

Abstract
Despite mounting genetic evidence implicating a recent origin of modern humans, the elucidation of early migratory gene-flow episodes remains incomplete. Geographic distribution of haplotypes may show traces of ancestral migrations. However, such evolutionary signatures can be erased easily by recombination and mutational perturbations. A 565-bp chromosome 21 region near the MX1 gene, which contains nine sites frequently polymorphic in human populations, has been found. It is unaffected by recombination and recurrent mutation and thus reflects only migratory history, genetic drift, and possibly selection. Geographic distribution of contemporary haplotypes implies distinctive prehistoric human migrations: one to Oceania, one to Asia and subsequently to America, and a third one predominantly to Europe. The findings with chromosome 21 are confirmed by independent evidence from a Y chromosome phylogeny. Loci of this type will help to decipher the evolutionary history of modern humans.
Paleoanthropologists have proposed two extreme versions of the origin of modern humans (Homo sapiens sapiens) from Homo erectus; one has multiple origins, one in almost every continent (1, 2), whereas the other has a recent origin in Africa, followed by rapid spread to all other continents (3, 4). The first hypothesis is based on morphological continuity in the fossil record; however, this hypothesis has been contested after a reexamination of the data (5). The second hypothesis is in agreement with almost all genetic data (6–12) obtained from mtDNA, the Y chromosome, and autosomes. However, the number of migrations from Africa is still unknown. A single emergence from Africa has been suggested, judging from haplotypes associated with the CD4 locus on chromosome 12 (6), whereas results from a study of the major-histocompatibility-complex locus imply a more intricate history (13). Recombination and recurrent mutation may impede the reliable inference of intact progenitor haplotypes, causing difficulties in tracing ancestral events. Only the comparison of multiple haplotype systems that contain preferentially densely spaced single-nucleotide polymorphisms, which have low mutation rates and essentially no recombination, will resolve this issue, which is significant in regard to the use of linkage disequilibrium for gene localization.
MATERIALS AND METHODS
DNA Samples.
The populations studied are Pygmy (Biaka and Mbuti), Bantu (Lisongo), Sudanese, South African, Central and South American (Mayan, Surui, and Karitiana), Chinese, Japanese, Taiwanese (Ami, Yami, and Atayal), Cambodian, Tibetan, Northern and Central European, Italian, Basque, Indo-European-speaking Pakistani (Pathan, Sindhi, and Baloochi), Brahui, Tamil (speaking Dravidian languages), Hunza (speaking Burushaski, a language isolate), Australian Aborigines, New Guinean highlanders, and Melanesians from Bougainville (total of 354 individuals). The numbers of chromosomes studied for each population are given in Table 1.
Table 1.
Global MX1 haplotype distribution
| Population | Haplotype
|
No. of chromosomes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ht1 | Ht2 | Ht3 | Ht4 | Ht5 | Ht6 | Ht7 | Ht8 | Ht9 | Ht10 | ||
| Africa | |||||||||||
| Pygmy | 5 | 3 | 0 | 0 | 21 | 16 | 20 | 4 | 0 | 3 | 72 |
| Bantu | 1 | 0 | 0 | 0 | 0 | 3 | 4 | 0 | 0 | 0 | 8 |
| Sudan | 3 | 0 | 0 | 0 | 1 | 0 | 5 | 3 | 0 | 0 | 12 |
| South Africa | 0 | 0 | 0 | 0 | 0 | 4 | 4 | 2 | 0 | 0 | 10 |
| Europe | |||||||||||
| North Europe | 0 | 4 | 0 | 0 | 0 | 0 | 8 | 8 | 0 | 0 | 20 |
| Italy | 0 | 3 | 0 | 0 | 1 | 0 | 25 | 20 | 1 | 0 | 50 |
| Basque | 4 | 3 | 0 | 0 | 0 | 0 | 30 | 29 | 0 | 0 | 66 |
| East Austria | 0 | 7 | 0 | 0 | 2 | 0 | 25 | 22 | 0 | 0 | 56 |
| Pakistan and India | |||||||||||
| Indo-European speakers | 0 | 2 | 0 | 0 | 2 | 0 | 11 | 19 | 0 | 0 | 34 |
| Hunza | 0 | 2 | 0 | 0 | 2 | 0 | 10 | 10 | 0 | 0 | 24 |
| Brahui | 0 | 1 | 0 | 0 | 2 | 0 | 6 | 5 | 0 | 0 | 14 |
| Tamil | 1 | 1 | 0 | 0 | 3 | 0 | 3 | 0 | 0 | 0 | 8 |
| East Asia | |||||||||||
| China | 0 | 1 | 0 | 0 | 9 | 0 | 18 | 0 | 0 | 0 | 28 |
| Japan | 0 | 1 | 0 | 0 | 12 | 3 | 8 | 0 | 0 | 0 | 24 |
| Taiwan | 0 | 1 | 0 | 0 | 29 | 7 | 13 | 0 | 0 | 0 | 50 |
| Cambodia | 0 | 0 | 0 | 0 | 8 | 1 | 3 | 0 | 0 | 0 | 12 |
| Tibet | 0 | 0 | 0 | 0 | 1 | 0 | 3 | 0 | 0 | 0 | 4 |
| America | |||||||||||
| Central and South | 0 | 14 | 0 | 0 | 78 | 0 | 2 | 26 | 0 | 0 | 120 |
| Oceania | |||||||||||
| Australia | 1 | 6 | 7 | 0 | 10 | 0 | 6 | 4 | 0 | 0 | 34 |
| New Guinea | 1 | 4 | 9 | 2 | 17 | 0 | 9 | 0 | 0 | 0 | 42 |
| Bougainville | 0 | 1 | 0 | 0 | 7 | 0 | 2 | 10 | 0 | 0 | 20 |
| Total | 16 | 54 | 16 | 2 | 205 | 34 | 215 | 162 | 1 | 3 | 708 |
The designation of haplotypes follows the descriptions in Fig. 1.
PCR.
A 248-bp fragment containing eight polymorphic sites in humans was amplified with primers 5′-TGAGCACTACTCTACCATGG and 5′-GGAGACGTTTTCACCATTAC. The ninth human site occurred at position 146 in a 227-bp fragment that was amplified with primers 5′-AGCCTGTCCTGTTGGAGAGG and 5′-CCACAAACTCTCTCCCTGCC. This site was genotyped by restriction analysis with StuI enzyme, yielding fragments 148 and 79 bp in length, respectively. Amplicons were obtained by means of a touchdown PCR regime.
Denaturing High-Performance Liquid Chromatography (DHPLC) Analysis.
DHPLC was conducted on an automated HPLC instrument (Rainin Instruments). The stationary phase consisted of 2-μm alkylated poly(styrene-divinylbenzene) particles (DNASep, Transgenomics, Santa Clara, CA). The mobile phase was 0.1 M triethylammonium acetate buffer, pH 7.0 (PE Applied Biosystems, Foster City, CA), containing 0.1 mM Na4 EDTA (Sigma). Elution of PCR products was accomplished with an acetonitrile gradient of 0.45% per min. The principle and detailed procedures of DHPLC have been reported elsewhere (7, 14, 15).
Sequencing.
Sequencing was used to genotype the cluster of eight polymorphisms found in the first amplicon. PCR products were purified by solid-phase extraction (QIAquick, QIAGEN, Chatsworth, CA) and sequenced with the Perkin–Elmer Applied Biosystems Dye Terminator Cycle Sequencing Kit and a model 373A DNA sequencer.
Haplotype Inference.
Haplotypes for each individual were inferred by a maximum-parsimony approach. Polymorphic sites from a group of haplotypes or sequences are considered congruent if they can be accommodated by the same phylogenetic topology. Any incongruence between loci indicates recombination or recurrent mutation. In the construction of the tree by maximum parsimony, haplotypes of each individual with multiple polymorphisms were inferred, assuming the least number of mutational steps. This process was made possible by an efficient algorithm developed to detect the possible presence of recombination and recurrent mutation and then by inferring haplotypes of each individual (L.J., unpublished work). Subsequently, the validity of the inferences was confirmed empirically by cloning representative PCR amplicons and sequencing transformants.
Data Analysis.
The log-likelihood ratio test (G test) was employed to test the statistical significance of the nonrandom distribution of chromosome 21 haplotypes. The extent of genetic variation was determined by FST analysis. Entropy was computed as
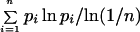 |
where n refers to the number of haplotypes or haplogroups, and pi is the frequency of haplotypes or haplogroups. The similarities between chromosome 21 haplotypes and Y chromosome haplogroups were tested by a measure of correlation suitable for relative-frequency data (16, 17): the quantity
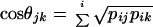 |
where the sum is extended to the six continental regions studied, pijand pik are the relative frequencies of the jth haplotype of chromosome 21 and of the kth Y chromosome haplogroup in the ith continental region.
RESULTS AND DISCUSSION
Discovery of Single-Nucleotide Polymorphisms.
The polymorphic segment was detected while assessing autosomal nucleotide-substitution variation by DHPLC. The genetic variation of a 62-kb segment in chromosome 21 was studied in a panel of 10 DNA samples formed by two individuals from each of the five continents. Overlapping segments of 300–500 bp in length were amplified by PCR, and each amplicon was subjected to a final denaturation and reannealing step before DHPLC analysis. Double- or multiple-peak patterns in a chromatogram indicated the presence of polymorphic site(s). The nucleotide nature of each polymorphism was identified by dye-terminator sequencing.
One segment, 5 kb downstream of the MX1 gene (GenBank accession no. L35674), was found to contain 11 substitution sites clustered within a 94-bp interval. Of these positions, three are polymorphic in primates but monomorphic in humans. The positions of the eight human polymorphic sites are given with the codes of the International Union of Biochemistry for base substitutions (in bold) as 5′-GTCWTGCAATCCCATTTGCAGGATCYGTCTGTGCACRTGCCTCYGTAGAGAGCRGCATTCCCAGGGACCWTGGAAACAGTTGRCACTGTAAGGTGCYTGC-3′.
Haplotype Determination.
The genotypes, determined from sequence data of 354 human individuals, could be arranged into nine haplotypes. The inclusion of a nearby A → T transversion located 485 bp upstream provided a 10th haplotype. The inference of haplotypes was confirmed empirically by cloning representative PCR amplicons and sequencing transformants. The phylogeny of the 10 haplotypes (labeled Ht1–Ht10) is presented in Fig. 1. Arrows indicate the direction of mutations. The hollow letters indicate the type and position of the 11 or 12 mutations postulated (it is not clear whether the difference between chimpanzees and humans involving two adjacent nucleotide substitutions is caused by one or two mutational events). All other mutations are single-nucleotide substitutions. The 10 human haplotypes can be classified further, based on phylogenetic structure: Ht1, Ht2, haplotype group A (Ht3 and Ht4), haplotype group B (Ht5 and Ht6), and haplotype group C (Ht7, Ht8, Ht9, and Ht10). Ht1 is apparently the oldest haplotype in human populations, whereas the latter four (i.e., Ht2 and groups A, B, and C) were derived individually from Ht1.
Figure 1.

Phylogenetic relationship among haplotypes. The arrows indicate the direction of the mutations (hollow letters). The phylogeny includes all equally parsimonious topologies by allowing multifurcation. All sites are congruent to each other, indicating the lack of recombination and recurrent mutation events within the 565-bp fragment encompassing 12 polymorphic sites in hominoids. Chimpanzee and gorilla sequences were used to determine the root of the phylogeny as well as the ancestral state of the sequences.
Geographic Distribution of Haplotypes.
Haplotype distribution in globally dispersed populations is given in Table 1. The majority of humans are parceled into 6 haplotypes (Ht1, Ht2, Ht5, Ht6, Ht7, and Ht8); 4 additional haplotypes, namely Ht3, Ht4, Ht9, and Ht10, are rare. Overall diversity is greatest in Africans, in whom seven haplotypes are represented. This diversity is consistent with many other observations leading to the hypothesis of an African origin of modern humans. Of the 10 haplotypes, 4 (Ht7, Ht5, Ht8, and Ht2) are present in all continents, with world frequencies of 30%, 29%, 23%, and 8%, respectively. All are likely to have originated in Africa before the great diaspora of H. sapiens sapiens. Only three rare haplotypes (Ht3, Ht4, and Ht9) were not found in Africa; the first two were exclusively detected in Oceania, and the third in Europe. It is likely that the mutations leading to them occurred in the continents where the mutants were found.
The distribution of the haplotypes is highly heterogeneous (G = 546.56; df = 45, where df is degree of freedom). Further, the 10 haplotypes tend to group themselves as follows: Ht1–Ht4, Ht5–Ht6, and Ht7–Ht10. The variation of the frequencies of these three haplogroups in the six continental regions is highly significant (with their degrees of freedom given in parentheses). Between haplogroups (Ht1–Ht4, Ht5 and Ht6, and Ht7–Ht10), G = 293.30 (df = 10). For Ht1–Ht4 vs. the rest, G = 42.44 (df = 5). For Ht5 and Ht6 vs. Ht7–Ht10, G = 250.90 (df = 5). All G values have probabilities lower than 10−7, indicating major differences in the geographic distribution of haplotypes.
Inference of Migratory Events.
If the variations observed were caused by drift entirely, generating local variation in the course of different migratory expansions from Africa to the rest of the world, at least three major migration streams or routes would be suggested from the data presented in Table 1.
Ht1 and Ht2.
Ht2 originated in Africa and is present everywhere, though at a relatively low frequency compared with other globally present haplotypes, and probably comigrated with its rarer progenitor Ht1. Ht1 is the original human haplotype from which four other haplotypes, Ht2, Ht3, Ht5, and Ht7, were generated, but Ht1 now seems to be on its way to extinction. Humans with Ht2 were likely the early settlers of Oceania, because their descendants accumulated two continent-specific mutations that gave rise to Ht3 and Ht4 in sequence. Of the three continents settled from Asia, Oceania was probably colonized earlier than the other two, Europe and America. A recent article suggests that Oceania was settled 40 thousand or more years ago (18); others suggest even earlier dates, 50 to 60 thousand years ago (9, 19). It has been suggested that Europe was settled 40 thousand years ago or slightly earlier (20) and that America was settled between 15 and 30 thousand years ago (9).
Ht2 is also found in South Asia (especially in Pakistan), on the most direct route from Africa to Oceania, and in Europe. The migration indicated by Ht2 may be the same one suggested by the analysis of β-globin (21), but no precise correlation analysis was done.
All other haplotypes of some importance were found in Oceania, and there is independent evidence that there were several migrations (summarized in ref. 9). This evidence is in agreement with the observation that Oceania has the greatest variety of haplotypes after Africa. The fact that the only mutants likely to have originated in Oceania stem from Ht2 suggests that Oceania was settled by the earliest migration.
Ht5 and Ht6.
Ht5 is found in all continents but is very rare in Europe. Its high frequency in East Asia suggests a migration independent from that postulated for Ht2. However, given its presence on the Indian subcontinent, this migration also might have taken place along the southern coast of Asia to Southeast Asia and then proceeded northwards to Northeast Asia and America and southwards to Oceania. The practical absence of Ht5 in Europe and its rarity in South Asia, except in the Tamils, are striking and indicate that Europe and the Indus valley were colonized to a large extent by a separate migration by Ht7 and Ht8 populations.
Ht6, a derivative of Ht5 found only in Japan, Taiwan, Cambodia, and Africa (Pygmy, Bantu, and Khoisan), has an intriguing geographic distribution, reminiscent of YAP, a Y chromosome Alu insertion believed to have originated in Asia and returned to Africa (22). The limited Asian distribution of Ht6 may reflect the settlement of aboriginal Jomon in Japan from Southeast Asia (23, 24).
Ht7–Ht10.
Ht7 and Ht8 should have a common history; Ht7 probably originated in Africa, and Ht8 descended from it. Their geographic distribution suggests that Ht7 and Ht8 spread via West and Central Asia to Europe, America, and the Indian subcontinent but somehow not to East Asia. Apart from the lack of Ht8 in East Asia and the rarity of Ht7 in America, these haplotypes have the most homogeneous global distribution.
Selection vs. Genetic Drift.
It is always possible that natural selection is responsible for at least some of these phenomena. The expectations of drift, migration, and natural selection differ enough that one may hope, at least in theory, to distinguish their effects provided that data from a sufficient number of independent genes are available.
It is also possible that natural selection does not affect any of the polymorphisms being studied directly but does affect those of neighboring genes that partially sweep away other selectively neutral, closely linked mutants. The chromosome 21 region that we have studied is about 5 kb from the MX1 gene, which encodes an interferon-inducible protein p78 endowed with antiviral activities against influenza viruses (25). The FST values calculated among the six regions of Table 1, averaged over the 10 haplotypes, vary between 0.13 and 0.18, depending on the method of computation, with a standard error of ±0.03. FST is the proportion of the total variation that can be understood as the variance of allele frequencies between populations. These values are similar to the general world average FST: 0.12 for restriction fragment length polymorphisms and 0.14 for protein markers (9). Naturally, the world FST values averaged over all genes are not necessarily equal to neutral expectation. They may be somewhat above it, being inflated by high values caused by variation among environments for Igs and HLA genes, which respond to variable epidemiological conditions around the world. Of the 10 haplotypes studied here, Ht5 shows the highest value (FST = 0.32) and is the haplotype most likely to be suspected of variable natural selection among places. If this suspicion were true, then Europe is the continent in which the fitness of Ht5 is smallest. But current observed values do not give enough reason for abandoning the neutral hypothesis. The choice between selection and major migration routes (and/or episodes) can be made only on the basis of possible environmental correlations that would favor selection or of clearly repeated patterns seen for several unrelated genes that would support migratory explanations. In this context, we were impressed by the similarities of the chromosome 21 data described here with those of an entirely different system, the Y chromosome. Clearly, any similarity between observations of totally independent genetic systems strengthens conclusions on migrations. We have accumulated a total of 160 biallelic markers, which define over 100 haplotypes falling into eight major groups (P.A.U., P.J.O., P.S., R.W.D., and L.L.C.-S., unpublished work). We give a summary of these data in Table 2.
Table 2.
Geographic distribution of eight Y chromosome haplogroups (unpublished) from populations similar to those examined at the MX1 locus
| Region | Haplogroup
|
|||||||
|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | VIII | |
| Africa | 51 | 50 | 186 | 0 | 29 | 0 | 3 | 9 |
| South and Central Asia | 0 | 2 | 7 | 26 | 64 | 21 | 42 | 128 |
| East Asia | 0 | 0 | 9 | 3 | 3 | 106 | 11 | 2 |
| Europe | 1 | 0 | 6 | 0 | 33 | 0 | 0 | 78 |
| America | 0 | 0 | 3 | 1 | 1 | 0 | 3 | 95 |
| Oceania | 0 | 0 | 0 | 7 | 1 | 0 | 19 | 1 |
| Total | 52 | 52 | 211 | 30 | 130 | 127 | 59 | 312 |
Similarities Between Loci on Chromosome 21 and the Y Chromosome.
There are definite similarities between the chromosome 21 and Y chromosome data shown in Tables 1 and 2, strengthening the hypothesis that the differences observed are caused by different migration streams. The cos θ values between chromosome 21 haplotypes and Y chromosome haplogroups are shown in Table 3, with their standard errors calculated by the bootstrap method. The larger similarities observed in every column of Table 3 are printed in bold. They confirm that both genetic systems agree in supporting at least the three expansions from Africa discussed above. It is also interesting to note that the chromosome 21 data seem to support the hypothesis of a return migration from Asia to Africa put forward by Hammer et al. (26).
Table 3.
Similarities between MX1 haplotypes and Y chromosome haplogroups
| Similarity
|
||||||
|---|---|---|---|---|---|---|
| MX1-Ht1 | MX1-Ht2 | MX1-Ht5 | MX1-Ht6 | MX1-Ht7 | MX1-Ht8 | |
| Y-I | 0.81 ± 0.08 | 0.31 ± 0.09 | 0.34 ± 0.03 | 0.81 ± 0.05 | 0.48 ± 0.06 | 0.33 ± 0.07 |
| Y-II | 0.78 ± 0.08 | 0.30 ± 0.08 | 0.36 ± 0.04 | 0.81 ± 0.05 | 0.46 ± 0.04 | 0.32 ± 0.05 |
| Y-III | 0.83 ± 0.07 | 0.49 ± 0.07 | 0.55 ± 0.04 | 0.89 ± 0.04 | 0.65 ± 0.04 | 0.47 ± 0.05 |
| Y-IV | 0.36 ± 0.14 | 0.63 ± 0.08 | 0.61 ± 0.08 | 0.16 ± 0.06 | 0.58 ± 0.04 | 0.58 ± 0.05 |
| Y-V | 0.81 ± 0.10 | 0.75 ± 0.07 | 0.53 ± 0.06 | 0.47 ± 0.04 | 0.87 ± 0.03 | 0.84 ± 0.04 |
| Y-VI | 0.10 ± 0.06 | 0.35 ± 0.08 | 0.58 ± 0.03 | 0.52 ± 0.07 | 0.57 ± 0.03 | 0.19 ± 0.02 |
| Y-VII | 0.51 ± 0.14 | 0.70 ± 0.06 | 0.74 ± 0.05 | 0.37 ± 0.06 | 0.68 ± 0.03 | 0.61 ± 0.04 |
| Y-VIII | 0.56 ± 0.10 | 0.86 ± 0.04 | 0.66 ± 0.04 | 0.18 ± 0.03 | 0.73 ± 0.03 | 0.92 ± 0.02 |
Entropy.
We have evaluated further the genetic variation in each continental region for the data sets shown in Tables 1 and 2 by calculating the entropy of gene frequencies relative to their maximum, a quantity that has a formal negative relationship with FST. Fig. 2 shows the correlation between entropy values of chromosome 21 and the Y chromosome for six continental regions. By averaging the entropy values over the two genetic systems, we find that Africa has the greatest entropy value, followed closely by South and Central Asia and Oceania, whereas America has the lowest value, especially for the Y chromosome. The poverty of genetic variation in America may be caused in part by the small number of tribes represented here, but our results are in agreement with previous reports of relative genetic homogeneity in Native Americans (27, 28), including Y chromosome data that suggest that most males descended from a single individual (29, 30). Some of the haplotypes have idiosyncratic frequencies in isolated populations, at times because of high drift and at other times because of admixture. Idiosyncratic frequencies were identified for Ht8, which was found in Nasioi Melanesians in Bougainville and exclusively in Karitiana in America.
Figure 2.

Entropies of subcontinents relative to the maximum value they can take for the two systems of haplotypes of MX1 (abscissa) and Y chromosome (ordinate) given in Tables 1 and 2. Standard errors indicated by horizontal and vertical segments are calculated by the bootstrap method.
The descending entropy values from Africa to America reflect the likely order of dates of settlement (Africa, then Asia, then Oceania, then Europe, and finally America), as expected if every new settlement is a genetic subset of its parental set. This scheme does not necessarily reflect where continent colonizers originated. However, subsequent gene flows of unknown magnitude between continental regions prevent accurate dating of settlement.
A comparison of the ratios of entropy values between chromosome 21 and the male-specific Y chromosome provides some insight into what proportion of genetic diversity may have been contributed to each continental region as a function of gender. For instance, genetic diversity on the Y chromosome in Oceania and America is roughly half of that observed on the autosome, whereas they are nearly equivalent in East Asia, Europe, and Africa. Whether the difference in entropy in South and Central Asia indeed reflects a greater contribution by males requires further study, because Central Asians were not analyzed at the MX1 locus.
In conclusion, the present data support at least three distinct dispersals from Africa. It is expected that several such loci will be found eventually by the systematic screening of chromosomes for single-nucleotide polymorphisms. Evaluation of their similarities may provide insight into population history, particularly with regard to the role of selection.
Acknowledgments
We thank the individuals who donated their DNA, as well as the investigators who provided the DNA samples: M. E. Ibrahim, T. Jenkins, J. Bertranpetit, P. Moral, T. Wagner, J. Kidd, M. Hsu, J. Chu, S. Q. Mehdi, P. Srinivasan, and P. Parham. Marc Feldman and an anonymous editor kindly provided constructive criticism. This work was supported by National Institutes of Health Grants HG00205 and GM28428.
ABBREVIATION
- DHPLC
denaturing high-performance liquid chromatography
- Htn
haplotype n
References
- 1.Wolpoff M H. Science. 1996;274:704–706. doi: 10.1126/science.274.5288.704d. [DOI] [PubMed] [Google Scholar]
- 2.Wolpoff M H. In: The Human Revolution: Behavioral and Biological Perspectives on the Origins of Modern Humans. Mellars P, Stringer C, editors. Edinburgh: Edinburgh Univ. Press; 1989. pp. 123–154. [Google Scholar]
- 3.Cann R L, Stoneking M, Wilson A C. Nature (London) 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- 4.Stringer C B. Nature (London) 1989;331:565–566. doi: 10.1038/331565a0. [DOI] [PubMed] [Google Scholar]
- 5.Lahr M M. The Evolution of Human Diversity: A Study of Cranial Variation. Cambridge, MA: Cambridge Univ. Press; 1996. [Google Scholar]
- 6.Tishkoff S A, Dietzsch E, Speed W, Pakstis A J, Kidd J R, Cheung K, Bonne-Tamir B, Santachiara-Benerecetti A S, Moral P, Krings M, et al. Science. 1996;271:1380–1387. doi: 10.1126/science.271.5254.1380. [DOI] [PubMed] [Google Scholar]
- 7.Underhill P A, Jin L, Lin A, Mehdi S Q, Jenkins T, Vollrath D, Davis R W, Cavalli-Sforza L L, Oefner P J. Genome Res. 1997;7:996–1005. doi: 10.1101/gr.7.10.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Underhill P A, Jin L, Lin A, Chan C, Shen P, Kauffman E, Choi L, Hurlbut A, Cavalli-Sforza L L, Medhi S Q, et al. Am J Hum Genet. 1997;61:A18. [Google Scholar]
- 9.Cavalli-Sforza L L, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton: Princeton Univ. Press; 1994. [Google Scholar]
- 10.Bowcock A M, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd J R, Cavalli-Sforza L L. Nature (London) 1994;368:455–457. doi: 10.1038/368455a0. [DOI] [PubMed] [Google Scholar]
- 11.Deka R, Jin L, Shriver M D, Yu L M, DeCroo S, Hundrieser J, Bunker C H, Ferrell R E, Chakraborty R. Am J Hum Genet. 1995;56:461–474. [PMC free article] [PubMed] [Google Scholar]
- 12.Shriver M D, Jin L, Ferrell R E, Deka R. Genome Res. 1997;7:586–591. doi: 10.1101/gr.7.6.586. [DOI] [PubMed] [Google Scholar]
- 13.Ayala F J, Escalante A, O’Huigin C, Klein J. Proc Natl Acad Sci USA. 1994;91:6787–6794. doi: 10.1073/pnas.91.15.6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oefner, P. J. & Underhill, P. A. (1998) U.S. Patent No. 5,795,976.
- 15.Oefner, P. J. & Underhill, P. A. (1998) Curr. Protoc. Hum. Genet.19, Suppl., 7.10.1–7.10.12.
- 16.Bhattacharrya A. Sankhya. 1956;7:401–406. [Google Scholar]
- 17.Cavalli-Sforza L L, Edwards A W F. Am J Hum Genet. 1967;21:550–570. [Google Scholar]
- 18.Connell J F, Allen J. Evol Anthropol. 1998;6:132–146. [Google Scholar]
- 19.Roberts R G, Jones R, Smith M A. Nature (London) 1990;345:153–156. [Google Scholar]
- 20.Klein R. J World Prehist. 1995;9:167–198. [Google Scholar]
- 21.Harding R M, Fullerton S M, Griffiths R C, Bond J, Cox M J, Schneider J A, Moulin D S, Clegg J B. Am J Hum Genet. 1997;60:772–789. [PMC free article] [PubMed] [Google Scholar]
- 22.Altheide T K, Hammer M F. Am J Human Genet. 1997;61:462–466. doi: 10.1016/S0002-9297(07)64077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammer M F, Horai S. Am J Hum Genet. 1995;56:951–962. [PMC free article] [PubMed] [Google Scholar]
- 24.Stoneking M, Fontius J J, Clifford S L, Soodyall H, Arcot S S, Saha N, Jenkins T, Tahir M A, Deininger P L, Batzer M A. Genome Res. 1997;7:1061–1071. doi: 10.1101/gr.7.11.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horisberger M A. Am J Respir Crit Care Med. 1995;57:A266. doi: 10.1164/ajrccm/152.4_Pt_2.S67. [DOI] [PubMed] [Google Scholar]
- 26.Hammer M F, Karafet T, Rasanayagam A, Wood E T, Altheide T K, Jenkins T, Griffiths R C, Templeton A R, Zegura S L. Mol Biol Evol. 1998;15:427–441. doi: 10.1093/oxfordjournals.molbev.a025939. [DOI] [PubMed] [Google Scholar]
- 27.Kidd J R, Black F L, Weiss K M, Balazs I, Kidd K K. Hum Genet. 1991;63:775–794. [PubMed] [Google Scholar]
- 28.Black F L. Science. 1992;258:1739–1740. doi: 10.1126/science.1465610. [DOI] [PubMed] [Google Scholar]
- 29.Underhill P A, Jin L, Zemans R, Oefner P J, Cavalli-Sforza L L. Proc Natl Acad Sci USA. 1996;93:196–200. doi: 10.1073/pnas.93.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santos F R, Rodrigues-Delfin L, Pena S D, Moore J, Weiss K M. Am J Hum Genet. 1996;58:1369–1370. [PMC free article] [PubMed] [Google Scholar]