

Abstract
Phosphorylation is thought to be an essential first step in the prompt deactivation of photoexcited rhodopsin. In vitro, the phosphorylation can be catalyzed either by rhodopsin kinase (RK) or by protein kinase C (PKC). To investigate the specific role of RK, we inactivated both alleles of the RK gene in mice. This eliminated the light-dependent phosphorylation of rhodopsin and caused the single-photon response to become larger and longer lasting than normal. These results demonstrate that RK is required for normal rhodopsin deactivation. When the photon responses of RK−/− rods did finally turn off, they did so abruptly and stochastically, revealing a first-order backup mechanism for rhodopsin deactivation. The rod outer segments of RK−/− mice raised in 12-hr cyclic illumination were 50% shorter than those of normal (RK+/+) rods or rods from RK−/− mice raised in constant darkness. One day of constant light caused the rods in the RK−/− mouse retina to undergo apoptotic degeneration. Mice lacking RK provide a valuable model for the study of Oguchi disease, a human RK deficiency that causes congenital stationary night blindness.
Photoexcited rhodopsin must be deactivated in a timely manner for effective rod vision (1). This is because rhodopsin plays a catalytic role in activating the cyclic GMP (cGMP) cascade that transduces incident photons into macroscopic neural signals (2, 3). In its active state, rhodopsin promotes GTP/GDP exchange on the G protein transducin, which in turn activates a cGMP phosphodiesterase (PDE). Active PDE hydrolyzes cGMP, allowing cGMP-gated cationic channels at the surface membrane to close and hyperpolarize the cell. If rhodopsin is quenched too slowly, the temporal resolution of vision will suffer, whereas if quenching is too fast, sufficient amplification may not be achieved and photons may go undetected.
Biochemical experiments have suggested that rhodopsin deactivation is initiated by phosphorylation of rhodopsin, followed by the binding of arrestin (4). Rhodopsin phosphorylation occurs at one or more serine or threonine residues located in rhodopsin’s C-terminal domain (5). Transgenic mouse rods that expressed rhodopsin molecules lacking the C-terminal phosphorylation sites gave prolonged photoresponses, further supporting the idea that light-dependent rhodopsin phosphorylation is necessary for rhodopsin deactivation (6). However, in vitro experiments indicate that the phosphorylation of rhodopsin, like that of other G-protein-coupled receptors, can be catalyzed by either a substrate-regulated kinase, rhodopsin kinase (RK), or a second messenger-regulated kinase, protein kinase C (PKC) (7–9). Since both of these kinases are present in the rod outer segment (8, 10) and phosphorylate rhodopsin’s C-terminal residues equally well in vitro (11), it is unclear which kinase is mainly responsible for rhodopsin deactivation in vivo. We determined the role of RK in rhodopsin deactivation in intact rods by deactivating both alleles of the RK gene. We found that RK is required for the normal deactivation of rhodopsin and that in its absence, dramatic functional and structural changes occurred.
MATERIALS AND METHODS
Transgenic Mice.
RK knockout mice were generated by standard procedures as described (12). Transfected mouse embryonic stem cell clones were screened by PCR using a primer inside the MC1neopA cassette (neo-4) and a primer outside the 0.8-kb short arm (RK-ko1) (Fig. 1A). Positive clones were microinjected into blastocysts derived from C57/B6 to generate chimeric mice, which were then mated with C57/B6 to produce hemizygous knockouts (RK+/−). The homozygous knockouts (RK−/−) were produced by inbreeding of the RK+/− mice. All experimental procedures were done in compliance with National Institutes of Health guidelines as approved by the Institutional Animal Care and Use Committee of California Institute of Technology.
Figure 1.

Inactivation of the RK gene. (A) Genomic structure of RK gene, targeting vector, and altered RK locus. The targeting vector contained 10 kb (long arm) of the 3′ downstream and 0.8 kb (short arm) of the 5′ upstream homologous sequence. The MC1neopA cassette (hatched bars) replaced the 2.7-kb BamHI fragment that contained the exon bearing the translation initiating codon (filled box). Restriction sites: B, BamHI; A, ApaI; K, KpnI. (B) Immunoblot analysis of RK from mice at 1 month of age; the analysis used a polyclonal antibody (8585) raised against the first 50 amino acids of bovine RK. The RK content of the hemizygous (+/−) retina was approximately 50% of that of the RK+/+. No detectable RK was found in the RK−/− retina.
Immunoblots.
Proteins were detected by Western blotting using a single retinal homogenate at a dilution of 1/10, except for detecting rhodopsin, when a dilution of 1/800 to 1/3200 was used. Antibodies for detecting photoreceptor-specific proteins were gifts from colleagues: anti-RK (8585, R. Lefkowitz, Duke University, 1:3000 dilution), anti-RGS9 (T. Wensel, Baylor College of Medicine, 1:5000 dilution), anti-rhodopsin (1D4, R. Molday, University of British Columbia, 1:5000 dilution), anti-guanylyl cyclase activating protein-2 (GCAP-2) (A. Dizhoor, Wayne State University, 1:2000 dilution), anti-phosducin (Gerti; R. W. Lee, University of California at Los Angeles, 1:3000 dilution), and anti-arrestin (I. Gary, National Institutes of Health, 1:5000 dilution).
Rhodopsin Phosphorylation.
RK+/+ mice were dark-adapted overnight prior to measurements, and RK−/− mice were reared in the dark. Detailed experimental procedures were followed as previously described (13). Briefly, two retinas were dissected into 100 μl of buffered solution (130 mM NaCl/2.2 mM KCl/1.2 mM MgCl2/1.3 mM KH2PO4/5.4 mM Na2PO4/10 mM Hepes/1.3 mM CaCl2/10 mM glucose) in the dark and exposed for 1.5 s to white light (7.7 mW/cm2), which bleached about 12% of rhodopsin. Two minutes after the exposure the reaction was stopped, the membranes were washed, and rhodopsin C termini were cleaved with endoproteinase Asp-N (Boehringer Mannheim). The phosphorylation state of the C-terminal peptide of rhodopsin (DDDASATASKTETSQVAPA) was determined by liquid chromatography-coupled mass spectrometry (LC-MS).
Electrophysiology.
All mice were kept in 24-hr darkness, and the retinas were isolated under infrared light. Suction electrode recording procedures were the same as previously described (14). Briefly, the retina was chopped and small pieces were placed into the recording chamber, which was perfused with bicarbonate-buffered Locke’s solution [112.5 mM NaCl/3.6 mM KCl/2.4 mM MgCl2/1.2 mM CaCl2/10 mM Hepes, pH 7.4/0.02 mM EDTA/20 mM NaHCO3/3 mM disodium succinate/0.5 mM sodium glutamate/10 mM glucose and 0.1% vitamin and amino acids supplement solution (Sigma)] bubbled with 95% O2/5% CO2 and warmed to 34–37°C. The outer segments of single rods were drawn into a suction electrode connected to a current-measuring amplifier (Axopatch; Axon Instruments, Foster City, CA). The electrode contained 140 mM NaCl, 3.6 mM KCl 2.4 mM MgCl2, 1.2 mM CaCl2, 3 mM Hepes (pH 7.4), 0.02 mM EDTA, 10 mM glucose, and 0.1% vitamin and amino acid supplement (Sigma). The responses were low-pass filtered at 20 Hz with an 8-pole Bessel filter and digitized at 100 Hz using an igor-based acquisition program written by Fred Rieke (University of Washington, Seattle). Brief flashes (10 ms) of 500-nm light were used for stimulation. The intensity of the light source was calibrated with a silicon detector (UDT350; Graseby Optronics, Orlando, FL) and the flash strength was controlled with calibrated neutral density filters.
Morphology.
Eyecups were fixed overnight in 2.5% glutaraldehyde/2% paraformaldehyde in 0.1 M NaH2PO4, pH 7.4, and processed in Epon. Sections 1 μm thick from the central retina were stained with 0.5% methylene blue/0.5% azure II in 0.5% borax.
Cell Death Assay.
Eyecups were fixed overnight in 4% paraformaldehyde in phosphate-buffered saline, pH 7.4. They were then embedded and frozen in OCT (Sakura FineTek, Torrance, CA). Frozen sections 15 μm thick were cut and stained with a cell death detection kit (Boehringer Mannheim), following the manufacturer’s procedures. Genomic DNA was isolated by digesting the retina in digestion buffer (0.25 mg/ml proteinase K in 10 mM Tris, pH 7.5/10 mM EDTA/0.5% N-lauroylsarcosine) followed by extraction with phenol/chloroform. Samples (20 μg) of DNA were resolved by 1% agarose gel electrophoresis.
RESULTS
RK Knockout Mice.
To inactivate the RK gene, we isolated and sequenced the murine RK cDNA (GenBank accession number AF085240) by degenerate reverse transcription–PCR and used the cDNA to screen a mouse genomic library for the RK gene. The exon containing the translation initiation codon was deleted and replaced by homologous recombination with a neomycin-resistance marker (Fig. 1A). Embryonic stem cells from 129SvJ mice were used to host the deletion and were injected into blastocysts of C57/B6 mice (Charles River). Chimeric mice were mated with C57/B6 to produce the hemizygous knockout (RK+/−). Homozygous knockout (RK−/−) mice were produced by inbreeding of the RK+/− animals. Western blot analysis showed that retinas of RK+/− mice contained half of the normal amount of RK. No RK was detectable in RK−/− retinas (Fig. 1B).
Light-Dependent Rhodopsin Phosphorylation.
To determine whether protein kinase C or other cellular kinases might phosphorylate photoactivated rhodopsin, we measured the light-dependent phosphorylation of rhodopsin in RK−/− mice. In the dark-adapted state, 98% of the rhodopsin C termini were unphosphorylated and 2% were monophosphorylated in both RK+/+ and RK−/− retinas. In RK+/+ retinas, a bright flash caused 14.4% ± 0.6% (mean ± SD) of the rhodopsin C termini to become monophosphorylated, 7.8% ± 0.7% to become diphosphorylated, and 4.5% ± 2.6% to become triphosphorylated. In contrast, no increase in rhodopsin phosphorylation was observed in RK−/− retinas (Fig. 2). Thus, under these conditions, phosphorylation of photoexcited rhodopsin did not occur in the absence of RK.
Figure 2.

Absence of light-dependent phosphorylation of rhodopsin in RK−/− retina. Light caused about 43 phosphates to be incorporated per 100 rhodopsins in RK+/+ retinas; no light-dependent phosphorylation was observed in RK−/− retinas. The number of phosphates incorporated per 100 rhodopsins was calculated as follows: 1 × (% monophosphorylated rhodopsin) + 2 × (% di-phosphorylated rhodopsin) + 3 × (% triphosphorylated rhodopsin). Bars equal SD, n = 2 except for RK+/+ dark, where n = 3.
Flash Responses of Transgenic Rods.
To evaluate the effect of RK deletion on phototransduction, we recorded membrane currents from single rods of transgenic and control mice that were born and raised in darkness. The flash responses of RK−/− rods were greatly prolonged, suggesting slowed rhodopsin deactivation (Fig. 3A). In RK−/− rods, the single-photon response (15) consisted of a long plateau (Fig. 3B), about 2-fold larger in amplitude than the peak of the control response (Table 1). The prolonged response decayed suddenly and rapidly at widely variable times. The distribution of response durations was fitted by a single-exponential function with a time constant of about 3 s (Fig. 3C), which suggests that in the absence of RK, rhodopsin was deactivated by a memoryless, first-order process. Responses of RK+/− rods to dim flashes recovered slightly more slowly than those of control rods (Fig. 3B), and the time spent in saturation in response to a bright flash was longer than that of RK+/+ (Fig. 3A Inset) at all flash strengths examined (data not shown). This finding suggests that deactivation of rhodopsin was slightly slowed when RK expression was halved. The dark current of all three lines was similar (Table 1), suggesting that RK depletion did not affect the cGMP-gated channels, guanylyl cyclase, or basal PDE activity.
Figure 3.

Flash responses from rods lacking rhodopsin kinase. (A) Saturating responses from RK+/+ and RK+/− rods (Inset), and an RK−/− rod to a flash that elicited about 300 photoisomerizations in each case. Unlike the +/+ and +/− responses (see Inset), the RK−/− response was greatly prolonged, and it recovered in a series of stepped transitions that represent the deactivation and occasional reactivation of individual rhodopsin molecules. The dark currents of the three cells were 15.0 pA (+/+), 16.7 pA (+/−), and 16.6 pA (−/−). Flashes were delivered at t = 0. (B) Form of the single photon response from rods of each of the three lines. The RK−/− trace is the average of 14 responses; the RK+/− trace is the average of 80 responses; and the RK+/+ trace is the average of 57 responses. The dark current of the three cells was 13.8, 18.9 and 11.3 pA, respectively. (C) Distribution of the durations of 198 RK−/− quantal events measured at half-maximal amplitude from three RK−/− cells. The smooth curve is an exponential function with a time constant of 3.3 s. The Inset is an example of a quantal event from an RK−/− rod whose dark current was 7.0 pA. (D) Dependence of the rate of change of PDE activity on the number of flash-induced photoisomerizations in RK+/+ and −/− rods. Slope of the linear regression is 0.99.
Table 1.
Characteristics of dim flash responses from RK-deficient and normal rods
| Strain* | Single-photon response amplitude, pA/photoisomerization | Time to peak,† ms | Integration time, ms | Dark current, pA |
|---|---|---|---|---|
| RK +/+ | 0.527 ± 0.022 | 109 ± 2 | 213 ± 14 | 12.5 ± 3.4 |
| RK +/− | 0.574 ± 0.028 | 115 ± 4 | 329 ± 11 | 14.1 ± 0.5 |
| RK −/− | 0.910 ± 0.024 | 337 ± 7 | 7060 ± 17 | 10.8 ± 0.2 |
All values are mean ± SEM.
Numbers of rods examined: RK+/+, 11; RK+/−, 11; RK−/−, 19.
For RK−/−, time to reach the plateau amplitude.
Retinal Morphology and Light-Induced Photoreceptor Death.
How does the retina tolerate the prolonged photoresponses? In RK−/− retinas, gross morphological changes were confined to the photoreceptor layer and depended strongly on the history of light exposure. In retinas of RK−/− mice born and raised in darkness, the morphology of the photoreceptor was indistinguishable from that in RK+/+ retinas. However, in retinas of RK−/− mice born and raised in cyclic illumination (12 hr of 50 lux, followed by 12 hr of darkness) the rod outer segments were 50% shorter than normal at 6 weeks of age (Fig. 4A). Less shortening was evident in outer segments of RK−/− mice transferred from cyclic illumination to complete darkness (data not shown). Severe degeneration was observed when dark-raised RK−/− mice were kept in continuous room light (approximately 450 lux) for 24 hr, whereas the retinas of wild-type mice showed no defects after this exposure. The light-induced degeneration of dark-raised RK−/− retinas was apparently the result of apoptosis, as evidenced by positive terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) (Fig. 4B) and characteristic DNA degradation (Fig. 4C).
Figure 4.

Altered morphology of RK−/− retina. (A) Morphology of the central retina of cyclic-light reared and dark-reared mouse retinas at 6 weeks of age. (1) Control (+/+), raised in cyclic illumination (50 lux). (2) Hemizygous knockout (+/−), raised in cyclic illumination (50 lux). (3) Homozygous knockout (−/−), born and raised in the dark. (4) Homozygous knockout (−/−), reared in cyclic illumination (50 lux). The outer segments in 4 lacked the characteristic orderly arrangement seen in 1, 2, and 3 and were approximately 50% shorter. (Scale bar = 16 μm.) (B) Light-induced photoreceptor apoptosis of dark-reared RK−/− mice after 24 hr in continuous room light (450 lux). (Upper) Results of TUNEL staining photographed in the presence of dim background light. Positive nuclei were in the outer nuclear layer. (Scale bar = 100 μm.) (Lower) Corresponding morphological changes shown in Upper. (Scale bar = 16 μm.) (C) DNA degradation. Lane 1, control (+/+); lane 2, RK−/− after 24 hr of continuous illumination; lane 3, RK−/− after 48 hr of continuous illumination. DNA size markers are shown in base pairs as indicated at right. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Expression of Other Photoreceptor Proteins.
In addition to changing the rod morphology, light exposure also changed the expression level of several phototransduction proteins. Whereas the expressions of rhodopsin, transducin, arrestin, recoverin, GCAP-2, phosducin, and RGS-9 in the retina were similar in dark-raised RK−/− and +/+ mice as judged by Western blot analysis, RK−/− mice raised in cyclic illumination showed a 2- to 4-fold reduction in rhodopsin, transducin, RGS-9, and GCAP-2 levels, as one might expect from the shortened outer segments. The reduction in opsin level was not accompanied by a reduction in steady-state levels of opsin mRNA (data not shown), suggesting that transcription of the rhodopsin gene was not affected. In contrast, the expression of arrestin, recoverin, and phosducin was indistinguishable from normal, despite the shortened outer segments. The relationship between the shortening of the outer segments and the relative rates of transport, synthesis, and degradation of these proteins remains to be determined.
DISCUSSION
The amplitude, shape, and duration of the single-photon responses of RK−/− rods were similar to those of the anomalous photon responses in transgenic mouse rods that expressed truncated rhodopsin molecules lacking the C-terminal phosphorylation sites (S334ter) (6). The results from both studies are consistent with the idea that the fundamental defect is a lack of light-dependent rhodopsin phosphorylation. These data suggest that RK is solely responsible for normal rhodopsin deactivation in the dark-adapted rod. The physiological role of protein kinase C-catalyzed rhodopsin phosphorylation, if any (16), remains to be determined.
In the absence of RK, the single-photon response turned off abruptly, at times that varied widely in different trials (Fig. 3C). The average response duration of about 3 s is much shorter than the time required for the thermal decay of metarhodopsin II, the catalytically active form of rhodopsin (17, 18). The exponential distribution of response durations (Fig. 3C) suggests that this back-up mechanism is a memoryless, first-order process. The stochastic nature of the RK−/− single-photon responses contrasts strongly with the highly reproducible nature of the normal single-photon response (19). This fact indicates that rhodopsin phosphorylation by RK is an essential requirement for reproducibility. The molecular basis of the stochastic backup mechanism remains to be determined.
We compared the rising phases of RK+/+ and −/− responses to determine when RK normally begins to quench rhodopsin’s catalytic activity. The initial gain of the phototransduction cascade was determined by the rate of change of the PDE activity (dPDE*/dt) at early times after the flash (20):
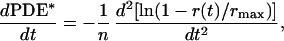 |
where r(t) is the time course of the flash response, rmax is the maximal response amplitude, and n is the cooperativity of the cGMP-gated channels, which we took to be 3 (21, 22). The initial rate of change of the PDE activity after the flash was the same in RK+/+ and RK−/− rods (Fig. 3D). This finding provides further evidence that the amplifying proteins in the phototransduction cascade were unaffected by the absence of RK. However, in RK−/− rods the responses continued to rise for a longer time, diverging from RK+/+ responses at about 100 ms after the flash (Fig. 3B). This suggests that RK binding and/or phosphorylation normally begins to reduce rhodopsin’s activity around this time.
The susceptibility to damage by dim light is unusual for retinas of pigmented mice. However, other transgenic mice with prolonged photoresponses, such as those lacking arrestin, also show light-dependent morphological abnormalities (23). Thus, a plausible hypothesis is that prolonged activation of the phototransduction cascade induces outer segment shortening (24) and eventual photoreceptor death (25, 26). If so, such light effects should be suppressed by deletion of protein(s) responsible for activation of the cascade. This idea is currently being tested by crossing RK−/− mice with animals that have deletions in other components of the phototransduction cascade (27).
In humans, mutations in both copies of the RK (28) or arrestin gene (29, 30) cause Oguchi disease, a stationary form of congenital night blindness in which there is a prolonged insensitivity of rod vision after light exposure. Electroretinograms (ERGs) of Oguchi patients lacking functional RK exhibit slowed recovery kinetics (31), consistent with the prolonged active lifetime of rhodopsin that we observed in the RK−/− mice. These patients also do not adapt normally to background light. Our results suggest that this failure may occur because the longer time integral of the single-photon response in RK−/− rods (Table 1) increases the sensitivity to steady background light, thus causing the rod’s response to saturate at abnormally low light intensities (data not shown). Mice lacking RK provide a valuable model for human Oguchi disease. In addition, they may prove useful for studying the mechanism of photoreceptor apoptosis that occurs in various retinal degenerations.
Acknowledgments
We thank members of the Caltech Transgenic Core Facility for their technical support. We also thank Wolfgang Baehr for the mouse retinal cDNA library. This work was supported by grants from the National Institute of Aging (M.I.S.), the National Eye Institute (D.A.B. and J.B.H.), the McKnight Foundation (D.A.B.), and the Ruth and Milton Steinbach fund (D.A.B.).
ABBREVIATIONS
- RK
rhodopsin kinase
- PDE
phosphodiesterase
- GCAP
guanylyl cyclase activating protein
- TUNEL
terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF085240).
References
- 1.Baylor D A, Burns M E. Eye. 1998;12:521–525. doi: 10.1038/eye.1998.140. [DOI] [PubMed] [Google Scholar]
- 2.Scott K, Zuker C. Trends Biochem Sci. 1997;22:350–354. doi: 10.1016/s0968-0004(97)01100-6. [DOI] [PubMed] [Google Scholar]
- 3.Palczewski K, Saari J C. Curr Opin Neurobiol. 1997;7:500–504. doi: 10.1016/s0959-4388(97)80029-3. [DOI] [PubMed] [Google Scholar]
- 4.Wilden U, Hall S W, Kuhn H. Proc Natl Acad Sci USA. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohguro H, Palczewski K, Ericsson L H, Walsh K A, Johnson R S. Biochemistry. 1993;32:5718–5724. doi: 10.1021/bi00072a030. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Makino C L, Peachey N S, Baylor D A, Simon M I. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- 7.Lorenz W, Inglese J, Palczewski K, Onorato J J, Caron M G, Lefkowitz R J. Proc Natl Acad Sci USA. 1991;88:8715–8719. doi: 10.1073/pnas.88.19.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams D S, Liu X, Schlamp C L, Ondek B, Jaken S, Newton A C. J Neurochem. 1997;69:1693–1702. doi: 10.1046/j.1471-4159.1997.69041693.x. [DOI] [PubMed] [Google Scholar]
- 9.Greene N M, Williams D S, Newton A C. J Biol Chem. 1997;272:10341–10344. doi: 10.1074/jbc.272.16.10341. [DOI] [PubMed] [Google Scholar]
- 10.Kelleher D J, Johnson G L. J Biol Chem. 1990;265:2632–2639. [PubMed] [Google Scholar]
- 11.Udovichenko I P, Newton A C, Williams D S. J Biol Chem. 1997;272:7952–7959. doi: 10.1074/jbc.272.12.7952. [DOI] [PubMed] [Google Scholar]
- 12.Ramirez-Solis R, Davis A C, Bradley A. Methods Enzymol. 1993;225:855–878. doi: 10.1016/0076-6879(93)25054-6. [DOI] [PubMed] [Google Scholar]
- 13.Hurley J B, Spencer M, Niemi G A. Vision Res. 1998;38:1341–1352. doi: 10.1016/s0042-6989(97)00459-8. [DOI] [PubMed] [Google Scholar]
- 14.Tsang S H, Burns M E, Calvert P D, Gouras P, Baylor D A, Goff S P, Arshavsky V Y. Science. 1998;282:117–121. doi: 10.1126/science.282.5386.117. [DOI] [PubMed] [Google Scholar]
- 15.Baylor D A, Lamb T D, Yau K-W. J Physiol (London) 1979;288:613–634. [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong W, Nakatani K, Ye B, Yau K. J Gen Physiol. 1997;110:441–452. doi: 10.1085/jgp.110.4.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebrey T G. Vision Res. 1968;8:965–982. doi: 10.1016/0042-6989(68)90071-0. [DOI] [PubMed] [Google Scholar]
- 18.Cone R A, Cobbs W H. Nature (London) 1969;221:820–822. doi: 10.1038/221820a0. [DOI] [PubMed] [Google Scholar]
- 19.Rieke F, Baylor D A. Biophys J. 1998;75:1836–1857. doi: 10.1016/S0006-3495(98)77625-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pugh E N, Lamb T D. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- 21.Zimmerman A L, Baylor D A. Nature (London) 1986;321:70–72. doi: 10.1038/321070a0. [DOI] [PubMed] [Google Scholar]
- 22.Haynes L W, Kay A R, Yau K–W. Nature (London) 1986;321:66–70. doi: 10.1038/321066a0. [DOI] [PubMed] [Google Scholar]
- 23.Makino C L, Flannery J G, Chen J, Dodd R L. In: Photostasis and Related Phenomena. Williams T P, Thistle A B, editors. New York: Plenum; 1998. pp. 129–151. [Google Scholar]
- 24.Penn J S, Williams T P. Exp Eye Res. 1986;43:915–928. doi: 10.1016/0014-4835(86)90070-9. [DOI] [PubMed] [Google Scholar]
- 25.Lisman J E, Fain G L. Nat Med. 1995;1:1254–1255. doi: 10.1038/nm1295-1254. [DOI] [PubMed] [Google Scholar]
- 26.Fain G L, Lisman J E. Exp Eye Res. 1993;57:335–340. doi: 10.1006/exer.1993.1132. [DOI] [PubMed] [Google Scholar]
- 27.Lem J, Sidman R, Kosaras B, Calvert P, Makino C, Nicolo M, Margolskee R, Wong G, Gannon K, Krasnoperova N. Invest Ophthalmol Visual Sci. 1998;39:2995. (abstr.). [Google Scholar]
- 28.Yamamoto S, Sippel K C, Berson E L, Dryja T P. Nat Genet. 1997;15:175–178. doi: 10.1038/ng0297-175. [DOI] [PubMed] [Google Scholar]
- 29.Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A. Nat Genet. 1995;10:360–362. doi: 10.1038/ng0795-360. [DOI] [PubMed] [Google Scholar]
- 30.Maw M, Kumaramanickavel G, Kar B, John S, Bridges R, Denton M. Hum Mutat Suppl. 1998;1:S317–S319. doi: 10.1002/humu.1380110199. [DOI] [PubMed] [Google Scholar]
- 31.Cideciyan A V, Zhao X, Nielsen L, Khani S C, Jacobson S G, Palczewski K. Proc Natl Acad Sci USA. 1998;95:328–333. doi: 10.1073/pnas.95.1.328. [DOI] [PMC free article] [PubMed] [Google Scholar]