

Abstract
Aberrations in methylation profile of the genome occur in human cancers induced by folate deficiency. To elucidate the underlying mechanism, male F344 rats were fed a diet deficient in L-methionine and devoid of folic acid and choline (FMD diet), which is known to induce hepatocellular carcinomas. We investigated changes in the DNA methylation machinery, namely, de novo DNA methyltransferases (Dnmt3a and 3b), maintenance DNA methyltransferase (Dnmt1), and methyl CpG binding proteins (MBDs), in rat livers during early stages of tumorigenesis. RT-PCR and Western blot analyses revealed differential expression of these proteins in the livers of rats fed the FMD diet. Although the hepatic Dnmt1 mRNA level declined with age (P < 0.001), it was elevated (P < 0.001) in deficient rats compared with controls. The changes in hepatic Dnmt1 protein level with the diet correlated with its mRNA levels (r = 0.60, P = 0.002). Similarly, the Dnmt3a mRNA level was elevated in rats fed the FMD diet (P < 0.001), whereas the Dnmt3b level (mRNA and protein) was not affected by diet or age. Compared with controls, hepatic MBD1–3 RNA levels increased (P < 0.001) and the protein levels of MBD1, 2, and 4 were elevated (P < 0.001) in the deficient rats. In both diet groups, hepatic MBD2 protein decreased (P < 0.001), whereas MeCP2 protein increased (P < 0.001) with age. These results demonstrate that a combined folate and methyl deficiency alters components of the DNA methylation machinery by both transcriptional and posttranscriptional mechanisms during early stages of hepatocarcinogenesis.
Keywords: hepatocarcinogenesis, folate-and methyl-deficient diet, Dnmt1/3a/3b, MBD1–4
Epidemiological studies indicate that inadequate dietary intake of folate may predispose humans to increased cancer risk [for review, see (1)]. Many micronutrients and vitamins are indispensable for metabolic pathways (2,3). Folate, an important mediator in the transfer of methyl groups, is essential for the de novo biosynthesis of purines and thymidylate and plays a crucial role in DNA synthesis, stability, and integrity. Aberrations in any or all of these processes have been implicated in colorectal (4,5) and hepatocellular carcinogenesis (6,7). Folic acid is metabolized to 5-methyltetrahydrofolate that acts as a methyl donor in the conversion of homocysteine to L-methionine. L-Methionine is subsequently metabolized to S-adenosyl-L-methionine (SAM)4, the principal methyl donor in the majority of biochemical reactions, including methylation of macromolecules. Folate deficiency causes depletion of SAM, resulting in genome-wide DNA hypomethylation, activation of oncogenes, and malignant transformation (1). Paradoxically, recent studies show that limited folate intake also results in hypermethylation of the CpG islands of many tumor suppressor genes (4,8).
Methylation at 5-position of cytosine is the predominant epigenetic modification in the mammalian genome that is essential for development [for review, see (9,10)]. It is heritable due to the activity of DNA methyltransferase (Dnmt) (see Fig. 1A). DNA methylation is initiated by de novo DNA methyltransferases Dnmt3a and Dnmt3b, which is propagated in the newly replicated DNA strand by the maintenance DNA methyltransferase, Dnmt1 (11,12). The C-terminal catalytic domains of these enzymes contain motifs that are homologous to bacterial methyltransferase. Their N-terminal regions harbor several domains that interact with DNA and protein. The major function of DNA methylation is the silencing of retroviral promoters, transposable elements in the genome, and regulating expression of imprinted genes (13). Apart from methyltransferase activity, these enzymes also act as transcriptional repressors by recruiting corepressors like histone deacetylase and histone methyltransferase (Fig. 1B) (14,15).
FIGURE 1.
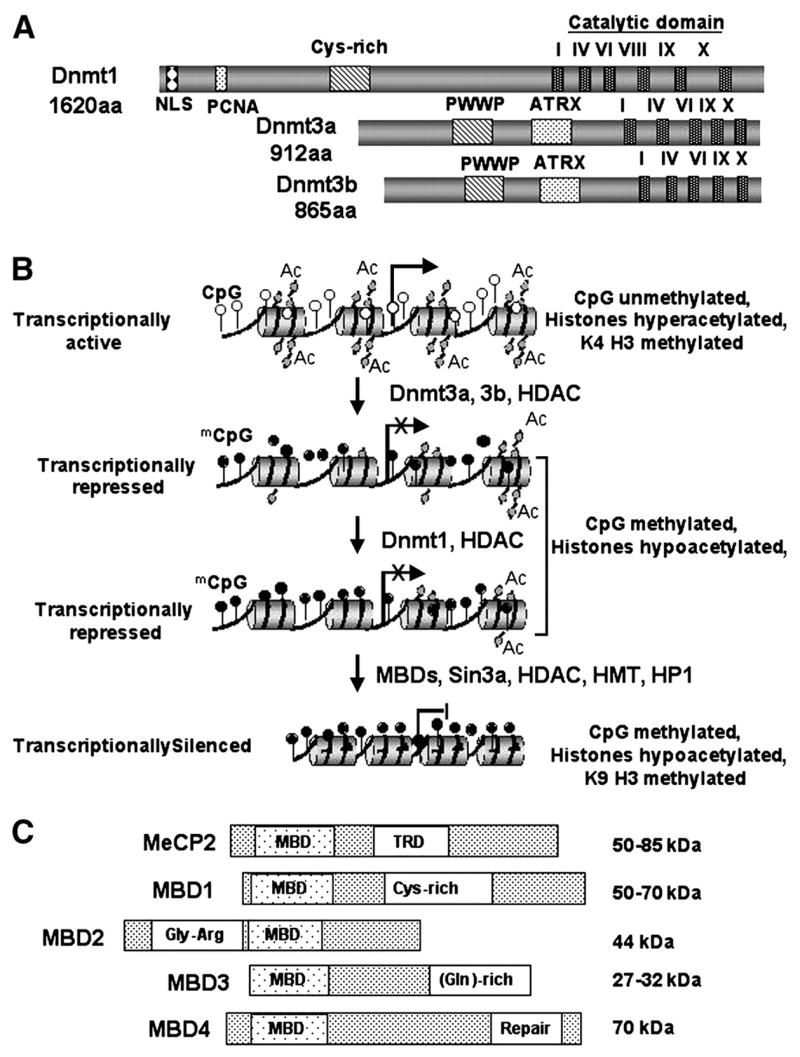
Schematic representation of Dnmts and MBDs involved in epigenetic silencing of genes. Three functional Dnmts highly conserved in mammals (A). Simplified mechanism of methylation mediated silencing (B). Nucleosomes wrapped around transcriptionally active promoters are relaxed where CpG base pairs are unmethylated and core nucleosomal histones are acetylated. Dnmt3a or Dnmt3b (de novo methyl transferase) initiate methylation of CpG base pairs, which are maintained postreplication by Dnmt1. Methyl CpG binding proteins then bind to the methylated CpGs and recruit different corepressors, resulting in nucleosomal condensation and epigenetic silencing. Open and filled lollipops denote methylated and unmethylated CpG, respectively. Schematic diagram of MBD family members (C). MBD and TRD stand for methyl CpG binding domain and transcriptional repressor domain, respectively.
Methyl CpG binding domain proteins (MBDs) are the interpreters of the DNA methylation signal. Five MBDs with signature methyl CpG binding domain have been identified in mammals (Fig. 1C). These proteins bind symmetrically methylated 5-methyl-CpG through their conserved methyl CpG binding domain. The majority of the MBDs can mediate transcriptional repression of methylated promoters by recruiting a silencing complex. Methylated DNA acquires a compact, condensed, inactive heterochromatin configuration, inaccessible to transcriptional machinery that results in gene silencing (Fig. 1B) (16).
Previous studies in rodents have shown that a diet deficient in methionine and devoid of folic acid and choline (FMD diet) induces preneoplastic nodules in the livers of male F344 rats after wk 36 and hepatocellular carcinomas after wk 54 (7,17). Recent studies have shown that this diet induces global hypomethylation specifically in the liver, the target tissue susceptible to neoplastic transformation (17). This diet causes the depletion of SAM and a decrease in the SAM/S-adenosyl homocysteine (SAH) ratio, leading to genome-wide hypomethylation (18). Interestingly, this diet also induces gene-specific hypermethylation (7). We used restriction landmark genomic scanning (RLGS) analysis to demonstrate genome-wide alteration in the methylation profile in preneoplastic nodules and carcinomas (19). One of the hypermethylated genes was identified as the receptor-type protein, tyrosine phosphatase (PTPRO), which was subsequently found to be methylated in different types of human cancer, including those of the liver, lung (20), and colon (21).
To elucidate the molecular mechanisms by which the FMD diet induces de novo methylation and the silencing of specific genes, we made a systematic analysis of the expression profile of the components of DNA methylation machinery that include Dnmts and MBDs at early stages of hepatocarcinogenesis.
MATERIALS AND METHODS
Rats, diet, and specimen
Male weanling F344 rats were obtained from the National Center for Toxicological Research (NCTR) breeding facility, housed 2/cage in a temperature-controlled (24°C) room with a 12 h light/dark cycle and given free access to water and NIH-31 pelleted diet (Purina Mills). All rats were handled humanely and the study was approved by the NCTR (protocol number E-0712801). At 4 wk of age, the rats (body weight 50 g) were allocated randomly to receive either a diet low in L-methionine (0.18%), devoid of choline and folic acid (methyl deficient) (Supplemental Tables S1 and S2), or the methyl-adequate diet (deficient diet supplemented with 0.4% L-methionine, 0.3% choline, and 2 mg/kg folic acid) (Dyets) (7,22). To increase the severity of methyl deficiency, folate was not included in the standard low L-methionine, choline-devoid methyl-deficient diet. The diets were stored at 4°C and the rats were given fresh feed biweekly. Four rats/group were killed at wk 9, 18, or 36 of methyl deficiency. Body weights and food consumption were recorded weekly and these values did not differ between the 2 diet groups. To confirm that the rats used in the present study were significantly deficient in L-methionine, hepatic SAM level was measured in the liver of each rat. FMD fed rats had a lower hepatic SAM concentration and SAM/SAH ratio than age-matched controls (18). The livers were excised, frozen immediately in liquid nitrogen, and stored at −80°C for subsequent analyses.
Measurement of SAM and SAH levels
SAM and SAH in the liver were measured by HPLC, as described previously (23).
RNA isolation and reverse transcription
Frozen livers were pulverized and used for RNA, DNA, and nuclei isolation. Total RNA was isolated with the guanidinium isothiocyanate-acid phenol method, as described earlier (24). Reverse transcription was carried out with random hexamers and M-MuLV reverse transcriptase from 3 μg of total RNA in 20 μL of total volume, following the protocol provided in the GeneAmp RNA PCR kit (Applied Biosystems).
Real time RT-PCR analysis
An aliquot of the cDNA (equivalent to 100 ng of RNA for Dnmt1, 250 ng for each of Dnmt3A or 3B, and 10 ng for 18S rRNA) was used for real-time RT-PCR analysis as described (15,25). The optimum primer concentration was 150 nmol/L. A standard curve for each cDNA was first generated using 10-fold serial dilutions (108–102 copies) of the respective cDNAs as a template. To create the standard curve, rat 18S rRNA, Dnmt1, Dnmt3a, and Dnmt3b, MBD1–4, and MeCP2 cDNAs were amplified by RT-PCR. The copy number of each cDNA expressed in rat liver was calculated from the standard curve and normalized to that of 18S rRNA. The primer sequences and conditions for RT-PCR are described in Supplemental Table S3.
Isolation of tissue nuclei and preparation of nuclear extract
The nuclei were isolated from individual rat livers by homogenization in hypotonic buffer containing 2 mol/L sucrose, which was followed by sucrose density gradient centrifugation, as described (26,27). The nuclear pellet from each sample was resuspended in the lysis buffer (50 mmol/L Tris-HCl, pH 8.10; 1 mmol/L EDTA, pH 8; and 10 g/L SDS) containing a protease inhibitor cocktail (Sigma Chemical) and was followed by brief sonication to shear viscous DNA. The supernatant obtained after centrifuging the extract at 20,000 × g for 10 min was subjected to Western blot analysis with anti-Dnmt1, Dnmt3a, Dnmt3b, MBD1–4, and MeCP2 antibodies, using protocols described earlier (24,27,28). Antibodies against all MBDs, Dnmt3a, and Dnmt3b were raised in our laboratory (24,27,28). Antibodies against Dnmt3a and 3b were raised against their N-terminal domains that lack a conserved catalytic site (27). MBDs were raised against the recombinant protein fragments that lacked a highly conserved methyl CpG binding domain at the N-terminus. Antibody against Dnmt1 was a generous gift from Dr. Shoji Tajima (29). β-Tubulin antibody was from Santa Cruz Biotechnology. An aliquot of nuclear extract (200 μg for Dnmts and MBD1, 2, and 4, and 50 μg for MBD3) was used for immunoblot analysis.
Statistical analysis
The data were analyzed by a 2-way ANOVA (diet × age) using the SPSS statistical package (SPSS, version 13.0). When the assumption of homogeneity of variances was violated with Levene’s test showing a P ≤ 0.01, we log transformed the data prior to analysis. Due to multiple comparisons and resulting α-inflation, overall main effects and interaction P-value were required to be ≤0.001 to be considered significant, whereas pairwise tests were required to have P < 0.01. If overall P-values were ≤0.001 for treatment and age, pairwise t tests with adjusted α were used to differentiate between control and FMD treatments at each age. Linear correlation was performed using Pearson correlation with P ≤ 0.01 considered significant. Values presented are means ± SD.
RESULTS
Dnmt1 and Dnmt3a are upregulated in the livers of rats fed the FMD diet
Feeding the FMD diet upregulated (P < 0.001) glutathione S-transferase-π RNA, a marker for preneoplastic transformation of hepatocytes, as early as wk 9; this level was maintained until wk 36 (Table 1). Similarly, the hepatic SAM concentration, a marker for methionine level, was reduced to 50–60% (P < 0.002) of controls in rats fed the FMD diet at wk 9, wk 18, and wk 36 without significant changes in hepatic SAH (data not shown) (18). These results confirmed that these rats were indeed methyl deficient, and preneoplastic changes in hepatocytes occurred in all rats fed the FMD diet.
TABLE 1.
Hepatic mRNA levels of GST-π and Dnmt1, 3a, and 3b in Fisher rats fed control or FMD diet for 9, 18, or 36 wk1
| 2-Way ANOVA
|
|||||||
|---|---|---|---|---|---|---|---|
| Gene | Diet | 9 wk | 18 wk | 6 wk | Diet | Age | Diet × age |
| copy number/105 copies of 18S rRNA | |||||||
| GST-π2 | Control | 50.74 ± 5.32 | 53.08 ± 6.26 | 49.26 ± 4.64 | <0.001 | 0.169 | 0.38 |
| FMD | 136.97 ± 16.32* | 152.36 ± 7.44* | 96.52 ± 51.01* | ||||
| Dnmt1 | Control | 14.13 ± 1.56a | 10.16 ± 3.08b | 7.19 ± 1.48c | <0.001 | <0.001 | 0.004 |
| FMD | 32.06 ± 6.68*a | 16.34 ± 4.18*b | 11.61 ± 1.86*c | ||||
| Dnmt3a | Control | 1.37 ± 0.31 | 0.83 ± 0.58 | 1.00 ± 0.31 | <0.001 | 0.004 | 0.06 |
| FMD | 3.86 ± 0.88* | 2.62 ± 0.76* | 1.95 ± 0.59* | ||||
| Dnmt3b | Control | 4.56 ± 2.76 | 5.22 ± 3.64 | 4.47 ± 3.07 | 0.047 | 0.056 | 0.12 |
| FMD | 4.21 ± 2.17 | 12.23 ± 5.52 | 6.78 ± 2.30 | ||||
Values are means ± SD, n = 4. Means in a row without a common letter differ, P < 0.01.
Different from the control group at that time, P < 0.01.
GST-π, glutathione S-transferase π.
The hepatic Dnmt1 mRNA level was elevated (P < 0.001) in rats fed the FMD diet compared with controls (Table 1). The maximal increase occurred at wk 9 (a 1.2-fold increase) and the increased level was maintained at wk 18 and wk 36 (60 and 50% greater than controls, respectively). Interestingly, the Dnmt1 mRNA level decreased with age to 72 and 51% of wk 9 controls at wk 18 and wk 36, respectively, whereas its reduction was more pronounced in the livers of rats fed the FMD diet. In that group, the levels were 51 and 36% of wk 9 controls at wk 18 and wk 36, respectively (Table 1).
The hepatic Dnmt1 protein level was also elevated (P < 0.001) in rats fed the FMD diet (Fig. 2, a representative Western blot; and Table 2). Although Dnmt1 protein was not affected by age and the interaction was not significant, the diet-induced increase was 2.8-fold at wk 9, 1-fold at wk 18, and 1.2-fold at wk 36. The correlation between RNA and protein levels (r = 0.60, P < 0.01) indicates that folate and methyl deficiency induces Dnmt1 gene expression at a transcriptional or posttranscriptional level by stabilizing its mRNA. The FMD diet-induced increase in hepatic Dnmt3a mRNA level was 1.8-, 2.1-, and 1-fold at wk 9, wk 18, and wk 36, respectively (Table 1), whereas its protein level was not affected by diet or age (Table 2). In contrast, the Dnmt3b RNA and protein levels were not affected by diet or age (Tables 1 and 2). These results demonstrate that upregulation of both de novo (Dnmt3a) and maintenance (Dnmt1) methyltransferases is an early event in FMD diet–induced hepatocarcinogenesis.
FIGURE 2.
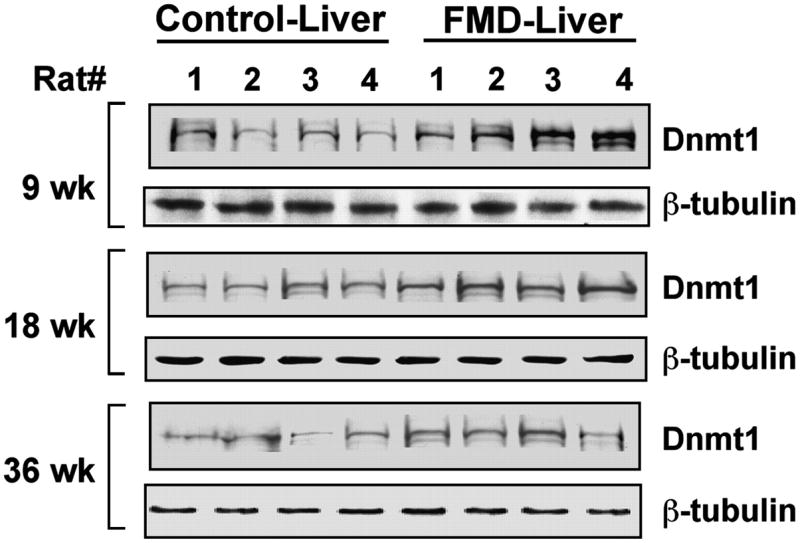
Hepatic Dnmt1 protein levels in rats fed the FMD diet or a methyl-adequate diet for different times. The nuclear extracts were separated by SDS-polyacrylamide (7.5% acrylamide) gel electrophoresis and subjected to immunoblot analysis with respective antibodies. The blot was reprobed with β-tubulin antibody. HRP-conjugated anti-rabbit was used as a secondary antibody and the signal was developed using ECL reagent; the scanned X-ray film was quantified by Kodak Imaging software.
TABLE 2.
Hepatic Dnmt1, 3a, and 3b protein levels in Fisher rats fed control or FMD diet for 9, 18, or 36 wk1
| 2-Way ANOVA
|
|||||||
|---|---|---|---|---|---|---|---|
| Gene | Diet | 9 wk | 18 wk | 36 wk | Diet | Age | Diet × age |
| arbitrary units2 | |||||||
| Dnmt1 | Control | 1.77 ± 0.66 | 2.36 ± 0.47 | 1.86 ± 1.27 | <0.001 | 0.233 | 0.139 |
| FMD | 6.70 ± 2.25* | 4.73 ± 2.03* | 3.99 ± 1.27* | ||||
| Dnmt3a | Control | 3.94 ± 0.64 | 2.55 ± 1.37 | 2.42 ± 2.07 | 0.003 | 0.108 | 0.658 |
| FMD | 7.70 ± 0.47 | 6.11 ± 1.00 | 4.34 ± 4.58 | ||||
| Dnmt3b | Control | 2.95 ± 1.96 | 2.46 ± 1.21 | 2.21 ± 1.35 | 0.130 | 0.325 | 0.233 |
| FMD | 2.58 ± 1.89 | 5.89 ± 3.84 | 3.28 ± 1.27 | ||||
Values are means ± SD, n = 4.
Different from the control group at that time, P < 0.01.
Normalized to β-tubulin.
Differential expression of the methyl CpG-binding proteins in the liver of rats fed the FMD diet
Because MBDs mediate silencing of methylated genes, we also studied their alteration in expression profile as a response to dietary deficiency. The mRNA levels of MBD1–3 were greater (P ≤ 0.001) in the livers of rats fed the FMD diet than in controls (Table 3). The increases in hepatic MBD1 mRNA were comparable at wk 9 (70%), wk 18 (120%), and wk 36 (100%). The FMD diet induced a 2.2-fold increase in hepatic MBD2 mRNA at wk 9, which was greater than those at wk 18 (1.8-fold) and wk 36 (1.4-fold). The upregulation in MBD3 mRNA was maximum (150%) at wk 9, after which it declined at wk 18 and wk 36 to 120% (Table 3). The expression of MBD2 and MBD3, like that of Dnmt1, decreased (P < 0.001) with age. The MBD2 mRNA level was highest at wk 9, after which it decreased with age to 66 and 47% of the wk 9 level at wk 18 and wk 36, respectively (Table 3). In the FMD diet-fed rats, the MBD2 mRNA level declined to 58 and 35% of the wk 9 level at wk 18 and wk 36, respectively. In the controls, the decrease in MBD3 level was 29% at wk 18 followed by a 43% decrease at wk 36. The decreases were more robust (by 65 and 71% at wk 18 and wk 36, respectively) in the FMD fed rats. Neither diet nor age affected the mRNA level of MeCP2 and MBD4.
TABLE 3.
Hepatic mRNA level of methyl CpG binding proteins in Fisher rats fed control or FMD diet for 9, 18, or 36 wk1
| 2-Way ANOVA
|
|||||||
|---|---|---|---|---|---|---|---|
| Gene | Diet | 9 wk | 18 wk | 36 wk | Diet | Age | Diet × age |
| copy number/105 copies of 18S rRNA | |||||||
| MeCP2 | Control | 10.49 ± 3.30 | 4.90 ± 3.07 | 5.14 ± 4.56 | 0.086 | 0.108 | 0.582 |
| FMD | 14.77 ± 12.50 | 6.40 ± 2.39 | 13.18 ± 5.31 | ||||
| MBD1 | Control | 6.81 ± 1.70 | 5.62 ± 1.42 | 4.12 ± 2.09 | 0.001 | 0.112 | 0.676 |
| FMD | 11.65 ± 3.10* | 12.28 ± 6.32* | 7.99 ± 0.97* | ||||
| MBD2 | Control | 20.90 ± 3.59a | 13.91 ± 3.04b | 9.82 ± 1.59c | <0.001 | <0.001 | 0.006 |
| FMD | 66.09 ± 16.47*a | 38.61 ± 9.21*b | 23.35 ± 8.22*c | ||||
| MBD3 | Control | 4.72 ± 0.77*a | 3.32 ± 0.94*b | 2.72 ± 0.74*c | 0.001 | <0.001 | 0.001 |
| FMD | 11.45 ± 3.56*a | 3.95 ± 0.33*b | 3.36 ± 0.23*c | ||||
| MBD4 | Control | 6.26 ± 3.29 | 4.66 ± 4.64 | 4.84 ± 1.33 | 0.016 | 0.922 | 0.509 |
| FMD | 7.45 ± 1.82 | 8.31 ± 2.10 | 9.27 ± 2.65 | ||||
Values are means ± SD, n = 4. Means in a row without common letter differ, P < 0.01.
Different from the control group at that time, P < 0.01.
To determine whether the upregulated mRNAs are indeed translated into proteins, we measured the hepatic protein levels. The levels of MBD1, 2, and 4 levels were greater in the livers of FMD-fed rats than in controls (Table 4). The MBD1 protein level was greater in livers of the deficient rats than the controls at each time point (1.9-, 1.1-, and 1.5-fold increase at wk 9, wk 18, and wk 36, respectively). The MBD2 level was elevated, compared with controls, by 1.6-, 4.6-, and 3.4-fold at wk 9, wk 18, and wk 36, respectively. In control rats, it decreased to 36 and 13.6% of the wk 9 level at wk 18 and wk 36, respectively (Table 4). In contrast, MBD2 was downregulated in rats fed the FMD diet to 70 and 22.5% of wk 9 level at wk 18 and wk 36, respectively. The MBD4 level in the livers of FMD diet–fed rats, compared with controls, was ~1-, 3-, and 2.4-fold greater at wk 9, wk 18, and wk 36, respectively (Table 4). Among the MBDs, MeCP2 protein was unique because its level increased with age in both groups compared with the wk 9 controls (P < 0.001). Among the controls, MeCP2 protein increased 4.4- and 9-fold at wk 18 and wk 36, respectively, compared with wk 9 (Table 4). In the deficient group, the increase in MeCP2 level was comparable at wk 18 (90%) and wk 36 (100%). Because MBD4 and MeCP2 mRNA levels were not affected by age (Table 3), these data suggest that the age-dependent increase in these proteins is probably due to translational or posttranslational mechanisms.
TABLE 4.
Hepatic protein level of methyl CpG binding proteins in Fisher rats fed control or FMD diet for 9, 18, or 36 wk1
| 2-Way ANOVA
|
|||||||
|---|---|---|---|---|---|---|---|
| Gene | Diet | 9 wk | 18 wk | 36 wk | Diet | Age | Diet × age |
| arbitrary units2 | |||||||
| MeCP2 | Control | 9.74 ± 3.58c | 52.15 ± 17.13b | 96.88 ± 21.47a | 0.027 | <0.001 | 0.013 |
| FMD | 19.99 ± 5.40b | 38.46 ± 5.12a | 39.89 ± 41.04a | ||||
| MBD1 | Control | 2.00 ± 0.37 | 3.67 ± 0.96 | 1.74 ± 0.53 | <0.001 | 0.013 | 0.82 |
| FMD | 5.80 ± 3.41* | 7.78 ± 1.62* | 4.39 ± 0.55* | ||||
| MBD2 | Control | 2.80 ± 0.43a | 1.00 ± 0.18b | 0.38 ± 0.09c | <0.001 | <0.001 | <0.001 |
| FMD | 7.32 ± 1.50*a | 5.15 ± 0.67*a | 1.65 ± 0.26*b | ||||
| MBD3 | Control | 3.24 ± 0.92 | 4.14 ± 1.11 | 2.16 ± 0.82 | 0.009 | 0.563 | 0.13 |
| FMD | 4.20 ± 1.56 | 5.20 ± 0.78 | 6.40 ± 3.55 | ||||
| MBD4 | Control | 2.60 ± 0.79 | 1.36 ± 0.89 | 3.31 ± 1.36 | <0.001 | 0.102 | 0.08 |
| FMD | 5.05 ± 1.23* | 7.69 ± 1.65* | 8.00 ± 2.87* | ||||
Values are means ± SD, n = 4. Means in a row without common letter differ, P < 0.01.
Different from the control group at that time, P < 0.01.
Normalized to β-tubulin.
DISCUSSION
To understand the role of epigenetic changes in folate-induced carcinogenesis in humans we used a rat model of multistage hepatocarcinogemesis in which consuming a diet low in L-methionine and devoid of folic acid and choline results in the induction of hepatoma in the absence of exogenous carcinogens. Significant reductions in the SAM concentration and the SAM/SAH ratio in the liver at the earliest time point tested (9 wk), and their persistence at 18 wk and 36 wk, demonstrate that these rats were indeed methionine deficient (18). Previous studies in our laboratory using the FMD diet–induced hepatocarcinogenesis rat model demonstrated alterations in the methylation profiles of ~50 CpG among ~1500 islands analyzed by RLGS (19). As a first step to elucidate the underlying mechanism, we studied the effect of this dietary regimen on the expression of proteins involved in DNA methylation-mediated gene silencing. To our knowledge, this is the first comprehensive study of the alteration in the DNA methylation machinery at early stages of tumor development. The mRNA and protein levels of some Dnmts and MBDs in the liver were altered in response to methyl deficiency for 36 wk, when preneoplastic nodules became visible. Significant upregulation of specific Dnmts and MBDs at early stages of this nutritional deficiency suggests their potential role in the neoplastic process. Knock-out or hypomorphic mice will be useful in addressing the functional implication of the differential expression of some of these factors in the carcinogenic process.
Global hypomethylation, particularly of the repetitive elements and oncogenes, is a common epigenetic event during early stages of carcinogenesis (30,31). The intriguing observation is that, in addition to DNA hypomethylation, folate and methyl deficiency can lead to localized or regional hypermethylation and suppression of a few genes that include tumor suppressor genes (4,19). This finding is consistent with a profound and progressive increase in the transcript and protein levels of Dnmt1 at the early stage of hepatocarcinogenesis. Interestingly, the mRNA levels of all 3 Dnmts were upregulated in human hepatocellular carcinoma (8,32,33). However, in those studies, the protein levels of these enzymes were not measured. Recent study has shown that global DNA hypomethylation in rats fed the FMD diet occurs specifically in livers that undergo neoplastic changes (17). The limiting SAM concentration in the deficient liver is probably responsible for global DNA hypomethylation.
Analysis of the transcript and protein levels of the MBDs revealed many striking features. In agreement with the previous observation (34), the mRNA and protein levels of MBD2 were upregulated in the methyl-deficient liver. Upregulation of MBD2 expression in response to methyl deficiency is of particular importance in view of similar observations made in other experimental systems. Thus, MBD2 has been shown to play a role in the methylation-mediated suppression of human ribosomal RNA (35) and mouse metallothionein-I genes (36). Unlike the promoters of tumor suppressor genes transcribed by RNA polymerase II, the ribosomal gene, transcribed by RNA polymerase I, was hypomethylated in human hepatocellular carcinomas relative to matching liver tissue (35). In another example, MBD2 was markedly upregulated in benign breast tumors compared with normal breast tissues, whereas there was no significant change in MeCP2 expression in the breast tumor (37).
The expression of 2 other members, namely, MBD1 and MBD4, followed a pattern similar to that of Dnmt1, exhibiting an increased expression of folate and methyl deficiency at wk 9. This observation indicates that there are changes in the expression of these components of epigenetic regulation at the early stages of hepatocyte transformation. Elevated expression of MBDs and the associated corepressor complex with the methylated promoter are likely involved in the methylation-mediated silencing of p16 (38) and PTPRO (the gene encoding the receptor-type protein tyrosine phosphatase-type O) (19) in the livers of rats fed the FMD diet.
Earlier studies reveal DNA damage in exon 5 of the p53 gene in the rat liver following 9 wk of folate deficiency (22). Folate deficiency causes uracil misincorporation into DNA, which, in turn, leads to single and double strand breakages in DNA. MBD4, the methyl CpG binding protein, is likely to be responsible for DNA mismatch repair. MBD4 exhibits strong DNA glycosylase activity and preference for G:T mismatches (39) that probably occurs as a result of deamination of 5-methyl cytosines in a symmetrically methylated CpG base pair. Moreover, MBD4 also functions as transcriptional repressor for methylated DNA (40). Upregulation of MBD4 protein in the livers of folate-deficient rats probably facilitates the elimination of CpG: TpG mismatches in DNA caused by methyl deficiency.
It is noteworthy that the mammalian Dnmts can act as transcriptional repressors because of their relatively large N-terminal regions, which are absent in bacterial methyltransferasess (Fig. 1) (41,42). We have recently shown that differentiation of PC12 (pheochromocytoma) cells to neuronal cells results in upregulation of Dnmt3b and inhibition of Dnmt3b expression by RNA interference and leads to continuous proliferation of the cells and failure to generate neurites (15). The striking observation was that Dnmt3b-mediated neuronal differentiation occurred in the absence of its catalytic activity. It is possible that Dnmt1 and Dnmt3a act in concert initially to repress target gene promoters that are subsequently hypermethylated and permanently silenced. Additional experiments are needed to test this hypothesis.
Switching rats to normal diets after consuming the FMD diet can replete the hepatic SAM level and the SAM/SAH ratio only in those that had consumed the FMD diet for 9 wk, but not for 18 wk or 36 wk (18). The inability to recover the hepatic SAM level and the DNA methylation profile in rats consuming the diet for 18 wk or 36 wk before shifting them to a folate- and methyl-adequate diet suggests that irreversible changes had occurred in the livers undergoing preneoplastic transformation. The inability to reverse mRNA profiles of Dnmts and MBDs toward normal levels (data not shown) further supports the notion that prolonged dietary deficiency induces permanent genetic and epigenetic changes in the liver that cannot be reversed by consuming a normal diet at a later stage.
In conclusion, the present study shows that substantial alterations in the expression of DNA methyltransferases and methyl CpG binding proteins occur at early stages of hepatocarciniogenesis induced by folate and methyl deficiency in rats. These changes may be one of the important contributing factors in the formation of epigenetically reprogrammed cells that become neoplastic.
Supplementary Material
Supplemental Material can be found at: http://jn.nutrition.org/cgi/content/full/136/6/1522/DC1
Footnotes
Supported by grant CA 86978 from the NIH.
Supplemental Tables 1–3 are available with the online posting of this paper at www.nutrition.org.
Abbreviations used: Dnmt, DNA methyltransferase; FMD diet, folate- and methyl-deficient diet; MBD, methyl CpG binding domain protein; PTPRO, receptor type protein tyrosine phosphatase; RLGS, restriction landmark genomic scanning; SAH, S-adenosyl homocysteine; SAM, S-adenosyl methionine.
LITERATURE CITED
- 1.Duthie SJ, Narayanan S, Sharp L, Little J, Basten G, Powers H. Folate, DNA stability and colorectal neoplasia. Proc Nutr Soc. 2004;63:571–8. doi: 10.1079/pns2004. [DOI] [PubMed] [Google Scholar]
- 2.Friso S, Choi SW. Gene-nutrient interactions in one-carbon metabolism. Curr Drug Metab. 2005;6:37–46. doi: 10.2174/1389200052997339. [DOI] [PubMed] [Google Scholar]
- 3.Ulrey CL, Liu L, Andrews LG, Tollefsbol TO. The impact of metabolism on DNA methylation. Hum Mol Genet. 2005;14:R139–147. doi: 10.1093/hmg/ddi100. [DOI] [PubMed] [Google Scholar]
- 4.Kim YI. Folate and DNA methylation: a mechanistic link between folate deficiency and colorectal cancer? Cancer Epidemiol Biomarkers Prev. 2004;13:511–9. [PubMed] [Google Scholar]
- 5.Ulrich CM. Nutrigenetics in cancer research–folate metabolism and colorectal cancer. J Nutr. 2005;135:2698–702. doi: 10.1093/jn/135.11.2698. [DOI] [PubMed] [Google Scholar]
- 6.Davis CD, Uthus EO. DNA methylation, cancer susceptibility, and nutrient interactions. Exp Biol Med (Maywood) 2004;229:988–95. doi: 10.1177/153537020422901002. [DOI] [PubMed] [Google Scholar]
- 7.James SJ, Pogribny IP, Pogribna M, Miller BJ, Jernigan S, Melnyk S. Mechanisms of DNA damage, DNA hypomethylation, and tumor progression in the folate/methyl-deficient rat model of hepatocarcinogenesis. J Nutr. 2003;133:3740S–7S. doi: 10.1093/jn/133.11.3740S. [DOI] [PubMed] [Google Scholar]
- 8.Park HJ, Yu E, Shim YH. DNA methyltransferase expression and DNA hypermethylation in human hepatocellular carcinoma. Cancer Lett. doi: 10.1016/j.canlet.2005.03.017. Epub 2005 May 7. [DOI] [PubMed] [Google Scholar]
- 9.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 10.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23:5594–605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim GD, Ni J, Kelesoglu N, Roberts RJ, Pradhan S. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 2002;21:4183–95. doi: 10.1093/emboj/cdf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsieh CL. The de novo methylation activity of Dnmt3a is distinctly different than that of Dnmt1. BMC Biochem. 2005;6:6. doi: 10.1186/1471-2091-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bird AP, Wolffe AP. Methylation-induced repression–belts, braces, and chromatin. Cell. 1999;99:451–4. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 14.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–12. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai S, Ghoshal K, Datta J, Majumder S, Yoon SO, Jacob ST. DNA methyltransferase 3b regulates nerve growth factor-induced differentiation of PC12 cells by recruiting histone deacetylase 2. Mol Cell Biol. 2005;25:751–66. doi: 10.1128/MCB.25.2.751-766.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Santini V, Kantarjian HM, Issa JP. Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications. Ann Intern Med. 2001;134:573–86. doi: 10.7326/0003-4819-134-7-200104030-00011. [DOI] [PubMed] [Google Scholar]
- 17.Pogribny IP, James SJ, Jernigan S, Pogribna M. Genomic hypomethylation is specific for preneoplastic liver in folate/methyl deficient rats and does not occur in non-target tissues. Mutat Res. 2004;548:53–9. doi: 10.1016/j.mrfmmm.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 18.Pogribny IP, Ross SA, Wise C, Pogribna M, Jones EA, Tryndyak VP, James SJ, Dragan YP, Poirier LA. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat Res. 2006;593:80–7. doi: 10.1016/j.mrfmmm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 19.Motiwala T, Ghoshal K, Das A, Majumder S, Weichenhan D, Wu YZ, Holman K, James SJ, Jacob ST, Plass C. Suppression of the protein tyrosine phosphatase receptor type O gene (PTPRO) by methylation in hepatocellular carcinomas. Oncogene. 2003;22:6319–31. doi: 10.1038/sj.onc.1206750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motiwala T, Kutay H, Ghoshal K, Bai S, Seimiya H, Tsuruo T, Suster S, Morrison C, Jacob ST. Protein tyrosine phosphatase receptor-type O (PTPRO) exhibits characteristics of a candidate tumor suppressor in human lung cancer. Proc Natl Acad Sci USA. 2004;101:13844–9. doi: 10.1073/pnas.0405451101. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Mori Y, Yin J, Sato F, Sterian A, Simms LA, Selaru FM, Schulmann K, Xu Y, Olaru A, et al. Identification of genes uniquely involved in frequent microsatellite instability colon carcinogenesis by expression profiling combined with epigenetic scanning. Cancer Res. 2004;64:2434–8. doi: 10.1158/0008-5472.can-03-3508. [DOI] [PubMed] [Google Scholar]
- 22.Pogribny IP, Miller BJ, James SJ. Alterations in hepatic p53 gene methylation patterns during tumor progression with folate/methyl deficiency in the rat. Cancer Lett. 1997;115:31–8. doi: 10.1016/s0304-3835(97)04708-3. [DOI] [PubMed] [Google Scholar]
- 23.Wise CK, Fullerton FR. Analytical procedure for determination of S-adenosylmethionine, S-adenosylhomocysteine and S-adenosylethionine in the same isocratic HPLC run, with a procedure for preparation and analysis of the analog, S-adenosylhomocysteine sulfoxide. J Liq Chromatogr. 1995;18:2005–17. [Google Scholar]
- 24.Ghoshal K, Datta J, Majumder S, Bai S, Dong X, Parthun M, Jacob ST. Inhibitors of histone deacetylase and DNA methyltransferase synergistically activate the methylated metallothionein I promoter by activating the transcription factor MTF-1 and forming an open chromatin structure. Mol Cell Biol. 2002;22:8302–19. doi: 10.1128/MCB.22.23.8302-8319.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Azadeoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25:4727–41. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Ghoshal K, Majumder S, Li Z, Dong X, Jacob ST. Suppression of metallothionein gene expression in a rat hepatoma because of promoter-specific DNA methylation. J Biol Chem. 2000;275:539–47. doi: 10.1074/jbc.275.1.539. [DOI] [PubMed] [Google Scholar]
- 27.Majumder S, Ghoshal K, Datta J, Bai S, Dong X, Quan N, Plass C, Jacob ST. Role of de novo DNA methyltransferases and methyl CpG-binding proteins in gene silencing in a rat hepatoma. J Biol Chem. 2002;277:16048–58. doi: 10.1074/jbc.M111662200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Datta J, Ghoshal K, Sharma SM, Tajima S, Jacob ST. Biochemical fractionation reveals association of DNA methyltransferase (Dnmt) 3b with Dnmt1 and that of Dnmt 3a with a histone H3 methyltransferase and Hdac1. J Cell Biochem. 2003;88:855–64. doi: 10.1002/jcb.10457. [DOI] [PubMed] [Google Scholar]
- 29.Vilkaitis G, Suetake I, Klimasauskas S, Tajima S. Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J Biol Chem. 2005;280:64–72. doi: 10.1074/jbc.M411126200. [DOI] [PubMed] [Google Scholar]
- 30.Yamada Y, Jackson-Grusby L, Linhart H, Meissner A, Eden A, Lin H, Jaenisch R. Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc Natl Acad Sci USA. 2005;102:13580–5. doi: 10.1073/pnas.0506612102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ehrlich M. The controversial denouement of vertebrate DNA methylation research. Biochemistry (Mosc) 2005;70:568–75. doi: 10.1007/s10541-005-0150-z. [DOI] [PubMed] [Google Scholar]
- 32.Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Over-expression of a splice variant of DNA methyltransferase 3b, DNMT3b4, associated with DNA hypomethylation on pericentromeric satellite regions during human hepatocarcinogenesis. Proc Natl Acad Sci USA. 2002;99:10060–5. doi: 10.1073/pnas.152121799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi MS, Shim YH, Hwa JY, Lee SK, Ro JY, Kim JS, Yu E. Expression of DNA methyltransferases in multistep hepatocarcinogenesis. Hum Pathol. 2003;34:11–7. doi: 10.1053/hupa.2003.5. [DOI] [PubMed] [Google Scholar]
- 34.Esfandiari F, Green R, Cotterman RF, Pogribny IP, James SJ, Miller JW. Methyl deficiency causes reduction of the methyl-CpG-binding protein, MeCP2, in rat liver. Carcinogenesis. 2003;24:1935–40. doi: 10.1093/carcin/bgg163. [DOI] [PubMed] [Google Scholar]
- 35.Ghoshal K, Majumder S, Datta J, Motiwala T, Bai S, Sharma SM, Frankel W, Jacob ST. Role of human ribosomal RNA (rRNA) promoter methylation and of methyl-CpG-binding protein MBD2 in the suppression of rRNA gene expression. J Biol Chem. 2004;279:6783–93. doi: 10.1074/jbc.M309393200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Majumder S, Huban K, Datta J, Summers D, Jacob ST, Ghoshal K. Epigenetic regulation of Metallothionein-I Gene Expression: Differential regulation of methylated and unmethylated promoters by DNA methyltransferases and methyl CpG Binding Proteins. J Cell Biochem. 2005 doi: 10.1002/jcb.20738. in press. [DOI] [PubMed] [Google Scholar]
- 37.Billard LM, Magdinier F, Lenoir GM, Frappart L, Dante R. MeCP2 and MBD2 expression during normal and pathological growth of the human mammary gland. Oncogene. 2002;21:2704–12. doi: 10.1038/sj.onc.1205357. [DOI] [PubMed] [Google Scholar]
- 38.Pogribny IP, James SJ. De novo methylation of the p16INK4A gene in early preneoplastic liver and tumors induced by folate/methyl deficiency in rats. Cancer Lett. 2002;187:69–75. doi: 10.1016/s0304-3835(02)00408-1. [DOI] [PubMed] [Google Scholar]
- 39.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–4. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 40.Kondo E, Gu Z, Horii A, Fukushige S. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol Cell Biol. 2005;25:4388–96. doi: 10.1128/MCB.25.11.4388-4396.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–42. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 42.Bachman KE, Rountree MR, Baylin SB. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem. 2001;276:32282–7. doi: 10.1074/jbc.M104661200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material can be found at: http://jn.nutrition.org/cgi/content/full/136/6/1522/DC1