

Abstract
BH1115 is a gene from Bacillus halodurans strain C-125 that hypothetically encodes a rhamnogalacturonan acetyl esterase (RGAE) of the CE-12 family. As confirmation, this gene was cloned, and the product was expressed in Escherichia coli strain Rosetta (DE3) cells and purified. The enzyme obtained was monomeric, with a molecular mass of 45 kDa, and exhibited alkaliphilic properties. A study of the inhibition of the activity by some modulators confirmed that the catalytic triad for the esterase activity was Ser-His-Asp. This enzyme also presents broad substrate specificity and is active toward 7-aminocephalosporanic acid, cephalosporin C, p-nitrophenyl acetate, β-naphthyl acetate, glucose pentaacetate, and acetylated xylan. Moreover, RGAE from B. halodurans achieves a synergistic effect with xylanase A toward acetylated xylan. As a member of the SGNH family, it does not adopt the common α/β hydrolase fold. The homology between the folds of RGAE from Aspergillus aculeatus and the hypothetical YxiM precursor from Bacillus subtilis, which both belong to the SGNH family, illustrates the divergence of such proteins from a common ancestor. Furthermore, the enzyme possesses a putative substrate binding region at the N terminus of the protein which has never been described to date for any RGAE.
Rhamnogalacturonan acetyl esterase (RGAE; EC 3.1.1.-) is a carbohydrate esterase which belongs to the CE-12 family inside the carbohydrate esterase classification (CAZy family) (6). Regarding this classification, this enzyme catalyzes the deacetylation of rhamnogalacturonan I, which is one of the most complex pectic polysaccharides present in the wall of higher plants (13). The polysaccharide rhamnogalacturonan I is composed of alternating rhamnose and galacturonic acid residues. The latter can have acetylations at the C-2 and C-3 positions (13), and the removal of such acetyl groups facilitates the action of lyases and hydrolases, since the acetylation sterically hinders the cleavage of the glycosyl linkages (21).
Only a few enzymes have been characterized within the CE-12 family. The importance of the CE-12 family lies, for instance in the industrial application of some of its members for the production of β-lactam antibiotics or bleaching of paper. Moreover, Bacillus halodurans RGAE (BhRGAE) belongs to the SGNH hydrolase family, which has unique structural features. However, this family is not well-known, since not all its members have been characterized. Thus, the characterization of these members could help in understanding some of the features of this family, which is closely related to the well-known α/β hydrolase family.
One member of the SGNH hydrolase family is a cephalosporin C deacetylase (CCD) from Bacillus sp. KCCM10143 which has been used for the industrial transformation of 2-methoxyimino-2-furylacetyl cephalosporanic acid (MIFACA) and 7-aminocephalosporanic acid (7-ACA) into their corresponding 3-deacetyl compounds, which are advanced intermediates in the production of semisynthetic β-lactam antibiotics (4, 5). Another characterized enzyme is an RGAE from Aspergillus aculeatus (AaRGAE) which has been studied to determine its kinetic (15) and crystallographic structure (20). This enzyme has not been characterized with derivatives of the cephalosporins, but sequence alignment reveals that CCD and AaRGAE belong to the SGNH superfamily (20). The SGNH family is characterized by the following features: (i) four blocks of conserved residues, (ii) a central five-stranded parallel β-sheet, (iii) a lack of the nucleophilic elbow motif, (iv) nucleophilic Ser placed on a type I β-turn at the carboxyl end of β-strand 1, and (v) Asp and His of the catalytic triad are much closer at the same loop region than in the α/β hydrolase fold (20). Other proteins have been included inside this structural family (SCOP superfamily c.23.10), even though they do not belong to the carbohydrate esterase family CE-12, e.g., platelet-activating factor acetylhydrolase, isoform Ib, the alpha subunit from Bos taurus, and esterase A from Streptomyces scabiei (20).
An unknown conserved protein inside the CE-12 family, BH1115 from Bacillus halodurans C-125 (NCBI accession no. BAB04834.1), is interesting because it came from a microorganism which can grow well in alkaline environments (pH 7 to 10.5) (27, 30). This feature predicts an enzyme with alkaliphilic properties, which is often required in industrial applications where harsh conditions exist (8, 29). In addition, its sequence not only reveals the highest identity with enzymes from other microorganisms, like beta-tubulin (51%) from Bacillus licheniformis DSM 13 and a putative pectin acetyl esterase (51%) from Clostridium beijerinck NCIMB8052, but also with YesT (50%) from Bacillus subtilis, CCD (41%), and AaRGAE (39%). Thus, it seems to be a new member of the SGNH family.
The aims of this paper were to describe the cloning and overexpression, for the first time, of the BH1115 protein derived from B. halodurans C-125, in Escherichia coli Rosetta (DE3) cells, to characterize it, and to obtain its three-dimensional model and its topology scheme.
MATERIALS AND METHODS
Strains, plasmid, enzymes, and chemicals.
Bacillus halodurans C-125 genomic DNA was prepared by the method described previously (28). E. coli Rosetta (DE3) cells and plasmid pET28a were from Novagen (United States). Restriction enzymes were obtained from New England BioLabs (Beverly, MA). T4 DNA ligase was from Roche Diagnostics. Pfu Turbo DNA polymerase and its 10× reaction buffer were from Stratagene (La Jolla, CA). The DNeasy system for genomic DNA isolation, QIAprep spin plasmid miniprep kit, and QIAquick PCR purification kit were from QIAgen (United States). Substrates, antibiotics, and broths were from Sigma. Ultrafiltration and microfiltration membranes, His-Trap FF chelating, PD10, phenyl sepharose and Superdex 200 prepacked columns were from GE Healthcare (Barcelona, Spain). Acetylated xylan was prepared following the method described by Johnson et al. (14). All other chemicals were of reagent grade and obtained from commercial sources.
Isolation of the BH1115 gene from B. halodurans.
The cloning and transformation techniques used were essentially those described by Sambrook et al. (24). PCR amplification of the BH1115 gene was obtained using Pfu Turbo DNA polymerase (Stratagene) and the following oligonucleotide sequence: BhRGAE-up (5′ GGAATTCATGGAATTGGTGGTGCAAAATCGAG 3′), derived from the 5′, and BhRGAE-down (5′ GGCGGCCGCTCATCGCTGTTCCTCCTTCAATGT 3′), derived from the 3′, of the nucleotide sequence of the BH1115 gene. The sequences recognized by restriction enzyme EcoRI in BhRGAE-up and restriction enzyme NotI in BhRGAE-down are underlined. This amplification yielded a product of 1,112 bp, which represents the entire BH1115 gene plus the sequences recognized by the restriction enzyme. The PCR product was digested with EcoRI and NotI, purified by using a QIAquick PCR purification kit (Qiagen), and inserted into the expression vector pET28a downstream of the T7 RNA polymerase promoter, which had been previously digested with the same restriction enzymes as the PCR product. This produced the recombinant plasmid pET28a6His-BhRGAE, which encodes an additional 36-amino-acid N-terminal sequence containing a six-histidine tag. Plasmids were transferred to E. coli Rosetta (DE3) electrocompetent cells (Novagen) and plated onto LB-kanamycin-chloramphenicol agar plates overnight. Then, cells were picked and transferred into 50-ml shake flasks containing LB-kanamycin-chloramphenicol supplemented with 1 mM isopropyl thio-β-d-galactoside (IPTG). The correct sequence was checked by DNA sequencing.
Gene expression and protein purification of recombinant BhRGAE.
Recombinant Rosetta (DE3) cells were grown at 37°C in a TB medium containing kanamycin 50 μg·ml−1 and chloramphenicol 34 μg·ml−1 and induced at selected values of the optical density measured at 600 nm by adding IPTG at different concentrations to determine the optimum amount of inductor. After induction, the cells were grown at 30°C and collected at intervals (from 1 to 24 h) by centrifugation. A 100-ml LB culture was used to inoculate 1 liter of Terrific broth-kanamycin-chloramphenicol medium, which was grown in a 2.5-liter flask. Crude extracts were lysed with a mill homogenizator (Minizeta IIE; Netzsch, Germany) from a cell suspension obtained by diafiltration of the broth, which had been previously concentrated by microfiltration with a 500-kDa-cutoff membrane on a QuixStand system (GE Healthcare) with 100 mM potassium phosphate buffer, pH 7.3. The insoluble fraction of the lysate was removed by centrifugation at 5,500 × g for 15 min at 4°C. The supernatant was cleaned of nucleic acids with 3 U·ml−1 DNase I (Sigma) for 30 min at room temperature, followed by centrifugation at 5,500 × g for 15 min at 4°C. The extract was concentrated by ultrafiltration with a 10-kDa cutoff membrane on a QuixStand system (GE Healthcare) and diafiltrated twice with 100 mM potassium phosphate buffer, pH 7.3. The enzyme solution was applied to a Resource Q ionic column (GE Healthcare). The protein was eluted, desalted on PD10 columns (GE Healthcare), and applied to a HisTrap FF chelating affinity column (1 ml) (GE Healthcare) equilibrated with 0.5 M NaCl and 5 mM imidazole using an ÁKTA-FPLC system (GE Healthcare). The bound enzyme was eluted with 100 mM potassium phosphate buffer, pH 7.3, in a gradient from 5 to 500 mM imidazole. The fractions containing RGAE activity were pooled, desalted on PD10 columns (GE Healthcare), and stored at −80°C in 100 mM potassium phosphate buffer, pH 7.3, containing 10% glycerol.
Protein determination and enzyme assays.
The protein concentration was determined by the bicinchoninic acid dye-binding procedure, using bovine serum albumin as the standard (26). Protein elution from the columns during purification was monitored as the absorbance at 280 nm.
The molecular weight of the recombinant protein was determined by gel filtration onto a Superdex 200 column (GE Healthcare) which had been previously calibrated using standard proteins of known molecular weight. The molecular mass was also determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (32) using 12% acrylamide resolving gel. Samples were heated at 100°C for 5 min in 2% SDS, 5% 2-mercaptoethanol and run in comparison with molecular weight standards. The protein percentage and size were determined using an image scanner (Amersham Biosciences) to analyze the image obtained with the Image Quant TL (Amersham Biosciences) software.
The enzyme was assayed spectrophotometrically by measuring the conversion of 1 mM p-nitrophenyl acetate (pNPA) to p-nitrophenol at 37°C for 5 min at 405 nm. The standard reaction mixture contained (per milliliter) 1 mM of substrate and 50 μg of enzyme in 100 mM potassium phosphate buffer, pH 7.3 (2). One unit of activity was defined as the amount of enzyme required to produce 1 μmol product per min under the standard assay conditions. The increase in absorbance at 405 nm was also followed using 96-well plates in a Synergy HT spectrophotometer (Biotek, United States). The catalysis toward glucose pentaacetate, 7-ACA, cephalosporin C, and acetylated xylan was quantified by the determination of the acetic acid released, using a colorimetric assay based on a pH indicator adapted to the microplates (17), also at pH 7.3, due to the intrinsic chemical instability of the three former substrates. Naphthyl derivatives were analyzed by using 2 mM α- or β-naphthyl acetate and 5.13 mM fast red salt in 100 mM potassium phosphate buffer, pH 7.3. The reaction was followed at 480 nm, as previously described (2).
Inhibition studies and effect of metal ions and chelating agents.
The enzyme (50 μg) was incubated at 25°C in 100 mM potassium phosphate buffer, pH 7.3, for 5 min with different concentrations of each inhibitor. The reactions were stopped by chilling on ice, and aliquots (100 μl) were measured by pNPA assay. The influence of metal ions and the chelating agent EDTA on the activity was investigated by the addition of these compounds to final concentrations of 2 and 10 mM in the same reaction mixture used for the pNPA assay.
Thermostability and thermophilicity studies.
Enzyme stability was studied over the temperature range of 20 to 70°C. The enzyme (50 μg) was incubated in 100 mM potassium phosphate buffer, pH 7.3, for 1 h at different temperatures. Aliquots were withdrawn at regular intervals and assayed at 25°C by pNPA assay. The dependence of enzymatic activity on temperature was studied over the range of 20 to 50°C with pNPA. The activity was calculated, taking into account the autohydrolysis of the substrate with the temperature.
pH stability and determination of optimum pH.
In the pH stability study, the residual enzyme activity was measured after the incubation of aliquots of the enzyme in 100 mM buffer from pH 6.5 to 11 at 25°C for 1 h. Aliquots (100 μl) were measured by pNPA assay, with a final enzyme concentration of 50 μg·ml−1. Potassium phosphate buffer was used for pH 6.5 to 8, sodium pyrophosphate buffer for pH 8 to 9.5, and boric acid for pH 9.5 to 11.
The determination of the optimum pH was performed with the same buffers as described above. The concentration of the enzyme in the reaction mixture was 50 μg·ml−1, and the substrate concentration was 1 mM pNPA. The molar absorption of the product of the reaction (p-nitrophenol) changes with different pHs, so the activity was corrected using the appropriate value. In addition, in the case of 7-ACA, cephalosporin C, and glucose pentaacetate, the activities were not determined above pH 8.0, due to their chemical instability above this pH.
Synergy with xylanase.
Per milliliter, the reaction mixture contained 100 mM phosphate buffer, pH 7.3, 10 mg of acetylated xylan, 25 U of recombinant xylanase A from Trichoderma viridae (Sigma), and 13 U of BhRGAE product (measured with pNPA) (0.84 mg of enzyme). The incubation was carried out at 25°C. The release of acetate was determined by the acetic acid kit assay (Boehringer Mannheim, Germany).
Sequence analysis and structure homologies.
The protein similarity searches and alignment were performed using the data from CLUSTAL-W (31) and from other programs, such as Gapped BLAST and PSI-BLAST (1). Model building was developed by using SWISS-MODEL and the Swiss-PdbViewer (10) programs (http://www.expasy.ch/spdbv) using Protein Data Bank accession no. 2o14 (hypothetical YxiM precursor) as template, which has a sequence identity of 26% with BhRGAE. The ESPript output was used to render the analysis of multiple sequence alignments (9). Topology diagrams were prepared using TOPS (19).
RESULTS AND DISCUSSION
Heterologous gene expression and purification of recombinant His6-BhRGAE.
The locus tag BH1115 was amplified by PCR from the genomic DNA from B. halodurans C-125. The fusion protein produced also had the 30 additional residues added at the N terminus of the protein, introduced by the pET28a vector, before the starting methionine of the original BhRGAE (MGSSHHHHHHSSGLVPRGSHMASMTGGQQMGRGSEF). The recombinant plasmid pET28a6His-BhRGAE was used to transform electrocompetent E. coli Rosetta (DE3) cells. Optimization of the IPTG concentration and induction time were performed (data not shown), with the highest specific activity being achieved after induction with 1 mM IPTG for 14 h at 30°C. BhRGAE was expressed totally in soluble form (data not shown) and represented 24% of the total protein when the results of SDS-PAGE were analyzed (Fig. 1, lane 2). Protein production for the following steps was obtained in 1 liter of TB medium under the optimized conditions.
FIG. 1.
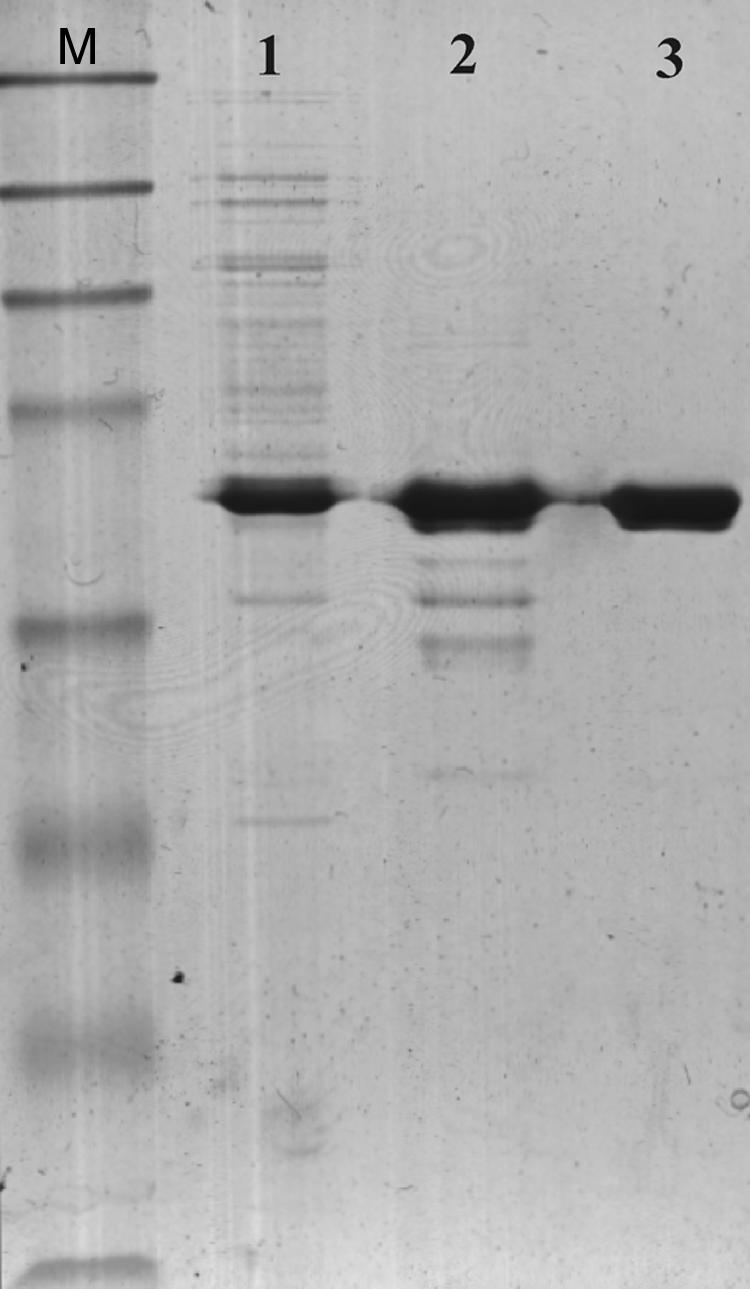
SDS-PAGE of the BH1115 gene product (BhRGAE) after 14 h of induction with 1 mM IPTG. Each lane contained 5 mg protein. Lane M, molecular mass markers (maltose-binding protein β-galactosidase, 175 kDa; maltose-binding protein paramyosin, 83 kDa; glutamic dehydrogenase, 62 kDa; aldolase, 47.5 kDa; triosephosphate isomerase, 32.5 kDa; β-lactoglobulin A, 25 kDa; lysozyme, 16.5 kDa; aprotinin, 6.5 kDa); lane 1, crude extract; lane 2, Resource Q column pooled fraction; lane 3, His-trap FF column pooled fraction.
The soluble recombinant BhRGAE expressed into E. coli Rosetta (DE3) cells was unable to bind onto a Ni2+-chelating column (His-Trap FF; GE Healthcare), maybe because of the binding of the enzyme to other components of the crude extract. Therefore, partial purification of the extract with a strong ion exchange column (Resource Q) was necessary before it was applied to a Ni-chelating column. As a result of this procedure, a homogeneous enzyme was obtained, as shown by the results of SDS-PAGE (Fig. 1, lane 3), with 7.6-fold purification and 60% yield (data not shown). The overall BhRGAE expression achieved a specific activity of 15.43 U·mg−1 protein (measured toward 1 mM pNPA) (Table 1), which represents 15- and 2.6-fold increases in specific activity compared with the published data for CCD (1.03 U·mg−1) (4) and AaRGAE (15) (6 U·mg−1), respectively.
TABLE 1.
Kinetic parameters of recombinant BhRGAEa
| Substrate | Km (mM) | Vmax (μM/min) | Vmax/Km |
|---|---|---|---|
| pNPA | 14.1 | 5,635 | 391 |
| α-Naphthyl acetate | 0.9 | 11 | 12 |
| β-Naphthyl acetate | 1.2 | 36 | 30 |
| Glucose pentaacetateb | 9.1 | 9,356 | 1,028 |
| 7-ACAb | 51.4 | 1,021 | 20 |
| Cephalosporin Cb | 78.7 | 670 | 9 |
Reactions were performed at 37°C and with 50 μg of protein in the standard reaction medium.
The experiment was carried out at pH 7.3, due to the substrate's chemical instability.
The molecular mass of the purified enzyme was 45 kDa by SDS-PAGE (Fig. 1, lane 3). This value was the same as that determined by gel permeation chromatography on a Superdex 200 column (data not shown), indicating the monomeric nature of BhRGAE. The overexpressed protein represented 24% of the total protein when the results of SDS-PAGE were analyzed by densitometry. Thus, the anomalous degree of purification (7.6-fold) achieved could be due to the removal of a possible inhibitor in the crude extract of E. coli cells. The deviation of the measured molecular mass from the theoretical value calculated for the sequence is explained by the expression of BhRGAE as a fusion protein with an additional 36-amino-acid tag at the N terminus. Thus, BhRGAE is a monomer, as occurs in AaRGAE (15), but different from CCD, which is a dimer (5).
Kinetic determinations.
The kinetic parameters were determined at pH 7.3 using the purified recombinant BhRGAE toward several acetylated substrates and showing that the best substrates for the enzyme were glucose pentaacetate and pNPA, according to the highest maximum initial velocity (Vmax)/Km values (Table 1). Moreover, BhRGAE was active toward 7-ACA and cephalosporin C. Although the Km values for the last substrates were higher than those reported for CCD, the Vmax values were similar (4). This represents an improvement from the industrial point of view, since deacetyl derivatives could be obtained in a higher volumetric yield (g·liter−1).
In addition, BhRGAE was active toward acetylated xylan, as measured by the acetate released (data not shown). This release increased 10-fold in the presence of commercial xylanase A from Tricoderma ressei, showing a clear synergistic effect, as occurs in nature. This could be explained by the fact that BhRGAE removes the acetyl groups from small xylooligosaccharides more easily than from the complete polysaccharide. This behavior has been previously reported for acetyl xylan esterase from Orpinomyces sp. (3) and clearly distinguishes BhRGAE from AaRGAE, since the latter does not release acetate from acetylated xylan (15).
The thermal stability of BhRGAE and its optimum temperature were determined by carrying out enzymatic assays at different temperatures from 20 to 70°C. The enzyme was mesophilic, with a maximum activity at 40°C under the conditions used. In addition, the enzyme was temperature sensitive above 50°C, with a half-life of less than 5 min.
BhRGAE was sensitive to pH, showing the highest catalytic efficiency around pH 8.0 and with pNPA as substrate. This clearly differs from the results obtained with AaRGAE (15), which presents an optimum pH of 6.0, at which the activity of BhRGAE is almost zero. However, this optimum pH was similar to that found in CCD from Bacillus sp. (4). The enzyme showed its maximum pH stability above pH 8.0 (data not shown), as expected from an enzyme that comes from an alkaliphilic microorganism.
BhRGAE was inhibited by diethylpyrocarbonate at 25 mM, showing the importance of the His residue in the catalytic triad. Incubation with a Ser-modifying-mechanism-based inhibitor, phenylmethylsulfonyl fluoride, checked the presence of the nucleophilic Ser at the active site (12). Phenylmethylsulfonyl fluoride completely inhibited the enzyme at 30 mM; dimethyl phosphite achieved only a 67% inhibition with a high concentration (900 mM), which reveals BhRGAE's insensitivity to phosphonate esters compared with previously reported data for a cephalosporin esterase (10 mM) (22). In addition, BhRGAE was hardly inhibited by metal ions. Only a small inhibiting effect was achieved with Ni2+ and Co2+ ions, whereas the enzyme suffered no effect or little activation with the rest of the ions tested (data not shown). These results differ from others previously described, such as for acetyl xylan esterase from Schizophyllum commune, which is drastically inhibited by 1 mM Mn2+ (85%) (11), but they are in agreement with those from some lipases that are activated by some ions (16).
In silico analysis of BH1115.
Sequence analysis of the B. halodurans C-125 genome (30) allowed the identification of a 1,098-bp open reading frame (annotated as BAB04834.1) which potentially coded for a putative protein inside the carbohydrate esterase family CE-12. The derived amino acid sequence predicted a polypeptide consisting of 366 residues with a calculated molecular mass of 41 kDa and a pI of 6.04. All nonredundant databases were screened for entries. They showed high levels of similarity for parts of the protein with a beta-tubulin from B. licheniformis DSM 13 and a putative pectin acetyl esterase from Clostridium beijerinck NCIMB8052 but, also, similarity with the RGAE characterized from A. aculeatus and CCD from Bacillus sp. KCCM10143, using the Gapped BLAST and PSI-BLAST programs (1). Using the HMMER website (http://selab.janelia.org/), it was also possible to distinguish that the first 135 residues of the protein are similar to the fibronectin type III domain (FnIII) and the rest are similar to RGAE from Aspergillus aculeatus. Fibronectins are multidomain glycoproteins found in a soluble form in plasma and in an insoluble form in loose connective tissue and basement membranes. They contain multiple copies of three repeat regions (types I, II, and III) which bind to a variety of substances, including heparin, collagen, DNA, actin, fibrin, and fibronectin receptors on cell surfaces (7). The 10th FnIII repeat contains an RGD cell recognition sequence in a flexible loop between two strands. One of its functions is to bind it to glycoproteins (7). However, there are no RGD sequences throughout the BhRGAE sequence, although there are up to three GD sequences among the first 135 residues (Fig. 2). By multiple sequence alignment with hierarchical clustering with ESPript output (9), similarities of BhRGAE to some proteins of the SGNH family were found, since BhRGAE has the four conserved blocks which are a feature of the SGNH family (Fig. 2). Block I has the characteristic GDS sequence motif. Block II has a Gly as the only conserved residue in the members of this family. GXND is the consensus sequence in block III. Finally, block V has the DXXHP conserved sequence that is placed in a variable loop, whereas the other blocks are found at the C-terminal ends of the central β-sheet (20).
FIG. 2.

Multiple alignment of some members of the SGNH family. ESPript (9) outputs were obtained from the BhRGAE, AaRGAE (Protein Data Bank accession no. 1deo), and hypothetical YxiM precursor (Protein Data Bank accession no. 2o14) sequences from the SWISSPROT databank (10) and aligned with CLUSTAL-W (32). Block I has the characteristic GDS sequence motif. Block II has a Gly as the only conserved residue in the members of this family. GXND is the consensus sequence in block III. Finally, block V has the DXXHP conserved sequence that is placed in a variable loop, whereas the other blocks are found at the C-terminal ends of the central β-sheet (20). Sequences are grouped according to similarity. Rectangles below the sequences show accessible residues in black, intermediate ones in gray, and buried ones in white. Residues strictly conserved among groups are shown in white font on a dark background. Residues conserved within a group but showing significant differences between groups are in gray font. The symbols above the blocks of sequences represent the secondary structure. Triangles represent the locations of the active sites.
In particular, alignment with AaRGAE reveals that the residues of the catalytic site (Ser, Asp, and His) are completely conserved and located at the same position as in this protein.
Homology modeling and topology diagram.
When the three-dimensional analysis was rendered using the AaRGAE structure as template (Protein Data Bank accession no. 1deo), the first 135 residues were not modeled. However, using a recent unpublished crystallization of a protein classified as a hypothethical YxiM precursor from B. subtilis strain 168 (Protein Data Bank accession no. 2o14), this region was represented as an immunoglobulin-like β sandwich fold. The overall structure is thus represented as having two domains (Fig. 3A), where one contains the catalytic triad and is similar to that from AaRGAE. This domain revealed the presence of a central β-sheet consisting of five parallel strands surrounded by α-helices in an αβα sandwich fold (Fig. 3B) (20). Therefore, BhRGAE could be considered a full-blooded member of the SGNH family, with all of the features that define the family but with a special N-terminal domain of unknown function. On investigation, sequence analysis revealed an FnIII-like domain located at this position, which could represent a substrate binding domain similar to the cellulose binding domains and the xylan binding domains found in nature. In the cases of the cellulose and xylan binding domains, a hydrophobic strip of aromatic residues (mainly tyrosines and tryptophans) is required for such interaction (25). Although there is no consensus sequence conserved for such binding domains, the N-terminal region of BhRGAE shows three tyrosine residues and one tryptophan residue. The possibility that the N-terminal domain might be a substrate binding domain is reinforced by structural similarity to the hemagglutinin-esterase fusion glycoprotein of influenza C virus, since the domain contains the receptor binding sites for sialosides (23). This immunoglobulin-like β sandwich fold that is present in both proteins is characterized in the SCOP database as containing seven strands folded in two sheets, although some members of the fold have additional strands. In the case of BhRGAE, it seems to have the same structure but with additional structural elements: a β-strand, two-stranded β-sheets, and an α-helix (Fig. 3A and 3). This unique α-helix links strands 2 and 3. This helical segment has been recently reported as a critical site for polysialylation of N-glycans on the adjacent Ig5 domain of neural cell adhesion molecules (18) and is a unique feature among known FnIII repeats and, as described for the first time in this paper, in BhRGAE.
FIG. 3.

Ribbon diagram of the monomeric structure of recombinant BhRGAE. (A) Overview of the whole modeled structure. (B) Representation of the catalytic domain. (C) Representation of the putative substrate binding domain. The amino acids of the active site in panels A and B are represented in the same shade. Dark-gray parts of the three representations indicate the presence of β-turns. These figures were rendered using SWISS-MODEL and Swiss-PdbViewer (10).
In the case of BhRGAE acting toward complex polysaccharides from plant cell walls, the synergistic effect with xylanase seems to reinforce the substrate binding motif role. Thus, this motif could allow BhRGAE to transform the oligosaccharides released by xylanase more efficiently than if these oligosaccharides move in a Brownian way through the reaction medium.
On the other hand, the topology diagrams of BhRGAE, AaRGAE, and the hypothetical YxiM precursor were also performed (Fig. 4A, B, and C). If we focus on the strands in the central β-sheet without taking into account the N-terminal region of BhRGAE and YxiM and the additional antiparallel β-sheet of AaRGAE, it is possible to distinguish a similar order (2-1-3-4-5) (Fig. 4). Such evidence suggests that these proteins could have undergone a divergence from a common ancestral version. Moreover, this hypothetical YxiM precursor from B. subtilis could show the same substrate specificity as BhRGAE, since it belongs to the same CE-12 family. In consequence, these kinds of proteins (BhRGAE and YxiM) could also represent conserved functional proteins in some Bacillus species.
FIG. 4.

Topology diagrams. (A) Diagram of recombinant BhRGAE. (B) Representation of RGAE (Protein Data Bank accession no. 1deo). (C) Representation of hypothetical YxiM precursor (Protein Data Bank accession no. 2o14). Helices are represented by circles, and β-strands are represented by triangles. White circles represent 310 or other small helices in the structures. White triangles represent those strands not contained in the central β-sheet. The diagrams were made using the TOPS program (19).
In conclusion, these results make this enzyme an interesting new member of the SGNH and CE-12 families, with important, hitherto undescribed, biotechnological and structural properties.
Acknowledgments
This work was partially supported by MEC and FEDER (BIO2004-00439), Fundación Séneca (00608/PI/04), and DGI Consejería Educación CARM (09BIO2005-01-6470). J.N.-F. is the holder of a predoctoral research (FPI) grant from MEC, Spain. I.M.-M. is the holder of a predoctoral research grant from CAM, Spain. S.M.-G. is the holder of a predoctoral research (FPI) grant from Fundación Séneca, Spain.
Footnotes
Published ahead of print on 14 December 2007.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhan, Z. Zhan, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 253389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biely, P., J. Puls, and H. Schneider. 1985. Acetyl xylan esterases in fungal cellulolytic system. FEBS Lett. 18680-84. [Google Scholar]
- 3.Blum, D. L., X. Li, H. Chen, and L. G. Ljungdahl. 1999. Characterization of an acetyl xylan esterase from the anaerobic fungus Orpinomyces sp. strain PC-2. Appl. Environ. Microbiol. 653990-3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi, D. H., K. S. Han, I. S. Jeong, S. H. Lee, B. S. Lim, and Y. D. Kim. 1999. Cephalosporin deacetylase, gene coding for it, and preparation method of deacetylated cephalosporin compounds using it. WO patent (WO/1999/055881). Chem. Abstr. 131332993. [Google Scholar]
- 5.Choi, D. H., Y. D. Kim, I. S. Chung, S. H. Lee, S. M. Kang, T. J. Kwon, and K. S. Han. 2000. Gene cloning and expression of cephalosporin-C deacetylase from Bacillus sp. KCCM10143. J. Microbiol. Biotechnol. 10221-226. [Google Scholar]
- 6.Coutinho, P., and B. Henrissat. 1999. Carbohydrate-active enzymes: an integrated database approach, p. 3-12. In H. J. Gilbert, (ed.), Recent advances in carbohydrate bioengineering. The Royal Society of Chemistry, Cambridge, United Kingdom.
- 7.DeSimone, D. W., P. A. Norton, and R. O. Hynes. 1992. Identification and characterization of alternatively spliced fibronectin mRNAs expressed in early Xenopus embryos. Dev. Biol. 149357-369. [DOI] [PubMed] [Google Scholar]
- 8.Gessesse, A., and B. H. Gashe. 1997. Production of alkaline xylanase by an alkaliphilic Bacillus sp. isolated from an alkaline soda lake. J. Appl. Microbiol. 83402-406. [Google Scholar]
- 9.Gouet, P., E. Courcelle, D. I. Stuart, and F. Metoz. 1999. ESPript: multiple sequence alignments in PostScript. Bioinformatics 15305-308. [DOI] [PubMed] [Google Scholar]
- 10.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modelling. Electrophoresis 182714-2723. [DOI] [PubMed] [Google Scholar]
- 11.Hagasova, N., E. Kutejova, and J. Timko. 1994. Purification and some characteristics of the xylan esterase from Schizophyllum commune. Biochem. J. 298751-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heyman, E. 1980. Carboxylesterases and amidases, p. 291-323. In W. B. Jakoby (ed.), Enzymatic basis of detoxification, vol. 2. Academic Press, New York, NY. [Google Scholar]
- 13.Ishii, T. 1997. O-acetylated oligosaccharides from pectins of potato tuber cell walls. Plant Physiol. 1131265-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson, K. G., J. D. Fontana, and C. R. MacKenzie.1988. Measurement of acetylxylan esterase in Streptomyces. Methods Enzymol. 160551-560. [Google Scholar]
- 15.Kaupinnen, S., S. Christgau, L. V. Kofod, T. Halkier, K. Dörreich, and H. Dalbøge. 1995. Molecular cloning and characterization of a rhamnogalacturonan acetylesterase from Aspergillus aculeatus. J. Biol. Chem. 27027172-27178. [DOI] [PubMed] [Google Scholar]
- 16.Lee, D. W., Y. S. Koh, K. J. Kim, B. C. Kim, H. J. Choi, D. S. Kim, M. T. Suhartono, and Y. R. Pyun. 1999. Isolation and characterization of thermophilic lipase from Bacillus thermoleovorans ID-1. FEMS Microbiol. Lett. 179393-400. [DOI] [PubMed] [Google Scholar]
- 17.Martínez-Martínez, I., S. Montoro-García, J. D. Lozada-Ramírez, A. Sánchez-Ferrer, and F. García-Carmona. 2007. A colorimetric assay for the determination of acetyl xylan esterase or cephalosporin C acetyl esterase activities using 7-amino cephalosporanic acid, cephalosporin C or acetylated xylan as substrates. Anal. Biochem. 369210-217. [DOI] [PubMed] [Google Scholar]
- 18.Mendiratta, S. S., N. Sekulic, F. G. Hernandez-Guzman, B. E. Close, A. Lavie, and K. J. Colley. 2006. A novel α-helix in the first fibronectin type III repeat of the neural cell adhesion molecule is critical for N-glycan polysialylation. J. Biol. Chem. 28136052-36059. [DOI] [PubMed] [Google Scholar]
- 19.Michalopoulos, I., G. M. Torrance, D. R. Gilbert, and D. R. Westhead. 2004. TOPS: an enhanced database of protein structural topology. Nucleic Acids Res. 32251-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mølgaard, A., S. Kaupinnen, and S. Larsen. 2000. Rhamnogalacturonan acetylesterase elucidates the structure and function of a new family of hydrolases. Structure 8373-383. [DOI] [PubMed] [Google Scholar]
- 21.Petersen, T., S. Kaupinnen, and S. Larsen. 1997. The crystal structure of rhamnogalacturonase A from Aspergillus aculeatus: a right-handed parallel β helix. Structure 5533-544. [DOI] [PubMed] [Google Scholar]
- 22.Politino, M., S. M. Tonzi, W. V. Burnett, G. Romancik, and J. J. Usher. 1997. Purification and characterization of a cephalosporin esterase from Rhodosporidium toruloides. Appl. Environ. Microbiol. 634807-4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenthal, P. B., X. Zhang, F. Formanowski, W. Fitz, C. H. Wong, H. Meier-Ewert, J. J. Skehell, and D. C. Wiley. 1998. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 39692-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sambrook, J., E. P. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Simpson, P. J., D. N. Bolam, A. Cooper, A. Ciruela, G. P. Hazlewood, H. J. Gilbert, and M. P. Williamson. 1999. A family IIb xylan-binding domain has a similar secondary structure to a homologous family IIa cellulose-binding domain but different ligand specificity. Structure 7853-864. [DOI] [PubMed] [Google Scholar]
- 26.Smith, P. K., R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson, and D. C. Klenk. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 15076-85. [DOI] [PubMed] [Google Scholar]
- 27.Takami, H., K. Nakasone, C. Hirama, Y. Takaki, N. Masui, F. Fuji, Y. Nakamura, and A. Inoue. 1999. An improved physical and genetic map of the genome of alkaliphilic Bacillus sp. C-125. Extremophiles 321-28. [DOI] [PubMed] [Google Scholar]
- 28.Takami, H., K. Nakasone, N. Ogasawara, Y. Nakamura, C. Hirama, F. Fuji, Y. Takaki, N. Masui, A. Inoe, and K. Horikoshi. 1999. Sequencing of three lambda clones from the genome of alkaliphilic Bacillus sp. C-125. Extremophiles 329-34. [DOI] [PubMed] [Google Scholar]
- 29.Takami, H., and K. Horikoshi. 2000. Genome analysis of alkaliphilic Bacillus strain from an industrial point of view. Extremophiles 499-108. [DOI] [PubMed] [Google Scholar]
- 30.Takami, H., K. Nakasone, Y. Takaki, G. Maeno, R. Sasaki, N. Masui, F. Fuji, C. Hirama, Y. Nakamura, N. Ogasawara, S. Kuhara, and K. Horikoshi. 2000. Complete genome sequence of alkaliphilic bacterium, Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res. 284317-4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 224673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber, K., J. R. Pringle, and M. Osborne. 1972. Measurement of molecular weight by electrophoresis on SDS-acrylamide gel. Methods Enzymol. 260 3-27. [DOI] [PubMed] [Google Scholar]