

Summary
DNA transposases use a single active center to sequentially cleave the transposable element DNA and join this DNA to a target site. Recombination requires controlled conformational changes within the transposase to ensure that these chemically distinct steps occur at the right time and place, and that the reaction proceeds in the net forward direction. Mu transposition is catalyzed by a stable complex of MuA transposase bound to paired Mu DNA ends (a transpososome). We find that Mu transpososomes efficiently catalyze disintegration when recombination on one end of the Mu DNA is blocked. The MuB activator protein controls the integration vs. disintegration equilibrium. When MuB is present, disintegration occurs slowly and transpososomes that have disintegrated catalyze subsequent rounds of recombination. In the absence of MuB, disintegration goes to completion. These results together with experiments mapping the MuA-MuB contacts during DNA joining suggest that MuB controls progression of recombination by specifically stabilizing a concerted transition to the ‘joining’ configuration of MuA. Thus, we propose that MuB's interaction with the transpososome actively promotes coupled joining of both ends of the element DNA into the same target site and thus may provide a mechanism to antagonize formation of single-end transposition products.
Keywords: transposition target site, phage Mu, transposon, integrase
Introduction
The multiple steps of genetic recombination are often carried out by a single nucleoprotein complex1; 2; 3. To prevent formation of anomalous products—and thus potential damage to the genome—DNA processing within these complexes is carefully orchestrated. Recombinases of the transposase/retroviral integrase superfamily (the DDE-motif transposases) have an extra challenge as these proteins use a single active site to catalyze the two distinct chemical steps of DNA cleavage and DNA joining (a reaction called DNA strand transfer) 2; 4; 5. Successful recombination requires that each active site promotes the proper chemistry at the proper time and that the reaction progresses forward, such that the substrate DNA molecules are efficiently converted to the product configuration. How transposase-DNA complexes change throughout recombination to alter the active site and ensure this forward progress is largely unknown.
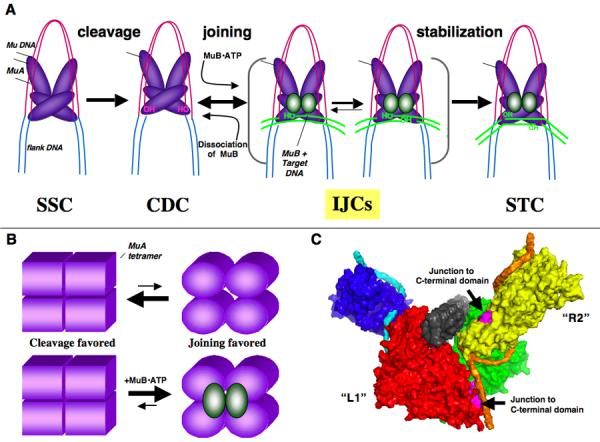
Mu encodes an extremely active DDE-transposase, the MuA protein, responsible for transposition of the phage genome6; 7. To initiate transposition, a tetramer of MuA assembles on specific DNA recognition sites (L1, L2, R1, and R2) located at the left and right ends of the Mu DNA (called the donor DNA). These assembled complexes are known as Mu transpososomes (for schematic of their organization see Figure 1A).
Figure 1. Overview of Mu Transposition Pathway.
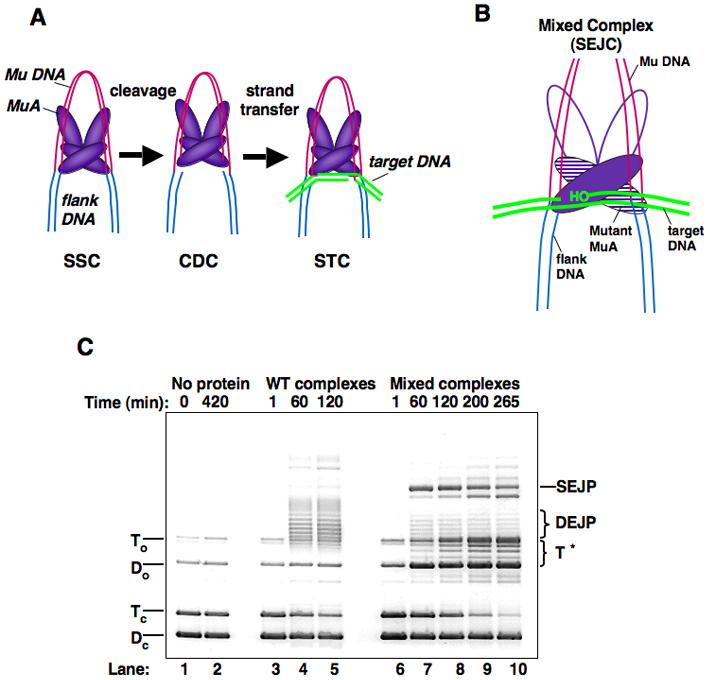
A) The stable synaptic complex (SSC) is formed upon assembly of the MuA tetramer at the ends of the Mu genome. MuA hydrolyzes the 3' end of each strand of the genome, resulting in a cleaved donor complex (CDC). After target DNA is delivered to the transpososome, the majority of the joined complexes undergo stabilizing changes to form final strand transfer complexes (STC). “Strand transfer” encompasses reaction steps between the CDC and STC, and the branched DNA structure stabilized within the STC is known as “strand transfer product” (STP). B) Cartoon of a strand transfer complex formed with a mixture of WT MuA and catalytically inactive MuA (DE/NQ). C) Agarose gel of plasmid recombination products. Reactions contained WT MuA or a mixture of MuA and MuA DE/NQ. Over long time courses (6 hours), the MuA reactions form double-end joined products (DEJP) (lanes 4-5.) During the same incubation period, mixed reactions form single-end joined product (SEJP) when complexes assemble with one WT MuA catalytic monomer and one MuA(DE/NQ) catalytic monomer (lanes 7-10). These reactions also yield a series of target-only products (T*) that migrate between To and Do. Reactions were stopped at times indicated in 0.2 x vol STOP solution. Dc: supercoiled donor, Tc: supercoiled target, Do: nicked donor, To: nicked target.
After some early assembly steps8; 9; 10, the Mu transpososome forms a stable synaptic complex (SSC or type 0 complex)11; MuA then cleaves the DNA at the junction of the Mu genome and flanking DNA to generate 3'OH nicked ends12. This form of the transpososome is called the cleaved donor complex (CDC or type 1 complex)12; 13; 14. With the help of the ATP-dependent activator protein, MuB, the transpososome associates with the new DNA segment that will serve as the site for transposon insertion (the target DNA)15; 16. The 3' hydroxyl groups generated by the cleavage step then attack and join to opposite strands of the target DNA in a reaction called DNA strand transfer; this step generates the strand transfer complex (STC or type II complex)17; 18; 19. Although the details of the structural changes that occur in the transpososome are not yet clear, recent biochemical work2; 20, a structural model of the transpososome based on EM image analysis21, as well as high-resolution structures of individual domains of MuA7; 21; 22, provide an excellent foundation for molecular studies.
The MuB activator plays several roles during transposition. MuB promotes assembly of the MuA tetramer, selects target DNA, and stimulates conversion of the transpososome from the CDC to the STC. MuB interacts with MuA via direct protein-protein contact with MuA's C-terminal domain16; 23. How MuA and MuB work together to carry out recombination has been difficult to analyze. MuB forms large, RecA-like polymers on DNA, and MuB's interaction with DNA is controlled by ATP-binding and hydrolysis24; 25. Solution and single-molecule fluorescence studies reveal that MuB binds DNA relatively non-specifically and with a high affinity when bound to ATP24; 25. In contrast, the ADP-bound MuB has a lower affinity for DNA. Thus, with each cycle of ATP-hydrolysis by MuB, the protein is thought to dissociate from the DNA24; 25. To direct recombination into a distant target site, MuB must be in the ATP- and DNA-bound form. However, both the ATP and ADP-bound forms of MuB can interact with MuA and stimulate recombination. Therefore, either the ATP- or ADP-bound form can promote recombination into a DNA site within the Mu genome, a site likely to be chosen by MuA without assistance from MuB23; 26. How MuA and MuB physically interact during transposition is not well understood, although regions of each protein that are likely to be involved in contacting the other protein have been identified23; 27; 28; 29. In addition, MuB has been suggested to act as an allosteric activator of MuA, and this model is supported and extended by the results of this study.
What drives the DNA joining process forward? For each strand transfer reaction, a phosphodiester bond in the target DNA is broken, while the new bond between the Mu DNA and the target DNA is formed. Thus, DNA joining is isoenergetic with respect to phosphodiester bonds. However, the transposition reaction proceeds in the forward direction because the STC is more stable than the CDC13. This increased stability must be due to changed contacts within the transposase-DNA complex that serve to ensure that most CDCs progress forward to complete strand transfer. In fact, the STC is such a stable complex that the host-encoded AAA+ ATPase ClpX is required to destabilize its protein structure prior to phage DNA replication30; 31; 32. Despite this evidence that the STC is an irreversible product of Mu transposition, little is known about specific changes that occur within the transpososome to generate the STC. Understanding this transition is essential to elucidating the overall mechanism of recombination and how transposition is regulated.
Here, we describe a state of the transpososome (the Initial Joined Complex, IJC) in which DNA joining has occurred at one end, but the active site is still in a configuration that can readily reverse this chemistry. These complexes are abundant when processing of one end of the Mu DNA is blocked by the presence of an inactive MuA subunit within the transpososome, and we propose that these complexes are transient intermediates in the transposition pathway promoted by the wild-type MuA transposase. Our results demonstrate that DNA joining and generation of the final, stable strand transfer complex are separable events, usually dependent upon the ability of the transpososome to complete recombination of both Mu DNA ends. Furthermore, we find that MuB, by binding to a subset of the MuA subunits in the transpososome , has a substantial controlling influence on whether the transpososome favors integration into, or disintegration from, the target. Thus, we propose that one function of MuB is to stabilize the transient IJC long enough for both ends of the Mu DNA to complete strand transfer. In this manner the same contacts between MuA and MuB can promote DNA strand transfer and antagonize disintegration. Once these strand transfer reactions are complete, the transpososome undergoes the final stabilizing conformational change to render joining effectively irreversible, in the presence or absence of MuB.
Results
Mixed mutant transpososomes efficiently generate relaxed target DNA
To probe the interactions within the Mu transpososome responsible for stabilizing final strand transfer products, we studied the properties of complexes that were only able to process one end of the Mu DNA due to the presence of a subunit (or subunits) carrying an active-site mutation (Figure 1B). These mixed transpososomes were assembled using a 1:1 ratio of wild-type MuA (MuA) and MuADE/NQ. MuADE/NQ carries two mutations in catalytic residues (D269N and E392Q). These mutations abolish the cleavage and joining activities of the transposase, but not its ability to assemble into stable transpososomes6.
As observed previously, transpososomes comprised of only wild-type MuA efficiently generated products that have both ends of the Mu donor plasmid (donor DNA) joined to the target DNA (DEJP) (Figure 1C, lanes 3-5). In contrast, transpososomes carrying a mixture of wild-type MuA and MuADE/NQ (mixed complexes) generated substantial amounts of the single-end joined product (SEJP), as well as cleaved donor DNA (Do) (Figure 1C, lanes 7-10)33. Reactions with mixed complexes also generated a family of novel products, apparently absent from reactions carrying only MuA. We refer to these products as T* (Figure 1C). The mobility of the T* products, as well as hybridization results, indicated that they contain only target DNA34 (see below). They are, however, formed by a process requiring MuA, MuB, Mg2+, and donor DNA. Mixed complexes made with MuA and either MuAD269N or MuAE392Q also formed the T* products, revealing that the both mutations in MuA's active site were not necessary for their production (data not shown).
To investigate the structure of the T* products, the DNA species generated by mixed transpososomes were analyzed by native two-dimensional gel electrophoresis followed by Southern hybridization with probes specific to either the target or the donor DNA (Figure 2A and data not shown). The second gel dimension was run in the presence of ethidium bromide and intercalation of this reagent into closed circular DNA molecules induces positive supercoiling, leading to rapid migration of this DNA through the gel matrix. By contrast, the conformation of nicked or linear DNA products is not greatly affected by ethidium bromide and therefore these molecules migrate slowly in the second dimension compared to their covalently closed counterparts.
Figure 2. Identification of novel products by 2D gel and Southern analysis.
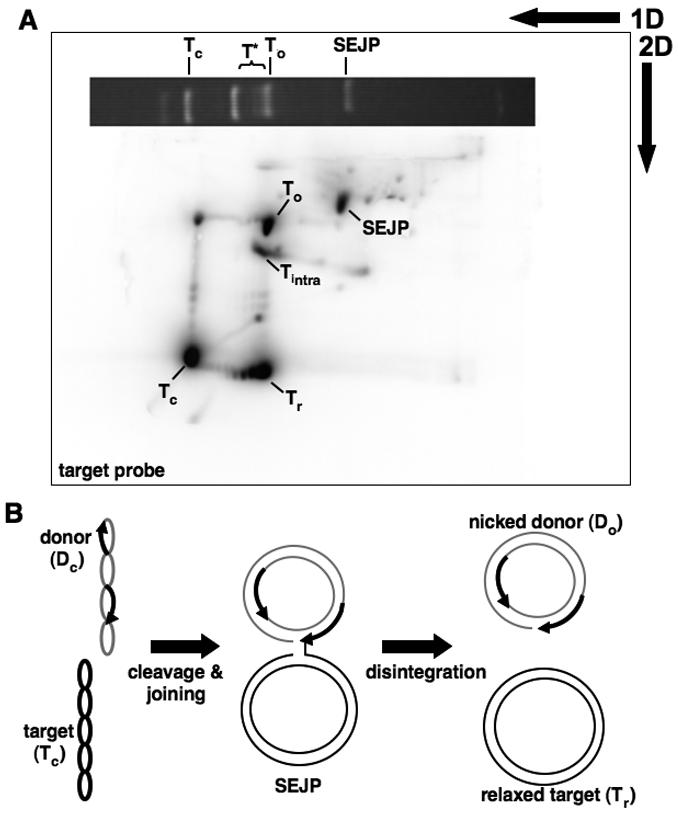
A) Southern blot hybridized with radiolabeled target DNA. A photo of the first dimension gel slice separating the mixed reaction products is positioned horizontally across the top of the blot to illustrate where the reaction products migrated. Tc: supercoiled target, To: nicked target, T* and Tr: closed, relaxed target, Tintra: intramolecular recombination product, SEJP: single-end joined product. B) Model for disintegration from SEJP. Supercoiled donor and target DNA molecules are joined to form SEJP. The free 3' OH group on the end of the target DNA attacks the Mu end, separating the SEJP into closed, relaxed target and nicked donor DNA.
Hybridization of a target probe revealed that the T* products migrated slowly, near the position of nicked target DNA, during the first dimension. However, in the second dimension the majority of these molecules migrated rapidly, in fact slightly more rapidly than the unreacted, supercoiled target DNA (Figure 2A). Hybridization with the donor DNA probe confirmed that these products contained only target DNA sequences (data not shown). We conclude that the majority of the T* DNA product is relaxed, covalently closed target DNA (marked Tr). In addition to this relaxed fraction, a portion of the T* DNA migrated between the nicked target (To) and nicked donor DNA (Do) in both gel dimensions (marked Tintra). These molecules are the transposition product described by Goldhaber-Gordon et al. (2002) in which the target DNA undergoes an intramolecular rearrangement. These products are generated when mixed transpososomes assemble using one Mu DNA end and one “pseudo end” present on the target DNA34.
The analysis presented above reveals that closed but relaxed target DNA is an abundant product of transposition by complexes containing a mixture of active and inactive MuA. Other abundant products include nicked donor DNA and the single-end joined product. Formation of relaxed target DNA suggests that during recombination the originally supercoiled target DNA is sometimes nicked and then re-closed. A distribution of supercoils (near zero) are trapped at the time of reclosure, therefore the Tr products appear as a ladder of topoisomers. Given the requirements for both MuA and MuB in generating these products, we hypothesized that disintegration of single-end joined complexes is likely responsible for generating the relaxed target (Figure 2B). Attack of the free 3' hydroxyl of the target DNA on the junction between the donor and target portions of the single-end joined product would lead to production of both relaxed target and nicked donor DNA.
Transpososomes with one joined Mu DNA end efficiently reverse strand transfer
To test for disintegration by transpososomes, we needed to establish a direct relationship between single-end joined complexes (SEJCs) and the relaxed (Tr) products. Therefore, we assembled and purified SEJCs and then asked if the DNA from these complexes could be “chased” into the Tr products upon further incubation (Figure 3A). The experiment was set up as follows: SEJCs were generated as before using a 1:1 mixture of MuA:MuADE/NQ, the Mu donor DNA plasmid, the target DNA, and MuB. These complexes were isolated by native gel electrophoresis from free DNA and protein. The purified complexes were then reactivated by the addition of Mg2+ to the gel slice (the 1st electrophoresis buffer contained EDTA, which blocks transposase activity), and incubated at 37°C (the “chase” step). Either immediately after Mg2+ addition, or after a 4 hour “chase”, the reactions were stopped, and the DNA was extracted from the complexes and run on a second gel to observe the distribution of DNA products (Figure 3B, lanes 3 and 4). Complexes containing only wild-type MuA were treated identically for comparison (Figure 3B, lanes 1 and 2). Likewise, reactions with either no MuA (lane 5), wild-type MuA (lane 6), or the MuA mixture (lane 7) were incubated for 10 hours without the first gel-purification step, as a control for the pattern of DNA products made during these long incubation/processing steps.
Figure 3. Isolated transpososomes chase into reversal products.

A) Description of the experimental procedure. B) Denaturing gel of transposition products after second incubation. In samples 1-4, purified complexes were incubated at 37°C in the presence of Mg2+ for the indicated amount of time. In samples 5-7, the entire reaction was incubated at 37° for 10hrs. Small amounts of Tc, Dc in lanes 1-4 are due to the method used to excise complexes from the native gel.
As expected, a major DNA species extracted from the purified SEJC prior to the second incubation was the SEJP. (Contamination by DEJP, and free donor and target DNA was also observed; this is not surprising, as there are also non-covalent donor-target DNA complexes that migrate in a similar position as the SEJC35 [data not shown]). However, after incubating the isolated SEJC for four hours, all detectable SEJP disappeared whereas the amount of relaxed target DNA (Tr) and cleaved donor DNA (Do) in this sample clearly increased. This experiment therefore strongly supports the hypothesis that the SEJC is the precursor of the relaxed target DNA and cleaved donor DNA. We conclude that mixed transpososomes that have joined one Mu DNA end to the target DNA efficiently catalyze disintegration from this target DNA.
In contrast to the results observed with the mixed complex SEJCs, transpososomes that contained exclusively wild-type MuA made only the double-end joined complexes (DEJCs) (Figure 3B, lanes 1, 2, and 6) and these complexes were not affected by the second incubation; little change in the distribution of DNA products was observed (lanes 1 and 2). These data indicate that disintegration is infrequent after both Mu DNA ends have completed DNA strand transfer.
In addition to supporting the reversal of joining hypothesis, this chase experiment also revealed that the isolated SEJCs promote disintegration more completely than do non-isolated complexes (compare Figure 1C, lane 10 to Figure 3B, lane 4). Without isolation, the SEJP was clearly present even after 7 and 10 hours. In contrast, the isolated complexes had completed reversal in less than 4 hours. Therefore, we looked for factors in the transposition reaction that influence the partitioning between SEJCs and complexes that have undergone disintegration.
The MuB activator promotes joining and antagonizes reversal
Because MuB is a strong stimulator of strand transfer complex formation23; 35; 36, we considered that it might have a specific role in preventing disintegration. To address this question, we developed a two-stage assay to monitor MuB's role in the fate of complexes (Figure 4A). In the first stage, mixed complexes were incubated in the presence of MuB and ATP to generate SEJCs. These complexes were then separated from free protein and nucleotide by gel filtration. Western blotting confirmed that all detectable MuB was removed from the fractions containing the protein-DNA complexes (data not shown). The persistence of the SEJCs was then measured (by the amount of SEJP present) as a function of time during a second incubation, with or without the addition of MuB and ATP.
Figure 4. Effect of MuB on reversal equilibrium.
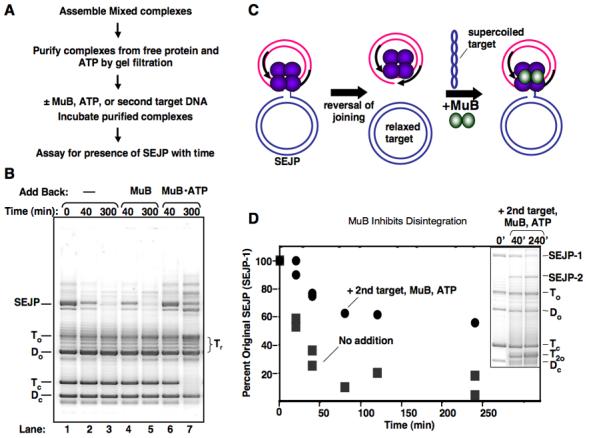
A) Description of experimental procedure. B) Agarose gel showing the presence of single-end joined product among complexes purified by gel filtration. “Time” refers to the time of 37°C incubation following purification of complexes. Nothing was added back to the sample in lanes 1-3. Lanes 4&5 had MuB (650nM) added back, and lanes 6&7 had MuB (650 nM) and ATP (2 mM) added back. Samples from reactions were stopped at times indicated in 0.2 x vol STOP solution. C) Illustration of a disintegration product undergoing a second round of strand transfer into a new molecule of target DNA. D) Sustained presence of SEJP with MuB-ATP addition is not entirely due to re-formation of SEJP. The original SEJP (SEJP-1) still persists in the presence of added back MuB-ATP and an excess of a second target DNA. An inset of the DNA products following gel filtration and incubation with added MuB and second target is also shown. Supercoiled second target is run off the end of the gel. No add back: dark grey squares; Add back of 2.6 uM MuB, 2 mM ATP, and 90 ug/ml pUC19: black circles.
When MuB was not added back to the purified complexes, more than half of the SEJC originally present disappeared after 40 min (Figure 4B lane 2). By five hours, less than 15% of these joined complexes remained (lane 3). Coincident with this disappearance in the SEJP was the appearance of the relaxed target (Tr), as expected for disintegration. By contrast, when MuB and ATP were added back to the purified complexes, the SEJP remained for much longer time periods; after five hours approximately half of the initial level of this product was still present (lane 7). ATP was required for this effect, indicating that nucleotide-bound MuB was responsible (lanes 5 and 7). ADP and ATPγS also increased the lifetime of the SEJC, although these nucleotides were not as effective as ATP (data not shown).
We considered two possible mechanisms to explain how MuB•ATP influenced the level of the single-end joined product: (1) MuB could interact with the Mu transpososomes that had catalyzed single-end joining in a manner that favors the joined configuration and thereby antagonizes the disintegration reaction (SEJP persistence); or (2) MuB could deliver target DNA molecules to free cleaved donor complexes (CDCs) and thereby promote the continued formation of new joined products (SEJP re-formation, Figure 4C).
Inspection of the product distribution from reactions containing MuB suggested that it was very likely that MuB was promoting re-formation of SEJPs. For example, in the reactions containing MuB in the second incubation, the supercoiled target DNA decreased throughout the time course, and new high-molecular weight DNA products accumulated (Figure 4B, compare lane 3 to lane 7). Continued transposition requires the presence of active CDCs among the purified complexes. New CDC formation via transpososome assembly is prevented under these conditions because the free MuA was removed by the gel filtration. (Also note that the amount of supercoiled donor DNA (Dc) did not change significantly during the second incubation.) Therefore, the most likely source of active CDCs is the single-end joined complexes that undergo disintegration. These complexes participate in re-formation of SEJPs.
We then sought to determine if MuB-stimulated re-formation of SEJPs was sufficient to account for the higher levels of these joined products or if MuB also influences the persistence of the SEJCs. To distinguish between the MuB-stimulated persistence and re-formation of SEJPs, we purified SEJCs and added back MuB•ATP along with a four-fold excess of a second target DNA. Because it is present at a higher concentration, the second target is more likely to be used by the CDCs and DNA joining should result in a new type of SEJP, distinguishable by gel electrophoresis since the two target DNAs are different sizes. Therefore, the probability of re-forming the original type of SEJP (SEJP-1) was decreased substantially in this experiment, and the presence of SEJP-1 would largely be a measure of the influence of MuB on the stability/lifetime of the pre-existing single-end joined complexes.
In the presence of MuB, ATP, and a second target DNA, the original single-end joined complexes (SEJC-1) persisted longer than in the absence of MuB (Figure 4D). Over four hours of incubation, SEJP-1 only decreased to 55% of its initial levels when both second target and MuB were added. Although some disintegration takes place over the time course, we conclude that the prolonged presence of SEJP-1 was due to MuB-stimulated persistence of SEJC-1s. Addition of the second target also prevented disappearance of the original supercoiled target DNA and formation of a second type of SEJP was observed (SEJP-2) (Figure 4D, inset). These findings lend further support to the conclusion that SEJCs that undergo disintegration give rise to active CDCs, which can then attack a second target DNA molecule.
Based on this analysis, we conclude that MuB has a substantial influence on the transpososome even after the first DNA joining reaction has been completed. MuB serves both to extend the lifetime of the initial joined complexes, as well as to promote the turnover of transpososomes that have completed disintegration.
MuB promotes a cooperative change to influence the activity of the Mu transpososome
Previous experiments20; 23; 26; 37 together with the analysis presented above reveal that MuB is a multifaceted activator of MuA and that it is especially important in driving the recombination reaction forward to the final strand transfer complex. To understand how MuB functions to control the activity of the transpososome, we sought to determine if it makes preferential contacts with specific MuA subunits within the transpososome. The transpososome is a homotetramer of MuA. This tetramer contains two types of MuA subunits: (1) those bound near the cleavage sites (to the R1 and L1 binding sites) that donate their catalytic domains for the DNA cleavage and joining reactions; and (2) those bound to the distal sites (R2 and L2) that do not contribute as directly to the reaction chemistry (see Figure 1A).
To test which subunits of the transpososome interact with MuB, we modified a method developed previously which involves “marking” MuA subunits bound to specific sites using UV-induced protein-DNA crosslinking38; 39. This crosslinking approach is achieved by using synthetic DNA fragments as the donor DNA. These fragments carry the first 50bp from the right end of the Mu DNA, with 5-Iodouracil (IdU) base substitutions at the R1 and R2 sites (Figure 5A). Upon exposure to UV light, the IdU bases crosslink to the MuA subunits bound directly to either the R1 or R2 sites. Since our donor DNA fragments contain nicks on either side of the IdU bases, denaturation of the DNA will yield a 10 base oligonucleotide covalently attached to a MuA subunit. By radiolabeling the 5' end of the IdU-containing oligonucleotide at either the R1 or the R2 position, it is possible to distinguish MuA subunits that bind to one site or the other. Those subunits that bind to the radiolabeled site, upon crosslinking, will become covalently attached to the 32P label via the DNA linkage. MuA subunits bound to the non-labeled site will still crosslink to the DNA, but as this DNA is not radiolabeled, the subunits will be invisible in subsequent autoradiography steps
Figure 5. Characterizing MuB's Interaction with the Transpososome.
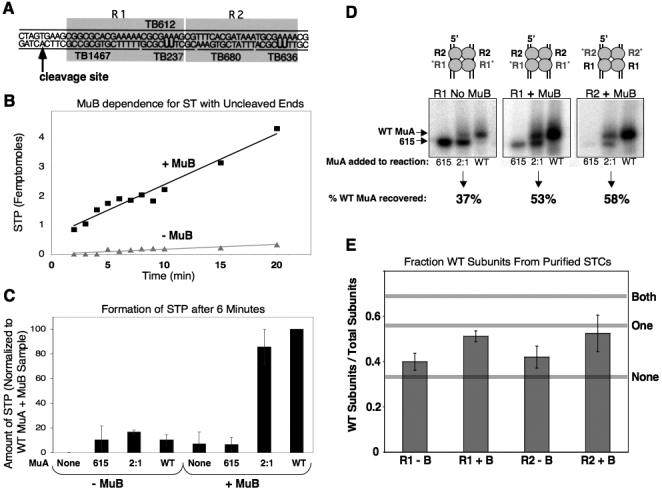
A) Diagram of crosslinkable Mu end fragment. Fragment is composed of four small oligonucleotides annealed to one long oligonucleotide. B) Accumulation of strand transfer products using un-cleaved Mu end fragments with or without MuB. C) Accumulation of strand transfer product after 6 minutes with un-cleaved Mu end fragments. Reactions contained either MuA 615, a 2:1 ratio of MuA 615 to WT MuA, or all WT MuA, in the presence or absence of MuB. Amount of strand transfer product was normalized to reactions containing all wild-type MuA in the presence of MuB. D) Autoradiograph of polyacrylamide gel from crosslinking experiment. Samples are purified STCs from reactions containing either MuA 615, a 2:1 ratio of MuA 615 to WT MuA, or all WT MuA, in the presence or absence of MuB. Oligonucleotides in this experiment were radiolabeled at either the R1 or the R2 position, as indicated. Percentages indicate the relative amount of WT MuA recovered from purified STCs from samples initially containing a 2:1 ratio of MuA 615 to WT MuA. E) Fraction of wild-type MuA subunits recovered from reactions containing a 2:1 ratio of MuA 615 to WT MuA (i.e. an initial fraction of WT MuA subunits equal to 0.333). Reactions were done in both the presence and absence of MuB, with either the R1 or R2 position radiolabeled. Horizontal grey lines correspond to the expected recovery of wild-type MuA under three possible scenarios: neither, one, or both MuA subunits at a given position are required to interact with MuB for efficient recombination.
To determine which subunits within the transpososome interact with MuB, we established conditions in which efficient recombination was highly dependent on the presence of MuB. As shown in Figure 5B, strong MuB-dependence was observed with donor fragments in which the transferred strand extended substantially past the cleavage site (here 28 nucleotides). In contrast, MuB had little effect on the reaction rate or efficiency when the donor DNA was pre-cleaved. Control experiments confirmed that MuB principally affected a post-cleavage step, rather than complex assembly or donor DNA cleavage20; 36 (data not shown).
To decipher whether MuB stimulates transposition by interacting with MuA subunits bound to the R1 and/or R2 positions, we performed in vitro transposition using the crosslinkable Mu end DNA fragments described above. Transpososomes were assembled, purified and assayed as follows. The modified Mu end fragments were incubated with a 2:1 ratio of MuA1-615 to full-length MuA and allowed to form mixed complexes. MuA1-615 carries a C-terminal truncation that renders it unable to interact with MuB, but fully functional in transpososome assembly and catalysis of recombination23. Target DNA, MuB and ATP were added to these mixed complexes and recombination was allowed to proceed. The reactions were then UV-irradiated and those complexes that had successfully recombined with the target DNA were isolated on a native agarose gel. Because the reaction conditions are such that recombination is strongly stimulated by MuB (Figure 5C), this step selects for tetramers that were able to interact with MuB. Finally, the purified complexes were run on a denaturing polyacrylamide gel that separates full-length MuA from MuA1-615. The autoradiograph of the gel revealed the ratio of MuA1-615 to full-length MuA bound to the radiolabeled site (either R1 or R2) in the purified, active complexes. If MuB is absent from the reaction, then we would expect to recover radiolabeled MuA from the purified complexes in approximately the same ratio as the starting conditions (2:1 MuA1-615:full-length MuA). Likewise, if MuB is present in the reaction but does not interact with the MuA subunits at a given site, we expect to recover a 2:1 ratio of MuA1-615:full-length MuA. However, if the MuA subunits at the assayed position do interact with MuB for stimulation of recombination, then there should be an enrichment of full-length subunits bound to that position in the purified complexes.
Full-length MuA was modestly enriched at both the R1 and R2 positions in the purified recombination complexes when MuB was present (Figure 5D). As a control, reactions were also performed in the absence of MuB. Although recombination was much less efficient without MuB, a small amount of product was generated under this condition (Figure 5C). As expected, there was less enrichment for full-length MuA at either the R1 or R2 position in these purified complexes (Figure 5D). Thus, we conclude that MuB-can contact both R1- and R2-bound MuA subunits to stimulate recombination.
In the experiments shown in Figure 5, samples that contained a 2:1 ratio of MuA1-615 to full-length MuA recombined at a rate that was ∼85% that seen in reactions containing only full-length MuA (Figure 5C). Since only 1.2% of the transpososomes that assembled with this 2:1 mixture of MuA are expected to contain four full-length MuA subunits (assuming random assortment during assembly, as our group has established previously38), it is very unlikely that MuB must interact with all four MuA subunits to stimulate efficient recombination.
We therefore further analyzed our data to investigate whether MuB must interact with either both R1-bound or both R2-bound subunits to stimulate transposition or whether fewer MuA-MuB contacts were sufficient for stimulation. Using the experimentally observed rates of STP formation in the presence or absence of MuB and basic probability, we were able to model the expected recovery of full-length MuA subunits under three possible reaction scenarios (Figure 5E, see grey horizontal lines). These scenarios were: (1) MuB must interact with both MuA subunits at a given position (either both R1 subunits or both R2 subunits) to stimulate recombination. (2) MuB must interact with only one MuA subunit at a given position (one R1 subunit or one R2 subunit) to stimulate recombination; or (3) MuB is not required to interact with either subunit at a given position to stimulate recombination. The bar graphs in Figure 5E indicate the experimentally observed recovery for full-length MuA in reactions either with or without MuB at either the R1 or R2 position. In reactions without MuB, the fraction of full-length MuA recovered from the purified STPs was close to that of the initial reaction conditions (e.g 0.33). When MuB was present, enrichment of full-length MuA was observed at both the R1 and R2 sites. However, the observed level of enrichment was significantly lower than would be expected if both subunits at a given position were required to interact with MuB for efficient recombination. Therefore, these data indicate that MuB can stimulate recombination by interacting with MuA subunits bound to either the R1 or R2 positions, but MuB need not interact with both of the subunits at either position to be effective.
That MuB does not need to contact both MuA subunits at either position is interesting, as >80% of recombination events resulted in successful strand transfer of both Mu end fragments (data not shown). Taken with previous experiments27; 40; 41, these results support the conclusion that MuB can interact with the MuA transpososome to stimulate recombination of one Mu DNA end without interacting with the MuA subunit that directly catalyzes recombination of that end. Thus, this analysis argues against a single architecture of the MuA-MuB-DNA complex that is essential for MuB-stimulated transposition. Rather, our data support a model in which MuB, by interacting with any of the MuA subunits in the complex, acts to stabilize a conformation of the transpososome that favors DNA joining (see Discussion). By this model, MuB could stimulate the DNA strand transfer step of recombination and antagonize the disintegration reaction using a common mechanism.
Discussion
Successful transposition requires the sequential processing of multiple DNA sites bound by a single transposase complex. The DNA strand transfer reaction is not intrinsically energetically favorable. Therefore, to drive transposition toward recombinant products, DNA joining must be coupled to a conformational change in the transposase-DNA complex that stabilizes the recombined DNA and thus effectively prevents reversal of DNA joining. By studying reactions catalyzed by transpososomes that can process only one of the two Mu DNA ends bound within the complex, we find evidence for a new state of the transpososome, the initial joined complex (IJC), in which MuA must be in a conformation that favors the DNA joining reaction, but the final stabilization process has not yet occurred (Figure 6A). These complexes, unlike strand transfer complexes, can efficiently promote disintegration to generate relaxed, covalently closed target DNA and active cleaved donor complexes. Thus, we conclude that STC stabilization is not obligatorily coupled to DNA joining, but a separable molecular event that normally occurs only after both Mu DNA ends are successfully joined. Furthermore, our data illustrate the need for this final stabilizing conformational change, as we find that the joined DNA products that cannot undergo this transition can efficiently revert to an unjoined state.
Figure 6. Model of How MuB Affects the MuA Tetramer.
A) Model for progression of Mu transposition. This model includes the IJC, a newly-defined state of the transpososome in which Mu DNA is joined to target DNA, but the final stable STC conformation is not yet achieved. MuB promotes the “joined” state of the transpososome and antagonizes disintegration. B) Model of MuB's allosteric effect on the cleavage-favored vs. joining-favored states of the MuA tetramer. Note, in both these pictures MuB is depicted as a dimer, as it behaves as a kinetic dimer in ATPase studies56. However, it is clear that the normal form of MuB that participates in recombination is a large, DNA-bound polymer56; 57. It remains to be determined how many subunits in the polymer interact with MuA to regulate transposition, although from the results presented here, fewer than four subunits are likely needed. C) Proposed structure of the transpososome based on the image analysis of Yuan et al (2005). The structure of the C-terminal domain of MuA is unknown, and therefore could not be included in this analysis. The pink areas in the “L1” (red) and “R2” (yellow) subunits show where the C-terminal (MuB-interaction) domain should connect with the rest of the protein. The Mu DNA backbone is light blue and orange and the target DNA is grey.
Analysis of reactions in which the IJC is well populated also revealed a new activity for the transposase-activator protein MuB. Previously, MuB was known to deliver target DNA to MuA16; 35, stimulate DNA strand transfer23, and protect transpososomes from premature destabilization by ClpX28. We find that MuB also antagonizes disintegration, thereby prolonging the lifetime of the IJCs. Thus, a possible role for MuB in the normal transposition pathway is to slow disintegration of the first Mu DNA end to be joined, so as to allow the second Mu DNA end a chance to also join to the target DNA. It is difficult to know how important this role of MuB might be in the normal reaction pathway. However, we have observed for many years that certain types of Mu DNA end fragments yield a high fraction of single-end joined products, and that this behavior is suppressed by MuB protein (see 38 and unpublished observations); these observations thus provide independent evidence indicating that MuB plays a general role in promoting double-end transposition.
In this study, disintegration was only convincingly detected with transpososomes containing one inactive catalytic center due to the presence of a mutant subunit. The amino acid substitutions in the mutant subunit may play a role in favoring disintegration. These active-site residues are undoubtedly in intimate contact with the substrate and product DNA molecules, and could therefore participate in important contacts that stabilize the final STC. We assume that the IJC is a short-lived state of the transpososome with wild-type MuA. Nonetheless, analysis of the artificially “trapped” IJCs provides mechanistic clues as to the conformational states of the transpososome and activities of MuB that are likely to contribute to the normal pathway but are difficult to analyze because the IJC to STC transition with the WT protein is so efficient.
The retroviral integrases42; 43; 44, as well as several DNA transposases45; 46; 47; 48, and the Rag recombinases49 have been shown to reverse DNA strand transfer. In the majority of these studies, the transposase/integrase was assembled on a synthetic DNA substrate designed to mimic the normal DNA product of this joining process. As a consequence of this experimental design, it has been difficult to determine if the protein assembles on these substrates in the same way that it would be bound if it had just made the new DNA junction. In fact, in some cases the products generated by transposase acting on these disintegration substrates clearly suggest alternative modes of transposase-DNA interaction45; 49; 50 (M. Mizuuchi, P.A. Rice and K Mizuuchi, personal communication). In contrast, we observe that transpososomes that have just completed DNA cleavage and joining of one Mu DNA end also catalyze the disintegration of that DNA end. Reversal is efficiently observed because, by blocking catalysis on one DNA end, we “trap” the transpososomes before they undergo the stabilizing events that normally accompany the final steps of recombination. Another study reports the reversal of DNA joining by MuA using transpososomes that had joined both DNA ends to a target site45. In this study reversal of joining on one Mu DNA end was observed after incubating STCs at high temperature (75 °C). Similar to the conclusions of our work, these authors deduce that after DNA joining, the Mu transpososome undergoes an important stabilizing conformational change that functions to prevent reversal. By incubating at high temperature, this conformation is destabilized, making reversal a detectable reaction.
Characterization of IJC transposomes provides new insight into how MuB modifies the activity of the Mu transpososome. When MuB is present, transpososomes reside for longer times in the state where the donor DNA is joined to the target site. In contrast, when MuB is removed from the mixed transpososomes, disintegration is efficient, and goes to completion. These observations, together with our data indicating that MuB needs to interact with only a subset of MuA subunits in the tetramer to stimulate recombination, are consistent with a simple two-state model for Mu transpososomes in which the complex exists in distinct conformations: the “cleavage favored” and the “joining favored” states (Figure 6B). Protein-protein contact between MuB and MuA is one means to tip the balance toward the joining-favored configuration. Our data indicate that MuB interacts with MuA subunits at both the R1 and the R2 positions. Furthermore, MuB does not need to interact with both subunits at either position to effectively stimulate recombination of both Mu ends. Therefore, MuB can stimulate recombination of a particular Mu end without directly interacting with the MuA subunit that is responsible for catalysis of that end. Previous studies suggested that MuB allosterically activates MuA20; 23; 38. Our new data extend these studies and support a model in which MuB functions to influence the conformational state of the transpososome in a concerted manner. Thus, we hypothesize that by favoring joined complexes, MuB acts both to stimulate strand transfer and to inhibit disintegration using a common mechanism. This model is still speculative and is proposed based on the activity of complexes; clearly, direct structural data will be required to validate this mechanism. We hope that this working model stimulates further analysis of the mechanism of MuB-dependent activation of transposition, as control of transposition by protein-protein contacts is increasingly being recognized as an important, but poorly understood, aspect of recombination.
That MuB appears to promote a concerted conformational change in the MuA tetramer by interacting with subunits at either the R1 or the R2 position is especially interesting given recent insight into the structure of the transpososome. The structures indicate that the MuA subunits at the R1 and R2 positions are in different relative orientations, and suggest that the two R2 subunits do not contact each other (Figure 6C)21. Therefore, it is not obvious how a signal needed for the conformational change would be transmitted between these subunits; a reasonable solution is that the signal is mediated through the R1-R2 interfaces. Furthermore, the proposed target DNA binding site within the transpososome (grey cylinder in Figure 6C) is near the C-terminal (MuB-interacting) domains of the R2 but not the R1 MuA subunits. How, then, do MuB molecules bound to the target DNA contact the MuA subunits at the R1 position? Must the target DNA wrap around to contact these subunits? How does the MuB polymer interact with this complex and how many MuB subunits contact MuA for optimal stimulation? Obviously, more experiments are necessary to understand the elemental steps involved in delivering the target DNA and promoting conversion to the “joining favored” state of the transpososome prior the generation of the final strand transfer complex.
How do other transposases prevent disintegration? Recent in vitro experiments suggest that the intrinsic efficiency of disintegration by different transposases varies widely. For example, the retroviral integrases and Rag recombinases are comparitively efficient at disintegration, whereas the Tn10 transposase promotes this reaction only feebly. It has been argued that efficient disintegration by the Rag proteins may be stabilizing to the genome, as it will prevent generation of Rag-induced genomic rearrangements49. In contrast, the Tn10 and Mu transposases are inefficient at catalyzing disintegration once the final strand transfer product is formed, thus driving the reaction toward transposon movement and the associated genome rearrangements. The cost is that the strand transfer complex is very stable, and requires energy-dependent disassembly by protein-unfolding enzymes to resolve these structures51. However, based on the results presented here, we find it attractive to consider that recombination pathways promoted by transposase/retro-viral integrase family members may have transient intermediate complexes in which disintegration is a robust reaction; these complexes would be well positioned to be regulated by interactions with cellular or element-encoded accessory proteins. Many transposases interact with other DNA binding proteins, and these interactions are increasingly found to have regulatory consequences. For example, Tn7 transposase is regulated by TnsC/D52, Ty5 integrase is regulated by the chromatin factor Sir4p53, and Ty3 integrase is regulated by promoter-bound transcription factor TFIIIB54. Controlling the joining versus disintegration choice may provide an important additional checkpoint to ensure the balance between successful transposition and healthy genomic integrity.
Materials and Methods
DNA
ϕX174 RFI was purchased from either New England Biolabs or Invitrogen. The mini-Mu plasmid was pMK58619, and was purified by CsCl/ethidium bromide ultracentrifugation. Oligonucleotides for fragment assays described were synthesized by Invitrogen and purified by denaturing PAGE.
Proteins
HU was purified as described by Baker and Luo (1994). Wild-type MuA, MuA (1-615), MuA(DE/NQ), MuA(E392Q), and MuA(D269N) were purified as in Baker et al. (1993). MuA and HU were diluted into 25 mM HEPES-KOH (pH 7.6), 300 mM NaCl, 0.1 mM dithiothreitol, and 10% glycerol23.
Wild-type MuB was purified by the method described in Yamauchi and Baker (1998). MuB dilutions were done in 1 M NaCl, 25 mM HEPES-KOH (pH 7.6), 0.1 mM EDTA, 20% glycerol, and 1 mM dithiothreitol.
Transposition Assays
Reversal products were observed following a two-step plasmid transposition assay. Blocking one end of the transpososome was achieved by a 1:1 ratio of WT MuA:MuA(DE/NQ) in the assay. The first incubation at 30°C was as described by Baker et al. (1994). Briefly, reactions included: 25 mM Tris-HCl pH8, 156 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol, 10 ug/ml BSA, 15% glycerol, 2 mM ATP, 10 ug/ml mini-Mu donor, 10 ug/ml target plasmid (usually ϕX174 RFI), 130 nM HU, 130 nM MuB, 26 nM wild-type MuA, and 26 nM MuA(DE/NQ). Addition of 0.02 vol 50% glycerol prior to a second incubation at 37°C34 enhanced the production of reversal products. Time points were stopped in 0.2 vol STOP solution (2.5% SDS, 50 mM EDTA, 30% glycerol, bromophenol blue). Reaction products were run on a 1.0% HGT-agarose (Cambrex) gel in 1 X TAB (40 mM Tris-HCl pH 8.0, 3.6 mM EDTA, 27 mM sodium acetate) at 70V for 2.5 hours. Gels were stained using Vistra Green (1:10000 dilution) and visualized on a Molecular Dynamics fluorimager.
The fragment assay in Figure 5B contained 25 mM Tris-HCl at pH8, 30 mM dithiothreitol, 2 mM ATP, 100 ug/ml bovine serum albumin, 15% glycerol, 100 mM NaCl, 0.1% Triton X-100, 10 mM MgCl2 , 48 nM donor fragment, 30 ug/ml ϕX174 RFI, 20 ug/ml MuA, 24 ug/ml MuB. Reactions were incubated at 30°, stopped in STOP solution (see above), and run on a 0.9% HGT-agarose gel. Gels were stained in Vistra Green (1:10000 dilution) and visualized on a Molecular Dynamics fluorimager.
Fragment assays in Figures 5C-E were performed as in 5B, except that the concentrations of donor fragment, ϕX174 RFI, MuA, and MuB were all increased 5 fold. The crosslinking experiments in Figures 5D and 5E were performed as described in Aldaz et al (1996).
2D Gel Analysis
To analyze the topologies and sequences of reversal products, two-dimensional gel electrophoresis was followed by Southern analysis. Products of mixed transposition reactions (described above) were run on a 0.4% HGT-agarose in 1X TBE (89 mM Tris, 89 mM boric acid, 2 mM EDTA) at 1 V/cm for 24 hours. The gel was stained in 0.3 ug/ml ethidium bromide and lanes of interest were excised over a UV transilluminator. Gel slices were embedded horizontally across the top of a 1.1% HGT-agarose gel with 5 ug/ml EtBr. The second dimension was run in 1xTBE and 5 ug/ml EtBr with recirculation of buffer. Products were transferred to a Gene-Screen Plus membrane in 10 X SSC (3 M NaCl, 0.3 M sodium citrate.) Products were visualized by hybridizing blots with 32P labeled fragments randomly primed off either target or donor plasmid. Blots were exposed to phosphorimager plates overnight and scanned using a Molecular Dynamics Phosphorimager.
Chase Experiment
SEJCs, formed by incubating plasmid transposition reactions at 30°C for 2 hours, were isolated by native gel electrophoresis (0.85% SeaPlaque agarose (Cambrex), 80 ug/ml BSA) in 1X TAB and 1 mM DTT with recirculation of the buffer. Half of the native gel was stained in 0.3 ug/ml EtBr, and complexes were visualized on a UVtransilluminator. Unstained complexes were then excised by aligning the two halves of the gel. Gel slices were submerged in plasmid assay buffer (25 mM Tris-HCl pH8, 156 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol, 10 ug/ml BSA, 15% glycerol) to a volume that was 3x their mass and incubated at 37°C. Products were extracted from gel slices at various time points using a Qiagen gel extraction kit. Complexes were denatured by addition of 0.2 vol SDS buffer and products were run on 0.9% HGT-agarose gel.
SEJC Purification by Gel Filtration
For purification of complexes by gel filtration, 5x assembly reactions were prepared (130 nM MuA and DE/NQ, 650 nM MuB, 650 nM HU, 50 ug/ml donor, 50 ug/ml target). Complexes were purified on a 1 mL column of either BioGel A15m beads (BioRad) or ABT 4% plain agarose beads (Iberagar)55. Peak fractions were pooled and incubated at 37°C for up to 5 hours with addition of one or more of the following components: ATP (2 mM), MuB (650 nM or 2.6 uM), pUC19 DNA (90 ug/ml). Samples of reactions were stopped at various intervals with 0.2 vol 5x STOP buffer. DNA products were analyzed by 1.0% HGT-agarose gel and visualized as described above. Presence of SEJP was analyzed using ImageQuant.
Acknowledgements
We are grateful to Tanya Sokolsky and Elizabeth S.C. Oakes for insights on this manuscript and technical suggestions on experiments, and we thank all Baker lab members for helpful discussions related to this project. We acknowledge early experiments done by Ilana Goldhaber-Gordon that led to this work. We thank George Chaconas for providing the structure files used for the model in Figure 6C. T.A.B is an employee of the Howard Hughes Medical Institute. This work was supported by NIH grant GM-49224.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Curcio MJ, Derbyshire KM. The outs and ins of transposition: from mu to kangaroo. Nat Rev Mol Cell Biol. 2003;4:865–77. doi: 10.1038/nrm1241. [DOI] [PubMed] [Google Scholar]
- 2.Gueguen E, Rousseau P, Duval-Valentin G, Chandler M. The transpososome: control of transposition at the level of catalysis. Trends Microbiol. 2005;13:543–9. doi: 10.1016/j.tim.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Grindley ND, Whiteson KL, Rice PA. Mechanisms of Site-Specific Recombination. Annu Rev Biochem. 2006 doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- 4.Rice PA, Baker TA. Comparative architecture of transposase and integrase complexes. Nat Struct Biol. 2001;8:302–7. doi: 10.1038/86166. [DOI] [PubMed] [Google Scholar]
- 5.Mizuuchi M, a. B. TA. Chemical mechanisms for mobilizing DNA. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. ASM Press; Washington, DC: 2002. pp. 12–23. [Google Scholar]
- 6.Baker TA, Luo L. Identification of residues in the Mu transposase essential for catalysis. Proc Natl Acad Sci U S A. 1994;91:6654–8. doi: 10.1073/pnas.91.14.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rice P, Mizuuchi K. Structure of the bacteriophage Mu transposase core: a common structural motif for DNA transposition and retroviral integration. Cell. 1995;82:209–20. doi: 10.1016/0092-8674(95)90308-9. [DOI] [PubMed] [Google Scholar]
- 8.Yin Z, Harshey RM. Enhancer-independent Mu transposition from two topologically distinct synapses. Proc Natl Acad Sci U S A. 2005;102:18884–9. doi: 10.1073/pnas.0506873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harshey RM, Jayaram M. The mu transpososome through a topological lens. Crit Rev Biochem Mol Biol. 2006;41:387–405. doi: 10.1080/10409230600946015. [DOI] [PubMed] [Google Scholar]
- 10.Pathania S, Jayaram M, Harshey RM. Path of DNA within the Mu transpososome. Transposase interactions bridging two Mu ends and the enhancer trap five DNA supercoils. Cell. 2002;109:425–36. doi: 10.1016/s0092-8674(02)00728-6. [DOI] [PubMed] [Google Scholar]
- 11.Mizuuchi M, Baker TA, Mizuuchi K. Assembly of the active form of the transposase-Mu DNA complex: a critical control point in Mu transposition. Cell. 1992;70:303–11. doi: 10.1016/0092-8674(92)90104-k. [DOI] [PubMed] [Google Scholar]
- 12.Craigie R, Mizuuchi K. Transposition of Mu DNA: joining of Mu to target DNA can be uncoupled from cleavage at the ends of Mu. Cell. 1987;51:493–501. doi: 10.1016/0092-8674(87)90645-3. [DOI] [PubMed] [Google Scholar]
- 13.Surette MG, Buch SJ, Chaconas G. Transpososomes: stable protein-DNA complexes involved in the in vitro transposition of bacteriophage Mu DNA. Cell. 1987;49:253–62. doi: 10.1016/0092-8674(87)90566-6. [DOI] [PubMed] [Google Scholar]
- 14.Lavoie BD, Chan BS, Allison RG, Chaconas G. Structural aspects of a higher order nucleoprotein complex: induction of an altered DNA structure at the Mu-host junction of the Mu type 1 transpososome. Embo J. 1991;10:3051–9. doi: 10.1002/j.1460-2075.1991.tb07856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maxwell A, Craigie R, Mizuuchi K. B protein of bacteriophage mu is an ATPase that preferentially stimulates intermolecular DNA strand transfer. Proc Natl Acad Sci U S A. 1987;84:699–703. doi: 10.1073/pnas.84.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adzuma K, Mizuuchi K. Target immunity of Mu transposition reflects a differential distribution of Mu B protein. Cell. 1988;53:257–66. doi: 10.1016/0092-8674(88)90387-x. [DOI] [PubMed] [Google Scholar]
- 17.Mizuuchi K. Mechanism of transposition of bacteriophage Mu: polarity of the strand transfer reaction at the initiation of transposition. Cell. 1984;39:395–404. doi: 10.1016/0092-8674(84)90018-7. [DOI] [PubMed] [Google Scholar]
- 18.Mizuuchi K, Adzuma K. Inversion of the phosphate chirality at the target site of Mu DNA strand transfer: evidence for a one-step transesterification mechanism. Cell. 1991;66:129–40. doi: 10.1016/0092-8674(91)90145-o. [DOI] [PubMed] [Google Scholar]
- 19.Mizuuchi M, Baker TA, Mizuuchi K. DNase protection analysis of the stable synaptic complexes involved in Mu transposition. Proc Natl Acad Sci U S A. 1991;88:9031–5. doi: 10.1073/pnas.88.20.9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams TL, Baker TA. Reorganization of the Mu transpososome active sites during a cooperative transition between DNA cleavage and joining. J Biol Chem. 2004;279:5135–45. doi: 10.1074/jbc.M308156200. [DOI] [PubMed] [Google Scholar]
- 21.Yuan JF, Beniac DR, Chaconas G, Ottensmeyer FP. 3D reconstruction of the Mu transposase and the Type 1 transpososome: a structural framework for Mu DNA transposition. Genes Dev. 2005;19:840–52. doi: 10.1101/gad.1291405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clubb RT, Schumacher S, Mizuuchi K, Gronenborn AM, Clore GM. Solution structure of the I gamma subdomain of the Mu end DNA-binding domain of phage Mu transposase. J Mol Biol. 1997;273:19–25. doi: 10.1006/jmbi.1997.1312. [DOI] [PubMed] [Google Scholar]
- 23.Baker TA, Mizuuchi M, Mizuuchi K. MuB protein allosterically activates strand transfer by the transposase of phage Mu. Cell. 1991;65:1003–13. doi: 10.1016/0092-8674(91)90552-a. [DOI] [PubMed] [Google Scholar]
- 24.Greene EC, Mizuuchi K. Visualizing the assembly and disassembly mechanisms of the MuB transposition targeting complex. J Biol Chem. 2004;279:16736–43. doi: 10.1074/jbc.M311883200. [DOI] [PubMed] [Google Scholar]
- 25.Greene EC, Mizuuchi K. Direct observation of single MuB polymers: evidence for a DNA-dependent conformational change for generating an active target complex. Mol Cell. 2002;9:1079–89. doi: 10.1016/s1097-2765(02)00514-2. [DOI] [PubMed] [Google Scholar]
- 26.Surette MG, Chaconas G. Stimulation of the Mu DNA strand cleavage and intramolecular strand transfer reactions by the Mu B protein is independent of stable binding of the Mu B protein to DNA. J Biol Chem. 1991;266:17306–13. [PubMed] [Google Scholar]
- 27.Mariconda S, Namgoong SY, Yoon KH, Jiang H, Harshey RM. Domain III function of Mu transposase analysed by directed placement of subunits within the transpososome. J Biosci. 2000;25:347–60. doi: 10.1007/BF02703788. [DOI] [PubMed] [Google Scholar]
- 28.Levchenko I, Yamauchi M, Baker TA. ClpX and MuB interact with overlapping regions of Mu transposase: implications for control of the transposition pathway. Genes Dev. 1997;11:1561–72. doi: 10.1101/gad.11.12.1561. [DOI] [PubMed] [Google Scholar]
- 29.Coros CJ, Sekino Y, Baker TA, Chaconas G. Effect of mutations in the C-terminal domain of Mu B on DNA binding and interactions with Mu A transposase. J Biol Chem. 2003;278:31210–7. doi: 10.1074/jbc.M303693200. [DOI] [PubMed] [Google Scholar]
- 30.Levchenko I, Luo L, Baker TA. Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes Dev. 1995;9:2399–408. doi: 10.1101/gad.9.19.2399. [DOI] [PubMed] [Google Scholar]
- 31.Nakai H, Kruklitis R. Disassembly of the bacteriophage Mu transposase for the initiation of Mu DNA replication. J Biol Chem. 1995;270:19591–8. doi: 10.1074/jbc.270.33.19591. [DOI] [PubMed] [Google Scholar]
- 32.Kruklitis R, Welty DJ, Nakai H. ClpX protein of Escherichia coli activates bacteriophage Mu transposase in the strand transfer complex for initiation of Mu DNA synthesis. Embo J. 1996;15:935–44. [PMC free article] [PubMed] [Google Scholar]
- 33.Baker TA, Kremenstova E, Luo L. Complete transposition requires four active monomers in the mu transposase tetramer. Genes Dev. 1994;8:2416–28. doi: 10.1101/gad.8.20.2416. [DOI] [PubMed] [Google Scholar]
- 34.Goldhaber-Gordon I, Williams TL, Baker TA. DNA recognition sites activate MuA transposase to perform transposition of non-Mu DNA. J Biol Chem. 2002;277:7694–702. doi: 10.1074/jbc.M110341200. [DOI] [PubMed] [Google Scholar]
- 35.Naigamwalla DZ, Chaconas G. A new set of Mu DNA transposition intermediates: alternate pathways of target capture preceding strand transfer. Embo J. 1997;16:5227–34. doi: 10.1093/emboj/16.17.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldhaber-Gordon I, Early MH, Baker TA. MuA transposase separates DNA sequence recognition from catalysis. Biochemistry. 2003;42:14633–42. doi: 10.1021/bi035360o. [DOI] [PubMed] [Google Scholar]
- 37.Yamauchi M, Baker TA. An ATP-ADP switch in MuB controls progression of the Mu transposition pathway. Embo J. 1998;17:5509–18. doi: 10.1093/emboj/17.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams TL, Jackson EL, Carritte A, Baker TA. Organization and dynamics of the Mu transpososome: recombination by communication between two active sites. Genes Dev. 1999;13:2725–37. doi: 10.1101/gad.13.20.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aldaz H, Schuster E, Baker TA. The interwoven architecture of the Mu transposase couples DNA synapsis to catalysis. Cell. 1996;85:257–69. doi: 10.1016/s0092-8674(00)81102-2. [DOI] [PubMed] [Google Scholar]
- 40.Yang JY, Kim K, Jayaram M, Harshey RM. A domain sharing model for active site assembly within the Mu A tetramer during transposition: the enhancer may specify domain contributions. Embo J. 1995;14:2374–84. doi: 10.1002/j.1460-2075.1995.tb07232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizuuchi M, Baker TA, Mizuuchi K. Assembly of phage Mu transpososomes: cooperative transitions assisted by protein and DNA scaffolds. Cell. 1995;83:375–85. doi: 10.1016/0092-8674(95)90115-9. [DOI] [PubMed] [Google Scholar]
- 42.Chow SA, Vincent KA, Ellison V, Brown PO. Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science. 1992;255:723–6. doi: 10.1126/science.1738845. [DOI] [PubMed] [Google Scholar]
- 43.Jonsson CB, Donzella GA, Roth MJ. Characterization of the forward and reverse integration reactions of the Moloney murine leukemia virus integrase protein purified from Escherichia coli. J Biol Chem. 1993;268:1462–9. [PubMed] [Google Scholar]
- 44.Gerton JL, Herschlag D, Brown PO. Stereospecificity of reactions catalyzed by HIV-1 integrase. J Biol Chem. 1999;274:33480–7. doi: 10.1074/jbc.274.47.33480. [DOI] [PubMed] [Google Scholar]
- 45.Au TK, Pathania S, Harshey RM. True reversal of Mu integration. Embo J. 2004;23:3408–20. doi: 10.1038/sj.emboj.7600344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stewart BJ, Wardle SJ, Haniford DB. IHF-independent assembly of the Tn10 strand transfer transpososome: implications for inhibition of disintegration. Embo J. 2002;21:4380–90. doi: 10.1093/emboj/cdf425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beall EL, Rio DC. Transposase makes critical contacts with, and is stimulated by, single-stranded DNA at the P element termini in vitro. Embo J. 1998;17:2122–36. doi: 10.1093/emboj/17.7.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polard P, Ton-Hoang B, Haren L, Betermier M, Walczak R, Chandler M. IS911-mediated transpositional recombination in vitro. J Mol Biol. 1996;264:68–81. doi: 10.1006/jmbi.1996.0624. [DOI] [PubMed] [Google Scholar]
- 49.Melek M, Gellert M. RAG1/2-mediated resolution of transposition intermediates: two pathways and possible consequences. Cell. 2000;101:625–33. doi: 10.1016/s0092-8674(00)80874-0. [DOI] [PubMed] [Google Scholar]
- 50.Mazumder A, Engelman A, Craigie R, Fesen M, Pommier Y. Intermolecular disintegration and intramolecular strand transfer activities of wild-type and mutant HIV-1 integrase. Nucleic Acids Res. 1994;22:1037–43. doi: 10.1093/nar/22.6.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burton BM, Baker TA. Remodeling protein complexes: insights from the AAA+ unfoldase ClpX and Mu transposase. Protein Sci. 2005;14:1945–54. doi: 10.1110/ps.051417505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stellwagen AE, Craig NL. Avoiding self: two Tn7-encoded proteins mediate target immunity in Tn7 transposition. Embo J. 1997;16:6823–34. doi: 10.1093/emboj/16.22.6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie W, Gai X, Zhu Y, Zappulla DC, Sternglanz R, Voytas DF. Targeting of the yeast Ty5 retrotransposon to silent chromatin is mediated by interactions between integrase and Sir4p. Mol Cell Biol. 2001;21:6606–14. doi: 10.1128/MCB.21.19.6606-6614.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirchner J, Connolly CM, Sandmeyer SB. Requirement of RNA polymerase III transcription factors for in vitro position-specific integration of a retroviruslike element. Science. 1995;267:1488–91. doi: 10.1126/science.7878467. [DOI] [PubMed] [Google Scholar]
- 55.Burton BM, Baker TA. Mu transpososome architecture ensures that unfolding by ClpX or proteolysis by ClpXP remodels but does not destroy the complex. Chem Biol. 2003;10:463–72. doi: 10.1016/s1074-5521(03)00102-9. [DOI] [PubMed] [Google Scholar]
- 56.Adzuma K, Mizuuchi K. Steady-state kinetic analysis of ATP hydrolysis by the B protein of bacteriophage mu. Involvement of protein oligomerization in the ATPase cycle. J Biol Chem. 1991;266:6159–67. [PubMed] [Google Scholar]
- 57.Greene EC, Mizuuchi K. Dynamics of a protein polymer: the assembly and disassembly pathways of the MuB transposition target complex. Embo J. 2002;21:1477–86. doi: 10.1093/emboj/21.6.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]