

Summary
Adoptive cell transfer therapy using tumor-infiltrating lymphocytes for patients with metastatic melanoma has demonstrated significant objective response rates. One major limitation of these current therapies is the frequent inability to isolate tumor-reactive lymphocytes for treatment. Genetic engineering of peripheral blood lymphocytes with retroviral vectors encoding tumor antigen-specific T-cell receptors (TCRs) bypasses this restriction. To evaluate the efficacy of TCR gene therapy, a murine treatment model was developed. A retroviral vector was constructed encoding the pmel-1 TCR genes targeting the B16 melanoma antigen, gp100. Transduction of C57BL/6 lymphocytes resulted in efficient pmel-1 TCR expression. Lymphocytes transduced with this retrovirus specifically recognized gp100-pulsed target cells as measured by interferon-γ secretion assays. Upon transfer into B16 tumor-bearing mice, the genetically engineered lymphocytes significantly slowed tumor development. The effectiveness of tumor treatment was directly correlated with the number of TCR-engineered T cells administered. These results demonstrated that TCR gene therapy targeting a native tumor antigen significantly delayed the growth of established tumors. When C57BL/6 lymphocytes were added to antigen-reactive pmel-1 T cells, a reduction in the ability of pmel-1 T cell to treat B16 melanomas was seen, suggesting that untransduced cells may be deleterious to TCR gene therapy. This model may be a powerful tool for evaluating future TCR gene transfer-based strategies.
Keywords: murine, gene therapy, T-cell receptor, gp100 tumor antigen
Cellular adoptive immunotherapy has been shown to mediate the regression of large solid tumors in 50% of patients with metastatic melanoma.1,2 A limitation of this treatment is the need to isolate and expand tumor-reactive lymphocytes that preexist in the patient. Those requirements can be overcome by transduction of genes encoding antitumor T-cell receptor (TCR), thus providing an alternative source of tumor-specific lymphocytes.3–7 We recently reported that this approach led to objective cancer regression in 2 patients with metastatic melanoma, a response rate of 13%.8
The pmel-1 mouse model was developed as a system to model treatment of malignant melanoma using adoptive cell therapy (ACT).9 The target antigen, pmel-17, is an ortholog of the melanocyte differentiation antigen gp100, which is often overexpressed in human melanomas. Adoptive transfer of transgenic T cells expressing the anti-gp100 TCR from pmel-1 mice can effectively mediate the regression of large established tumors when administered in combination with a lymphodepleting pretreatment regimen, γc cytokine administration, and vaccination.9–12 In this study, we further adapted the pmel-1 system to model TCR gene transfer therapy by retroviral vector-mediated gene transfer of the pmel-1 TCR genes into normal lymphocytes and the use of these genetically engineered cells to treat established B16 melanomas.
MATERIALS AND METHODS
Mice and Tumor Lines
C57BL/6 and pmel-1 transgenic mice (Jackson Laboratory, Bar Harbor, ME) were housed at the National Institutes of Health (NIH). The Phoenix Eco (ATCC, Rockville, MD) retroviral vector packaging line and the methylcholanthrene-induced sarcoma MCA-205 cell line (National Cancer Institute Tumor Repository, Bethesda, MD) were maintained in Iscove’s 5% fetal calf serum (PCS) and RPMI 10% PCS, respectively. B16 (H-2b), a spontaneous gp100+ murine melanoma, was maintained in RPMI 10% PCS.
Retrovirus Production
The pmel-1 TCR-α and TCR-β chain cDNA genes were inserted into vector pMX13 where the expression was linked by an IRES sequence to yield pMX-pmel TCR. Phoenix Eco cells/well (1.5 × 105) were placed in 6 well plates with 2-mL/well Iscove’s 5% PCS. FuGENE 6 transfection reagent (6.25 μL) (Roche, Indianapolis, IN) was added to 200 μL of serum-free Iscove’s medium for each well and incubated for 5 minutes at room temperature. In all, 2.5μg of pMX-pmel TCR retroviral vector or pMSGIN (GFP transgene) and 1.75 μg of pCL-Eco (Imgenex, San Diego, CA) were added to the FuGENE for each well, incubated for 15 minutes at room temperature and added to the Phoenix Eco cells in culture. The following day, the cell medium was removed and the transfected Phoenix Eco cells were incubated overnight in fresh prewarmed Iscove’s 5% FCS. The following day, the retroviral supernatant was collected from the transfected Phoenix Eco cells, spun for 10 minutes at 1000g and the supernatant removed. Recombinant human interleukin-2 (rhIL-2, Chiron, Emeryville, CA) of 60 IU/mL was added to the viral supernatant before transduction.
Retroviral Transduction
C57BL/6 spleens were harvested, macerated over a filter, and resuspended in ACK Lysing Buffer (Biosource, Rockville, MD). In all, 3 × 106 splenocytes per milliliter were placed in RPMI 10% with 1-ng/mL IL-7 and 2-μg/mL Concanavalin A (Calbiochem, La Jolla, CA), and incubated at 37°C. Two days later, the splenocytes were collected and where indicated, CD8+ cells were enriched using mouse CD8 Negative Isolation Kit (Dynal Biotech, Oslo, Norway). Splenocytes were spun for 7 minutes at 486g, resuspended in retroviral supernatant with rhIL-2 at 1 × 106cells/mL, plated on RetroNectin-coated plates (Takara Bio, Otsu, Japan), and spun at 1000g for 90 minutes at room temperature. The transduced cells were incubated at 37°C overnight. The second transductions were performed the following day using the same method. Transduced cells were in vitro stimulated with 1 μM of hgp10025–33-pulsed, irradiated C57BL/6 splenocytes 1 to 2 days after the final transduction, cultured as described,9,14 and used in adoptive cell transfer experiments 1 to 3 days later.
Adoptive Cell Transfer
C57BL/6 mice at 6 to 12 weeks of age were injected with 2×105 to 5 × 105 B16 melanoma cells and sublethally irradiated with 500 cGy 7 to 8 days later. The following day, groups of mice (n = 5) underwent tail vein injections of transduced C57BL/6 or pmel-1 splenocytes. Mice were vaccinated on the day of transfer with 2 × 107 PFU of recombinant fowlpox virus expressing human gp100 (rFPhgp100; Therion Biologics, Cambridge, MA) and 100,000 IU rhIL-2 administered intraperitoneally twice daily for 3 days. The perpendicular diameters of the tumors were subsequently measured with a caliper by a blinded investigator. The NCI Animal Ethics Committee of the NIH approved all animal experiments.
Flow Cytometry and TCR Activity Assay
All antibodies were purchased from BD Bioscience (San Jose, CA). The H2-Db-mgp10025–33 tetramer was purchased from Beckman Coulter (Fullerton, CA). FACSCalibur flow cytometer and CellQuest software were used to analyze the samples. Cytokine release assays and cytolytic assays were performed as previously described.15
RESULTS
Retroviral Vector Transduction of pmel-1 TCR
We previously described a murine model of human ACT based on the pmel-1 mouse.9 A bicistronic retroviral vector was constructed, which expressed the α and β chains of the anti-gp100 TCR expressed in pmel-1 T cells. The pmel-1 TCR retroviral vector was produced by transient transfection of an ecotropic packaging cell line and fresh supernatant was used to transduce normal T cells. One million fresh C57/B6 splenocytes were cultured for 2 days in ConA and IL-7 and then transduced with retrovirus encoding the pmel-1 TCR in the presence or absence of IL-2. At 24 hours posttransduction, both transduced cultures were analyzed by flow cytometry after staining for the pmel-1 TCR chain Vβ13. A representative example of the flow cytometry is shown in Figure 1A. The C57/B6 splenocyte population transduced in the absence of IL-2 contracted approximately 20% in cell number during the transduction and approximately 12% (as measured by Vβ13 staining) of the cells expressed the transgene. In comparison, C57/B6 splenocytes transduced in the presence of IL-2 demonstrated an approximate mean expansion of 1.8-fold and 29% of the cells expressed Vβ13 at 24 hours posttransduction. The increased cell proliferation in IL-2 containing cultures likely accounted for the higher transduction efficiency as retroviral vectors can only productively transduce actively dividing cells. From this point, all transductions were performed with the addition of IL-2 to the retroviral supernatant.
FIGURE 1.
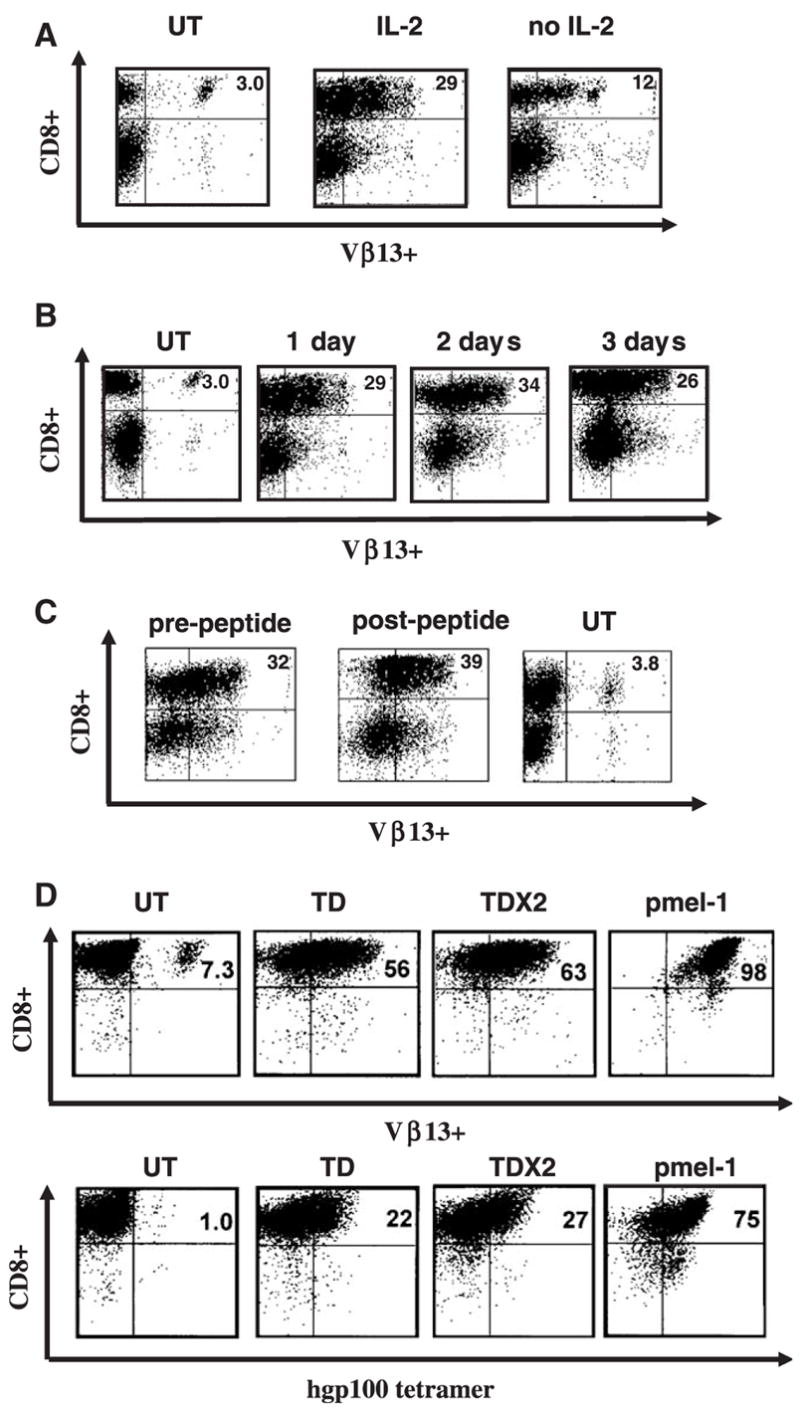
TCR transduction of mouse splenocytes. A, Murine C57BL/6 splenocytes were left untransduced (UT), transduced with retrovirus encoding the pmel-1 TCR in the presence or absence of IL-2 and 24 hours later were analyzed by flow cytometry for TCR Vβ13 staining. B, Following TCR transduction, samples of in vitro IL-2 cultured splenocytes were analyzed on 3 consecutive days for TCR gene expression (measured by Vβ13 staining). C, Transduced cells were stimulated with gp100-specific peptide 2 days posttransduction, and analyzed 3 days poststimulation for Vβ13 expression by FACS. D, After CD8+ enrichment through negative selection murine splenocytes were transduced with pmel-1 TCR vector (TD). Twenty-four hours later, a second retroviral TCR transduction was performed (TD × 2). The levels of Vβ13+ transgene expression and gp100 tetramer binding in comparison to pmel-1 T cells was evaluated by flow cytometry. Data shown are representative of 3 independent experiments.
Stable gene transfer efficiency of > 25% was consistently observed (with endogenous background staining of 1% to 5%) up to 3 days posttransduction (representative FACS shown in Fig. 1B). Two days posttransduction, cells were stimulated with gp100-specific peptide in an attempt to induce TCR-dependent proliferation. Three days poststimulation, cells were analyzed for Vβ13 expression by FACS. As shown in Figure 1C, peptide stimulation resulted in a little increase in the number of Vβ13+ cells (from 32% to 39%).
To increase the percentage of vector-transduced lymphocytes, we evaluated multiple transductions of CD8+ T-cells (Fig. 1D). C57/B6 splenocytes were placed in medium with ConA and IL-7 for 2 days, and the CD8+ population of splenocytes was negatively selected before transduction with the pmel-1 TCR retrovirus. At 24 hours posttransduction, the transduced CD8+ splenocytes were transduced a second time. Enrichment for CD8+ T cells before transduction increased the efficiency of gene transfer, yielding > 50% transduction as measured by Vβ13+ staining (Fig. 1D). The levels of Vβ13 + transgene expression and gp100 tetramer binding were only modestly increased with 2 transductions (Fig. 1D).
The biologic activity of the TCR-transduced C57/B6 cells was measured by IFN-γ secretion assay. C57/B6-transduced cell populations were cocultured with MCA-205 cells pulsed with serially diluted amounts of rhgp100 peptide (Fig. 2). After 24 hours, the supernatant was analyzed for the presence of IFN-γ by ELISA. C57/B6 splenocytes transduced with the pmel-1 TCR released IFN-γ in response to concentrations of gp100 peptide down to 1 nM, but were significantly less active than native pmel-1 T cells. The phenotypes of the C57/B6-transduced cells were analyzed for the presence of several cell surface markers associated with T-cell function: CD25, CD44, CD62L, and CD69 (data not shown). Transduction resulted in an increased level of the activation/effector marker CD25, whereas CD44 and CD69 did not demonstrate any observed change in expression after transduction or selection. The lymphoid-homing molecule, CD62L, was slightly down-regulated after transduction and ex vivo growth in IL-2. Overall, the transduction procedure resulted in cells with an intermediate effector phenotype.15
FIGURE 2.
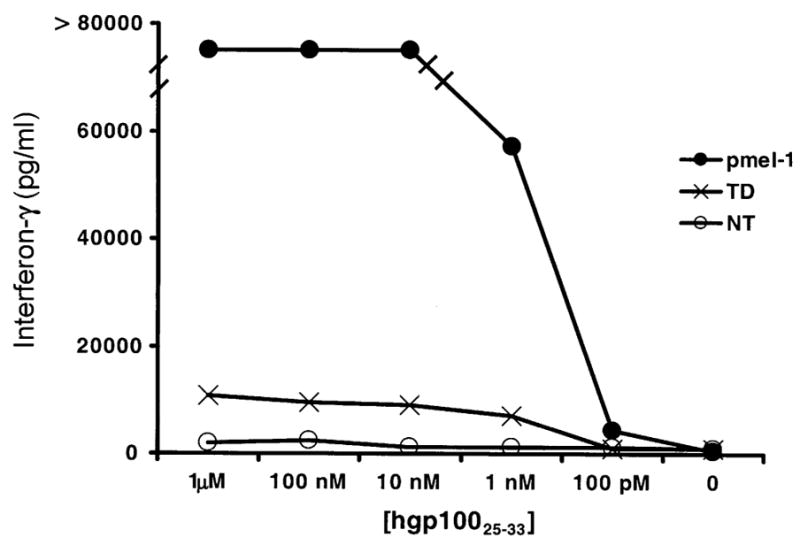
Function of transduced splenocytes. C57BL/6 splenocytes were transduced 1 time (TD) and co-cultured with hgp10025–33-pulsed MCA-205 cells for 24 hours. The amount of interferon-γ release was analyzed by ELISA. Untransduced (NT), and transgenic pmel-1 T cells (pmel) were used as negative and positive controls, respectively.
In Vivo Tumor Treatment
To test the in vivo efficacy of pmel-1 TCR-transduced PBL, we scaled up the gene transfer procedure to obtain large numbers of cells for multiple animal treatments. Seven to eight days before treatment, C57BL/6 mice were injected with 2 × 105 to 5 × 105 B16 melanoma cells and animals with well-established tumors were used for experimentation. The total number of transduced T cells administered was calculated and 1 × 106 or 7 × 106 gp100 tetramer + cells were transferred to 5 Gy irradiated, B16 tumor bearing C57/B6 mice with concurrent fowlpox virus expressing hgp100 vaccination and twice daily IL-2 administration for 3 days. Five days posttransfer, the peripheral blood of mice treated with pmel-1 TCR-transduced T cells was analyzed by flow cytometry. A representative example of the FACS data is shown (Fig. 3). The animals treated with higher amounts of transduced cells (7 × 106) had a larger percentage of Vβ13 + CD8+ cells in the peripheral blood than mice treated with 1 × 106 cells (63% vs. 46%), but this was less than that in mice that received 1 × 106 native pmel-1 cells (81% Vβ13 + CD8+).
FIGURE 3.
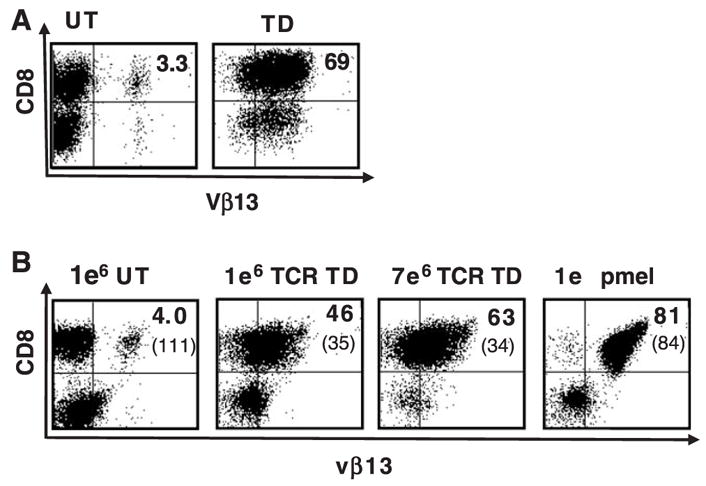
Persistence of TCR-transduced splenocytes after in vivo transfer. A, (In vitro) Murine C57BL/6 splenocytes were left untransduced (UT), or transduced (TD) with retrovirus encoding the pmel-1 TCR and 24 hours later analyzed by flow cytometry for TCR Vβ13 staining. B, (In vivo) The total number of CD8+gp100 tetramer+ transduced cells was calculated and 1 × 106 or 7 × 106 CD8+ gp 100 tetramer+ cells were transferred into 5 Gy irradiated, B16 tumor bearing C57BL/6 mice with concurrent fowlpox virus expressing hgp 100 vaccination and twice daily IL-2 administration for 3 days. Five days posttransfer, the peripheral blood of mice treated with pmel-1 TCR-transduced splenocytes was analyzed by flow cytometry. The mean fluorescence intensity of Vβ13 expression was as indicated in parentheses. A representative example of the FACS data is shown.
The tumor size following the transfer of pmel-1 TCR-transduced cells was measured 3 times weekly to evaluate response to treatment. The group of mice treated with 7 × 106 TCR-transduced CD8+ splenocytes demonstrated a statistically significant delay in tumor growth compared to those treated with the same number of GFP vector-transduced cells (P = 0.009) (Fig. 4A). Mice treated with 7 × 106 TCR-transduced cells achieved a greater delay in tumor growth compared to those treated with 1 × 106 TCR-transduced cells (P = 0.047). Similar results were obtained when the number of administered cells was calculated using Vβ13+ cells (Fig. 4B). Consistent delay in tumor growth was observed in 3 additional experiments where the infusion of higher numbers of cells (7 × 106 per animal) uniformly yielded a greater delay in tumor growth (data not shown).
FIGURE 4.
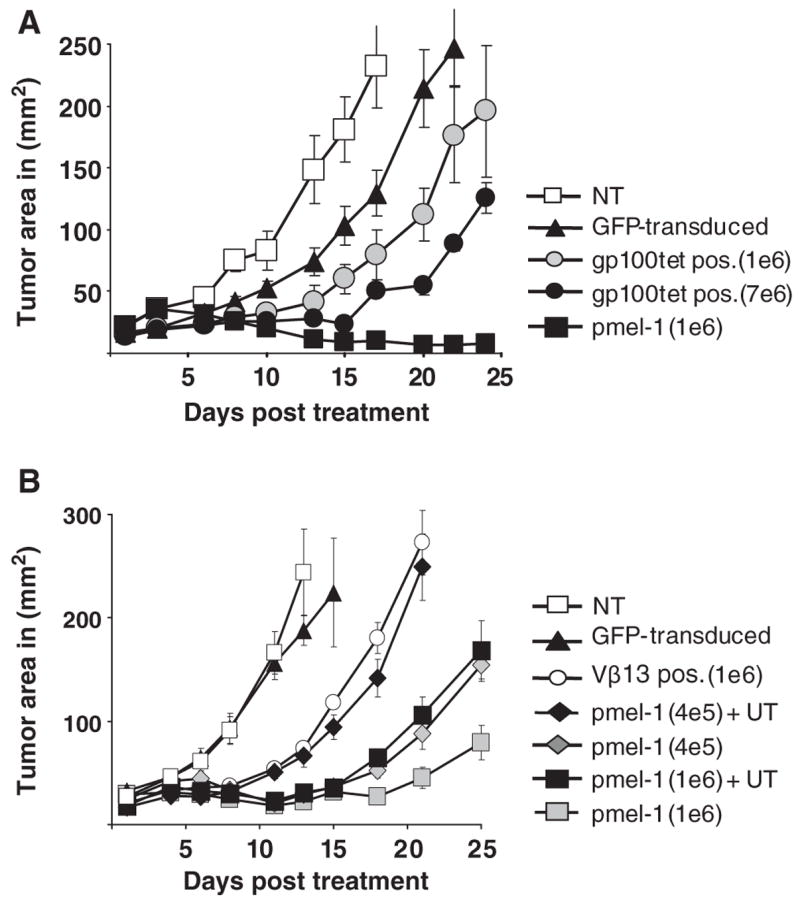
Delayed tumor growth following transfer of TCR-transduced splenocytes. A, C57BL/6 splenocytes were transduced and 1 × 106 or 7 × 106 CD8+ gp100 tetramer+ cells were transferred to B16 tumor bearing C57BL/6 mice. Animals received 5 Gy TBI with concurrent fowlpox virus expressing hgp100 vaccination and twice daily IL-2 administration for 3 days. As controls, animals were either left untreated (NT) or received GFP vector-transduced (GFP-transduced) splenocytes or transgenic pmel-1 T cells (pmel-1). Results of tumor area were the mean of measurement of 5 mice per group. The delay in tumor growth in mice that received 7 × 106 CD8+ gp100 tetramer+ cells versus the same number of GFP vector-transduced cells was statistically significant (P=0.009). B, A cell mixing experiment was performed to mimic the administration of 1 × 106 CD8+ Vβ13+-transduced cells. Using the same percentage of untransduced cells determined in the TCR-transduced population, untransduced C57BL/6 lymphocytes were added to 1 × 106 or 4 × 105 pmel-1 transgenic splenocytes (pmel-1+UT), and compared to pmel-1 without added untransduced cells (pmel-1). After 5 Gy TBI B16 tumor bearing C57BL/6 mice received cell transfer followed by fowlpox virus expressing hgp100 vaccination and twice daily IL-2 administration for 3 days. The addition of untransduced cells to pmel-1 splenocytes had a significant negative effect on tumor treatment (1 × 106 pmel-1, P=0.009 and with 4 × 105 pmel-1, P=0.014). Tumor area is presented as the mean of 5 mice per group.
After transduction, C57BL/6 lymphocytes contained a considerable percentage of untransduced cells, which could potentially inhibit the in vivo tumor treatment capacity. To investigate the inhibitory effect of untransduced cells, a cell mixing experiment using transgenic pmel-1 cells and normal lymphocytes was performed. Untransduced C57BL/6 CD8+ splenocytes were added to pmel-1 transgenic splenocytes to adjust for the total number of cells that were present when 1 × 106 (or 4 × 105) Vβ13+ CD8+-transduced cells were transferred (eg, 1 × 106 transgenic pmel-1 T-cells plus 2.5 × 106 C57BL/6 splenocytes were administered to mimic 1 × 106 Vβ13 + CD8+-transduced cells). The addition of transduced C57BL/6 CD8+ splenocytes to pmel-1 transgenic splenocytes resulted in a decrease in tumor response compared to pmel-1 transgenic cells without added splenocytes and was statistically significant (for 1 × 106 pmel-1, P = 0.009 and for 4 × 105, P = 0.014) (Fig. 4B).
On the basis of these observations, the positive selection of Vβ13 + CD8+ or gp100 tetramer positive cells might help to further increase the tumor treatment capacity. However, in our preliminary experiments we could not recover sufficient numbers of transduced CD8+ T cells after positive selection by Vβ13 or gp100 tetramer binding to perform animal experiments (usually performed using 5 animals per group). The reason for the low recovery might be the relative low expression of the retroviral transduced TCR compared to the endogenous pmel-1 TCR and/or the mispairing of the introduced TCR chains with the endogenous TCR chains.
DISCUSSION
In this paper, we demonstrated that retroviral transduction of murine splenocytes lead to the expression of the pmel-1 TCR receptor that recognized the native gp100 tumor antigen. Under optimal conditions, > 50% of normal mouse CD8+ T cells can be engineered to express the pmel-1 TCR (as measured by Vβ13 staining). When analyzed in parallel (eg, Fig. 1D), native pmel-1 T cells displayed 98% Vβ13+ cells and 75% of these bound tetramer, an indicator of specific TCR α and β chain pairing. In contrast, the transduced cells demonstrated a lower fraction of tetramer binding relative to Vβ13 + cells (less than 50% of Vβ13 + cells). The reduced fraction of tetramer positive cells in the transduced cell population was likely the result of random pairing between the introduced pmel-1 TCR and endogenous TCR subunits. This “mispairing” may lessen the overall biologic activity of engineered T cells as observed in Figure 2, where engineered cells released significantly less IFN-γ following coculture with peptide-pulsed target cells compared to the transgenic pmel-1 T cells. It is possible that mispairing may in part be overcome by appropriate protein engineering including codon optimization and manipulation of TCR constant regions to enhance specific chain pairing.16–20 Recently, the concept of “strong” or “dominant” TCRs has been proposed21 and thus, the choice of a naturally occurring “dominant” TCR may be a effective solution to TCR chain mispairing.
Significant dose-dependent retardation in B16 tumor growth was observed in adoptive cell transfer experiments using TCR-engineered cells. Considerable improvements in tumor treatment remain to be made in this gene therapy model and could include TCR protein engineering to increase affinity22,23 or chain pairing,17,19,20 increasing the amount and duration of vector expression,24 as well as, the potential use of hematopoietic stem cells to augment antitumor treatments.25 In addition to the molecular aspects of optimizing the gene transfer system used herein, the biology of ACT can also be manipulated to enhance antitumor treatment.26 The requirement for the stimulation of T cell growth by ConA and IL-7 is correlated with a change in cell surface markers similar to the natural shift in phenotype from a naive to a T cell with intermediate effector phenotype. Cells with this phenotype, although capable of mediating tumor regression, were demonstrated to be less active in mediating tumor regression than naive or early effector phenotype cells.15,26 It is possible that the use of lentiviral vectors may afford the ability to engineer minimally stimulated T cells,27 which retain more of a naive or early effector T-cell phenotype. Alternatively, TCR genes can be transferred into hematopoietic stem cells and these cells differentiated into naive T cells in vitro or in vivo.28,29
An unexpected observation in these studies was the significant reduction of tumor treatment when nonspecific bystander cells were added to the pmel-1 treatment cells (Fig. 4B). The transfer of antigen-specific T cells into a lymphopenic host results in homeostatic proliferation, and this can be inhibited by the cotransfer of an irrelevant population of T cells. This inhibition by bystander cells may reflect cytokine competition, the presence of inhibitory regulatory cells in the untransduced cell population, or the competition for antigen presenting cells.11,30,31 Enrichment for transduced cells before adoptive cell transfer was attempted with the current pmel-1 TCR vector but proved ineffective in yielding sufficient cell numbers. Our inability to purify TCR-engineered cells was possibly related to the lower level of protein expression of the transduced α and β chain genes. A lower lever of β chain expression was clearly observed in comparison to the native pmel-1 TCR as presented in Figure 3B, where the mean fluorescence intensity of Vβ13 was less than half of the endogenous Vβ13 protein. Further refinement in vector design to increase TCR expression or the addition of a coexpressed selectable marker gene may address this issue.
Finally, these observations extend and confirm our clinical experience demonstrating that antitumor antigen TCR gene transfer into mature lymphocytes when administered in combination with lymphodepletion, cytokine therapy, and vaccination can lead to the regression of large existing tumors.8 This murine adoptive immunotherapy model could be used in the future to further investigate and optimize in vivo the use of TCR encoding retroviruses and further reinforces the application of this approach for the treatment of cancer patients.
Acknowledgments
The authors would like to thank A. Mixon and S. Farid of the Flow Cytometry Unit (Surgery Branch of the NCI) for flow cytometry analyses.
Supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Financial Disclosure: All of the authors have declared there are no financial conflicts of interest related to this work.
References
- 1.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 4.Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol. 2002;2:512–519. doi: 10.1038/nri841. [DOI] [PubMed] [Google Scholar]
- 5.Hughes MS, Yu YY, Dudley ME, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA, Dudley ME, Yu YY, et al. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol. 2003;171:3287–3295. doi: 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Y, Zheng Z, Robbins PF, et al. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng R, Spolski R, Finkelstein SE, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. 2005;201:139–148. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klebanoff CA, Finkelstein SE, Surman DR, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Onishi M, Kinoshita S, Morikawa Y, et al. Applications of retrovirus-mediated expression cloning. Exp Hematol. 1996;24:324–329. [PubMed] [Google Scholar]
- 14.Palmer DC, Balasubramaniam S, Hanada K, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–7216. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jorritsma A, Gomez-Eerland R, Dokter M, et al. Selecting highly affine and well expressed TCRs for gene therapy of melanoma. Blood. 2007 doi: 10.1182/blood-2007-02-075010. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 17.Cohen CJ, Li YF, El-Gamil M, et al. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–3903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scholten KB, Kramer D, Kueter EW, et al. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin Immunol. 2006;119:135–145. doi: 10.1016/j.clim.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Cohen CJ, Zhao Y, Zheng Z, et al. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuball J, Dossett ML, Wolfl M, et al. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–2338. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sommermeyer D, Neudorfer J, Weinhold M, et al. Designer T cells by T cell receptor replacement. Eur J Immunol. 2006;36:3052–3059. doi: 10.1002/eji.200636539. [DOI] [PubMed] [Google Scholar]
- 22.Dunn SM, Rizkallah PJ, Baston E, et al. Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci. 2006;15:710–721. doi: 10.1110/ps.051936406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Moysey R, Molloy PE, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 24.Cooper LJ, Topp MS, Pinzon C, et al. Enhanced transgene expression in quiescent and activated human CD8+ T cells. Hum Gene Ther. 2004;15:648–658. doi: 10.1089/1043034041361217. [DOI] [PubMed] [Google Scholar]
- 25.Wrzesinski C, Paulos CM, Gattinoni L, et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J Clin Invest. 2007;117:492–501. doi: 10.1172/JCI30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gattinoni L, Powell DJ, Jr, Rosenberg SA, et al. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavalieri S, Cazzaniga S, Geuna M, et al. Human T lymphocytes transduced by lentiviral vectors in the absence of TCR activation maintain an intact immune competence. Blood. 2003;102:497–505. doi: 10.1182/blood-2003-01-0297. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y, Parkhurst MR, Zheng Z, et al. Extrathymic generation of tumor-specific T cells from genetically engineered human hematopoietic stem cells via Notch signaling. Cancer Res. 2007;67:2425–2429. doi: 10.1158/0008-5472.CAN-06-3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang L, Baltimore D. Long-term in vivo provision of antigen-specific T cell immunity by programming hematopoietic stem cells. Proc Natl Acad Sci USA. 2005;102:4518–4523. doi: 10.1073/pnas.0500600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kedl RM, Rees WA, Hildeman DA, et al. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med. 2000;192:1105–1113. doi: 10.1084/jem.192.8.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Antony PA, Piccirillo CA, Akpinarli A, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]