

Abstract
Retinoic acid (RA) is a signaling molecule synthesized from vitamin A that controls gene expression at the transcriptional level by functioning as a ligand for nuclear RA receptors. RA plays an essential role during embryonic development in higher animals by regulating key genes involved in pattern formation. RA is required for induction of several Hox genes involved in patterning of the hindbrain and spinal cord as neuroectoderm emerges from the primitive streak. Recent findings indicate that RA is also required to ensure bilaterally symmetrical generation of left and right somites as presomitic mesoderm emerges from the primitive streak. RA may control somitogenesis through its ability to repress posterior ectodermal expression of fibroblast growth factor-8 (Fgf8) for a short period of time during the late primitive streak stage when the somitogenesis clock initiates. During this tight temporal window, RA is required to limit Fgf8 expression to the most posterior ectoderm (epiblast), thus preventing ectopic Fgf8 expression in more anterior ectoderm including the node ectoderm and neuroectoderm. Although Fgf8 is required for the node to impart left–right asymmetry on specific tissues (heart, visceral organs, etc.), excess Fgf8 signaling following a loss of RA may stimulate the node to generate asymmetry also in presomitic mesoderm, leading to left–right asymmetry in the somitogenesis clock. These findings suggest that human vertebral birth defects such as scoliosis, an abnormal left–right bending of the vertebral column, may be caused by a defect in RA signaling during somitogenesis.
INTRODUCTION
Retinoic Acid Signaling
Retinoic acid (RA), the active form of vitamin A, plays a crucial role in stimulating nuclear receptor signaling during development. However, the developmental processes regulated by RA signaling are not well understood. This is pointed out by the recent discovery that RA controls left–right patterning during somitogenesis (Kawakami et al., 2005; Vermot et al., 2005; Vermot and Pourquié, 2005; Sirbu and Duester, 2006), a function that was totally unsuspected based upon what was known about RA function up to that point. These findings suggest that defective RA signaling may be responsible for human birth defects of the vertebral column, a topic which is discussed further below. Thus, rigorous assessment of RA target tissues and target genes in mammalian embryos is providing a better understanding of the role of this important signaling molecule in human embryonic development.
The ability to unravel the mechanism of RA action during mammalian development has been greatly enhanced by generation of mice carrying disruptions in enzymes essential for RA synthesis. RA synthesis is a two-step process in which enzymes catalyze metabolism of the alcohol form of vitamin A (retinol) first to an aldehyde (retinaldehyde) and then to a carboxylic acid (RA) (Duester, 2000). RA signaling initiates when retinaldehyde is metabolized by retinaldehyde dehydrogenase (RALDH) to RA, which serves as a ligand for nuclear RA receptors that directly regulate gene expression at the transcriptional level (Fig. 1).
Figure 1.
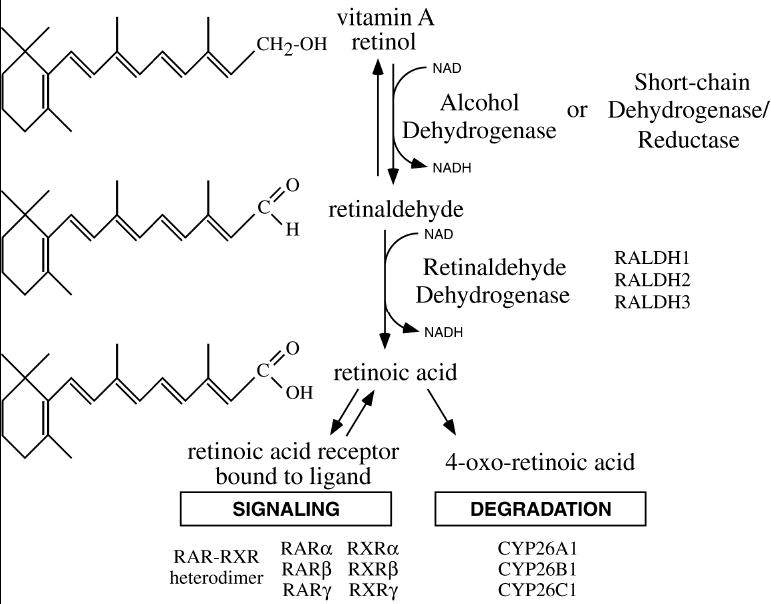
RA synthesis pathway. Conversion of vitamin A (retinol) to retinoic acid (RA) requires two sequential oxidation steps. First, retinol is oxidized to retinaldehyde by either alcohol dehydrogenase (ADH) or short-chain dehydrogenase/reductase (SDR). Second, retinaldehyde is oxidized to RA by retinaldehyde dehydrogenase (RALDH). Once synthesized, RA can regulate transcription by serving as a ligand for nuclear RA receptors (RAR), or RA can be further oxidized by P450 enzymes (CYP26) as part of a degradative pathway.
The first step of RA synthesis, oxidation of retinol to retinaldehyde, is catalyzed by several members of the alcohol dehydrogenase family (Adh1, Adh3, and Adh4), as shown by genetic loss-of-function studies in mice (Molotkov et al., 2002). The first step is also catalyzed by several members of the short-chain dehydrogenase/reductase (SDR) family (Napoli, 1999; Gallego et al., 2006). Mouse gene knockout studies have demonstrated that the second step of RA synthesis, oxidation of retinaldehyde to RA, is catalyzed by three members of the aldehyde dehydrogenase (ALDH) gene family known as RALDHs, Raldh1 (Fan et al., 2003), Raldh2 (Niederreither et al., 1999; Mic et al., 2002), and Raldh3 (Dupé et al., 2003; Molotkov et al., 2006); which are also referred to as Aldh1a1, Aldh1a2, and Aldh1a3, respectively. These three RALDH genes control most, if not all, RA synthesis during embryogenesis, and they have been conserved during vertebrate evolution (Table 1). Degradative oxidation of RA to 4-oxo-RA and other metabolites that are more easily excreted is initiated by several cytochrome P450 (CYP) genes known as Cyp26a1, Cyp26b1, and Cyp26c1, which display unique tissue-specific expression patterns during mouse embryogenesis (MacLean et al., 2001; Tahayato et al., 2003).
TABLE 1.
Retinaldehyde Dehydrogenases
| Common name | Official name | Other names | Orthologs identified in the following organisms |
|---|---|---|---|
| RALDH1 | ALDH1A1 | ALDH1, AHD2 | human, mouse, rat, chick, frog |
| RALDH2 | ALDH1A2 | human, mouse, rat, chick, frog, zebrafish, ascidian | |
| RALDH3 | ALDH1A3 | ALDH6 | human, mouse, chick, frog |
In vitro studies have shown that RA serves as a ligand for two families of nuclear receptors that bind DNA as heterodimers and directly regulate gene expression (Kastner et al., 1994; Mangelsdorf et al., 1994). Those studies demonstrated that RA functions as a high-affinity ligand for RA receptors (RAR-α, RAR-β, and RAR-γ) and a low-affinity ligand for their heterodimer partners, the retinoid X receptors (RXR-α, RXR-β, and RXR-γ). Recent in vivo studies demonstrated that ligand binding to just the RAR portion of the RAR/RXR heterodimers is sufficient and necessary to rescue a lethal defect in RA synthesis, and that ligand binding to RXR is unnecessary (Mic et al., 2003). When RA binds to the RAR partner of the RAR/RXR heterodimers that are bound to a regulatory DNA element, this stimulates a cascade of events resulting in displacement of transcriptional corepressors and recruitment of coactivators that induce transcription from a nearby promoter (Germain et al., 2002).
SPATIOTEMPORAL CONTROL OF RA SYNTHESIS DURING EARLY MOUSE EMBRYOGENESIS
In contrast to the great explosion of information regarding how RA receptors control transcription, we are just now beginning to understand what regulates synthesis of the ligand needed to trigger RA receptor activity. RA is not produced by all cells of the body at all stages of development, but is instead produced in a unique spatiotemporal pattern. Several classes of retinoid-binding proteins are involved in the control of RA synthesis and signaling (Noy, 2000) (Fig. 2). Retinol is transported in the blood at μM levels via serum retinol-binding protein (RBP) and is made available to all cells (including embryonic cells by maternal transfer) for potential conversion to RA (Soprano and Blaner, 1994). Some cells also express cellular RBP (CRBP), which facilitates uptake of retinol into cells and conversion to retinyl esters for storage (Yost et al., 1988; Molotkov et al., 2004). Some cells also express cellular RA-binding protein (CRABP), which shuttles RA from the cytoplasm to the nucleus, where RA receptors are located (Sessler and Noy, 2005).
Figure 2.

Control of RA signaling by retinoid-binding proteins. Retinol is carried in the bloodstream by serum retinol-binding protein (RBP), which facilitates entry of retinol into cells via a specific receptor. Upon entry, retinol binds cellular RBP (CRBP), which provides a sequestration function. Free retinol is oxidized to RA, some of which can diffuse out of the cell and enter nearby cells by passive diffusion facilitated by cellular RA-binding protein (CRABP), which provides a sequestration function. CRABP can also facilitate transport of RA to the nucleus, and upon dissociation from CRABP, RA can then bind the RA receptor (RAR). In the liganded state, RAR, bound to DNA as a heterodimer with RXR, recruits coactivator proteins that stimulate transcription. The DNA element to which RAR-RXR heterodimers bind is known as a retinoic acid response element (RARE).
RA synthesis is controlled both spatially and temporally. Oxidation of retinol to retinaldehyde in mammalian tissues appears not to be tissue-restricted, as it is catalyzed by several widely-expressed alcohol dehydrogenases and short-chain dehydrogenases/reductases (Molotkov et al., 2002). Also, retinol oxidation is reversible and multiple enzymes have been reported to participate in the reduction of retinaldehyde to retinol (Gallego et al., 2006). In contrast, oxidation of retinaldehyde to RA is irreversible, and all three RALDHs identified as catalysts for this reaction are expressed in dynamic nonoverlapping spatiotemporal patterns, indicating that this step is tissue-restricted and time-restricted (Haselbeck et al., 1999; Mic et al., 2002; Niederreither et al., 2002a; Molotkov et al., 2006). Thus, as all cells have access to retinol via the circulatory system, it is probable that all cells can establish an equilibrium between retinol and retinaldehyde via one or more of the first-step enzymes; but only cells expressing one of the RALDHs can oxidize the available retinaldehyde to RA. The unique expression patterns of the three RALDHs suggest that each enzyme controls distinct RA signaling functions.
Conversion of retinol to RA occurs at relatively low levels, but RA has been detected in whole-body extracts of mid-gestation mouse embryos at approximately 25 nM using HPLC analysis (Mic et al., 2003). RA activity has also been detected using sensitive RA-reporter assays, which can localize activity within the embryo. A transgenic RA-reporter mouse strain (RARE-lacZ), which contains lacZ linked to an RA response element (RARE), indicates that RA signaling activity is first observed in embryos at 7.5 days of embryonic development (E7.5; late primitive streak stage) and is localized to the trunk from E7.5–E8.5 (Rossant et al., 1991). Studies performed in this laboratory using a bioassay in which embryo explants are cultured on a lawn of F9 cells stably expressing RARE-lacZ have shown that RA signaling activity is undetectable in mouse embryos at E6.5, but that RA is detectable in the trunk from E7.5 to E8.5 (Ang et al., 1996). This suggests that endogenous RA synthesis initiates in the mouse at E7.5. The RA biosynthetic enzyme RALDH2 is first expressed at E7.5 in trunk paraxial mesoderm and, by E8.5, it displays expression in paraxial and lateral plate mesoderm that appears similar to the pattern of RA localization using the RARE-lacZ RA-reporter (Mic et al., 2002). Studies on Raldh2−/− embryos carrying RARE-lacZ have shown that Raldh2 is responsible for all RA signaling activity seen in the embryo up to E8.5 (Molotkova et al., 2005), but that at later stages Raldh1 and Raldh3 begin to generate RA in the eye, olfactory pit, and kidney (Mic et al., 2002; Molotkov et al., 2006).
LOSS OF RA AFFECTS PATTERNING OF EMBRYONIC TISSUES EMERGING FROM THE PRIMITIVE STREAK
Prior to genetic studies on RA-synthesizing enzymes, which allowed genetic elimination of RA, studies on the embryonic effects of a loss of RA were performed that used vitamin A deficiency to remove the precursor of RA (Maden et al., 1996; Dickman et al., 1997; White et al., 1998) or introduced inhibitors of RA synthesis such as disulfiram or citral (Marsh-Armstrong et al., 1994; Stratford et al., 1996). These early studies indicated that RA was essential for development of several organs including the hindbrain, spinal cord, heart, forelimb buds, and eye. Since then, genetic loss-of-function studies on RA-synthesizing enzymes have demonstrated that Raldh2−/− embryos are quite useful for studying the function of RA signaling during early mammalian embryogenesis, as they completely lack RA activity in posterior mesoderm, neuroectoderm, and endoderm during late primitive streak stages (Niederreither et al., 1999; Mic et al., 2002; Molotkova et al., 2005). Raldh1 and Raldh3 gene knockouts have been useful for investigating RA signaling in the eye, olfactory pit, and kidney (Dupé et al., 2003; Fan et al., 2003; Molotkov et al., 2006). These studies have demonstrated that Raldh2 is the only RA-generating enzyme in mammalian embryos during the primitive streak stage (gastrulation), and that Raldh1 and Raldh3 begin contributing to RA synthesis in specific tissues after gastrulation has ended.
The overall picture of RA function in early embryos is that it plays an essential role in several patterning events. The effects of RA on anteroposterior patterning of neuroectoderm have been well documented. Studies on hindbrain development indicate that RA induces Hox genes needed for rhombomeric segmentation of neuroectoderm emerging from the primitive streak (Marshall et al., 1994). Several studies have demonstrated that RA plays an essential role in anteroposterior patterning of the central nervous system (Niederreither et al., 2000; Begemann et al., 2001; Dupé and Lumsden, 2001; Maves and Kimmel, 2005). The mechanism of RA action in the hindbrain is designed to establish expression of Hoxb1 and vHnf1 (a repressor of Hoxb1) on opposite sides of the rhombomere 4/5 boundary in order to obtain rhombomere 4–specific expression of Hoxb1 (Hernandez et al., 2004; Sirbu et al., 2005). The hindbrain utilizes the RA-degrading function of Cyp26 genes to establish a boundary of RA activity at a certain point along the anteroposterior axis of the hindbrain that undergoes a positional shift as time proceeds (Sirbu et al., 2005). In the spinal cord, several studies have concluded that RA is needed to establish dorsoventral patterning of neuroectoderm emerging from the primitive streak (Del Corral et al., 2003; Novitch et al., 2003; Wilson et al., 2004; Molotkova et al., 2005). RA is thus required, along with sonic hedgehog (SHH), for expression of Pax6 and Olig2 that function to generate motor neurons in the ventral horns of the spinal cord.
RA also plays a role in patterning of mesodermal tissues emerging from the primitive streak. RA is required to initiate forelimb budding in mouse (Niederreither et al., 2002b; Mic et al., 2004) and zebrafish (Grandel et al., 2002). However, it is unclear what role RA signaling plays in limb bud development as its originally-proposed role in anteroposterior patterning (Tickle et al., 1982) has not been supported by other studies, which instead indicate that RA exists in a proximodistal gradient in the limb bud (Mic et al., 2004; Yashiro et al., 2004). Thus, the function of RA in limb bud initiation remains unclear, as do(es) the RA target gene(s) controlling limb development. It is also known that a loss of RA affects another mesodermal tissue, the somites, resulting in smaller somites compacted along the anteroposterior axis (Niederreither et al., 1999). Recent studies indicate that RA is also involved in patterning of somites as discussed below.
RA REGULATION OF THE SOMITOGENESIS CLOCK
Somitogenesis is the developmental process whereby trunk paraxial mesoderm exiting the primitive streak is sequentially segmented along the anteroposterior axis into left and right paired epithelial structures known as somites that later fuse to form vertebrae and skeletal muscles. Bilateral symmetry is evident in the arrangement of somites, which form as two symmetrical columns simultaneously on the left and right sides of the embryonic axis in a strict temporal cycle known as the somitogenesis clock. In mouse, the somitogenesis clock results in the formation of one new pair of somites approximately every 2 hr (Giudicelli and Lewis, 2004). Rhythmic somite formation has been proposed to rely on a “clock and wavefront” mechanism, in which a molecular oscillator dependent upon Notch and Wnt signaling controls rhythmic transcription of genes along the presomitic mesoderm (Pourquié 2003; Aulehla and Herrmann, 2004). In this model, as presomitic mesodermal cells migrate anteriorly away from the primitive streak (which is a source of Wnt ligands), they experience alternating cycles of high Notch and low Wnt signaling followed by low Notch and high Wnt signaling. Complementary cyclic expression of Notch signaling inhibitors (lunatic fringe; Lfng) and Wnt signaling inhibitors (Axin2) further enhance alternating cycles of Notch and Wnt signaling to generate the somitogenesis clock (Aulehla et al., 2003; Dale et al., 2003).
Recent studies demonstrated that a loss of RA leads to a loss of left–right bilateral symmetry in mouse, chick, or zebrafish embryos, such that one side has fewer somites than the other (Kawakami et al., 2005; Vermot et al., 2005; Vermot and Pourquié, 2005; Sirbu and Duester, 2006). The effect of a loss of RA on somite left–right patterning is depicted in Fig. 3. In contrast to wild-type embryos, presomitic mesoderm in RA-deficient embryos displays left–right asymmetric expression of Hes7 and Lfng required for Notch-dependent oscillator function during somitogenesis (Kawakami et al., 2005; Vermot et al., 2005; Vermot and Pourquié, 2005). Thus, a loss of RA allows left–right asymmetry to occur in presomitic mesoderm, where it normally does not occur. On the other hand, lateral plate mesoderm in Raldh2−/− mouse embryos still maintains normal left–right asymmetry of key genes, including Nodal and Pitx2 (Niederreither et al., 2001). Thus, RA acts as a buffer to prevent left–right asymmetry from occurring in presomitic mesoderm, while not interfering with the normal process of left–right asymmetry that is needed in lateral plate mesoderm for heart tube looping, gut looping, etc.
Figure 3.
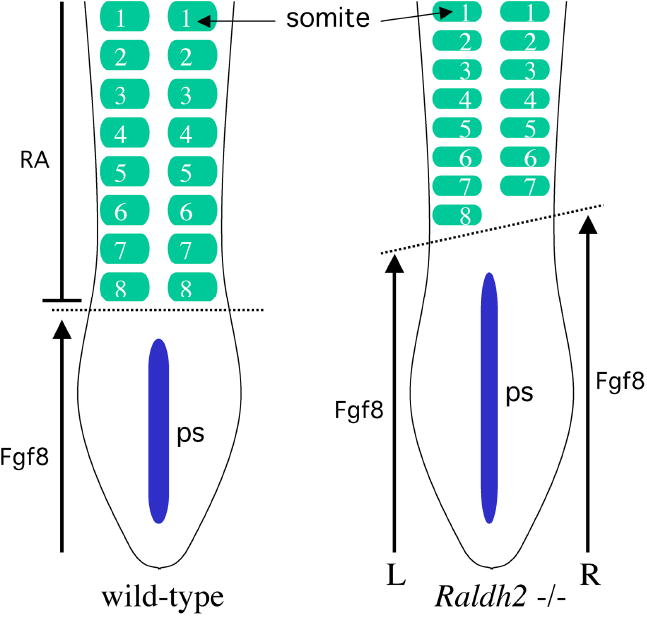
Disruption of somite left–right patterning in the absence of RA signaling. Depicted are embryos at the eight-somite stage in which the wild-type embryo has eight pairs of somites whereas the Raldh2−/− embryo has eight somites on the left side but only seven somites on the right. Thus, a loss of RA synthesis results in a loss of left–right bilateral symmetry during somite generation; plus, somites are smaller in size along the anteroposterior axis. RA is normally synthesized by Raldh2 expressed in the somites and RA released by somitic mesoderm functions to limit the anterior border of Fgf8 expression in ectodermal epiblast cells along the primitive streak (ps). Somites form in mesoderm just anterior to the Fgf8 expression domain. A loss of RA signaling results in an anterior advance of FGF8 signaling (advancing further on the right than on the left), which may be responsible for both the smaller size of somites and a failure to generate left–right somite pairs in synchrony.
Further insight into the mechanism of RA action during somitogenesis came from the discovery that RA generated by Raldh2 in the presomitic mesoderm acts in the adjacent neuroectoderm and node ectoderm to ensure that left and right somite pairs form in synchrony (Sirbu and Duester, 2006). This suggests that RA controls a signaling event that travels from neuroectoderm (and node ectoderm) back to the presomitic mesoderm. As fibroblast growth factor-8 (Fgf8) mRNA was found ectopically in the neuroectoderm and node ectoderm of Raldh2−/− embryos rather than being limited to more posterior ectoderm (epiblast) adjacent to the primitive streak, it was proposed that excessive FGF signaling from ectoderm to mesoderm may be responsible for the somite defect (Sirbu and Duester, 2006). This hypothesis is supported by two observations. First, the somite determination front normally occurs in presomitic mesoderm just anterior to the Fgf8 expression domain in the primitive streak, and perturbation of FGF signaling results in a shift in somite position along the anteroposterior axis (Dubrulle et al., 2001); as a conditional knockout of Fgf8 in the late primitive streak does not affect somitogenesis, other Fgf genes expressed in the primitive streak may also control the somite determination front (Perantoni et al., 2005). Second, as Fgf8 sends a signal to the node that is required for left–right asymmetry in the lateral plate mesoderm that is normally needed for heart tube looping, etc. (Meyers and Martin, 1999), it is possible that excessive FGF8 signaling to the node during RA deficiency may result in left–right asymmetry occurring in the presomitic mesoderm, where it should normally not occur (Fig. 4).
Figure 4.
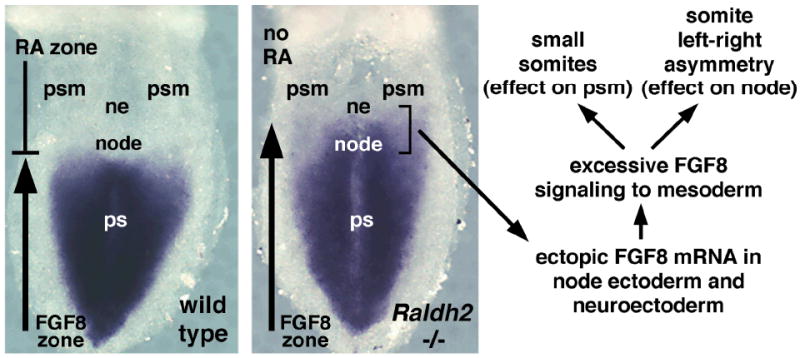
Anterior advance of Fgf8 expression following a loss of RA signaling. Fgf8 expression is normally confined to primitive streak (ps) tissues posterior to the node, a structure that plays a role in gastrulation and left–right patterning of lateral plate mesoderm. RA is first generated by Raldh2 expressed in the presomitic mesoderm (psm) during the late primitive streak stage, when it is required to limit the border of Fgf8 expression to a position just posterior to the node at the anterior end of the primitive streak. A loss of RA allows Fgf8 expression to enter the node and adjacent neuroectoderm (ne), which may lead to excessive FGF8 signaling to mesoderm. Excessive FGF8 signaling in the presomitic mesoderm could result in less cells condensing to form each somite (smaller somites), and excessive FGF8 signaling in the node could impart left–right asymmetry on presomitic mesoderm, which normally does not exhibit such asymmetry.
WHAT ARE THE RELEVANT TARGET TISSUES AND TARGET GENES DURING RA SIGNALING?
There have been numerous reports of potential RA target genes that may be relevant for development of various tissues, but many such genes may lack physiological relevance as they were based upon studies in cell lines or animals treated with unphysiologically high doses of RA (Balmer and Blomhoff, 2002). Indeed, many of these genes do not have recognizable RAREs in their promoter/enhancer regions, and of those that do, only a fraction of these RAREs are conserved among human, mouse, and rat homologous genes (Balmer and Blomhoff, 2005). A subset of Hox genes (genes on the 3′ ends of the Hox gene complexes) still represent the most convincing examples of genes directly regulated by RA, based on their conserved RAREs (Mainguy et al., 2003) as well as on their loss of expression in RA-deficient embryos (Maden, 2002). With the aid of loss-of-function studies, identification of other genes that are direct targets of RA signaling is still ongoing.
As RA is a secreted molecule, it is also essential to identify the tissues that are direct targets of RA action, as bona fide RA target genes must be expressed within those target tissues. In the various model systems used to study RA function, the mouse has been particularly useful for examining the location of RA signaling due to the existence of the RARE-lacZ RA-reporter transgene that marks tissues where transcriptional activity of RA is occurring, including the earliest activity of RA during the late primitive streak stages (Rossant et al., 1991). Transfer of RARE-lacZ into Raldh2−/− embryos has shown that this RA-reporter is indeed an accurate indicator of RA activity, as all expression is lost during the primitive streak stages (Molotkova et al., 2005; Sirbu and Duester, 2006). Also, those studies demonstrated that localized RA activity can be restored to Raldh2−/− embryos following in utero treatment with a low dose of RA; the RA dose used mimics the normal physiological level of RA previously measured in midgestation mouse embryos (Mic et al., 2003). In contrast, treatment of wild-type and Raldh2−/− embryos with a high dose of RA results in RARE-lacZ expression in all cells of the embryo at the late primitive streak stage (Mic et al., 2002). Thus, studies of Raldh2−/− embryos carrying RARE-lacZ can provide a more complete understanding of what tissues are RA target tissues at a particular time in development, what developmental processes RA affects, and what genes RA regulates. This information is essential for determining the mechanism of RA action during differentiation of progenitor cells during normal and disease states.
Through the use of Raldh2−/−embryos carrying RARE-lacZ, it has been demonstrated that a low maternal dose of exogenous RA (sufficient to rescue developmental defects) induces RA-reporter expression only in tissues adjacent to the normal sites of Raldh2 expression and not in those tissues themselves; in such rescue experiments, RA-reporter expression is observed in neuroectoderm and node ectoderm, but not in somites and presomitic mesoderm where Raldh2 is expressed (Molotkova et al., 2005; Sirbu and Duester, 2006). Interestingly, the RA target tissues marked by RARE-lacZ are the same tissues exhibiting ectopic Fgf8 expression in unrescued Raldh2−/− embryos (Sirbu and Duester, 2006), thus further strengthening the hypothesis that a loss of RA leads to deregulation of Fgf8 expression in the primitive streak. A direct role for RA in Fgf8 gene regulation is supported by studies showing that the human Fgf8 promoter contains a RARE (Brondani et al., 2002), and that this element is conserved in human, mouse, and rat Fgf8 genes (Balmer and Blomhoff, 2005).
It is unclear how maternal RA treatment of Raldh2−/− embryos leads to localized expression of RARE-lacZ in posterior neuroectoderm but not the adjacent posterior mesoderm. However, it is possible that the low dose of RA is preferentially sequestered by CRABP2, which is known to facilitate RA signaling (Noy, 2000). Crabp2 is expressed in neuroectoderm but not somitic or presomitic mesoderm in primitive streak stage mouse embryos (Sirbu and Duester, 2006). Also, within neuroectodermal target tissues RA activity is not distributed evenly, indicating that there are mechanisms in play such as RA-degradation by Cyp26 that determine where RA functions (Sirbu et al., 2005).
RA REGULATION OF FGF8 OCCURS ONLY DURING THE LATE PRIMITIVE STREAK STAGE WHEN THE SOMITOGENESIS CLOCK INITIATES
Recent studies suggest that Fgf8 expression in the primitive streak may be a direct target of RA repression, which prevents Fgf8 from being expressed in neuroectoderm and node ectoderm lying just anterior to the epiblast ectoderm (Sirbu and Duester, 2006). Such Fgf8 overexpression may be responsible for inducing left–right asymmetry in somites. It was also discovered that RA is required only during the late primitive streak stage (somites 1–6) to ensure that all subsequent somite pairs develop in synchrony to generate bilateral symmetry throughout somitogenesis (Sirbu and Duester, 2006). In those studies, it was found that the expression domains of Raldh2 (the RA source in the presomitic mesoderm) and Fgf8 (the RA target gene in the epiblast ectoderm) are initially located immediately adjacent to each other along the anteroposterior axis when the first somite pairs are formed. However, by the six-somite stage the Raldh2 expression domain (and RA activity) has shifted anteriorly, while the Fgf8 expression domain remains fixed, leaving a gap between the two. Thus, RA is required to set the anterior border of Fgf8 expression in the epiblast only during the late primitive streak stage when the first few somites are formed. As Fgf8 expression exists in the epiblast and subsequently in the tailbud until the end of somitogenesis (Crossley and Martin, 1995), RA regulation of Fgf8 is required only when the somitogenesis clock is being initiated.
The role of RA may be considered to be that of a regulator, which prevents excessive Fgf8 signaling when the somitogenesis clock is initiating. During this time, the last stages of gastrulation are still occurring and the node (which regulates left–right patterning in the lateral plate mesoderm) is still present. As gastrulation (Sun et al., 1999) and left–right signaling (Meyers and Martin, 1999) are exquisitely sensitive to Fgf8 signaling, RA is evidently needed to limit the extent of Fgf8 signaling during this time. By the 10-somite stage, gastrulation has ended, the node and primitive streak have regressed, and the posterior tissue of the embryo has become the tailbud (still expressing Fgf8). Somites that are generated from the tailbud after gastrulation has ended may not be as sensitive to Fgf8 signaling as earlier somites. Alternatively, the mechanism that limits Fgf8 signaling along the anteroposterior axis (now the tailbud) no longer requires RA.
DEFICIENT RA SIGNALING MAY CAUSE SPINAL COLUMN BIRTH DEFECTS
The discovery that RA is required for synchronous left–right development of somites indicates that RA is required to generate a bilaterally symmetrical spinal column (Kawakami et al., 2005; Vermot et al., 2005; Vermot and Pourquié, 2005; Sirbu and Duester, 2006). RA-deficient mouse embryos are unable to undergo axial turning to achieve the normal fetal position (Vermot et al., 2005; Sirbu and Duester, 2006). Administration of RA maternally to RA-deficient mouse embryos restores normal axial turning and normal spinal column development (Sirbu and Duester, 2006). These studies thus provide evidence that a loss of RA synthesis during gestation causes spinal column birth defects, but that normal development can be restored by supplying an exogenous source of RA.
These findings have implications for understanding the etiology of human spinal column birth defects. This may include birth defects such as scoliosis, an abnormal left–right bending of the vertebral column (Kane, 1977). Idiopathic scoliosis (scoliosis of unknown cause) is the most common type, accounting for 85% of all cases (Reamy and Slakey, 2001). Idiopathic scoliosis is present in 2–4% of children, and about 4 out of 1000 children develop spinal curves large enough to require treatment (Kane, 1977; Reamy and Slakey, 2001). Since scoliosis can run in families, some cases may be the result of a genetic defect (Miller, 1999; Sturm et al., 2001). Scoliosis may be caused by skeletal birth defects that result in hemivertebrae, whereby the left or right side of a vertebra fails to develop normally before birth (McMaster and David, 1986). As most individuals with hemivertebra have additional congenital abnormalities (cranial, cardiac, renal, intestinal, and skeletal) (Goldstein et al., 2005), the underlying defect could be a reduction in RA signaling, which is known to be required for development of all these tissues (Lohnes et al., 1994; Mendelsohn et al., 1994). Thus, either maternal vitamin A deficiency during pregnancy or an embryonic defect in metabolism of vitamin A to RA might be associated with increased risk of human vertebral defects.
References
- Ang HL, Deltour L, Hayamizu TF, et al. Retinoic acid synthesis in mouse embryos during gastrulation and craniofacial development linked to class IV alcohol dehydrogenase gene expression. J Biol Chem. 1996;271:9526–9534. doi: 10.1074/jbc.271.16.9526. [DOI] [PubMed] [Google Scholar]
- Aulehla A, Herrmann BG. Segmentation in vertebrates: clock and gradient finally joined. Genes Dev. 2004;18:2060–2067. doi: 10.1101/gad.1217404. [DOI] [PubMed] [Google Scholar]
- Aulehla A, Wehrle C, Brand-Saberi B, et al. Wnt3a plays a major role in the segmentation clock controlling somitogenesis. Dev Cell. 2003;4:395–406. doi: 10.1016/s1534-5807(03)00055-8. [DOI] [PubMed] [Google Scholar]
- Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- Balmer JE, Blomhoff R. A robust characterization of retinoic acid response elements based on a comparison of sites in three species. J Steroid Biochem Mol Biol. 2005;96:347–354. doi: 10.1016/j.jsbmb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Begemann G, Schilling TF, Rauch GJ, et al. The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development. 2001;128:3081–3094. doi: 10.1242/dev.128.16.3081. [DOI] [PubMed] [Google Scholar]
- Brondani V, Klimkait T, Egly JM, Hamy F. Promoter of FGF8 reveals a unique regulation by unliganded RARα. J Mol Biol. 2002;319:715–728. doi: 10.1016/S0022-2836(02)00376-5. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Martin GR. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development. 1995;121:439–451. doi: 10.1242/dev.121.2.439. [DOI] [PubMed] [Google Scholar]
- Dale JK, Maroto M, Dequeant ML, et al. Periodic notch inhibition by lunatic fringe underlies the chick segmentation clock. Nature. 2003;421:275–278. doi: 10.1038/nature01244. [DOI] [PubMed] [Google Scholar]
- Del Corral RD, Olivera-Martinez I, Goriely A, et al. Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extension. Neuron. 2003;40:65–79. doi: 10.1016/s0896-6273(03)00565-8. [DOI] [PubMed] [Google Scholar]
- Dickman ED, Thaller C, Smith SM. Temporally-regulated retinoic acid depletion produces specific neural crest, ocular and nervous system defects. Development. 1997;124:3111–3121. doi: 10.1242/dev.124.16.3111. [DOI] [PubMed] [Google Scholar]
- Dubrulle J, McGrew MJ, Pourquié O. FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell. 2001;106:219–232. doi: 10.1016/s0092-8674(01)00437-8. [DOI] [PubMed] [Google Scholar]
- Duester G. Families of retinoid dehydrogenases regulating vitamin A function: production of visual pigment and retinoic acid. Eur J Biochem. 2000;267:4315–4324. doi: 10.1046/j.1432-1327.2000.01497.x. [DOI] [PubMed] [Google Scholar]
- Dupé V, Lumsden A. Hindbrain patterning involves graded responses to retinoic acid signalling. Development. 2001;128:2199–2208. doi: 10.1242/dev.128.12.2199. [DOI] [PubMed] [Google Scholar]
- Dupé V, Matt N, Garnier J-M, et al. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc Natl Acad Sci USA. 2003;100:14036–14041. doi: 10.1073/pnas.2336223100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Molotkov A, Manabe S-I, et al. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol Cell Biol. 2003;23:4637–4648. doi: 10.1128/MCB.23.13.4637-4648.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego O, Belyaeva OV, Porte S, et al. Comparative functional analysis of human medium-chain dehydrogenases, short-chain dehydrogenases/reductases and aldo-keto reductases with retinoids. Biochem J. 2006;399:101–109. doi: 10.1042/BJ20051988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain P, Iyer J, Zechel C, Gronemeyer H. Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature. 2002;415:187–192. doi: 10.1038/415187a. [DOI] [PubMed] [Google Scholar]
- Giudicelli F, Lewis J. The vertebrate segmentation clock. Curr Opin Genet Dev. 2004;14:407–414. doi: 10.1016/j.gde.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Goldstein I, Makhoul IR, Weissman A, Drugan A. Hemivertebra: prenatal diagnosis, incidence and characteristics. Fetal Diagn Ther. 2005;20:121–126. doi: 10.1159/000082435. [DOI] [PubMed] [Google Scholar]
- Grandel H, Lun K, Rauch GJ, et al. Retinoic acid signalling in the zebrafish embryo is necessary during pre-segmentation stages to pattern the anterior-posterior axis of the CNS and to induce a pectoral fin bud. Development. 2002;129:2851–2865. doi: 10.1242/dev.129.12.2851. [DOI] [PubMed] [Google Scholar]
- Haselbeck RJ, Hoffmann I, Duester G. Distinct functions for Aldh1 and Raldh2 in the control of ligand production for embryonic retinoid signaling pathways. Dev Genet. 1999;25:353–364. doi: 10.1002/(SICI)1520-6408(1999)25:4<353::AID-DVG9>3.0.CO;2-G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez RE, Rikhof HA, Bachmann R, Moens CB. vhnf1 integrates global RA patterning and local FGF signals to direct posterior hindbrain development in zebrafish. Development. 2004;131:4511–4520. doi: 10.1242/dev.01297. [DOI] [PubMed] [Google Scholar]
- Kane WJ. Scoliosis prevalence: a call for a statement of terms. Clin Orthop Relat Res. 1977;126:43–46. [PubMed] [Google Scholar]
- Kastner P, Chambon P, Leid M. Role of nuclear retinoic acid receptors in the regulation of gene expression. In: Blomhoff R, editor. Vitamin A in health and disease. New York: Marcel Dekker; 1994. pp. 189–238. [Google Scholar]
- Kawakami Y, Raya A, Raya RM, et al. Retinoic acid signalling links left-right asymmetric patterning and bilaterally symmetric somitogenesis in the zebrafish embryo. Nature. 2005;435:165–171. doi: 10.1038/nature03512. [DOI] [PubMed] [Google Scholar]
- Lohnes D, Mark M, Mendelsohn C, et al. Function of the retinoic acid receptors (RARs) during development. (I) Craniofacial and skeletal abnormalities in RAR double mutants. Development. 1994;120:2723–2748. doi: 10.1242/dev.120.10.2723. [DOI] [PubMed] [Google Scholar]
- MacLean G, Abu-Abed S, Dollé P, et al. Cloning of a novel retinoic-acid metabolizing cytochrome P450, Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine development. Mech Dev. 2001;107:195–201. doi: 10.1016/s0925-4773(01)00463-4. [DOI] [PubMed] [Google Scholar]
- Maden M. Retinoid signalling in the development of the central nervous system. Nat Rev Neurosci. 2002;3:843–853. doi: 10.1038/nrn963. [DOI] [PubMed] [Google Scholar]
- Maden M, Gale E, Kostetskii I, Zile MH. Vitamin A-deficient quail embryos have half a hindbrain and other neural defects. Curr Biol. 1996;6:417–426. doi: 10.1016/s0960-9822(02)00509-2. [DOI] [PubMed] [Google Scholar]
- Mainguy G, Rieden PMJID, Berezikov E, et al. A position-dependent organisation of retinoid response elements is conserved in the vertebrate Hox clusters. Trends Genet. 2003;19:476–479. doi: 10.1016/S0168-9525(03)00202-6. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Umesono K, Evans RM. The retinoid receptors. In: Sporn MB, Roberts AB, Goodman DS, editors. The retinoids: biology, chemistry, and medicine. 2. New York: Raven Press; 1994. pp. 319–349. [Google Scholar]
- Marsh-Armstrong N, McCaffery P, Gilbert W, et al. Retinoic acid is necessary for development of the ventral retina in zebrafish. Proc Natl Acad Sci USA. 1994;91:7286–7290. doi: 10.1073/pnas.91.15.7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall H, Studer M, Pöpperl H, et al. A conserved retinoic acid response element required for early expression of the homeobox gene Hoxb-1. Nature. 1994;370:567–571. doi: 10.1038/370567a0. [DOI] [PubMed] [Google Scholar]
- Maves L, Kimmel CB. Dynamic and sequential patterning of the zebrafish posterior hindbrain by retinoic acid. Dev Biol. 2005;285:593–605. doi: 10.1016/j.ydbio.2005.07.015. [DOI] [PubMed] [Google Scholar]
- McMaster MJ, David CV. Hemivertebra as a cause of scoliosis: a study of 104 patients. J Bone Joint Surg Br. 1986;68:588–595. doi: 10.1302/0301-620X.68B4.3733836. [DOI] [PubMed] [Google Scholar]
- Mendelsohn C, Lohnes D, Décimo D, et al. Function of the retinoic acid receptors (RARs) during development. (II) Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994;120:2749–2771. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Martin GR. Differences in left-right axis pathways in mouse and chick: functions of FGF8 and SHH. Science. 1999;285:403–406. doi: 10.1126/science.285.5426.403. [DOI] [PubMed] [Google Scholar]
- Mic FA, Haselbeck RJ, Cuenca AE, Duester G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of Raldh2 null mutant mice. Development. 2002;129:2271–2282. doi: 10.1242/dev.129.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mic FA, Molotkov A, Benbrook DM, Duester G. Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoic acid synthesis. Proc Natl Acad Sci USA. 2003;100:7135–7140. doi: 10.1073/pnas.1231422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mic FA, Sirbu IO, Duester G. Retinoic acid synthesis controlled by Raldh2 is required early for limb bud initiation and then later as a proximodistal signal during apical ectodermal ridge formation. J Biol Chem. 2004;279:26698–26706. doi: 10.1074/jbc.M401920200. [DOI] [PubMed] [Google Scholar]
- Miller NH. Cause and natural history of adolescent idiopathic scoliosis. Orthop Clin North Am. 1999;30:343–352. doi: 10.1016/s0030-5898(05)70091-2. [DOI] [PubMed] [Google Scholar]
- Molotkov A, Fan X, Deltour L, et al. Stimulation of retinoic acid production and growth by ubiquitously-expressed alcohol dehydrogenase Adh3. Proc Natl Acad Sci USA. 2002;99:5337–5342. doi: 10.1073/pnas.082093299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkov A, Ghyselinck NB, Chambon P, Duester G. Opposing actions of cellular retinol-binding protein and alcohol dehydrogenase control the balance between retinol storage and degradation. Biochem J. 2004;383:295–302. doi: 10.1042/BJ20040621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkov A, Molotkova N, Duester G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development. 2006;133:1901–1910. doi: 10.1242/dev.02328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkova N, Molotkov A, Sirbu IO, Duester G. Requirement of mesodermal retinoic acid generated by Raldh2 for posterior neural transformation. Mech Dev. 2005;122:145–155. doi: 10.1016/j.mod.2004.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli JL. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochim Biophys Acta. 1999;1440:139–162. doi: 10.1016/s1388-1981(99)00117-1. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Subbarayan V, Dollé P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet. 1999;21:444–448. doi: 10.1038/7788. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Schuhbaur B, et al. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development. 2000;127:75–85. doi: 10.1242/dev.127.1.75. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Messaddeq N, et al. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development. 2001;128:1019–1031. doi: 10.1242/dev.128.7.1019. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Fraulob V, Garnier JM, et al. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech Dev. 2002a;110:165–171. doi: 10.1016/s0925-4773(01)00561-5. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Schuhbaur B, et al. Embryonic retinoic acid synthesis is required for forelimb growth and anteroposterior patterning in the mouse. Development. 2002b;129:3563–3574. doi: 10.1242/dev.129.15.3563. [DOI] [PubMed] [Google Scholar]
- Novitch BG, Wichterle H, Jessell TM, Sockanathan S. A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron. 2003;40:81–95. doi: 10.1016/j.neuron.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Noy N. Retinoid-binding proteins: mediators of retinoid action. Biochem J. 2000;348:481–495. [PMC free article] [PubMed] [Google Scholar]
- Perantoni AO, Timofeeva O, Naillat F, et al. Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development. 2005;132:3859–3871. doi: 10.1242/dev.01945. [DOI] [PubMed] [Google Scholar]
- Pourquié O. The segmentation clock: converting embryonic time into spatial pattern. Science. 2003;301:328–330. doi: 10.1126/science.1085887. [DOI] [PubMed] [Google Scholar]
- Reamy BV, Slakey JB. Adolescent idiopathic scoliosis: review and current concepts. Am Fam Physician. 2001;64:111–116. [PubMed] [Google Scholar]
- Rossant J, Zirngibl R, Cado D, et al. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991;5:1333–1344. doi: 10.1101/gad.5.8.1333. [DOI] [PubMed] [Google Scholar]
- Sessler RJ, Noy N. A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Mol Cell. 2005;18:343–353. doi: 10.1016/j.molcel.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Sirbu IO, Duester G. Retinoic acid signaling in node ectoderm and posterior neural plate directs left-right patterning of somitic mesoderm. Nat Cell Biol. 2006;8:271–277. doi: 10.1038/ncb1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu IO, Gresh L, Barra J, Duester G. Shifting boundaries of retinoic acid activity control hindbrain segmental gene expression. Development. 2005;132:2611–2622. doi: 10.1242/dev.01845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soprano DR, Blaner WS. Plasma retinol-binding protein. In: Sporn MB, Roberts AB, Goodman DS, editors. The retinoids: biology, chemistry, and medicine. 2. New York: Raven Press; 1994. pp. 257–281. [Google Scholar]
- Stratford T, Horton C, Maden M. Retinoic acid is required for the initiation of outgrowth in the chick limb bud. Curr Biol. 1996;6:1124–1133. doi: 10.1016/s0960-9822(02)70679-9. [DOI] [PubMed] [Google Scholar]
- Sturm PF, Chung R, Bomze SR. Hemivertebra in monozygotic twins. Spine. 2001;26:1389–1391. doi: 10.1097/00007632-200106150-00025. [DOI] [PubMed] [Google Scholar]
- Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999;13:1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahayato A, Dollé P, Petkovich M. Cyp26c1 encodes a novel retinoic acid-metabolizing enzyme expressed in the hindbrain, inner ear, first branchial arch and tooth buds during murine development. Gene Expr Patterns. 2003;3:449–454. doi: 10.1016/s1567-133x(03)00066-8. [DOI] [PubMed] [Google Scholar]
- Tickle C, Alberts BM, Wolpert L, Lee J. Local application of retinoic acid to the limb bud mimics the action of the polarizing region. Nature. 1982;296:564–565. doi: 10.1038/296564a0. [DOI] [PubMed] [Google Scholar]
- Vermot J, Pourquié O. Retinoic acid coordinates somitogenesis and left-right patterning in vertebrate embryos. Nature. 2005;435:215–220. doi: 10.1038/nature03488. [DOI] [PubMed] [Google Scholar]
- Vermot J, Llamas JG, Fraulob V, et al. Retinoic acid controls the bilateral symmetry of somite formation in the mouse embryo. Science. 2005;308:563–566. doi: 10.1126/science.1108363. [DOI] [PubMed] [Google Scholar]
- White JC, Shankar VN, Highland M, et al. Defects in embryonic hindbrain development and fetal resorption resulting from vitamin A deficiency in the rat are prevented by feeding pharmacological levels of all-trans-retinoic acid. Proc Natl Acad Sci USA. 1998;95:13459–13464. doi: 10.1073/pnas.95.23.13459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson L, Gale E, Chambers D, Maden M. Retinoic acid and the control of dorsoventral patterning in the avian spinal cord. Dev Biol. 2004;269:433–446. doi: 10.1016/j.ydbio.2004.01.034. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Zhao X, Uehara M, et al. Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing limb. Dev Cell. 2004;6:411–422. doi: 10.1016/s1534-5807(04)00062-0. [DOI] [PubMed] [Google Scholar]
- Yost RW, Harrison EH, Ross AC. Esterification by rat liver microsomes of retinol bound to cellular retinol-binding protein. J Biol Chem. 1988;263:18693–18701. [PubMed] [Google Scholar]