

Abstract
A modified 3-hydroxypropionate cycle has been proposed as the autotrophic CO2 fixation pathway for the thermoacidophilic crenarchaeon Metallosphaera sedula. The cycle requires the reductive conversion of 3-hydroxypropionate to propionyl-coenzyme A (propionyl-CoA). The specific activity of the 3-hydroxypropionate-, CoA-, and MgATP-dependent oxidation of NADPH in autotrophically grown cells was 0.023 μmol min−1mg protein−1. The reaction sequence is catalyzed by at least two enzymes. The first enzyme, 3-hydroxypropionyl-CoA synthetase, catalyzes the following reaction: 3-hydroxypropionate + ATP + CoA → 3-hydroxypropionyl-CoA + AMP + PPi. The enzyme was purified 95-fold to a specific activity of 18 μmol min−1 mg protein−1 from autotrophically grown M. sedula cells. An internal peptide sequence was determined and a gene encoding a homologous protein identified in the genome of Sulfolobus tokodaii; similar genes were found in S. solfataricus and S. acidocaldarius. The gene was heterologously expressed in Escherichia coli, and the His-tagged protein was purified. Both the native enzyme from M. sedula and the recombinant enzyme from S. tokodaii not only activated 3-hydroxypropionate to its CoA ester but also activated propionate, acrylate, acetate, and butyrate; however, with the exception of propionate, the affinities for these substrates were reduced. 3-Hydroxypropionyl-CoA synthetase is up-regulated eightfold in autotrophically versus heterotrophically grown M. sedula, supporting its proposed role during CO2 fixation in this archaeon and possibly other members of the Sulfolobaceae family.
A new autotrophic pathway, the 3-hydroxypropionate cycle, has been described for the phototrophic green nonsulfur bacterium Chloroflexus aurantiacus (9-12, 22, 23; Fig. 1). It represents the fourth known CO2 fixation pathway, the others being the reductive pentose phosphate cycle (Calvin-Bassham-Benson cycle), the reductive citric acid cycle, and the reductive acetyl coenzyme A (acetyl-CoA) pathway. However, additional pathways for the assimilation of CO2 may exist (14).
FIG. 1.

3-Hydroxypropionate cycle of autotrophic CO2 fixation in the phototrophic green nonsulfur eubacterium Chloroflexus aurantiacus. Step 1, acetyl-CoA carboxylase; step 2, malonyl-CoA reductase (bifunctional; see reference 16); step 3, propionyl-CoA synthase (trifunctional; see reference 1); step 4, propionyl-CoA carboxylase; step 5, methylmalonyl-CoA epimerase; step 6, methylmalonyl-CoA mutase; step 7, succinyl-CoA-l-malate CoA transferase; step 8, succinate dehydrogenase (electron acceptor unknown); step 9, fumarase; step 10, l-malyl-CoA/β-methylmalyl-CoA lyase (bifunctional; see reference 9).
Autotrophic members of the Sulfolobaceae appear to use a modified 3-hydroxypropionate cycle for CO2 fixation (6, 14, 17, 18, 21). The primary carboxylation steps are catalyzed by a bifunctional acetyl-CoA/propionyl-CoA carboxylase (7, 15). Malonyl-CoA, the carboxylation product of acetyl-CoA, is reduced to malonate semialdehyde by malonyl-CoA reductase (2) and is reduced further to the characteristic intermediate of the pathway, 3-hydroxypropionate (21). For C. aurantiacus, a single enzyme catalyzes both reduction steps (16; Fig. 1). However, the bifunctional enzyme from C. aurantiacus does not exhibit any significant sequence similarity with malonyl-CoA reductase from Metallosphaera sedula, indicating that the autotrophic pathways in members of the Chloroflexus and Sulfolobaceae families have evolved convergently. The further reductive conversion of 3-hydroxypropionate to propionyl-CoA requires three enzymatic steps: the activation of 3-hydroxypropionate to its CoA ester, dehydration to acrylyl-CoA, and reduction of acrylyl-CoA to propionyl-CoA. For C. aurantiacus, a single trifunctional enzyme called propionyl-CoA synthase catalyzes these steps (1; Fig. 1). This reaction sequence has not been studied for members of the Sulfolobaceae family in detail; however, reductive conversion of 3-hydroxypropionate to propionyl-CoA was catalyzed by cell extracts of M. sedula (21). Propionyl-CoA is carboxylated to methylmalonyl-CoA by acetyl-CoA/propionyl-CoA carboxylase, and the carbon skeleton is rearranged to form succinyl-CoA (14, 21). From here on the autotrophic pathways for Chloroflexus and members of the family Sulfolobaceae appear to differ. In the case of C. aurantiacus, succinyl-CoA is oxidatively converted to l-malyl-CoA (Fig. 1). l-Malyl-CoA lyase regenerates acetyl-CoA and releases glyoxylate as the primary CO2 fixation product (9). l-Malyl-CoA lyase activity was not detected in cell extracts of M. sedula (14). Therefore, the subsequent conversion of succinyl-CoA to acetyl-CoA and the formation of the initial CO2 fixation product in members of the Sulfolobaceae family are at issue.
In this work, the reductive conversion of 3-hydroxypropionate to propionyl-CoA for M. sedula was studied and was shown to involve at least two enzymes. This is in contrast to C. aurantiacus, for which all three reaction steps are catalyzed by propionyl-CoA synthase (1). The first enzyme of the reaction sequence, the 3-hydroxypropionyl-CoA synthetase, was purified and studied. A homologous protein is encoded by the genomes of Sulfolobus tokodaii. The corresponding gene was heterologously expressed in Escherichia coli, and the recombinant protein also catalyzed the CoA- and MgATP-dependent activation of 3-hydroxypropionate.
MATERIALS AND METHODS
Cell material.
Metallosphaera sedula TH2 (DSM 5348) was grown microaerobically and autotrophically on minimal salt medium with a gas phase of CO2-O2-H2 (19:3:78) at 75°C and pH 2.0 (13). As a control, cells grown microaerobically and heterotrophically with 0.05% yeast extract were used. The cells were harvested by centrifugation at an optical density at 578 nm of 0.2 to 0.3 and stored in liquid nitrogen until used. Sulfolobus tokodaii (DSMZ 16993) was grown aerobically under heterotrophic conditions, and chromosomal DNA was isolated as described previously (2).
Enzyme assays. (i) Reduction of 3-hydroxypropionate to propionyl-CoA.
ATP-, CoA-, and NADPH-dependent reduction of 3-hydroxypropionate was measured spectrophotometrically at 65°C using cell extracts. The standard reaction mixture (0.5 ml) contained 100 mM Tris-HCl (pH 7.8), 2 mM MgCl2, 6 mM ATP, 0.5 mM CoA, 0.5 mM NADPH, 2 mM dithioerythritol (DTE), and 0.5 to 1.7 mg cell extract protein. The reaction was started by the addition of 2 mM 3-hydroxypropionate and followed at 365 nm (ɛNADPH = 3,400 M−1 cm−1).
(ii) 3-Hydroxypropionyl-CoA synthetase.
Two spectrophotometric assays were used to measure 3-hydroxypropionyl-CoA synthetase activity. A discontinuous assay (assay a) was used to measure the ATP- and 3-hydroxypropionate-dependent disappearance of free CoA at 65°C. The standard reaction mixture (0.9 ml) contained 100 mM Tris-HCl (pH 8.4), 2 mM MgCl2, 3 mM ATP, 0.15 mM CoA, and protein. After 0 and 1 min at 65°C, 200 μl of the test mixture was removed and diluted in 200 μl of 200 mM HEPES-NaOH (pH 7.2)-0.5 mM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) (from 10 mM stock solution in 0.5 M potassium phosphate [pH 7.2]-1 mM EDTA) at 0°C. To the remaining test mixture 2 mM 3-hydroxypropionate was added to start the reaction. Samples (200 μl each) were taken after an additional 1 and 2 min and diluted as described above. Absorbance at 412 nm was measured to determine the amount of free CoA used during the reaction (ɛ412 nm = 13,600 M−1 cm−1). For the purified enzyme the formation of AMP was measured using a coupled assay (assay b) at 45°C. The reaction mixture (0.5 ml) contained 100 mM Tris-HCl (pH 8.5), 2 mM DTE, 2 mM MgCl2, 3 mM ATP, 0.1 to 0.5 mM CoA, 3 mM potassium phosphoenolpyruvate, 0.3 units of myokinase, 0.1 units of pyruvate kinase, 0.3 units of lactate dehydrogenase, 0.5 mM NADH, and 4 to 11 μg of purified 3-hydroxypropionate synthetase. The reaction was started by the addition of 3 mM 3-hydroxypropionate and followed at 365 nm (ɛNADH = 3,400 M−1 cm−1). For the purified recombinant enzyme the coupled assay was modified. The reaction mixture (0.5 ml) contained 200 mM MOPS (morpholinepropanesulfonic acid)-NaOH (pH 7.2), 2 mM DTE, 10 mM MgCl2, 9 mM ATP, 0.1 mM CoA, 9 mM phosphoenolpyruvate, 3.7 units of myokinase, 3.2 units of pyruvate kinase, 1.8 units of lactate dehydrogenase, 0.5 mM NADH, and protein. The reaction was started by the addition of 2 mM 3-hydroxypropionate and followed at 365 nm.
(iii) Malate dehydrogenase.
The oxaloacetate-dependent oxidation of NADH was measured spectrophotometrically at 65°C. The reaction mixture (0.5 ml) contained 100 mM Tris-HCl (pH 7.8), 2 mM MgCl2, 0.5 mM NADH, and protein. The reaction was started by the addition of 5 mM oxaloacetate and followed at 365 nm (ɛNADPH = 3,400 M−1 cm−1).
Protein concentrations were determined by the method of Bradford (4) using bovine serum albumin as the standard.
Purification of 3-hydroxypropionyl-CoA synthetase from M. sedula.
All procedures were done aerobically at 4°C. (i) For preparation of cell extract, thawed cell paste (10 g wet weight) of autotrophically grown cells was resuspended in 10 to 15 ml of 100 mM Tris-HCl (pH 8.5)-4 mM MgCl2 containing 0.2 mg DNase I and passed twice through a chilled French pressure cell at 138 kPa. The cell lysate was centrifuged at 20,000 × g for 10 min, and the supernatant was recentrifuged at 100,000 × g for 1 h. (ii) For ammonium sulfate precipitation, saturated ammonium sulfate solution was added to the cell extract to achieve a final saturation of 30% (NH4)2SO4 and the mixture was centrifuged at 100,000 × g for 1 h. (iii) For phenyl-Sepharose chromatography, the supernatant from step ii was loaded onto a 20-ml phenyl-Sepharose column (Amersham Bioscience, Inc.) equilibrated with 50 mM Tris-HCl (pH 7.9), 1 mM DTE, 5 mM MgCl2, and 1 M (NH4)2SO4. After a 20-ml wash with equilibration buffer, the column was developed with a 250-ml linear gradient at 1 ml min−1 from 1 M ammonium sulfate (equilibration buffer) to 0 M ammonium sulfate (2 mM Tris-HCl, pH 7.9). The enzyme eluted between 20 and 0 mM ammonium sulfate, and DTE was added to the pooled active fractions to achieve a final concentration of 2 mM. (iv) For MonoQ chromatography, the enzyme solution from step iii was applied to an 8-ml MonoQ HR 10/10 anion-exchange column (Amersham Bioscience, Inc.) equilibrated with buffer A (20 mM MOPS-KOH) (pH 7.2). The column was washed with 15 ml of buffer A and developed with a 100-ml linear gradient of 0 to 1 M KCl in buffer A at 0.8 ml min−1. 3-Hydroxypropionyl-CoA synthetase eluted between 50 and 120 mM KCl. (v) For reactive green chromatography, active pooled fractions from step iv were applied to a 10-ml reactive green 19 column (Sigma-Aldrich) equilibrated with buffer A. The column was washed with 25 ml buffer A containing 250 mM KCl and developed with a 100-ml increasing linear gradient of 0.25 to 1 M KCl in buffer A at 0.5 ml min−1. The enzyme eluted between 300 and 500 mM KCl. The combined active fractions were concentrated in dialysis tubing (cutoff, 3.5 kDa) embedded in dry polyethylene glycol 20,000 and dialyzed against 2 liters of 20 mM Tris-HCl (pH 8.1)-5 mM MgCl2. (vi) For gel filtration chromatography, aliquots of 1 ml from step v were applied onto a 120-ml Super 200 HiLoad 16/60 column (Amersham Biosciences) equilibrated with buffer B (20 mM MOPS-KOH [pH 7.2], 150 mM KCl). The column was developed at a flow rate of 0.4 ml min−1. Active fractions were pooled, concentrated as described for step v, and dialyzed against 1 liter of buffer B. DTE was added to achieve a final concentration of 1 mM, and the enzyme was stored at −20°C.
Heterologous expression of the lig gene from S. tokodaii and production of the enzyme in E. coli.
The gene encoding 3-hydroxypropionyl-CoA synthetase was amplified by PCR from S. tokodaii chromosomal DNA by using the primer combination ligNdeI_for (5′-CACGAGTCATATGACTGAAAAACTTCT-3′) and ligKpnI1_rev (5′-ATAGGTACCCATCTTCATCAATCATTG-3′) and the primer combination lig2KpnI_for (5′-ATAGGTACCTATGGGTAATGGGTAGA-3′) and lig2BamHI_rev (5′-GGAGGATCCTTAGAAAATCATTGG-3′), introducing an NdeI site (underlined) at the initiation codon, a KpnI site (underlined) in the middle of the gene (resulting in an I518L amino acid change in the protein), and a BamHI site (underlined) after the stop codon. The PCR products were isolated and cloned separately into pUC19 or pCRT7/CT TOPO, resulting in plasmids pAR1 and pAR2. Ligation of the KpnI/BamHI fragment of pAR2 into the likewise-restricted pAR1 resulted in plasmid pAR3. The sequence of the insert was determined to ensure that no errors (beside the purposely introduced changes) had occurred. pAR3 was digested with NdeI and BamHI, and the fragment containing the lig gene was ligated into pET16b, resulting in plasmid pAR5. Introduction of the NdeI/BamHI fragment into expression vector pET16b resulted in the expression of an extended 5′ coding region and in an N-terminal deca-His tag for the recombinant protein. Competent E. coli Rosetta 2 (DE3) cells were transformed with pAR5, grown in a 12-liter fermentor at 37°C in Luria-Bertani broth containing 100 μg ml−1 ampicillin and 34 μg ml−1 chloramphenicol and induced at an optical density of 0.5 with 0.4 mM isopropyl thiogalactopyranoside (IPTG). After additional growth for 6.5 h at 30°C, the cells (22 g wet weight) were harvested and stored in liquid nitrogen until use.
Purification of heterologously produced 3-hydroxypropionyl-CoA synthetase from S. tokodaii.
(i) For preparation of cell extract, thawed cell paste (11 g wet weight) was resuspended in 11 ml of 100 mM Tris-HCl (pH 8.0)-5 mM MgCl2 containing 0.2 mg DNase I and passed twice through a chilled French pressure cell at 138 kPa. The cell lysate was centrifuged at 20,000 × g for 15 min. (ii) For heat precipitation, an E. coli cell extract was incubated at 85°C for 15 min, cooled on ice for 15 min, and centrifuged at 100,000 × g at 4°C for 60 min. (iii) For nickel affinity chromatography, 12 ml of heat-precipitated cell extract was applied onto an Ni2+-chelating Sepharose affinity column (Amersham Biosciences, Freiburg, Germany) equilibrated with 20 mM Tris-HCl (pH 7.9)-250 mM KCl (buffer C). The column was washed with buffer C containing 200 mM imidazole and developed with a 75-ml increasing linear gradient of 0.2 to 0.5 M imidazole in buffer A at 1.0 ml min−1. Active fractions were pooled, washed with 20 mM Tris-HCl (pH 8.5), and concentrated to a final volume of 7 ml by ultrafiltration (Amicon YM 10 membrane; Millipore, Bedford, MA). The protein (18 mg) was stored at −20°C with 30% glycerol.
Characterization of the enzymes.
The apparent Km values of ATP, 3-hydroxypropionate, propionate, and acrylate for 3-hydroxypropionyl-CoA synthetase were determined using the coupled assay (assay b). The concentration of one substrate was adjusted while the concentrations of the other substrates were kept constant. The apparent Km value for ATP was also determined using the discontinuous assay (assay a). Both methods agreed well. The thermostability of the enzyme was determined by incubating the enzyme at 70, 80, 90, and 100°C for 15 min. The enzyme solution was cooled to 4°C before the activity levels were determined relative to those of samples kept on ice throughout the experiment. The specificity of the ATP- and CoA-dependent activation of 3-hydroxypropionate was determined by replacing 3-hydroxypropionate in the standard coupled assay (assay b) with propionate, acrylate, acetate, butyrate, (±)3-hydroxybutyrate, crotonate, glycolate, malonate, mesaconate, methylsuccinate, glycine, glycolate, succinate, β-alanine, 3-mercaptopropionate, or 3-chloropropionate. The specificity for ATP was determined by replacing ATP in the standard discontinuous assay (assay a) with 3 mM ADP, GTP, ITP, UTP, or CTP. Pyrophosphate was determined by a colorimetric method. Inorganic pyrophosphate was hydrolyzed by the use of inorganic pyrophosphatase (Sigma-Aldrich) to orthophosphate, which was quantified by the use of ammonium molybdate reagent (3). A sample blank without pyrophosphatase served as a control.
Molecular mass determination.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (10%) was performed as described previously (20). The native molecular mass was determined on a 300-ml Superdex 200 (Amersham Biosciences) gel filtration column calibrated with ovalbumin (43 kDa), bovine serum albumin (67 and 134 kDa), aldolase (158 kDa), and ferritin (440 kDa).
Peptide sequence.
An aliquot (20 μg) of the protein solution of the last purification step was precipitated with 8% trichloroacetic acid, separated by 9% SDS-PAGE, and transferred to an Immobilon-Psq transfer membrane (Millipore, Bedford, MA). Endoproteinase Lys-C digestion, high-pressure liquid chromatography separation of peptides, and N-terminal sequencing of one peptide was performed by TopLab (Martinsried, Germany).
Materials.
Chemicals were obtained from Fluka (Neu-Ulm, Germany), Sigma-Aldrich (Deisenhofen, Germany), VWR (Darmstadt, Germany), Genaxxon Biosciences (Biberach, Germany), MBI Fermentas (St. Leon-Rot, Germany), Invitrogen (Karlsruhe, Germany), Novagen (Schwalbach, Germany), MWG Biotech (Ebersberg, Germany), or Roth (Karlsruhe, Germany). 3-Hydroxypropionate was obtained by alkaline hydrolysis of 3-hydroxypropionitrile as previously described (10).
RESULTS AND DISCUSSION
Reductive conversion of 3-hydroxypropionate by cell extracts.
Extracts of autotrophically grown cells of M. sedula catalyzed the 3-hydroxypropionate-dependent oxidation of NADPH provided that Mg2+, ATP, and CoA were added. The specific activity at 65°C was 23 nmol min−1 mg−1 protein; this activity was threefold down-regulated in heterotrophically grown cells (Table 1). Malate dehydrogenase activity was used as a control, and its activity was about twice as high in heterotrophically as in autotrophically grown cells. These results indicate that the reductive conversion of 3-hydroxypropionate is involved in autotrophic CO2 fixation by M. sedula.
TABLE 1.
Regulation of some enzymes possibly relevant for autotrophic CO2 fixation by Metallosphaera sedula, as determined by activity measurements in cell extracts at 65°C
| Growth conditions | Sp act (nmol min−1 mg−1)
|
||
|---|---|---|---|
| 3-Hydroxy-propionate reduction | 3-Hydroxypropionyl-CoA synthetase | Malate dehydrogenase (control) | |
| Autotrophic | 23 | 91 | 1,400 |
| Heterotrophic | 8.5 | 12 | 3,200 |
The search for the enzyme activating 3-hydroxypropionate.
The reaction catalyzed by cell extracts forms propionyl-CoA (21). This requires first the activation of 3-hydroxypropionate to its CoA thioester. A discontinuous assay was developed which measured the 3-hydroxypropionate-, ATP-, and Mg2+-dependent consumption of CoA in the presence of cell extract. The specific activity at 65°C in cell extracts was 91 nmol min−1 mg−1 protein; this activity was eightfold down-regulated in heterotrophically grown cells (Table 1). In a first chromatographic step, protein was separated on a phenyl-Sepharose column. 3-Hydroxypropionate- and Mg2+/ATP-dependent CoA-consuming activity eluted between 0 and 20 mM ammonium sulfate. These pooled active fractions, however, were no longer able to catalyze the (entire) reductive conversion of 3-hydroxypropionate to propionyl-CoA but required the addition of protein fractions eluting from the same column at 400 mM ammonium sulfate. These results indicate that in contrast to C. aurantiacus (1), the reductive conversion of 3-hydroxypropionate to propionyl-CoA for M. sedula requires at least two enzymes, which could be separated by hydrophobic interaction chromatography.
Purification of the 3-hydroxypropionate-activating enzyme.
The enzyme catalyzing the activation of 3-hydroxypropionate with respect to its CoA ester was up-regulated in autotrophically versus heterotrophically grown cells (Table 1), indicating a function of the enzyme in autotrophic CO2 fixation. The enzyme was purified 95-fold from 10 g (wet weight) of autotrophically grown cells (Table 2) to apparent homogeneity, as indicated by the presence of a single protein band after electrophoresis in a denaturing gel (Fig. 2). The enzyme is tentatively referred to as 3-hydroxypropionyl-CoA synthetase.
TABLE 2.
Purification of 3-hydroxypropionyl-CoA synthetase from 10 g (wet cell mass) of autotrophically grown Metallosphaera sedula
| Purification step | Volume (ml) | Total activity (μmol min−1)a | Total protein (mg) | Specific activity (μmol min−1 mg−1) | Recovery (%) | Purification (fold) |
|---|---|---|---|---|---|---|
| Cell extract | 31 | 150 | 810 | 0.19 | (100)b | (1) |
| 30% (NH4)2SO4 | 25 | 108 | 550 | 0.20 | 72 | 1.1 |
| Phenyl-Sepharose | 35 | 60 | 160 | 0.38 | 40 | 2.0 |
| MonoQ | 12 | 42 | 37 | 1.1 | 28 | 5.8 |
| Reactive Green | 1.5 | 31 | 4.7 | 6.6 | 21 | 35 |
| Gel Filtration | 1.1 | 16 | 0.9 | 18 | 11 | 95 |
Activity was determined at 65°C using the discontinuous assay.
Parentheses around entries in row 1 signify set values.
FIG. 2.
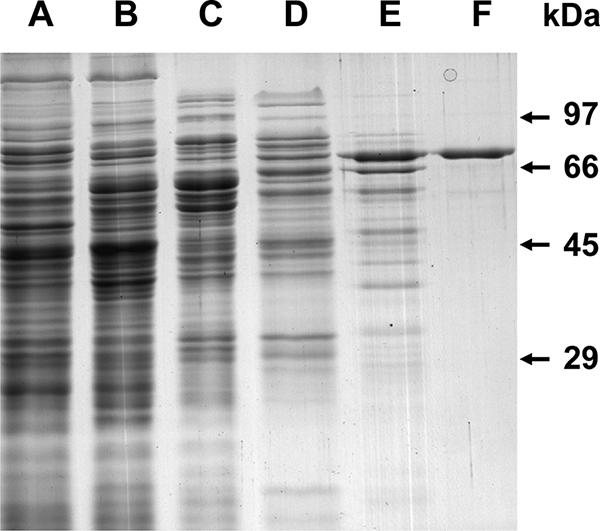
SDS-PAGE (10%) of the 3-hydroxypropionyl-CoA synthetase of Metallosphaera sedula at various steps during purification. Lane A, 20 μg of cell extract protein from heterotrophically grown M. sedula; lane B, 20 μg of cell extract protein from autotrophically grown M. sedula; lane C, 10 μg of protein from the phenyl-Sepharose column step; lane D, 7 μg of protein from the Mono Q column step; lane E, 3 μg of protein from the reactive green column step; lane F, 1 μg of protein from the gel filtration column step. The gel was stained with Coomassie brilliant blue.
Molecular properties of 3-hydroxypropionate-CoA synthetase.
Gel filtration chromatography of the native enzyme gave an estimated molecular mass of 340 kDa. SDS-PAGE produced one protein band of 78 kDa (Fig. 2). 3-Hydroxypropionyl-CoA synthetase, therefore, exists as a homotetramer. The enzyme was stable when incubated for 15 min at temperatures up to 80°C and during storage at −20°C for several weeks. After incubation for 15 min at 100°C, 50% of the activity remained relative to the activity seen with a sample kept at 4°C throughout the experiment. Acyl-CoA synthetases are usually described as monomers or homodimers composed of 70- to 75-kDa subunits. A homotetrameric structure is therefore unusual and may be a consequence of the thermostability of the enzyme (5).
Stoichiometry and products of the reaction.
High-pressure liquid chromatography analysis showed that 3-hydroxypropionyl-CoA was formed (data not shown). To determine whether AMP or ADP was formed, a coupled spectrophotometric assay was used. The stoichiometry of NADH oxidation per mole of 3-hydroxypropionate added was 1:1.8. This shows that ATP was hydrolyzed to AMP. Pyrophosphate was the second product of the reaction: the amount of pyrophosphate formed was determined in an assay using 0.4 mM coenzyme A (80% pure) and 2 mM MgATP; 0.3 mM pyrophosphate (the concentration of phosphate was determined to be 0.6 mM after addition of pyrophosphatase) was formed after equilibrium of the reaction was reached. This indicates that pyrophosphate and AMP were the products of the ligase reaction. Hence, 3-hydroxypropionyl-CoA synthetase catalyzes the following reaction: 3-hydroxypropionate + ATP + CoA → 3-hydroxypropionyl-CoA + AMP + PPi.
Catalytic properties of 3-hydroxypropionyl-CoA synthetase.
The dependency of the 3-hydroxypropionyl-CoA synthetase with respect to CoA did not follow Michaelis-Menten kinetics (Fig. 3). Maximal activity was obtained with 100 μM CoA, which was chosen to determine the apparent Km values for other substrates. The enzyme had a high affinity for ATP and 3-hydroxypropionate, with apparent Km values of 45 μM for ATP and 180 μM for 3-hydroxypropionate. ATP could be replaced by UTP or CTP; however, the specific activities were reduced (Table 3). 3-Hydroxypropionyl-CoA synthetase can also use propionate, acrylate, acetate, and butyrate as substrates (Table 3). At saturating substrate concentrations the turnover of propionate and acrylate was similar to the turnover for 3-hydroxypropionate (Table 4). The specificities (kcat/Km) of the enzyme with respect to 3-hydroxypropionate and propionate were similar but were decreased about 10-fold for acrylate (Table 4). We assume that the lower activities determined for the other substrates at a (nonsaturating) concentration of 4 mM (Table 3) were due to decreased affinities of 3-hydroxypropionyl-CoA synthetase toward these substrates rather than to a lower turnover number. The enzyme, therefore, is specific for 3-hydroxypropionate and propionate.
FIG. 3.

CoA dependency of the 3-hydroxypropionyl-CoA synthetase of Metallosphaera sedula. Coupled enzyme assays were used at 45°C with 3.7 μg of purified 3-hydroxypropionyl-CoA synthetase.
TABLE 3.
Molecular and catalytic properties of 3-hydroxypropionyl-CoA synthetase from M. sedula and the recombinant enzyme from S. tokodaii
| Species | Substrates | Products | Specific activity (μmol min−1 mg−1)a | Apparent Km value (μM)b | Native molecular mass (kDa) | Subunit molecular mass (kDa) | Suggested composition | Turnover (per subunit) (s−1) | Specificity (%)c |
|---|---|---|---|---|---|---|---|---|---|
| M. sedula | 3-Hydroxypropionate, CoA, ATP | 3-Hydroxypropionyl-CoA, AMP, PPi | 18 | 3-Hydroxypropionate, 180; ATP, 45; CoA, NA | 340 | 78 | α4 | 23 | ATP, 100; UTP, 32; CTP, 13; ADP, 5; ITP, 3; GTP, <3; 3-hydroxpropionate, 100; propionate, 103; acrylate, 77; acetate, 42, butyrate, 20; 3-hydroxybutyrate, <1; crotonate, <1; glycerate, <1; malonate, <1 |
| S. tokodaii | 3-Hydroxypropionate, CoA, ATP | 3-Hydroxypropionyl-CoA, AMP, PPi | 6.7 | 3-Hydroxypropionate, 190; ATP, 110; CoA, NA | 140 | 74 | α2 | 8 | 3-Hydroxypropionate, 100; propionate, 98; acrylate, 96; acetate, 65; butyrate, 27; glycolate, 3; 3-mercaptopropionate, 36; 3-chloropropionate, 64; 3-hydroxybutyrate, <1; crotonate, <1; glycerate, <1; malonate, <1; β-alanine, <1; glycine, <1; succinate, <1; mesaconate, <1; methylsuccinate, <1 |
Activities were determined using the discontinuous enzyme assay at 65°C.
NA, not applicable (CoA dependency does not follow Michaelis-Menten kinetics).
The final concentrations of nucleotides were 3 mM each; the final concentrations of other substrates were 4 mM each.
TABLE 4.
Substrate specificity of 3-hydroxypropionyl-CoA synthetase from Metallosphaera sedulaa
| Substrate | Apparent Km (mM) | kcat (s−1) | Apparent kcat/Km (10−4 M−1 s−1) |
|---|---|---|---|
| 3-Hydroxypropionate | 0.18 ± 0.03 | 5.7 ± 0.3 | 3.2 ± 0.2 |
| Propionate | 0.12 ± 0.02 | 5.6 ± 0.2 | 4.7 ± 0.2 |
| Acrylate | 1.42 ± 0.20 | 5.4 ± 0.2 | 0.4 ± 0.04 |
Activities were determined using a coupled spectrophotometric assay at 45°C.
Identification of a homologous protein for Sulfolobus tokodaii.
An internal peptide sequence of the purified enzyme from M. sedula, VVITADAYPRRGK, was determined. A search of protein databases revealed a match, with only one amino acid residue exchange (Y for P), to an internal sequence of a 659-amino-acid-long (74-kDa) hypothetical acetyl-CoA synthetase from the closely related crenarchaeon S. tokodaii 7 strain (GenBank accession number NP_376686) (19). The gene encoding this protein was amplified, cloned into pET16b expression vector, and heterologously expressed in the E. coli Rosetta (DE3) strain, which carries a plasmid with genes for rare tRNA species. A cell extract from this E. coli strain catalyzed the 3-hydroxypropionate-dependent oxidation of NADPH in the presence of MgATP and CoA, with a specific activity of 0.2 μmol min−1 mg−1 of protein (65°C). His-tagged 3-hydroxypropionyl-CoA synthetase was readily purified 34-fold by heat precipitation of cell extracts at 85°C and use of Ni2+ affinity chromatography, with almost 60% recovery. The catalytic properties of the purified recombinant enzyme from S. tokodaii are summarized in Table 3 and are compared to the properties of native enzyme from M. sedula. With the exception of the determined subunit composition, the two enzymes are almost indistinguishable. It is possible that the presence of the deca-His tag may prevent the recombinant enzyme from S. tokodaii from forming a homotetramer and might also explain the somewhat lower turnover number compared to the results seen with the enzyme from M. sedula.
3-Hydroxypropionyl-CoA synthetases from members of the Sulfolobaceae family.
Recently, the genome sequence for M. sedula was completed by researchers of the Joint Genome Institute and deposited in the NCBI databank (accession number CP000682). Based on the internal peptide sequence determined for purified 3-hydroxypropionyl-CoA synthetase, a gene was identified in the genome of M. sedula which codes for a protein with an internal sequence that is a 100% match for the determined peptide sequence. The protein (GenBank accession number YP_001191537) has a calculated molecular mass of 74 kDa and shows 76% sequence identity to the enzyme from S. tokodaii that was heterologously produced in E. coli in this work. A similar protein is also encoded by the genome of S. solfataricus (accession number NP_344510; 73% sequence identity) and S. acidocaldarius (accession number YP_255824; 69% sequence identity). It is assumed that all four proteins are 3-hydroxypropionyl-CoA synthetases. The genomic contexts of the genes encoding these 3-hydroxypropionyl-CoA synthetases, however, are different in each organism, and none of the genes appear to be cotranscribed with other genes. A putative acetyl-CoA synthetase from Thermofilum pendens (accession number YP_920297) represents the next-best match to the M. sedula enzyme, with overall sequence identity of 51%. The 3-hydroxypropionyl-CoA synthetase domain of the propionyl-CoA synthase of C. aurantiacus constitutes only the fourth best match among those of possible acyl-CoA synthetases in C. aurantiacus in comparison to that of the enzyme from M. sedula. The broad substrate specificity of 3-hydroxypropionyl-CoA synthetase (and of acyl-CoA synthetases in general) supports the idea that only small changes in the active site of these enzymes are necessary to increase catalytic efficiency of one substrate over another. Therefore, it cannot be ruled out that other 3-hydroxypropionate-specific acyl-CoA synthetases are present in the database that may play a role in a (modified) 3-hydroxypropionate cycle for other organisms.
A modified 3-hydroxypropionate cycle in Cenarchaeum symbiosum?
Yet another possibility may be realized for the marine chemolithoautorophic sponge symbiont C. symbiosum. Hallam et al. recently proposed that the organism uses the modified 3-hydroxypropionate cycle for autotrophic CO2 fixation based on genomic analyses (8). The genome, however, does not appear to encode any homologs of the 3-hydroxypropionyl-CoA synthetase described here. Instead, genes for ADP-forming acyl-CoA synthetases (COG database accession number COG1042) are found that show no significant sequence similarity to those encoding the AMP-forming enzymes (accession number COG0318). In C. symbiosum 3-hydroxypropionate may be activated to its CoA ester by means of an ADP-forming enzyme. The 3-hydroxypropioyl-CoA synthetase described here is the third enzyme of the modified 3-hydroxypropionate cycle for which the corresponding genes have been identified in M. sedula, the other two being acetyl-CoA/propionyl-CoA carboxylase and malonyl-CoA reductase (2, 15). Genes encoding the subunits of a biotin-dependent acyl-CoA carboxylase are present in the genome of C. symbiosum and are likely to encode acetyl-CoA/propionyl-CoA carboxylase. However, the homolog of malonyl-CoA reductase encoded by the C. symbiosum genome is probably an aspartate semialdehyde dehydrogenase involved in threonine biosynthesis. Although the idea of a modified 3-hydroxypropionate cycle for autotrophic CO2 fixation by C. symbiosum cannot be ruled out, different enzymes may have been recruited to catalyze individual steps during the process or parts of the pathway may be quite different.
In summary, the rate of reductive conversion of 3-hydroxypropionate to propionyl-CoA was up-regulated in autotrophically versus heterotrophically grown cells of M. sedula and the reaction sequence is catalyzed by at least two enzymes. In a first step, 3-hydroxypropionate is activated to its CoA ester. The gene encoding the 3-hydroxypropionyl-CoA synthetase has been identified in M. sedula and S. tokodaii. The gene(s) encoding the enzyme(s) responsible for dehydration of 3-hydroxypropionyl-CoA (enoyl-CoA hydratase) and further reduction of acrylyl-CoA to propionyl-CoA (enoyl-CoA reductase) could not be identified based on genomic analyses: (i) there are several candidates present in the genome of M. sedula that could encode proteins able to catalyze either set of reactions (no gene encoding a possible fusion protein was found, however); and (ii) genes encoding the enzymes catalyzing the consecutive steps in the conversion of 3-hydroxypropionate to propionyl-CoA appear not to be cotranscribed. In the genomic surroundings of the identified 3-hydroxypropionyl-CoA synthetase catalyzing the first step of the reaction sequence, genes possibly encoding enzymes catalyzing the second or third steps are absent. Therefore, in order to identify the enzyme(s) responsible for the reductive conversion of 3-hydroxypropionyl-CoA to propionyl-CoA during autotrophic growth of M. sedula, it will be necessary to purify the protein(s). The recombinant 3-hydroxypropionyl-CoA synthetase from S. tokodaii activates not only 3-hydroxypropionate but also acrylate to its CoA ester and, therefore, can be used to assay the reduction of acrylyl-CoA to propionyl-CoA independently from the dehydration reaction by use of a coupled assay.
Acknowledgments
This work was supported by the Deutsche Forschngsgemeinschaft, Bonn, Germany (Graduiertenkolleg Biochemie der Enzyme).
We thank A. Rieder for initial plasmid constructs, R. Teufel for quantification of pyrophosphate, M. Kies of TopLab, Munich, Germany, for peptide sequence analysis, and G. Igloi, Freiburg, Germany, for DNA sequencing. We especially thank N. Gad'on, Freiburg, Germany, for his expertise in growing cells. We also thank I. A. Berg for very helpful discussions and critical review of the manuscript.
Footnotes
Published ahead of print on 28 December 2007.
REFERENCES
- 1.Alber, B. E., and G. Fuchs. 2002. Propionyl-coenzyme A synthase from Chloroflexus aurantiacus, a key enzyme of the 3-hydroxypropionate cycle for autotrophic CO2 fixation. J. Biol. Chem. 27712137-12143. [DOI] [PubMed] [Google Scholar]
- 2.Alber, B., M. Olinger, A. Rieder, D. Kockelkorn, B. Jobst, M. Hügler, and G. Fuchs. 2006. Malonyl-coenzyme A reductase in the modified 3-hydroxypropionate cycle for autotrophic carbon fixation in archaeal Metallosphaera and Sulfolobus spp. J. Bacteriol. 1888551-8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anonymous. 1989. Methods of biochemical analysis and food analysis, p. 114-115. Boehringer Mannheim GmbH, Ingelheim, Germany.
- 4.Bradford, M. 1976. A rapid and sensitive method for quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72248-254. [DOI] [PubMed] [Google Scholar]
- 5.Bräsen, C., C. Urbanke, and P. Schönheit. 2005. A novel octameric AMP-forming acetyl-CoA synthetase from the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. FEBS Lett. 579477-482. [DOI] [PubMed] [Google Scholar]
- 6.Burton, N. P., T. D. Williams, and P. R. Norris. 1999. Carboxylase genes of Sulfolobus metallicus. Arch. Microbiol. 172349-353. [DOI] [PubMed] [Google Scholar]
- 7.Chuakrut, S., H. Arai, M. Ishii, and Y. Igarashi. 2003. Characterization of a bifunctional archaeal acyl coenzyme A carboxylase. J. Bacteriol. 185938-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hallam, S. J., T. J. Mincer, C. Schleper, C. M. Preston, K. Roberts, P. M. Richardson, and E. F. deLong. 2006. Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine Crenarchaeota. PLoS Biol. 4520-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herter, S., A. Busch, and G. Fuchs. 2002. l-Malyl-coenzyme A lyase/β-methylmalyl-coenzyme A lyase from Chloroflexus aurantiacus, a bifunctional enzyme involved in CO2 fixation. J. Bacteriol. 1845999-6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herter, S., J. Farfsing, N. Ga'don, C. Rieder, W. Eisenreich, A. Bacher, and G. Fuchs. 2001. Autotrophic CO2 fixation by Chloroflexus aurantiacus: glyoxylate formation and assimilation via the 3-hydroxypropionate cycle. J. Bacteriol. 1834305-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holo, H. 1989. Chloroflexus aurantiacus secretes 3-hydroxypropionate, a possible intermediate in the assimilation of CO2 and acetate. Arch. Microbiol. 151252-256. [Google Scholar]
- 12.Holo, H., and R. Sirevåg. 1986. Autotrophic growth and CO2 fixation in Chloroflexus aurantiacus. Arch. Microbiol. 145173-180. [Google Scholar]
- 13.Huber, G., C. Sinnler, A. Gambacorta, and K. O. Stetter. 1989. Metallosphaera sedula gen. and sp. nov. represents a new genus of aerobic, metal-mobilizing, thermoacidophilic archeaebacteria. Syst. Appl. Microbiol. 1238-47. [Google Scholar]
- 14.Hügler, M., H. Huber, K. O. Stetter, and G. Fuchs. 2003a. Autotrophic CO2 fixation pathways in archaea (Crenarchaeota). Arch. Microbiol. 179160-173. [DOI] [PubMed] [Google Scholar]
- 15.Hügler, M., R. S. Krieger, M. Jahn, and G. Fuchs. 2003. Characterization of acetyl-CoA/propionyl-CoA carboxylase in Metallosphaera sedula. Eur. J. Biochem. 270736-744. [DOI] [PubMed] [Google Scholar]
- 16.Hügler, M., C. Menendez, H. Schägger, and G. Fuchs. 2002. Malonyl-coenzyme A reductase from Chloroflexus aurantiacus, a key enzyme of the 3-hydroxypropionate cycle for autotrophic CO2 fixation. J. Bacteriol. 1842404-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishii, M., T. Miyake, T. Satoh, H. Sugiyama, Y. Oshima, T. Kodama, and Y. Igarashi. 1997. Autotrophic carbon dioxide fixation in Acidianus brierleyi. Arch. Microbiol. 166368-371. [DOI] [PubMed] [Google Scholar]
- 18.Ishii, M., S. Chuakrut, H. Arai, and Y. Igarashi. 2004. Occurrence, biochemistry and possible biotechnological application of the 3-hydroxypropionate cycle. Appl. Microbiol. Biotechnol. 64605-610. [DOI] [PubMed] [Google Scholar]
- 19.Kawarabayasi, Y., Y. Hino, H. Horikawa, K. Jin-no, M. Takahashi, M. Sekine, S. Baba, A. Ankai, H. Kosugi, A. Hosoyana, S. Fukui, Y. Nagai, K. Nishijima, R. Otsuka, H. Nakazawa, M. Takamiya, Y. Kato, T. Yoshizawa, T. Tanaka, Y. Kudoh, J. Yamazaki, N. Kushida, A. Oguchi, K. Aoki, S. Masuda, M. Yanagii, M. Nishimura, A. Yamagishi, T. Oshima, and H. Kikuchi. 2001. Complete genome sequence of an aerobic thermoacidophilic crenarchaeon, Sulfolobus tokodaii strain 7. DNA Res. 8123-140. [DOI] [PubMed] [Google Scholar]
- 20.Laemmli, U. K. 1970. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 227680-685. [DOI] [PubMed] [Google Scholar]
- 21.Menendez, C., Z. Bauer, H. Huber, N. Gad'on, K. O. Stetter, and G. Fuchs. 1999. Presence of acetyl coenzyme A (CoA) carboxylase and propionyl-CoA carboxylase in autotrophic Crenarchaeota and indication for operation of a 3-hydroxypropionate cycle in autotrophic carbon fixation. J. Bacteriol. 1811088-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strauss, G., W. Eisenreich, A. Bacher, and G. Fuchs. 1992. 13C-NMR study of autotrophic CO2 fixation pathways in the sulfur-reducing archaebacterium Thermoproteus neutrophilus and in the phototrophic eubacterium Chloroflexus aurantiacus. Eur. J. Biochem. 205853-866. [DOI] [PubMed] [Google Scholar]
- 23.Strauss, G., and G. Fuchs. 1993. Enzymes of a novel autotrophic CO2 fixation pathway in the phototrophic bacterium Chloroflexus aurantiacus, the 3-hydroxypropionate cycle. Eur. J. Biochem. 215633-643. [DOI] [PubMed] [Google Scholar]