

Abstract
Trinucleotide repeat instability is intrinsic to a family of human neurodegenerative diseases. The mechanism leading to repeat length variation is unclear. We previously showed that treatment with the demethylating agent 5-aza-2′-deoxycytidine (5-aza-CdR) dramatically increases triplet repeat instability in mammalian cells. Based on previous reports that demethylation increases homologous recombination (HR), and our own observations that HR destabilizes triplet repeats, we hypothesized that demethylation alters repeat stability by stimulating HR. Here, we test that hypothesis at the Aprt (adenosine phosphoribosyl transferase) locus in CHO cells, where CpG demethylation and HR have both been shown to increase CAG repeat instability. We find that the rate of HR at the Aprt locus is not altered by demethylation. The spectrum of recombinants, however, was shifted from the usual 6:1 ratio of conversions to crossovers to more equal proportions in 5-aza-CdR-treated cells. The subtle influences of demethylation on HR at the Aprt locus are not sufficient to account for its dramatic effects on repeat instability. We conclude that 5-aza-CdR promotes triplet repeat instability independently of HR.
Keywords: Recombination, APRT, DNA methylation, genome stability, trinucleotide repeats
INTRODUCTION
Expanded tracts of trinucleotide repeats cause at least 18 human diseases [1,2]. These repeat tracts are unstable and can further expand both in the germline, which causes more severe forms of disease in subsequent generations, and in somatic tissues, which probably contributes to disease progression and pathogenesis in the affected patient[1]. Defining the molecular mechanisms responsible for triplet repeat instability will be critical for development of therapies designed to restrict expansion or promote contraction. Studies in a variety of model organisms have shown that virtually any process that exposes single-stranded DNA—including DNA repair, replication, recombination, and transcription—allows triplet repeats to adopt aberrant secondary structures, which are often misprocessed, changing the length of the repeat [3].
We recently showed that genome-wide demethylation, induced by treatment with 5-aza-2′-deoxycytidine (5-aza-CdR), destabilized CAG triplet repeats in patient-derived cells, causing expansions and contractions [4]. Using a selectable assay for CAG repeat contractions at the Aprt (adenosine phosphoribosyl transferase) locus in CHO cells, we found that treatment with 5-aza-CdR, which leads to passive depletion of DNA methylation [5] and destruction of DNMT1 [6], or with hydralazine, which induces demethylation by a different pathway [7], increased repeat contractions by about 1000 and 50 fold, respectively [4]. Stimulation of repeat contraction by two mechanistically distinct agents, in rough proportion to their ability to decrease genome-wide methylation, argues for a causal link between CpG demethylation and CAG repeat instability [4]. In a separate investigation into the interactions between CAG repeats and homologous recombination (HR), we showed that HR significantly altered repeat stability at the Aprt locus in CHO cells [8]. Among homologous recombinants, 5% had lost large numbers of repeats, whereas no such changes were observed in more than 250 colonies that had not undergone HR [8]. These repeat contractions were evenly distributed between the two types of HR event—crossover and conversion—detected in our assay [8]. These two studies, coupled with the observations summarized below, led us to propose that 5-aza-CdR treatment destabilizes CAG repeats by stimulating HR [8]. Here, we test that hypothesis.
It has been suggested that a major role of DNA methylation—the addition of a methyl group to cytosine within CpG dinucleotides—is to inhibit HR between repeated sequences [9-11]. Support for this idea is strongest in fungi where crossovers between direct repeats during meiosis are inhibited more than 100-fold by CpG methylation [12]. In mammalian cells, the links between DNA methylation and HR are less direct, and in some cases, contradictory. Treatment with 5-aza-CdR, which induces demethylation, has been shown to stimulate sister-chromatid exchange [5], increase HR within a transgene array [13], and promotes double-minute chromosome dimerization [14]. In mouse ES cells, knockout of DNA methyltransferase 1 (DNMT1), the maintenance DNA methyltransferase, reduces CpG methylation and increases loss of heterozygosity (LOH) at the Dnmt1 locus, which was attributed to elevated levels of mitotic HR [15]. In the same study, knockout of Dnmt1 was also shown to increase genomic rearrangements at the Hprt (hypoxanthine phosphoribosyl transferase) locus, suggesting that CpG methylation may inhibit nonhomologous recombination, as well [15]. The view that CpG methylation normally inhibits both kinds of recombination can account for several additional observations: patients with mutations in Dnmt3a display increased rearrangements of centromeric repeats [16]; ES cells that lack DNMT1 show an increased efficiency of gene targeting [17]; and DNMT1-deficient human cancer cells have higher frequencies of chromosomal rearrangements [18].
In contrast to these studies, which generally support a role for CpG methylation in genome stability, other reports either failed to detect an effect, or showed that demethylation decreased HR. CpG-methylated and unmethylated DNA constructs, when transfected into COS1 monkey cells, gave identical frequencies of extrachromosomal recombination in an assay designed to detect single-strand annealing, one kind of HR [19]. Similarly, 5-aza-CdR treatment of HeLa human cells that were modified to carry a chromosomal recombination substrate designed to detect conversion events did not increase the frequency of HR [20]. Finally, DNMT1 deficiency in mouse ES cells, was shown to reduce the rate of loss of an array of selectable transgenes, suggesting that mitotic HR was decreased by demethylation [21].
The lack of agreement among these studies could arise for several reasons: differences between the cell lines, or the species from which they were derived; whether the recombination substrate is extrachromosomal, randomly integrated, or targeted to a specific locus; and the particular kind of event the substrate is designed to detect—crossover, conversion, or rearrangement. Because our original observations—5-aza-CdR treatment and HR destabilize repeats—were made at the Aprt locus in CHO cells, we chose to measure the effects of 5-aza-CdR treatment on HR at the same site, using a well-characterized recombination assay that can simultaneously detect crossovers, conversions, rearrangements, and mutations (Figure 1A) [22]. In this way we can measure the effects of 5-aza-CdR treatment on overall rates of recombination, as well as determine the differential effects, if any, on individual types of events.
Figure 1.
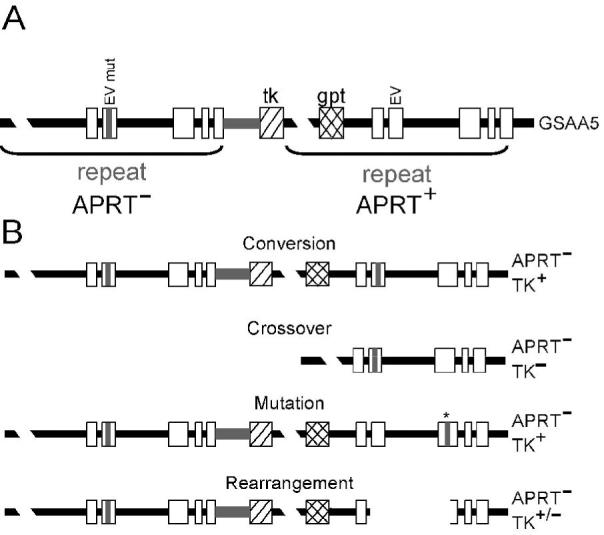
Recombination substrate and products. A. The duplication at the Aprt locus in GSAA5 cells. The upstream copy of Aprt contains a mutated EcoRV site (EV mut) in exon 2, as well as a truncated exon 5, rendering it nonfunctional. The downstream copy contains the normal EcoRV site (EV) and is functional, making GSAA5 cells APRT+. The herpes virus thymidine kinase (TK) and the guanine phosphoribosyl transferase (gpt) genes served as additional selective markers useful in generating the GSAA5 cell line and for characterizing the recombination products. B. Illustrations of the types of APRT- colonies that arise in GSAA5 cells. After selection against APRT function, it is possible to recover clones that underwent HR (conversions or crossovers), mutation of the wild-type copy of the Aprt gene (indicated by the * above exon 3), or rearrangement of the Aprt gene (shown here as a deletion of a portion of the Aprt gene). Each of these events can be distinguished by a combination of Southern blot and PCR analysis (see Materials and Methods).
By determining whether 5-aza-CdR treatment stimulates HR, we can distinguish between two alternatives for how genome-wide demethylation stimulates CAG repeat contraction. Because 5-aza-CdR treatment and HR each destabilize repeats, we proposed the hypothesis that they operate as part of a common pathway—with 5-aza-CdR stimulating HR, which in turn stimulates repeat contractions. A demonstration that 5-aza-CdR stimulates HR would cleanly distinguish this possibility from the alternative: that 5-aza-CdR and HR have their effects via independent pathways. Here we show that 5-aza-CdR treatment does not significantly alter the rate of HR at the Aprt locus, although it does shift the spectrum of events. We conclude that 5-aza-CdR treatment destabilizes triplet repeats by a mechanism that does not proceed via stimulation of HR.
MATERIALS AND METHODS
Cell lines and tissue culture conditions
Chinese hamster ovary (CHO) cells lines AT3-2, which contains a single copy of the wild type Aprt gene, and GSAA5, which was derived from AT3-2 cells and carries a duplication at the Aprt locus, have been described previously [23,24]. Both cell lines carry a functional Aprt gene and are phenotypically APRT+. These cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Gibco), nonessential amino acids, sodium pyruvate, penicillin, and streptomycin. For these experiments, we derived cell populations from individual colonies, and verified the genomic structure of the Aprt locus by Southern blotting and PCR (data not shown). To minimize spontaneous APRT- cells in the starting APRT+ cell populations, we propagated the parental cells at low density for 7 to 10 days in ALASA medium (30μM alanosine, 10μM azaserine, 1mM adenine), which does not support the proliferation of APRT- cells. Pre-selected populations of cells were frozen in aliquots, which were thawed at the start each experiment.
Recombination assay
To determine the frequencies of APRT- cells, we plated the untreated and 5-aza-CdR-treated cell populations at a density of 5×105 cells/100mm plates in complete medium supplemented with 400 μM 8-azaadenine, which kills APRT+ cells. After two weeks, we counted the APRT- colonies and used their frequencies to estimate rates, as described previously [4]. The number of APRT- colonies was adjusted for cell survival [APRT- colonies/(surviving colonies/plated cells)] and then divided by the number of population doublings that the cells underwent prior to applying selection for APRT- cells.
The spectra of events that led to the APRT- cells were analyzed by picking individual colonies from each independent treated (or untreated) cell population. Two colonies were initially isolated per experiment; if the two clones were identical, only one was counted to ensure independence. Southern blotting and PCR analysis were used to distinguish the four types of event that can give rise to APRT- cells (Fig. 1). After digestion of genomic DNA with BamH1, crossovers give rise to a single 4.1-kb band, whereas conversions and mutations give a two-band pattern: a 12.5-kb band from the upstream APRT copy, and a 4.1-kb band from the downstream copy. Rearrangements produce a pattern that is distinct from the above patterns; none were detected in the experiments in this paper. Conversions were distinguished from mutations by the presence of the mutant EcoRV site in both copies of the Aprt gene. Conversions gave a PCR product that was not cleaved by EcoRV, whereas the corresponding PCR product from mutations was cleaved [22]. If the PCR product was cleaved, indicating retention of the EcoRV site, the event was categorized as a mutation and was not analyzed further.
5-aza-CdR treatment and methylation analysis
Pre-selected cells were thawed and allowed to recover for 24 hours before being propagated at low density for 4 days. The exponentially growing cells were subsequently plated at densities of 2×105, 6×105, and 8×105 per 100mm plate for treatment with 0, 0.5, or 1μM 5-aza-CdR (Sigma), respectively. Higher numbers of cells were used in the treated plates to compensate for 5-aza-CdR toxicity [4]. 5-aza-CdR was added to the growth media 24 hr after plating and incubated for three days, with fresh media containing the drug substituted after 24 hrs. For the recombination assay, treated cells were allowed to recover for three days and then they were plated as described above. For global methylation analysis, the cells were harvested, genomic DNA was isolated using GenomicPrep (Amersham), digested with either MspI (NEB) or HpaII (NEB), and then subjected to electrophoresis on a 1% agarose gel. For analysis of methylation at the Aprt locus, 5μg of BamHI-cleaved DNA was digested with either MspI or HpaII, and the products were separated by electrophoresis on a 1% agarose gel and transferred to a nylon membrane (Amersham). The membrane was hybridized to the 2.1-kb XhoI-BamHI fragment from the 3′ end of Aprt, which was radiolabelled by random priming with alpha 32P-dCTP. The positions of the bands were visualized by autoradiography.
Real-Time RT-PCR
Aprt mRNA levels were measured by real-time RT-PCR using the Quantitech Qiagen SYBR green kit as described elsewhere [25]. Briefly, RNA was isolated using RNAeasy (Qiagen) and Aprt mRNA was amplified with two different pairs of primers: oVIN-219F (5′-ttg cag gag aga gaa gaa tgg) and oVIN-219R (5′-ggc cag aaa gtg gtt gtt gt), and oVIN-221F (5′-cga gag cat agg agg ctg ac) and oVIN-221R (5′-tgg cca gtc acc tta agt cc). For each sample, the Aprt mRNA levels were normalized to vimentin mRNA levels which were obtained using three different pairs of primers: oVIN-222F (5′- ggg cga cct cta tga gga g) and oVIN-222R (5′-gct ctc cgc ttc ctc tct ct), oVIN-223F (5′-aga gag agg aag cgg aga gc) and oVIN- 223R (5′-ctc ctg aat ctg ggc ctg ta), and oVIN-224F (5′-ttc taa gcc cga cct cac tg) and oVIN-224R (5′-ctc att tga ctc ctg ctt tgc).
Statistics
To compare APRT- rates, we used Student’s T-test. We used a Chi-square test with Yate’s correction to probe the significance of changes in recombination types between crossovers and conversions in treated versus untreated cells. Fisher’s exact test was used to obtain the probability values for the distribution of DNA methylation in recombinants.
RESULTS
Experimental design
To assay for the effects of demethylation on HR, we used a well-characterized recombination substrate that contains two direct repeats of the selectable Aprt gene at its endogenous locus in GSAA5 CHO cells (Fig.1). The upstream copy of the gene is truncated at the 3′ end and has a mutated EcoRV site in exon 2 [23]. The downstream copy is functional and contains the wild type EcoRV site. Selection for APRT- cells allows us to detect four types of event—conversion, crossover, rearrangement, and mutation—that inactivate the downstream copy (Fig. 1). As in our previous studies, these names refer to the products shown in Figure 1; they are not meant to imply (or exclude) any specific mechanism. We can distinguish these events by Southern blotting and PCR (see Materials and Methods). Spontaneous APRT- cells arise predominantly by HR, with conversions and crossovers accounting for 95% of all APRT- colonies [22,26,27]. To bring about CpG demethylation, we used the cytidine analog, 5-aza-CdR, which is incorporated into DNA and traps DNA methyltransferases. This leads to a passive demethylation of the genome during subsequent rounds of replication [5]. By comparing APRT- colony formation in treated and untreated cells, and by analyzing the spectrum of events, we can determine whether 5-aza-CdR treatment significantly affects HR, nonhomologous recombination, or mutation.
5-aza-CdR treatment reduces CpG methylation
To confirm our previous findings that 5-aza-CdR treatment of CHO cells reduces DNA methylation genome-wide [4], we grew GSAA5 cells for 3 days in the presence of 0 μM, 0.5 μM, or 1 μM of the drug. We tested 0.5 μM and 1 μM 5-aza-CdR because they had previously been shown to stimulate triplet repeat contraction 100- to 1000-fold [4]. After drug treatment, genomic DNA was isolated and digested with MspI, which cleaves unmethylated and methylated CCGG sites, or with HpaII, which only cleaves unmethylated CCGG sites. The digested DNA was displayed by electrophoresis on an agarose gel (Figure 2A). Consistent with our previous findings, 5-aza-CdR treatment makes the genomic DNA much more sensitive to HpaII digestion, indicating a substantial loss of CpG methylation. Similarly, we confirmed our previous results [4] that a defined CCGG site at the 3′ end of the Aprt gene (Figure 2B), which is 100% methylated in untreated cells, is only about 50% methylated in 5-aza-CdR-treated cells (data not shown).
Figure 2.
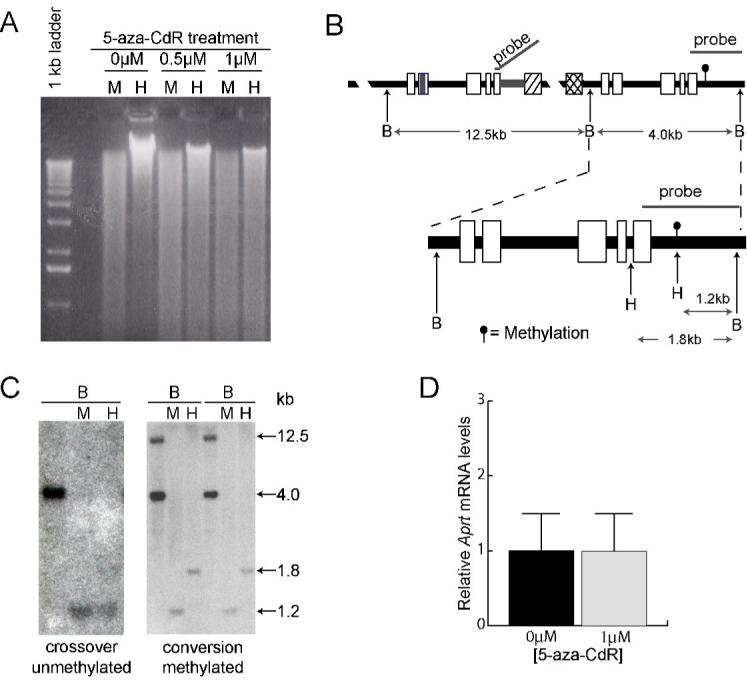
Methylation analysis of the Aprt locus. A. Analysis of global methylation in GSAA5 cells. Genomic DNA was isolated from untreated cells and from cells treated with 5-aza-CdR, digested with MspI (M) or its methyl-sensitive isoschizomer HpaII (H), and displayed by electrophoresis on an agarose gel. Decreased methylation in treated cells is shown by the more complete digestion of DNA by HpaII. A 1-kb marker ladder is shown on the left. B. Restriction maps and probe used for analysis of methylation at the Aprt locus. The complete recombination substrate is shown on top, with an expanded view of the 3′ copy of Aprt shown below. The methylated HpaII (H) site, which is indicated by the filled circle in the downstream copy of Aprt, is not present in the upstream copy. The probe hybridizes to the 12.5- and 4.0-kb BamHI (B) fragments, which contain the upstream and downstream copies of Aprt, respectively. C. Representative Southern blots showing the structure and methylation status of a crossover and two conversions. All samples were digested with BamHI (B) alone or in combination with MspI (M) or HpaII (H). Sizes of the digestion products (in kb) are shown on the right. D. Relative Aprt mRNA levels after 0 and 0.5μM 5-aza-CdR treatment are shown. Error bars represent one standard deviation.
Effects of 5-aza-CdR treatment on HR
To test whether CpG demethylation stimulates HR, we treated GSAA5 cells with 0 μM, 0.5 μM, or 1 μM 5-aza-CdR, and measured the rates at which APRT- colonies arose. As shown in Table 1, treated and untreated GSAA5 cells formed APRT- colonies at rates that were not significantly different, suggesting that HR was not altered by CpG demethylation. To verify this conclusion, we isolated several independent APRT- colonies to determine the spectrum of events that produced them. In our previous studies of spontaneous events [22,26,27], which are summarized in Table 2, we found that 95% of APRT- colonies arose by HR, with a 6:1 ratio of conversions to crossovers, 5% arose by mutation, and less than 0.5% arose by rearrangement. The eight independent APRT- colonies isolated from untreated GSAA5 cells, which were all conversions (Table 2), are consistent with the expected distribution of spontaneous events (P=0.53). The 15 independent APRT- colonies isolated from 5-aza-CdR-treated GSAA5 cells were also generated predominantly by HR (Table 2), but the spectrum of events is significantly different from that of spontaneous events (P=0.0055) with only 47% of the events being gene conversions and 40% crossovers. Thus, although 5-aza-CdR does not seem to stimulate HR overall, it apparently promotes crossovers at the expense of conversions.
Table 1.
Rates of APRT colony formation.
| Cell lines | 5-aza-CdR (μM) | Rates of APRT- colony formation (10-7)a | P value vs 0 μM | P value vs 1 μM |
|---|---|---|---|---|
| GSAA5 | 0 | 48 ± 7 | 0.7 | |
| 0.5 | 36 ± 2 | 0.15 | 0.4 | |
| 1 | 62 ± 24 | 0.7 | ||
| AT3-2 | 0 | 5.0 ± 0.5 | 0.8 | |
| 0.5 | 3.5 ± 0.2 | 0.06 | 0.6 | |
| 1 | 4.6 ± 2 | 0.8 |
Results represent at least three independent experiments. In each experiment, three to four different plates were treated in parallel and the mean rates for each experiment were averaged. The standard error is shown.
Table 2.
Distribution of events that gave rise to APRT- colonies.
| Cell line | 5-aza-CdR (μM) | Conversionb | Crossoverb | Mutationb | Rearrangement | Total |
|---|---|---|---|---|---|---|
| GSAA5 | 0 | 8 (100) | 0 (0) | 0 (0) | 0 | 8 |
| GSAA5 | 0.5 & 1 | 7 (47) | 6 (40) | 2 (13) | 0 | 15 |
| Othera | 0 | 172 (82) | 28 (28) | 10 (5) | 0 | 210 |
These numbers are the sum of published data for a variety of other CHO cell lines (GSB2, GSB5, GSC4, GSC6, GSE1, GSE3, GS21-15, and AK209) that each carries the duplication substrate at the Aprt locus [22,26,27]. These numbers define the expected distribution for spontaneous events at the duplicated Aprt locus.
The number of APRT- colonies identified in each category is shown, with the percentage of the total indicated in parentheses.
Methylation status of recombinants at the Aprt locus
Another way to assess the effects of 5-aza-CdR on HR is to determine whether the recombinants arose preferentially from the unmethylated or the methylated subpopulation. We showed previously that about 50% of 5-aza-CdR treated cells lose the methylation mark on the CCGG site at the 3′ end of the Aprt gene (Figure 2B) [4]. Because DNA methylation patterns are maintained during mitotic division, we can analyze the APRT- colonies to track the methylation status of their genomes near the time they underwent recombination. Therefore, we performed Southern blot analysis after digestion with BamHI, alone, and in combination with either MspI or HpaII to determine whether the clones were methylated at the Aprt locus. Analyses of one unmethylated crossover and two methylated conversion events are shown in Figure 2C. As expected, all eight recombinants from untreated cells were methylated. In contrast, nine of twelve recombinants from 5-aza-CdR-treated cells were not methylated (Table 3). These results confirm that the population of 5-aza-CdR-treated cells had in fact undergone demethylation. More importantly, the 1:3 ratio of methylated to unmethylated genomes is not significantly different (P=0.4) from the 6:6 ratio expected from the 50% 5-aza-CdR-induced demethylation observed previously at this site [4]. These results support our conclusion that 5-aza-CdR treatment does not stimulate HR.
Table 3.
Analysis of methylation in recombinants.
Mutagenesis of the APRT locus
In the spectrum of APRT- colonies that arose after 5-aza-CdR treatment, we observed more mutations than we had expected (Table 2). Although the difference is small (and not statistically significant), 5-aza-CdR is reported to be a weak mutagen [5]. Therefore, we asked whether 5-aza-CdR treatment increases the rate of mutation at the Aprt locus in CHO cells. AT3-2 cells, which carry a single wild type copy of Aprt [24], were treated with 0 μM, 0.5 μM, or 1 μM 5-aza-CdR and APRT- colonies were selected. As shown in Table 1, 5-aza-CdR treatment had no significant effect on mutation rates (P>0.05), consistent with our previous studies [4,28].
Transcription at the Aprt locus
Loss of DNA methylation can activate gene transcription [29], which has been shown to destabilize triplet repeats [25,30]. To determine whether 5-aza-CdR treatment might stimulate repeat contraction via effects on transcription, we examined Aprt mRNA levels using quantitative RT-PCR. We found that Aprt mRNA levels were unchanged by 5-aza-CdR treatment (Figure 2), suggesting that the drug does not induce repeat instability by increasing transcription through the repeat tract.
DISCUSSION
In previous studies, we showed that both 5-aza-CdR treatment and HR stimulate contraction of CAG repeat tract at the Aprt locus [4,8]. Because several observations in the literature suggest that demethylation increases HR, we proposed that 5-aza-CdR treatment promotes repeat instability via stimulation of HR [4]. As a critical test of this hypothesis, we measured the effects of 5-aza-CdR treatment on HR, using a well-characterized recombination substrate at the Aprt locus. We show here that 5-aza-CdR treatment does not alter the overall rate of HR. These results disprove the hypothesis. We conclude that treatment of cells with 5-aza-CdR increases contraction of CAG repeat tracts via a mechanism that does not involve HR. Thus, demethylation and HR destabilize repeat tracts by independent pathways.
Although 5-aza-CdR treatment did not increase the rate of HR, it did alter the proportions of conversions to crossovers, from the normal (spontaneous) 6:1 ratio to about a 1:1 ratio (Table 2). The increase in crossovers, which is likely to occur via the mechanism of single-strand annealing (SSA) (ref Sargent NAR 28:3771, 2000, plus some others), is intriguing because SSA within the CAG tract could readily generate repeat contractions. However, the magnitude of change observed here—a few fold—is not adequate to account for the 1000-fold effect of 5-aza-CdR on CAG repeat instability observed previously [4]. Moreover, a selective effect of 5-aza-CdR on crossovers does not jibe with our previous observation that HR stimulates CAG contractions about equally among crossovers and conversion events [8]. Thus, the subtle effects of 5-aza-CdR on HR observed cannot account for the dramatic effect 5-aza-CdR has on CAG repeat contraction [4].
Our results provide the first example of a bioactive molecule that changes the type of recombination event, without affecting the overall rate. Mammalian somatic cells are thought to be biased toward conversion events because they permit the beneficial exchange of genetic information during repair processes, without the potential for loss of heterozygosity (LOH) that can accompany crossover events [31]. Little is known, however, about how the cell imposes the preference for conversions over crossovers during HR. Our results, which indicate that small molecules can affect the outcome, suggest that screening chemical libraries could yield significant insights into this process.
This study also raises the possibility that DNA methylation may promote genome stability, not by preventing HR between repetitive sequences as previously suggested [9-12], but rather by helping to ensure that HR proceeds via conversion, which avoids several dangers: LOH by mitotic HR between homologues, deletion of information by HR between repeats on the same chromosome, and rearrangements by HR between repeats on different chromosomes. Previous studies, which all used assays that were specific for a single type of recombination event, would have missed the phenomenon reported here.
Demethylation-induced stimulation of crossovers at the expense of conversions could account for most of the observations in studies that have investigated the role of DNA methylation in chromosomal HR in mammalian cells. The one study that used conversion as an assay for HR detected no increase in DNMT1-deficient ES cells [20]. By contrast, with one exception [21], studies that used crossover as an assay detected a demethylation-induced stimulation of HR [5,13,15,16]. DNA demethylation has also been reported to increase rearrangements [18,21]; however, in neither study were the rearrangements analyzed to determine whether they were generated by HR between similar sequences or by nonhomologous recombination between unrelated sequences. If they arose by HR, then they would represent additional examples of crossover stimulation by demethylation.
Our studies do not define the mechanism of crossover stimulation by 5-aza-CdR treatment. There are several possibilities. First, at the Aprt locus, double-strand breaks (DSBs), which greatly increase overall HR frequencies, shift the bias from the 6:1 spontaneous ratio to about 3:1 [23]. Treatment with 5-aza-CdR, however, is unlikely to change the distribution of HR events by inducing DSBs because overall HR frequencies are not affected (Table 1). Second, 5-aza-CdR could act on HR indirectly by altering CpG methylation at distant promoters, thereby changing expression levels of genes such as Rad51 and Brca2, which are known to influence the competition between conversion and crossover [32], or of other DNA repair genes that might alter the ratio. Third, incorporation of 5-aza-CdR into the DNA at the Aprt locus, or its covalent attachment to DNMT1, could have direct effects on HR. For example, if 5-aza-CdR-modified DNA interfered with strand invasion or DNA synthesis primed by the invading strand, it would preferentially reduce conversions relative to crossovers, which are thought to occur predominantly by SSA and thus do not require strand invasion. Fourth, by sequestering or destroying DNMT1, 5-aza-CdR could alter the amounts or properties of proteins that are known to bind to DNMT1, including histone deacetylases 1 and 2 [33,34] and histone methyltransferases G9a [35] and SUV39H1 [36], thereby potentially altering the proportion of crossovers via effects on chromatin structure [37,38]. Fifth, local methylation changes at Aprt could, in principle, alter recombination via effects on chromatin structure or by influencing the formation or processing of recombination intermediates. It seems unlikely that the single site we have assayed—the only methylated HpaII site in the vicinity of the gene—could have such a profound effect, but there may be additional methylated CpG sites at the Aprt locus that we have not detected.
Although our studies rule out an intermediary role of HR in the pathway of 5-aza-CdR-induced stimulation of triplet-repeat instability, they do not further define the mechanism of demethylation-induced repeat instability. Treatment of CHO cells with 5-aza-CdR can increase contraction of a CAG repeat tract by more than 1000-fold, which is the largest effect on triplet-repeat instability observed to date [4]. Determining its mode of action, although challenging, may be especially relevant for developing therapeutics designed to stimulate contraction of triplet repeats. One well-defined effect of DNA methylation inhibitors, including 5-aza-CdR, is to cause changes in transcription [39]. Since transcription itself destabilizes CAG repeats [25,30], we analyzed transcription at the Aprt locus before and after treatment with 5-aza-CdR, but observed no differences (Figure 2). Thus, it seems unlikely that 5-aza-CdR treatment stimulates repeat instability via changes in transcription through the Aprt locus. It remains possible that 5-aza-CdR alters the expression of other genes—DNA repair genes, in particular—that modulate triplet repeat instability. Further experiments will be needed to define 5-aza-CdR’s mode of action leading to triplet repeat instability.
ACKNOWLEDGEMENTS
We would like to thank Fung Chan for technical help and members of the Wilson laboratory for helpful comments and discussions. We are also indebted to Fisun Hamaratoğlu and Murat B. Yaylaoğlu for critical reading of the manuscript. This work was supported by a postgraduate scholarship A and D from the Natural Sciences and Engineering Council of Canada to V.D., an NIH T32 training grant EY007102 to B.A.P. and an NIH grant GM38219 to J.H.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Pearson CE, Edamura KN, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat. Rev. Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- [2].Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- [3].Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- [4].Gorbunova V, Seluanov A, Mittelman D, Wilson JH. Genome-wide demethylation destabilizes CTG.CAG trinucleotide repeats in mammalian cells. Hum Mol Genet. 2004;13:2979–2989. doi: 10.1093/hmg/ddh317. [DOI] [PubMed] [Google Scholar]
- [5].Haaf T. The effects of 5-azacytidine and 5-azadeoxycytidine on chromosome structure and function: implications for methylation-associated cellular processes. Pharmacol Ther. 1995;65:19–46. doi: 10.1016/0163-7258(94)00053-6. [DOI] [PubMed] [Google Scholar]
- [6].Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25:4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [7].Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Mariscal I, Chavez A, Acuna C, Salazar AM, Lizano M, Duenas-Gonzalez A. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res. 2003;9:1596–1603. [PubMed] [Google Scholar]
- [8].Meservy JL, Sargent RG, Iyer RR, Chan F, McKenzie GJ, Wells RD, Wilson JH. Long CTG tracts from the myotonic dystrophy gene induce deletions and rearrangements during recombination at the APRT locus in CHO cells. Mol Cell Biol. 2003;23:3152–3162. doi: 10.1128/MCB.23.9.3152-3162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Colot V, Rossignol JL. Eukaryotic DNA methylation as an evolutionary device. Bioessays. 1999;21:402–411. doi: 10.1002/(SICI)1521-1878(199905)21:5<402::AID-BIES7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- [10].Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- [11].Kricker MC, Drake JW, Radman M. Duplication-targeted DNA methylation and mutagenesis in the evolution of eukaryotic chromosomes. Proc Natl Acad Sci U S A. 1992;89:1075–1079. doi: 10.1073/pnas.89.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Maloisel L, Rossignol JL. Suppression of crossing-over by DNA methylation in Ascobolus. Genes Dev. 1998;12:1381–1389. doi: 10.1101/gad.12.9.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].McBurney MW, Lau S, Jardine K, Yang X, Davies B. Reexpression of a cluster of silenced transgenes is associated with their rearrangement. Genes Chromosomes Cancer. 2001;32:311–323. doi: 10.1002/gcc.1196. [DOI] [PubMed] [Google Scholar]
- [14].Rizwana R, Hahn P. CpG methylation reduces genomic instability. Journal of Cell Science. 1999;112:4513. doi: 10.1242/jcs.112.24.4513. [DOI] [PubMed] [Google Scholar]
- [15].Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- [16].Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Pequignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- [17].Dominguez-Bendala J, McWhir J. Enhanced gene targeting frequency in ES cells with low genomic methylation levels. Transgenic Res. 2004;13:69–74. doi: 10.1023/b:trag.0000017176.77847.80. [DOI] [PubMed] [Google Scholar]
- [18].Karpf AR, Matsui S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005;65:8635–8639. doi: 10.1158/0008-5472.CAN-05-1961. [DOI] [PubMed] [Google Scholar]
- [19].Liang F, Jasin M. Studies on the influence of cytosine methylation on DNA recombination and end-joining in mammalian cells. J Biol Chem. 1995;270:23838–23844. doi: 10.1074/jbc.270.40.23838. [DOI] [PubMed] [Google Scholar]
- [20].Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Pardo AD, Messina S, Iuliano R, Fusco A, Santillo MR, Muller MT, Chiariotti L, Gottesman ME, Avvedimento EV. DNA Damage, Homology-Directed Repair, and DNA Methylation. PLoS Genet. 2007;3:e110. doi: 10.1371/journal.pgen.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [21].Chan MF, van Amerongen R, Nijjar T, Cuppen E, Jones PA, Laird PW. Reduced rates of gene loss, gene silencing, and gene mutation in Dnmt1-deficient embryonic stem cells. Mol Cell Biol. 2001;21:7587–7600. doi: 10.1128/MCB.21.22.7587-7600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sargent RG, Merrihew RV, Nairn R, Adair G, Meuth M, Wilson JH. The influence of a (GT)29 microsatellite sequence on homologous recombination in the hamster adenine phosphoribosyltransferase gene. Nucleic Acids Res. 1996;24:746–753. doi: 10.1093/nar/24.4.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sargent RG, Brenneman MA, Wilson JH. Repair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol Cell Biol. 1997;17:267–277. doi: 10.1128/mcb.17.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Adair GM, Carver JH, Wandres DL. Mutagenicity testing in mammalian cells. I. Derivation of a Chinese hamster ovary cell line heterozygous for the adenine phosphoribosyltransferase and thymidine kinase loci. Mutat Res. 1980;72:187–205. doi: 10.1016/0027-5107(80)90035-4. [DOI] [PubMed] [Google Scholar]
- [25].Lin Y, Dion V, Wilson JH. Transcription promotes contraction of CAG repeat tracts in human cells. Nat. Struct. Mol. Biol. 2006;13:179–180. doi: 10.1038/nsmb1042. [DOI] [PubMed] [Google Scholar]
- [26].Kilburn AE, Shea MJ, Sargent RG, Wilson JH. Insertion of a telomere repeat sequence into a mammalian gene causes chromosome instability. Mol Cell Biol. 2001;21:126–135. doi: 10.1128/MCB.21.1.126-135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sargent RG, Rolig RL, Kilburn AE, Adair GM, Wilson JH, Nairn RS. Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1. Proc Natl Acad Sci U S A. 1997;94:13122–13127. doi: 10.1073/pnas.94.24.13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lin Y, Dion V, Wilson JH. A novel selectable system for detecting expansion of CAG.CTG repeats in mammalian cells. Mutat Res. 2005;572:123–131. doi: 10.1016/j.mrfmmm.2005.01.013. [DOI] [PubMed] [Google Scholar]
- [29].Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- [30].Lin Y, Wilson JH. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2007;27:6209–6217. doi: 10.1128/MCB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Johnson RD, Jasin M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans. 2001;29:196–201. doi: 10.1042/0300-5127:0290196. [DOI] [PubMed] [Google Scholar]
- [32].Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- [34].Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- [35].Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang Z, Fan HY, Goldman JA, Kingston RE. Homology-driven chromatin remodeling by human RAD54. Nat Struct Mol Biol. 2007;14:397–405. doi: 10.1038/nsmb1223. [DOI] [PubMed] [Google Scholar]
- [38].Cummings WJ, Yabuki M, Ordinario EC, Bednarski DW, Quay S, Maizels N. Chromatin Structure Regulates Gene Conversion. PLoS Biol. 2007;5:e246. doi: 10.1371/journal.pbio.0050246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]