the kinesin-related motor ncd. Nature. 380:555–559. [DOI] [PubMed] [Google Scholar]
- Sheetz, M.P., and J.A. Spudich. 1983. Movement of myosin-coated fluorescent beads on actin cables in vitro. Nature. 303:31–35. [DOI] [PubMed] [Google Scholar]
- Smith, S.J., H.D. White, and R.C. Woledge. 1984. Microcalometric measurement of the enthalpy of binding of rabbit skeletal myosin subfragment 1 and heavy meromyosin to F-actin. J. Biol. Chem. 259:10303–10308. [PubMed] [Google Scholar]
- Stewart, R.J., J.P. Thaler, and L.S. Goldstein. 1993. Direction of microtubule movement is an intrinsic property of the motor domains of kinesin heavy chain and Drosophila ncd protein. Proc. Natl. Acad. Sci. USA. 90:5209–5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi, A.G. 2004. The early history of the biochemistry of muscle contraction. J. Gen. Physiol. 123:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, E.W. 1979. Mechanism of actomyosin ATPase and the problem of muscle contraction. CRC Crit. Rev. Biochem. 6:103–164. [DOI] [PubMed] [Google Scholar]
- Taylor, K.A., H. Schmitz, M.C. Reedy, Y.E. Goldman, C. Franzini-Armstrong, H. Sasaki, R.T. Tregear, K. Poole, C. Lucaveche, R.J. Edwards, et al. 1999. Tomographic 3D reconstruction of quick-frozen, Ca2+-activated contracting insect flight muscle. Cell. 99:421–431. [DOI] [PubMed] [Google Scholar]
- Toyoshima, Y.Y., S.J. Kron, E.M. McNally, K.R. Niebling, C. Toyoshima, and J.A. Spudich. 1987. Myosin subfragment-1 is sufficient to move actin filaments in vitro. Nature. 328:536–539. [DOI] [PubMed] [Google Scholar]
- Trentham, D.R., J.F. Eccleston, and C.R. Bagshaw. 1976. Kinetic analysis of ATPase mechanisms. Q. Rev. Biophysics. 9:217–281. [DOI] [PubMed] [Google Scholar]
- Turner, J., R. Anderson, J. Guo, C. Beraud, R. Fletterick, and R. Sakowicz. 2001. Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck-linker. J. Biol. Chem. 276:25496–25502. [DOI] [PubMed] [Google Scholar]
- Uemura, S., K. Kawaguchi, J. Yajima, M. Edamatsu, Y.Y. Toyoshima, and S. Ishiwata. 2002. Kinesin-microtubule binding depends on both nucleotide state and loading direction. Proc. Natl. Acad. Sci. USA. 99:5977–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeda, T.Q.P., P.D. Abramson, and J.A. Spudich. 1996. The neck region of the myosin motor domain acts as a lever arm to generate movement. Proc. Natl. Acad. Sci. USA. 93:4459–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, R.D., and R.A. Milligan. 2000. The way things move: looking under the hood of molecular motor proteins. Science. 288:88–95. [DOI] [PubMed] [Google Scholar]
- Vale, R.D., T.S. Reese, and M.P. Sheetz. 1985. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 42:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veigel, C., L.M. Coluccio, J.D. Jontes, J.C. Sparrow, R.A. Milligan, and J.E. Molloy. 1999. The motor protein myosin-I produces its working stroke in two steps. Nature. 398:530–533. [DOI] [PubMed] [Google Scholar]
- Veigel, C., F. Wang, M.L. Bartoo, J.R. Sellers, and J.E. Molloy. 2002. The gated gait of the processive molecular motor, myosin V. Nat. Cell Biol. 4:59–65. [DOI] [PubMed] [Google Scholar]
- Visscher, K., M.J. Schnitzer, and S.M. Block. 1999. Single kinesin molecules studied with a molecular force clamp. Nature. 400:184–189. [DOI] [PubMed] [Google Scholar]
- Volkmann, N., D. Hanein, G. Ouyang, K.M. Trybus, D.J. DeRosier, and S. Lowey. 2000. Evidence for cleft closure in actomyosin upon ADP release. Nat. Struct. Biol. 7:1147–1155. [DOI] [PubMed] [Google Scholar]
- Walker, M.L., S.A. Burgess, J.R. Sellers, F. Wang, J.A. Hammer III, J. Trinick, and P.J. Knight. 2000. Two-headed binding of a processive myosin to F-actin. Nature. 405:804–807. [DOI] [PubMed] [Google Scholar]
- Whittaker, M., E.M. Wilson-Kubalek, J.E. Smith, L. Faust, R.A. Milligan, and H.L. Sweeney. 1995. A 35-angstrom movement of smooth muscle myosin on ADP release. Nature. 378:748–751. [DOI] [PubMed] [Google Scholar]
- Yanagida, T. 1985. Angle of active site of myosin heads in contracting muscle during sudden length changes. J. Muscle Res. Cell Motility. 6:43–52. [DOI] [PubMed] [Google Scholar]
- Yildiz, A., J.N. Forkey, S.A. McKinney, T. Ha, Y.E. Goldman, and P.R. Selvin. 2003. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 300:2061–2065. [DOI] [PubMed] [Google Scholar]