

Abstract
The pregnane X receptor (PXR) regulates the metabolism and elimination of bile salts, steroids, and xenobiotics. The sequence of the PXR ligand-binding domain diverges extensively between different animals suggesting inter-species differences in ligands. Of the endogenous ligands known to activate PXR, biliary bile salts vary the most across vertebrate species, ranging from 27-carbon (C27) bile alcohol sulfates (early fish, amphibians) to C24 bile acids (birds, mammals). Using a luciferase-based reporter assay, human PXR was activated by a wide variety of bile salts. In contrast, zebrafish PXR was activated efficiently only by cyprinol sulfate, the major zebrafish bile salt, but not by recent bile acids. Chicken, mouse, rat, and rabbit PXRs were all activated by species-specific bile acids and by early fish bile alcohol sulfates. In addition, phylogenetic analysis using maximum likelihood demonstrated evidence for non-neutral evolution of the PXR ligand-binding domain. PXR activation by bile salts has expanded from narrow specificity for C27 bile alcohol sulfates (early fish) to a broader specificity for recent bile acids (birds, mammals). PXR specificity for bile salts has thus paralleled the increasing complexity of the bile salt synthetic pathway during vertebrate evolution, an unusual example of ligand-receptor co-evolution in the nuclear hormone receptor superfamily.
Keywords: Nuclear hormone receptor, xenobiotics, cholestasis, positive selection, metabolism, molecular evolution
INTRODUCTION
The pregnane X receptor (PXR; NR1I2), a member of the nuclear hormone receptor (NR) superfamily, is a key regulator of bile salt, steroid hormone, and xenobiotic metabolism and excretion. PXR binds a remarkably diverse array of compounds at low affinity, consistent with its hypothesized role as the initial step in the detoxification of a broad array of toxic compounds (1, 2). Although no high affinity endogenous ligands for PXR have been identified, pregnane steroids (e.g., the progesterone metabolite 5β-pregnane-3,20-dione) and bile acids activate PXR at micromolar concentrations (2-9). While the biological significance of PXR activation by steroid hormones is as yet unclear, the importance of PXR in regulating metabolism of bile acids has been established in mammals. Activation of mammalian PXRs by high concentrations of bile acids, which may accumulate in cholestasis (10), initiates a coordinated metabolic response to eliminate bile acids by increasing expression of metabolic enzymes and bile acid efflux pumps and decreasing expression of enzymes involved in bile acid biosynthesis and uptake (11-14). The protective role of PXR in regulating metabolism and elimination of toxic bile acids has been elegantly demonstrated in mouse models (4, 15-18). In contrast, the role of PXR in non-mammalian species is much less understood.
PXR genes have been cloned and functionally characterized from a variety of vertebrate species, including zebrafish, frog, chicken, rat, mouse, rabbit, dog, pig, rhesus monkey, and human (1, 7, 9, 19-21). PXR has the typical NR organization of an N-terminal DNA-binding domain (DBD) and a C-terminal ligand-binding domain (LBD); however, in comparing PXR sequences between vertebrate species, a striking feature is high cross-species sequence divergence in the LBD. The LBD of PXR shares amino acid identities of only 75% between human and rodent sequences and only 50% between human and chicken or fish sequences. These sequence identities are unusually low when compared to other NRs, for which the corresponding amino acid sequence identities tend to be at least 10-15% higher than the values above (7, 22).
The variation in the LBD of PXR is more striking when DNA sequences are analyzed, in particular by comparing the rate of non-synonymous (i.e., changes amino acid sequence) and synonymous (does not change amino acid sequence) nucleotide substitution rates. When comparing human and rodent sequences, the LBD of PXR has non-synonymous substitution rates (dN) 3.7 times higher than the average for all other NRs even though the synonymous substitution rates (dS) for PXR are average for NRs. The ratios of dN/dS for the PXR are 4 times higher than the average for other NR genes (22). Elevated dN/dS (ω) ratios suggest positive natural selection due to the accumulation of advantageous amino acid substitutions (23).
The high sequence divergence of the PXR LBD has been speculated to be an adaptation to xenobiotic ligands (7, 22), although the nature of such ligands is currently unknown. The xenobiotics currently known to activate PXR are all synthetic ligands or herbal products; dietary ligands for PXR have not yet been described. An alternative hypothesis is that cross-species variation in endogenous ligands has influenced PXR evolution (11). Of the endogenous ligands known to activate PXR, biliary bile salts vary the most across vertebrate species (Table 1) (24-26). For example, most mammals produce predominantly 24-carbon (C24) bile acids such as cholic acid and chenodeoxycholic acid, conjugated to either glycine or taurine, from end-metabolism of 27-carbon cholesterol. In contrast, cartilaginous fish and many early bony fish typically synthesize C27 bile alcohols, conjugated with sulfate, and, in fact, do not produce C24 bile acids (Table 1; Figs. 1 and 2).
Figure 1. Chemical structures of bile salts from various vertebrate species.
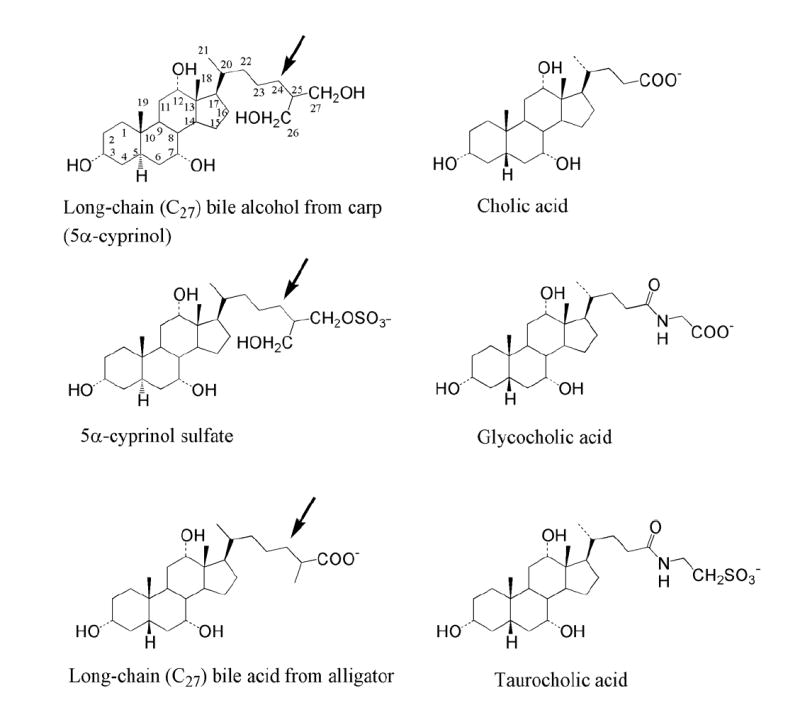
Cholic acid (upper right) is a 24-carbon bile acid typical of those found in birds and mammals. Cholic acid is commonly conjugated to either glycine or taurine (middle right and lower right, respectively). 5α-Cyprinol (upper left), a penta-hydroxylated bile alcohol, is found in a number of cypriniform fish species (including zebrafish) and was isolated in the current study from carp. This bile alcohol is typically conjugated with sulfate (middle left). The bottom left structure is a long-chain (27-carbon) bile acid isolated from alligator but typical of those found in reptiles and amphibians. Note the long side-chain (arrows) on the bile compounds from carp and alligator as compared to cholic acid, the more extensive hydroxylation of 5α-cyprinol as compared to cholic acid, and the differing orientations of the hydrogen atom at the 5 position between 5α-cyprinol and 5β-bile acids. The numbers for 5α-cyprinol indicate the numbering of the carbon atoms.
Figure 2. Comparative vertebrate pathways of bile salt biosynthesis.
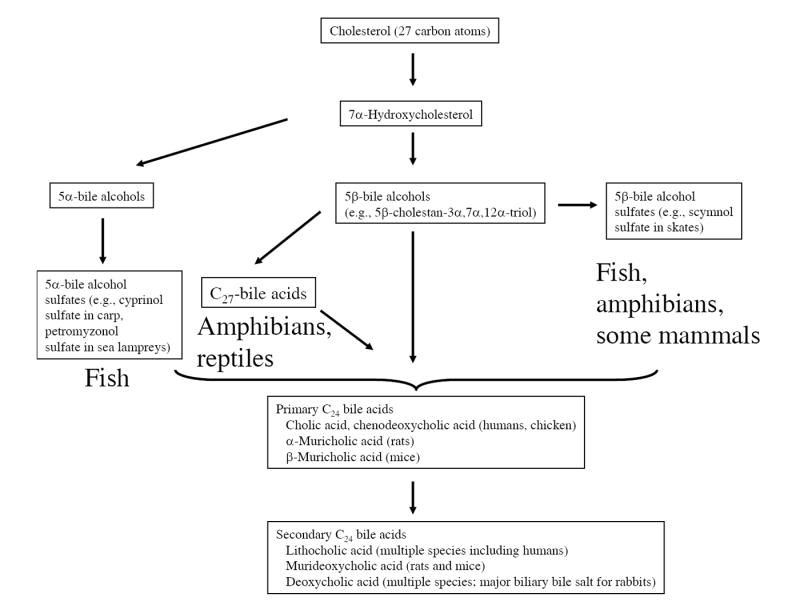
All bile salts are ultimately derived from end-metabolism of 27-carbon cholesterol with 7α-hydroxylation of cholesterol being the rate-limiting step. The evolutionarily earliest bile salts are 5α-bile alcohol sulfates, modern-day examples of which are 5α-cyprinol sulfate in Asiatic carp and 5α-petromyzonol sulfate in sea lampreys. C27 bile acids are found in reptiles and amphibians; some amphibians such as Xenopus laevis utilize both C27 bile acids and C27 bile alcohol sulfates. Mammals and birds predominantly use C24 bile acids as their biliary bile salts. Primary bile salts are synthesized in the liver while secondary bile salts are formed by the action of intestinal bacteria. Usually the predominant bile salts found in the bile are primary bile salts; an exception is the rabbit, for which the secondary bile salt deoxycholic acid is the predominant circulating bile salt.
In this study, we use an in vitro assay to examine activation of PXRs from a variety of vertebrate animals by species-specific bile salts, some of which were isolated from natural sources. For comparison, we also study bile salt activation of the vitamin D receptor (VDR; NR1I1), a receptor closely related to PXR that also plays a role in bile salt detoxification. We demonstrate that bile salt activation of PXRs is conserved from teleost fish to mammals, although PXR specificity for bile salts has expanded greatly during vertebrate evolution. In addition, using phylogenetic analysis we provide evidence of non-neutral evolution of the PXR LBD. We propose that evolution of the PXR LBD has been shaped by changes in vertebrate biliary bile salts, an unusual example in the NR superfamily of a receptor adapting to changing endogenous ligands.
RESULTS
Differing bile salt activation profiles for human PXR, zebrafish PXR, and human VDR
To study bile salt effects on a phylogenetically diverse set of vertebrate PXRs, the zebrafish PXR was chosen for a detailed comparison to human PXR and human VDR. Bony fish are the evolutionarily most distant organisms from mammals from which PXR genes has been cloned. Any function that is conserved between fish and mammals is likely to be fundamental for PXRs. Also, zebrafish biliary bile salts are quite different from those in mammals and thus provide a critical test to the hypothesis that PXRs are activated by species-specific bile salts. A previous report showed that although some pregnane and androstane steroids activated zebrafish PXR (like mammalian and chicken PXRs), mammalian bile acids such as cholic acid and lithocholic acid did not activate this receptor (7).
The bile salts of many non-mammalian species differ from the typical bile salts found in mammals and are generally commercially unavailable. To allow for the study of these compounds, the following chemically diverse bile salts were isolated from animals by extraction and Flash column chromatography (see Table 2 for chemical formulae): myxinol disulfate from the Atlantic hagfish (Myxine glutinosa); 5α-cyprinol sulfate from the Asiatic carp (Cyprinus carpio); 5β-scymnol sulfate from the Spotted eagle ray (Aetobatus narinari); and 3α,7α,12α-trihydroxy-5β-cholestan-27-oic acid, taurine conjugated, from the American alligator (Alligator mississippiensis). The isolation and purification of cyprinol and cyprinol sulfate has been previously described in detail (27). Bile alcohol sulfates were chemically deconjugated. Completeness of deconjugation and assessment of purity was performed by thin-layer chromatography using known standards.
We utilized an in vitro assay system in HepG2 human liver cells that allowed for detailed determination of the EC50 and relative efficacy of compounds that induce PXRs or VDRs (see Materials and Methods). Figs. 3A and 3C show that, as previously described (7, 8, 28), the mammalian bile acid lithocholic acid activated human PXR and VDR in the micromolar range. This activation had low efficacy relative to that produced by rifampicin and 1,25-(OH)2-vitamin D3, respectively (see Supplementary Table Ia for maximal activators used for each receptor). Lithocholic acid did not activate zebrafish PXR (Fig. 3B). In contrast, the major digestive detergent of zebrafish bile, 5α-cyprinol sulfate, was a robust activator of human and zebrafish PXRs, but not human VDR (Fig. 3). None of the three receptors were affected by unconjugated cyprinol, a poorly water-soluble compound that is present in fish mainly as a precursor to the secreted cyprinol sulfate (27). Both lithocholic acid and cyprinol sulfate also activated a GAL4-LBD fusion construct for human PXR. The EC50 values for activation of this fusion construct were similar to those determined for the full-length human PXR; the efficacies of lithocholic acid and cyprinol sulfate relative to rifampicin were higher than seen for the full-length receptor although in either case cyprinol sulfate was more efficacious than lithocholic acid as a human PXR activator (Table 2). These results indicate that the activation of the human PXR by cyprinol sulfate is a function of the LBD and not of another region of the receptor.
Figure 3. Concentration-response data for activation of human PXR, zebrafish PXR, and human VDR.
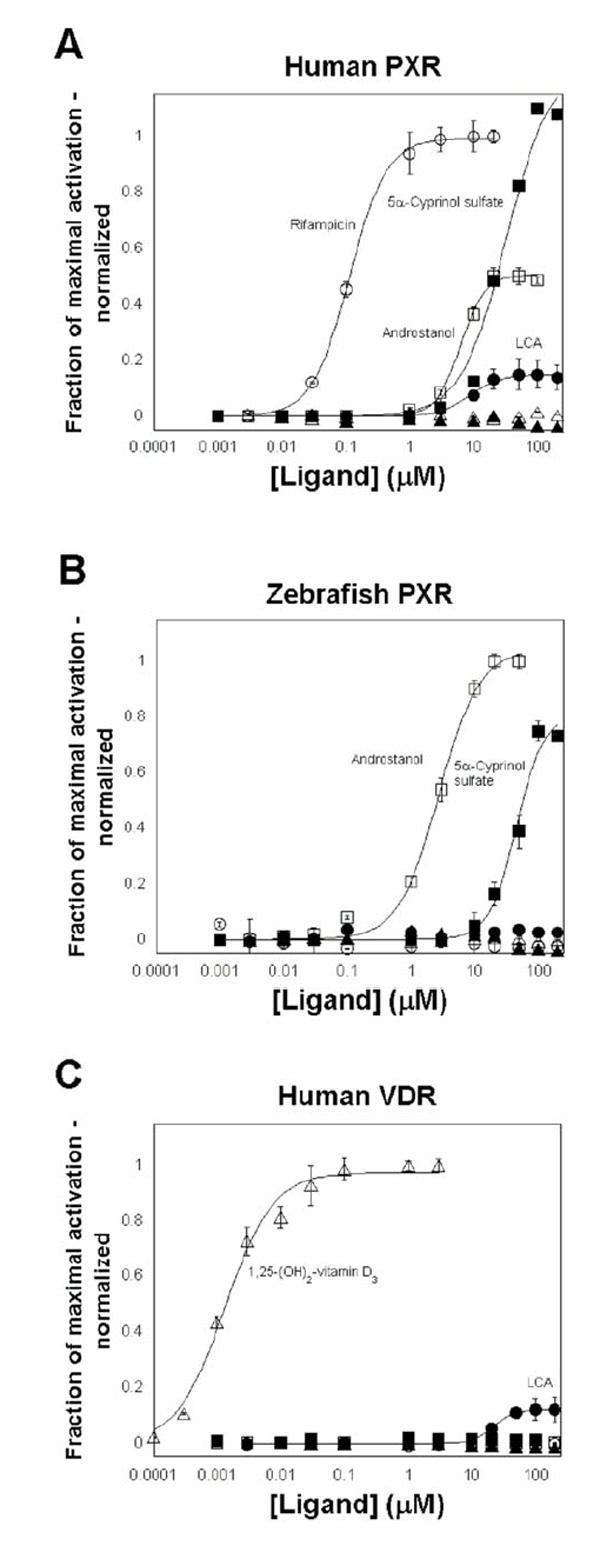
The ordinate represents activation of the PXR or VDR, relative to vehicle control, and normalized to the maximal activator (rifampicin for human PXR, 5α-androstan-3α-ol for zebrafish PXR, and 1,25-(OH)2-vitamin D3 for human VDR; see Supplementary Table Ia). The drugs tested were rifampicin (○), lithocholic acid (LCA, ●), androstanol (□), 5α-cyprinol sulfate (■), 5α-cyprinol (▲), and 1,25-(OH)2-vitamin D3 (Δ). (A) Human PXR is activated efficaciously by rifampicin, androstanol, and 5α-cyprinol sulfate. Micromolar concentrations of lithocholic acid activated human PXR but with low efficacy relative to rifampicin (approximately ε =0.15). Human PXR was not activated by 1,25-(OH)2-vitamin D3 or unconjugated cyprinol. (B) Zebrafish PXR is activated efficaciously by micromolar concentrations of androstanol and 5α-cyprinol sulfate but is not activated by lithocholic acid, unconjugated cyprinol, rifampicin, or 1,25-(OH)2-vitamin D3. (C) Human VDR is activated by nanomolar concentrations of 1,25-(OH)2-vitamin D3 and with low efficacy by micromolar concentrations of lithocholic acid. Human VDR is not activated by androstanol, rifampicin, cyprinol sulfate, or unconjugated cyprinol. In (A) and (B), full-length receptors for human PXR and VDR were used, with the reporter plasmid being CYP3A4-PXRE-Luc. In (C), a GAL4-LBD fusion was used for zebrafish PXR, with the reporter plasmid being tk-UAS-Luc.
A total of 47 structurally diverse bile salts or bile salt precursors were tested on human PXR, zebrafish PXR, mouse VDR, and human VDR. Human PXR had broad specificity for bile salts, being activated by 25 of the 47 compounds tested (Figs. 3A and 4; Table 2). Zebrafish PXR was only activated by 4 of 47 bile salts tested. In addition to cyprinol sulfate, zebrafish PXR was also activated by 5β-scymnol sulfate (a C27 hexa-hydroxylated bile alcohol sulfate derived from the Spotted eagle ray, a cartilaginous fish) and lithocholic acid 3-sulfate, indicating a preference for sulfated bile salts; zebrafish PXR was also activated weakly by the synthetic bile acid derivative lithocholic acid acetate. None of the unconjugated bile alcohols, glycine-conjugated bile acids, or taurine-conjugated bile acids tested activated zebrafish PXR (Figs. 3B and 4; Table 2). Human and mouse VDRs were activated by a narrow range of bile acids, mainly lithocholic acid and its metabolites or synthetic derivatives, and not by any of the C27 bile acids, sulfated bile alcohols, or unconjugated bile alcohols (Figs. 3C and 4; Table 2). Mouse VDR was not activated by specific rodent primary or secondary biliary bile acids including α-muricholic acid, β-muricholic acid (unconjugated and taurine-conjugated), ω-muricholic acid, and murideoxycholic acid (Table 2).
Figure 4. Patterns of bile salt activation of PXRs and VDRs.
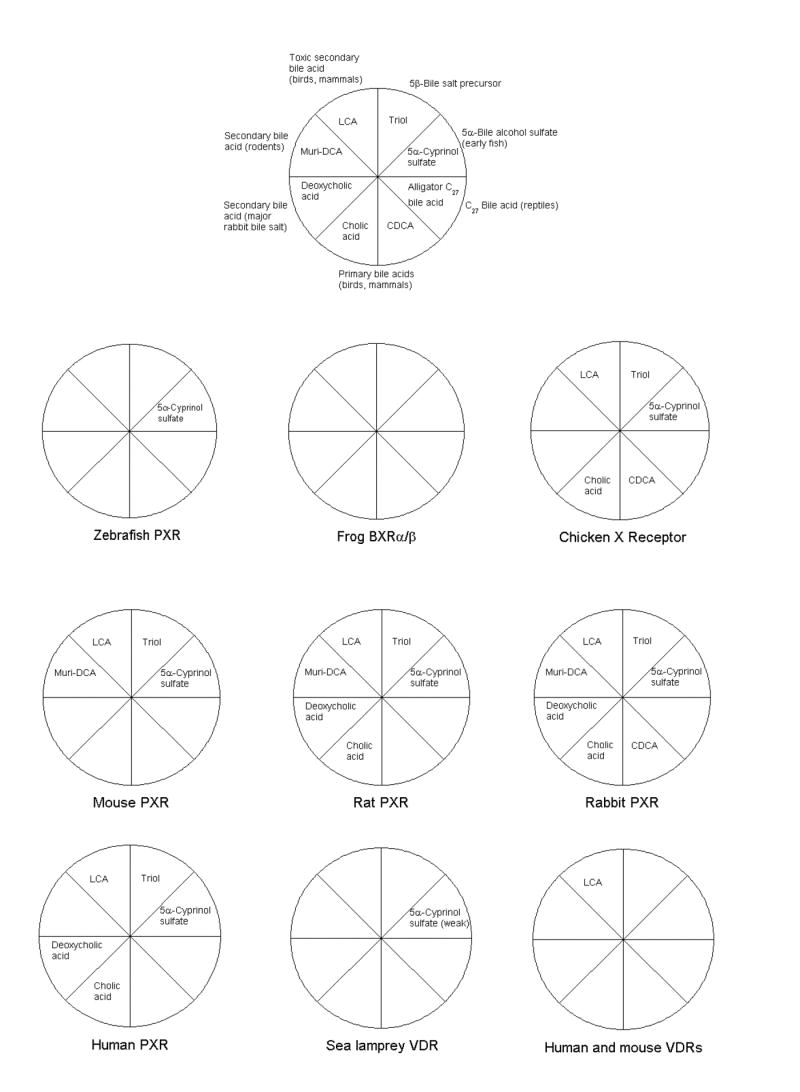
Eight compounds were chosen to encompass the chemical diversity of bile salts and their synthetic precursors across vertebrates. 5β-Cholestan-3α,7α,12α-triol (“Triol”) is a precursor to all bile salts studied in this report except the 5α-bile alcohol sulfates and uncommon 5α-bile acids (e.g., allocholic acid). The “alligator C27 bile acid’ represents taurine-conjugated 3α,7α,12α-trihydroxy-5β-cholestan-27-oic acid isolated from bile of the American alligator. Other abbreviations include: CDCA, chenodeoxycholic acid (chicken and mammalian primary bile acid); muri-DCA, murideoxycholic acid (rodent secondary bile acid); and LCA, lithocholic acid (toxic secondary bile acid of birds and mammals). Of the compounds represented, zebrafish PXR was only activated by 5α-cyprinol sulfate. Frog BXRα/β were not activated by any bile salts, consistent with these receptors having diverged from other PXRs to mediate specific developmental roles in amphibians. Chicken and mammalian PXRs are generally broadly activated by bile salts, with each species activated well by its own bile salts. Of the eight compounds depicted, mouse and human VDRs were only activated by lithocholic acid, a toxic secondary bile acid. Lamprey VDR was activated only very weakly by cyprinol (maximal effect approximately 5% that achieved with 1,25-(OH)2-vitamin D3).
Activity of the sea lamprey VDR
Recently, a VDR was cloned and functionally expressed from the sea lamprey (Petromyzon marinus), a jawless vertebrate (29). Expression of the full-length sea lamprey cDNA in a mammalian cell line resulted in a product able to transactivate a luciferase construct driven by a promoter containing the distal VDRE from the human CYP3A4 gene; expression was enhanced by co-transfection with a zebrafish RXR gene (29). In our experiments in HepG2 cells, 1,25(OH)2-vitamin D3 caused a robust increase in luciferase activity when lamprey VDR cDNA was co-transfected with CYP3A4-PXRE-Luc, a reporter plasmid containing the distal VDRE from the human CYP3A4 gene (Supplementary Table Ia). Expression was further enhanced by co-transfection with 10 ng/well of zebrafish RXRβ (Supplementary Table Ia). All subsequent experiments with lamprey VDR were thus performed with co-transfected zebrafish RXRβ.
Although the lamprey VDR was activated well by 1,25(OH)2-vitamin D3, no other compounds tested were able to activate this receptor to more than 10% of the maximal response achieved with 300 nM 1,25(OH)2-vitamin D3. Unlike the mouse and human VDRs, recent bile acids, particularly lithocholic acid, did not activate the lamprey VDR (Supplementary Table Ib). Several early bile salts, such as petromyzonol sulfate, 3-ketopetromyzonol sulfate, and cyprinol sulfate, produced weak but concentration-dependent and reproducible increases in reporter activity; however, for all three compounds, the maximal effects were only < 5-6% of the maximal effect seen with 300 nM 1,25(OH)2-vitamin D3. The EC50 value for activation of the lamprey VDR by petromyzonol sulfate and 3-ketopetromyzonol sulfate was approximately 1 μM while that for cyprinol sulfate was approximately 20 μM (Supplementary Table Ib). The small effects produced by these bile alcohol sulfates require correlation with in vivo findings in sea lampreys.
Activation of other vertebrate PXRs by bile salts
Rats and mice have a similar bile acid profile although they use slightly different primary bile acids (α- and β-muricholic acid, respectively; Table 1; Fig. 2). PXRs from both rodents were activated by micromolar concentrations of murideoxycholic acid (the secondary bile acid metabolite formed by bacterial 7-dehydroxylation of α- or β-muricholic acids), lithocholic acid, and α-muricholic acid (Table 3; Fig. 4).
Rabbits have an atypical circulating bile salt pool consisting mainly of deoxycholic acid. As in other mammals, deoxycholic acid is a secondary bile acid formed in the rabbit intestine by bacterial 7-dehydroxylation of primary bile acids such as chenodeoxycholic acid (30). For most mammalian species, however, deoxycholic acid is quite toxic and represents only a small fraction of the bile acid pool; in the rabbit, deoxycholic acid is > 95% of the circulating bile acids. Rabbit PXR was activated efficaciously in the mid-micromolar range by deoxycholic acid, glycodeoxycholic acid, taurodeoxycholic acid, and 7-ketodeoxycholic acid, and in general was activated by a variety of diverse bile salts (Table 4; Fig. 4).
The main primary bile acids in chickens are chenodeoxycholic acid and cholic acid (Table 1; Fig. 2). The chicken PXR was activated by chenodeoxycholic acid, glycochenodeoxycholic acid, cholic acid, and lithocholic acid in the micromolar range (Table 4; Fig. 4).
Interestingly, chicken, mouse, rabbit, rat, and human PXRs were all activated by 5α-cyprinol sulfate and 5β-scymnol sulfate, bile alcohol sulfates derived from carp and skate bile, respectively (Figs. 3A and 4; Tables 2-4). Because these or similar bile salts are not the major bile salts of chickens, rabbits, mice, rats, or humans (although they can be found in trace amounts in healthy animals or in higher levels in certain disease states; see Discussion), activation by these compounds likely represents an ancestral property conserved in chicken and mammalian PXRs.
BXRα and BXRβ are insensitive to all bile salts
Bile from Xenopus laevis frogs and some other amphibians contain a mixture of C27 bile alcohols and bile acids (L.R. Hagey, unpublished data) (24-26). In this study, we tested a variety of bile alcohol sulfates, unconjugated bile alcohols, and bile salts, including myxinol disulfate (from hagfish), cyprinol sulfate (from carp), scymnol sulfate (from skate), petromyzonol sulfate (from sea lamprey), and taurine-conjugated 3α,7α,12α-trihydroxy-5β-cholestan-27-oic acid (from alligator; same as the C27 bile acid found in Xenopus laevis), on BXRα and BXRβ. None of these compounds activated BXRα or BXRβ (Supplementary Table Ic; Fig. 4), suggesting that these receptors do not play a role in sensing bile salts. As previously reported (21), benzoate compounds robustly activated these two receptors (Supplementary Table Ic).
The effects of five benzoate compounds on zebrafish, chicken, and mammalian PXRs have been previously reported (7). Interestingly, two of the benzoate compounds (n-butyl-p-aminobenzoate and n-propyl-p-hydroxybenzoate) tested by Moore et al. activated a number of the PXRs (7). The endogenous benzoate isolated by Blumberg et al. from Xenopus laevis embryos (21), 3-aminoethylbenzoate, however, only activated chicken PXR in addition to BXRα (7). We confirmed the sensitivity of human PXR to n-butyl-p-aminobenzoate and also show activation of zebrafish PXR by n-propyl-p-hydroxybenzoate (Table 2). In contrast, n-butyl-p-aminobenzoate, n-propyl-p-aminobenzoate, and 3-aminoethylbenzoate all did not activate human, mouse, or lamprey VDRs (Tables 2 and 4; Supplementary Table Ib).
Activation of PXRs by the bile salt precursor 5β-cholestan-3α,7α,12α-triol
The bile salt biosynthetic pathways for all of the vertebrates considered in this study except zebrafish include 5β-bile alcohol precursors (zebrafish use an early 5α-bile alcohol pathway). One of these precursors is 5β-cholestan-3α,7α,12α-triol, a compound which accumulates in the rare disease cerebrotendinous xanthomatosis (CTX; OMIM # 213700), an inborn error of metabolism caused by deficiency of CYP27A1. In humans, CYP27A1 deficiency leads to pathologic accumulations of C27 bile alcohols and the hallmark symptoms of CTX, namely xanthomas, gallstones, and neurological dysfunction (31).
Tables 2-4 show that while human and zebrafish PXRs were not activated by 5β-cholestan-3α,7α,12α-triol, mouse, rabbit, rat, and chicken PXRs were all activated by low micromolar concentrations of this bile alcohol. Rat and mouse PXRs were activated particularly efficaciously by this bile alcohol (Table 3), as previously described for the mouse PXR (3, 4). BXRα and BXRβ were not activated by 5β-cholestan-3α,7α,12α-triol (Supplementary Table Ic). The lack of activation of zebrafish PXR by 5β-cholestan-3α,7α,12α-triol may be a result of these fish utilizing the earlier 5α-bile alcohol biosynthetic pathway also found in jawless fish; as a result, 5β-cholestan-3α,7α,12α-triol is not an intermediate in the synthesis of 5α-cyprinol sulfate, the major zebrafish bile salt (27). None of the PXRs or VDRs were activated by 7α-hydroxycholesterol, the earliest precursor in the biotransformation of cholesterol to bile salts (Tables 2-4).
The lack of activation of human PXR by the bile salt precursor 5β-cholestan-3α,7α,12α-triol detected in the transactivation assay is consistent with previous data by Dussault et al. showing that 5β-cholestan-3α,7α,12α-triol is a very low efficacy partial agonist of human PXR (3). In that report, 5β-cholestan-3α,7α,12α-triol weakly transactivated a human PXR construct but was unable to induce CYP3A4 mRNA expression in primary human hepatocytes; 5β-cholestan-3α,7α,12α-triol did, however, competitively displace [3H]-SR12813 from binding to human PXR, demonstrating that this bile alcohol does bind to human PXR (3). The significance of the lack of efficacy for 5β-cholestan-3α,7α,12α-triol for activating human PXR is uncertain, although it may be of interest that studies with primary human hepatocytes show that this bile alcohol precursor is practically non-existent in the formation of bile acids such as cholic or chenodeoxycholic acid, a finding that contrasts with the previously assumed pathways of bile acid biosynthesis in humans (32). Comparative studies in other species have not been performed to study whether this finding is unique to humans or common throughout mammals.
Evidence for non-neutral evolution of the PXR ligand-binding domain
Previous phylogenetic investigations of PXR genes have only looked at two-sequence comparisons in calculating dN/dS (ω) ratios - one study focused on human, mouse, and rat sequences (22) and the other on human, chimpanzee, and mouse sequences (33). Table 5 shows the results from PAML analysis of sequence data for PXR and VDR genes. A total of 6 analyses were performed for each gene: full-length sequence, restricted to DBD, and restricted to LBD for datasets of either all available vertebrate species or mammals only.
The analyses for the full-length sequence, DBD, and LBD for VDR, whether applied to all available species or mammals only, show low ω ratios, consistent with purifying (negative) selection as the dominant evolutionary force for this gene. The PXR analyses, on the other hand, for the full-length sequence and LBD shows a minority of codons (sites) with ω ratios approaching or even exceeding one (results in bold in Table 5). In contrast, the ω ratios for the DBD are low for the PXR analyses. These results are consistent with positive selection acting on the PXR LBD.
The analysis for the LBD of PXR restricted to mammalian species, where 7% of codons were associated with an ω ratio of 1.23, also highlighted 4 amino acids residues as likely targets of positive selection: 187S, 231S (H1-H3 insert), 312A (helix 6), and 360R (helix 9) (amino acid numbers for human PXR as in Moore et al., (7); complete sequence alignment of the LBD of VDRs and PXRs available as Supplementary Figure 1). Posterior probabilities for the positive selection of the individual codon sites are greater than 0.99 for 360R and 0.90-0.98 for 187S, 231S, and 312A.
We also utilized PAML to detect whether evidence of molecular adaptation occurs along specific lineages. With regard to ligand specificity, the most divergent of the PXRs are clearly the BXRs (Fig. 5). If the assignment of BXRα and BXRβ as true orthologs of other PXRs is correct, then these receptors have lost the ability to respond to endogenous steroids and bile salts and also the broad specificity required to bind structurally diverse xenobiotics. Using a PAML model that allows for two discrete classes of ω for all sequences in a phylogeny and an additional class of ω for defined lineage(s), (‘model B’ of ref. (34); see Materials and Methods for more details), we analyzed the entire set of available PXRs. For the full-length, DBD only, and LBD only sequences, we separately analyzed four specific sets of lineages for positive selection: the branch from all other PXRs to the BXRs (‘branch a’ of Fig. 5), the branch to BXRα (‘branch b’ of Fig. 5), the branch to BXRβ (‘branch c’ of Fig. 5), and branches a, b, c together.
Figure 5. Hypothetical phylogeny of PXRs and VDRs showing functional characteristics.
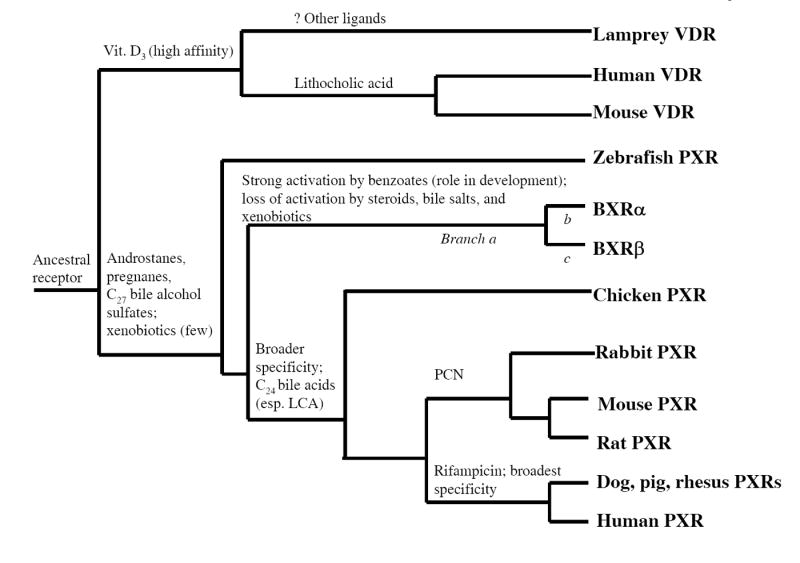
The phylogenetic tree is based on known phylogenetic relationships between the species. Features included are activation by endogenous compounds (bile acids, bile alcohol sulfates, androstane steroids, pregnane steroids, 1,25-(OH)2-vitamin D3, and benzoates) and xenobiotics (pregnenolone 16α-carbonitrile, PCN; rifampicin). The organization is one possible arrangement and may change with sequence data in additional species. Branches a, b, and c indicate specific branches of the phylogeny tested in the phylogenetic analysis by maximum likelihood (see Results and Supplementary Table II).
We find evidence for positive selection in individual branches a, b, and c (Supplementary Table II). The amino acid residues identified as likely targets of positive selection are all in the LBD (Supplementary Figure 1; Supplementary Table II). Three of the residues identified correspond to residues in human PXR shown to directly interact with ligand in the two crystal structures of the human PXR LBD complexed with ligand (35, 36). This suggests that if indeed BXRs are orthologs of other PXRs, then sequence changes that altered the ligand specificity and response of an ancestral PXR have been positively selected for in amphibian evolution.
DISCUSSION
Activation of PXRs by species-specific bile salts is conserved across vertebrates
The major finding of this study is that activation of PXR by bile salts is conserved across vertebrates, from teleost fish to mammals, an evolutionary time span of more than 400 million years. A previous study had shown no activation by bile salts in zebrafish PXR but only tested mammalian-type C24 bile acids (7). The exception to the general sensitivity of PXRs to bile salts is found in the frog. Xenopus laevis PXRs, the benzoate X receptors (BXRs), exhibit pharmacologic properties and tissue expression patterns markedly different from other PXRs (see below) (7, 21, 37).
PXR activation by bile salts parallels the evolution of complexity of the bile salt biosynthetic pathway. Unlike most forms of evolution, bile salt evolution has proceeded by elongation of an enzyme pathway. This long pathway is now shared by different hepatic organelles, with its start in the mitochondria, steroid ring modification in the endoplasmic reticulum and cytoplasm, and side-chain modifications in the peroxisomes (38). Jawless fish, cartilaginous fish, and some bony fish (e.g., zebrafish) utilize relatively simple bile alcohol pathways that generally leave the side-chain of cholesterol intact. The insensitivity of zebrafish PXR to ‘recent’ 5β-bile acids is perhaps not surprising, as these fish would never encounter such compounds (nor would their ancestors). PXR activation by bile salts has thus expanded from narrow specificity for C27 bile alcohol sulfates (early fish pattern) to a broader specificity for recent bile acids and their synthetic precursors (birds and mammals). PXR selectivity for bile salts has therefore paralleled the elongation and increasing complexity of the bile salt synthetic pathway during vertebrate evolution.
The evolution of the bile salt biosynthesis pathway and its relation to PXR evolution contrasts with that for the classical steroid hormones and their cognate receptors. In a detailed analysis of the vertebrate NR3 family (estrogen, androgen, progesterone, glucocorticoid, and mineralocorticoid receptors), Thornton has proposed that “…the terminal ligand in a biosynthetic pathway is the first for which a receptor evolves; selection for this hormone also selects for the synthesis of intermediates despite the absence of receptors, and duplicated receptors then evolve affinity for these substances (39).” Thornton also concluded that the estrogen receptor was likely the most ancient classical steroid receptor (39), an idea supported by an elegant reconstruction and functional expression of the ancestral estrogen receptor (40). The bile salt biosynthetic pathway has evolved in a considerably different fashion from the adrenocortical and sex steroid pathways, starting off from a relatively simple pathway to produce C27 bile alcohol sulfates and steadily elongating to a more complex pathway that results in the production of C24 bile acids. Therefore, the evolution of PXR in mammals and birds has adapted not to the intermediate products of bile salt biosynthesis but to the species-specific, specialized, terminal products of a biosynthetic pathway elongating over evolutionary time.
Biliary bile salts show a striking amount of diversity between different vertebrate species (24, 25). This contrasts with hormones or neurotransmitters, which tend to be well-conserved (41). The PXRs have adapted to cross-species variation in bile composition. The zebrafish PXR has narrow selectivity towards bile salts, being activated by only two sulfated bile alcohols, one sulfated bile acid, and one synthetic bile acid derivative out of a panel of 47 bile compounds tested. The most efficacious activation of zebrafish PXR was by its dominant bile detergent, 5α-cyprinol sulfate. In contrast, mammalian PXRs, in particular human and rabbit PXRs, are activated efficaciously by a wide variety of bile acids (conjugated or unconjugated) and sulfated bile alcohols. Rat and mice PXRs are activated efficaciously by α-muricholic acid as well as murideoxycholic acid, the secondary bile acid of rodent α- and β-muricholic acids. In contrast, murideoxycholic acid does not activate human or chicken PXRs, two animals where this bile acid would not be present. This suggests that PXRs have adapted to subtle species-specific differences in bile salt biosynthesis and metabolism.
An unexpected finding was that all PXRs except frog BXRs were activated by 5α-cyprinol sulfate and 5β-scymnol sulfate. The most ancient vertebrates, hagfish and lampreys (jawless fish), utilize 5α-bile alcohol sulfates similar to cyprinol sulfate as their major bile detergents; these compounds are assumed to be the first class of bile detergents synthesized by ancestral vertebrates (42, 43). Consequently, activation of PXR by the earliest bile constituents, 5α-bile alcohols sulfates, is an ‘ancestral’ property conserved in comparatively recent fish (like zebrafish), birds, and mammals. This pattern of conservation is remarkable considering the extensive divergence between the LBDs of zebrafish and mammalian PXRs (only 50% amino acid sequence identity) and the more than 400 million years of time since the last common ancestor of tetrapods and telost fish (44).
Interestingly, even though C24 bile acids are the dominant digestive detergents of most mammals, low concentrations of 5β-cyprinol or similar C27 bile alcohols are found in humans and other mammals (45-47). This is a reflection of the evolution of the bile salt biosynthetic pathway described above, in which the synthesis of a recent bile acid such as cholic acid results from extra processing and enzymatic steps ‘added onto’ the basic pathway that produces C27 bile alcohols (38). As a result, all mammals produce some C27 bile alcohols although the efficiency of conversion to C24 bile acids in healthy animals normally reduces bile alcohols to trace levels. Detailed analysis of urinary bile alcohols in healthy humans reveals a range of bile alcohols including 5β-cyprinol; in patients with cholestasis and certain inborn errors of bile metabolism, levels of urinary bile alcohols can rise dramatically (46, 48). In addition, a small number of mammals including elephants and rock hyraxes produce C27 bile alcohol sulfates as their only bile detergents; in essence, these mammals have retained the basic bile detergent synthetic pathway found in ancestral fishes (24, 25). The persistence of C27 bile alcohols even in healthy animals and their elevation in certain disease states may provide some selective advantage for mammalian PXRs to ‘maintain’ their ancestral sensitivity to early bile salts such as cyprinol sulfate and scymnol sulfate.
Physiological importance of bile salt activation of vertebrate PXRs
Detection of bile salts by PXR may not simply be a mechanism to increase removal of bile salts but part of a more general protective mechanism. In fact, with a few exceptions (e.g., lithocholic acid), bile salts are not highly toxic compounds (49). However, elevated intra-hepatic levels of bile acids are a ‘sentinel’ marker of cholestasis and herald impaired biliary elimination of other toxic compounds. Activation of PXRs by abnormally high bile salt concentrations would be beneficial by inducing alternative routes of metabolism and elimination of toxins normally cleared in the bile.
Lithocholic acid activated chicken PXR and all mammalian PXRs. Numerous studies have demonstrated the toxicity of this mono-hydroxylated, secondary bile acid, generated by bacterial 7-dehydroxylation of primary bile acids such as chenodeoxycholic acid in the intestine (49). Lithocholic acid and its metabolites were also activators of mouse and human VDRs, as previously reported (28). For animals using C24 bile acids such as chenodeoxycholic acid, mechanisms to detect and reduce the toxicity of lithocholic acid would be advantageous. In the case of mammals, two related NRs, PXR and VDR, both detect lithocholic acid and activate pathways to detoxify this compound (28, 49). The studies with sea lamprey VDR show that activation by lithocholic acid and its derivatives is not a conserved property of all VDRs.
While the physiologic importance of PXR activation by bile acids in mammals has been demonstrated, the situation is less clear in non-mammalian species. The metabolism and toxicology of fish bile salts has been little studied, and the genes affected by PXR activation in fish have not been defined. 5α-cyprinol sulfate, the bile detergent of zebrafish and other cypriniform fish (e.g., carp, goldfish, zebrafish), is quite toxic to a number of mammals (27, 50), including humans, and can cause renal and hepatic failure in individuals who ingest raw goldfish or carp gallbladders (51), a delicacy in East Asia.
Relation of PXR activation to physiologically relevant bile salt concentrations
The concentration of species-specific bile salts that activate the vertebrate PXRs in this study are within the range encountered in disease affecting the hepatobiliary system. In healthy humans, plasma concentrations of bile acids are generally low micromolar (see Materials and Methods). In subjects with cholestasis from a variety of etiologies, plasma bile acid concentrations can rise dramatically and exceed 100 μM, well within the levels that activate human PXR (10).
Plasma concentrations of bile acids in healthy chickens, mice, rats, and rabbits are similar to humans, with normal total plasma bile acid concentrations generally in the low micromolar range (see Materials and Methods). The concentrations required to activate PXRs in these species are generally about an order of magnitude or more higher than would be seen in healthy animals and would be encountered in animals with hepatobiliary disorders such as cholestasis. As in humans, diseases affecting the hepatobiliary systems can dramatically elevate plasma bile acid concentrations in other animals. Concentration of plasma bile acids exceeding 1 mM have been achieved in experimental animals, such as in mice following bile duct ligation (52). Unfortunately, at the present time intra-hepatocyte or plasma concentrations of bile salts, or their degree of binding to plasma proteins, have not been reported for fish or amphibians.
Lack of activation of frog BXRs by bile salts
BXRα and BXRβ, the putative PXRs from Xenopus laevis, are clearly different from other vertebrate PXRs. Although the complete genome of Xenopus laevis has not yet been sequenced (leaving open the possibility of discovering additional PXR-like genes), sequence similarity and phylogeny strongly supports the classification of these receptors as PXRs (7, 37, 53). Nevertheless, the tissue distribution and functional properties of these receptors are quite different from other PXRs. For example, BXRs are highly expressed in ovary and testis but poorly expressed in xenobiotic metabolizing organs such as liver, lung, or kidney (37). BXRα and BXRβ do not appear to function as xenobiotic sensors, and previous reports have shown these receptors to be insensitive to a wide range of steroidal compounds (7, 21, 37). The present study finds these two receptors to be completely insensitive to amphibian bile salts. BXRα and BXRβ are both activated well by benzoates, compounds that have unique roles in frog development (21). Assuming that these two receptors are indeed orthologous to other PXRs, they have evidently diverged extensively to perform specific benzoate-mediated developmental functions in frogs (21, 37).
In this report, we confirmed the findings of Moore et al. that certain benzoate compounds activate a variety of PXRs other than the BXRs (7), including those from zebrafish, chicken, and human, and also show that activation by benzoates is not shared by human, mouse, or lamprey VDRs. The significance of activation of PXRs other than BXRα and BXRβ by synthetic benzoates is unknown; physiologic actions of this class of endogenous compounds has only been described in amphibians (21, 37). Even if benzoates do not have a physiologic role in modern-day fish, birds, or mammals, it is possible that ancestors to these species may have used benzoate compounds and, thus, that sensitivity to these compounds is a shared ‘ancestral trait’ of PXRs. Alternatively, low-affinity activation of non-amphibian PXRs by benzoates, compounds with low molecular weight relative to other PXR ligands, may simply be a consequence of the broad specificity of PXRs and not have physiological or evolutionary significance. BXRs have not been cloned and characterized from any species other than Xenopus laevis (21, 37). Whether receptors similar to the Xenopus BXRs exist in other amphibians (e.g., the salamander axolotl), reptiles, or any other species remains to be discovered. Complete sequencing of the Xenopus genome and of other species will be critical in determining if BXRs are indeed orthologous to other PXRs and also whether amphibians have PXR-like xenobiotic receptors that have not yet been identified.
Evolution of the NR1I subfamily of nuclear hormone receptors
The NR1I subfamily includes VDR (NR1I1), PXR (NR1I2), and the constitutive androstane receptor (CAR; NR1I3) (6, 7, 54). VDR and PXR are found in diverse vertebrates from bony fish to mammals. CAR, on the other hand, appears restricted to mammals, although there is some uncertainty in classifying the chicken PXR (CXR), as this receptor has about equal sequence and functional similarity to both PXRs and CARs (7, 20). Recent evidence suggests that the CAR gene arose from a duplication of a pre-mammalian PXR gene (55). This is consistent with the demonstration of two xenobiotic-responsive NRs in mammals, but only one in chickens and fish (53, 55). The complete genome of the fugu fish contains a single PXR gene and shows no evidence for a CAR gene (53).
Based on current genetic data, distinct NR1I subfamily members have been found only in vertebrates. A VDR was cloned and functionally expressed from the sea lamprey (Petromyzon marinus), a surprising finding as this jawless vertebrate lacks calcified tissue (29). PXR has not yet been identified in jawless vertebrates, and our attempts to clone a PXR gene from the sea lamprey have been unsuccessful. The draft genome of the urochordate Ciona intestinalis, the closest invertebrate relative of vertebrates for which complete genome information is available, revealed a single NR gene equally related to VDR and a Drosophila NR (56). The functional properties of this invertebrate NR remain uncharacterized.
An excellent study of the evolutionary genomics of the NR superfamily provides convincing evidence that a single ancestral NR duplicated and diverged to form VDR and PXR. This paper also proposes that the NR complement in the ancestor to Bilateria (animals) possessed 9 genes in the NR1 family (57). Thus, distinct PXR and VDR genes are likely vertebrate innovations although complete genome information is not yet available for a cephalochordate such as amphioxus, an invertebrate chordate more closely related to vertebrates than Ciona.
Fig. 5 shows a hypothetical phylogenetic tree of the NR1I subfamily that incorporates functional characteristics of the subfamily members. Pharmacologic features shown are activation by endogenous compounds (bile acids, bile alcohol sulfates, steroid hormones, benzoates, and 1,25-(OH)2-vitamin D3) and xenobiotics (pregnenolone 16α-carbonitrile, PCN; rifampicin). Fig. 5 shows the progressive broadening of ligand specificity for the PXRs, with human, dog, and pig PXRs having the broadest ligand specificity, at least for the ligands which have been tested to date (7).
Several reports have suggested that the most ancient function of the NR1I family is to recognize toxic levels of exogenous and/or endogenous compounds (7, 11, 28, 29). The detoxification function of VDR in humans has only been recently recognized, and is exemplified by the ability of VDR to sense toxic levels of lithocholic acid in the intestine and upregulate metabolic enzymes to detoxify this secondary bile acid (17, 28). A further description of the NR1I subfamily origins awaits complete genome information in jawless fish and cephalochordates and functional characterization of the NR1I subfamily members in these organisms, if present.
Structural basis for differing ligand selectivities between the PXRs
The mechanism of ligand activation of human PXR has been demonstrated by high-resolution, crystallographic, three-dimensional structures of the LBD alone (35) and in combination with the hypocholesterolemic compound SR12813 (35, 58), hyperforin (active component of St. John’s wort) (36), or the human steroid receptor coactivator-1 (SRC-1) (58). The human PXR LBD shares structural features with other NR LBDs, including an ‘α-helical sandwich’ of helices α1/α3, α4/α5/α8, and α7/α10 in one hemisphere of the molecule and the ligand-binding cavity in the other hemisphere (35, 59). The human PXR ligand-binding cavity is smooth, hydrophobic, and large (1150 Å3), second in size for NRs only to peroxisome proliferator-activated receptor-γ (1500 Å3) (60). This is in contrast to most NRs, which have a compact ligand-binding cavity that closely resembles the shape of their specific ligand(s) (59). There is also considerable flexibility in the human PXR ligand-binding cavity, which increases in size by 250 Å3 following binding of hyperforin (36). There are a number of additional features of the human PXR LBD that contribute to its broad ligand specificity: a variable four-residue turn between helices α1 and α3, replacement of α6 by a large, flexible loop, and two additional β strands not observed in other NRs (35, 36, 58, 59).
A total of eighteen amino acid residues in the human PXR LBD have been identified as contacting either SR12813, hyperforin, or both (see Supplementary Figure 1 for sequence alignments and highlighting of ligand-binding residues); one residue contacts hyperforin only and five residues contact SR12813 only (35, 36). No three-dimensional structures have been reported of human PXR bound to an endogenous ligand. The generally low affinity of these compounds may make this difficult to achieve. In comparing human and zebrafish PXR sequences, 12 of the 18 amino acid residues identified in binding SR12813 and/or hyperforin differ between the two sequences, in some cases dramatically (Supplementary Figure 1A,B). In fact, alignment of human PXR, chicken PXR, BXRα, BXRβ, and zebrafish PXR shows that only 3 of these 18 ligand-contacting amino acid residues are completely conserved among these five sequences (Supplementary Figure 1B). A combination of molecular modeling and site-directed mutagenesis will probably be required to elucidate the structural basis of ligand selectivity differences between these PXRs.
Contrasting levels of PXR nucleotide diversity within humans and between vertebrates
The marked diversity of the PXR LBD between vertebrate species contrasts with a detailed re-sequencing study of the human PXR gene that showed very low nucleotide diversity and no non-synonymous substitutions in the PXR LBD in genomic DNA of approximately 100 individuals from diverse ethnic populations. The total nucleotide diversity was 1 in 21,607 bp for the LBD and 1 in 22,053 for the DBD (61). This low level of nucleotide diversity within humans differs markedly with the high levels of divergence of PXR genes between vertebrate species. This suggests that important ligand(s) for PXR do not vary significantly across human populations but do vary across vertebrate species. We propose biliary bile salts as a group of endogenous ligands that fit this qualification.
In summary, this study shows that activation by species-specific bile salts is a conserved property of PXRs from teleost fish to mammals and that recognizing high concentrations of bile salts is likely an ancestral function of PXRs. More strikingly, PXRs specificity to bile salts has expanded during vertebrate evolution, mirroring the complex evolution of bile salt biosynthetic pathways across vertebrate species. Given evidence for positive selection acting on the PXR LBD, we propose that differences in biliary bile salts between vertebrate species have been a selective force for PXR evolution.
MATERIALS AND METHODS
Isolation of bile salts from natural sources
Commercially unavailable bile salts were obtained by extraction and Flash column chromatography using bile from natural sources in which a single major bile salt predominates. The protocol had been approved by the Committee on Animal Studies of the University of California, San Diego. Bile salts isolated were (see Table 2 for chemical formulae): myxinol disulfate from the Atlantic hagfish (Myxine glutinosa); 5α-cyprinol sulfate from the Asiatic carp (Cyprinus carpio); 5β-scymnol sulfate from the Spotted eagle ray (Aetobatus narinari); and 3α,7α,12α-trihydroxy-5β-cholestan-27-oic acid, taurine conjugated, from the American alligator (Alligator mississippiensis). The isolation and purification of cyprinol and cyprinol sulfate has been previously described in detail (27). Bile alcohol sulfates were deconjugated using a solution of 2,2-dimethoxypropane:1.0 N HCl, 7:1 v/v, and incubating 2 hours at 37°C, followed by the addition of water and extraction into ether. Completeness of deconjugation and assessment of purity was performed by thin-layer chromatography using known standards.
Reagents
The sources of the chemicals were as follows: rifampicin, n-propyl-4-hydroxybenzoate, n-butyl-4-aminobenzoate, pregnenolone 16α-carbonitrile, nifedipine, 1,25-(OH)2-vitamin D3, glycocholic acid, taurocholic acid, glycodeoxycholic acid, taurodeoxycholic acid, lithocholic acid 3-sulfate, taurolithocholic acid 3-sulfate, and glycolithocholic acid 3-sulfate (Sigma, St. Louis, MO, USA); 5α-petromyzonol, petromyzonol sulfate, 3-ketopetromyzonol, 3-ketopetromyzonol sulfate, 3-keto-7α,12α-dihydro-5α-cholanic acid, and allocholic acid (Toronto Research Chemicals, Inc., North York, ON, Canada). All other bile salts as well as 5α-androstan-3α-ol and 5β-pregnane-3,20-dione were obtained from Steraloids (Newport, RI, USA).
Construction of HepG2 cell line stably expressing human NTCP
HepG2 (human liver) cell lines stably expressing human NTCP (62), which is required for effective uptake of conjugated bile compounds into cells, were used for the transactivation experiments. To construct this line, hNTCP cDNA was subcloned into pcDNA3. HepG2 cells (104 cells per P60 dish, plated on day 1) were transfected on day 2 by calcium phosphate co-precipitation with 5 μg of pcDNA3-hNTCP plasmid. HepG2 cells stably transfected with pcDNA3-hNTCP were grown in MEM-α medium (Invitrogen) containing 10% fetal bovine serum and 1% penicillin/streptomycin. The HepG2-hNTCP cells were grown under selective pressure with 1000 μg/ml G418. The cell lines were grown at 37°C in 5% CO2.
To determine the hNTCP expression, RNA was isolated from HepG2-hNTCP cells using Trizol reagent (Invitrogen). Total RNA (5 μg) was reverse-transcribed according to instructions. Primers used to amplify were 5’-gggacatgaacctcagcatt-3’ (forward) and 5’-cgtttggatttgaggacgat-3’ (reverse). PCR reactions contained 1 μL of cDNA, 5 pmol of primers, 1.5 mmol MgCl2, 0.2 mmol dNTPs and 2.5 U of Taq polymerase (Promega, Madison, WI, USA). PCR consisted of an initial denaturation at 92°C for 5 min followed by 34 cycles of denaturation at 92°C for 30 s, annealing at 60°C for 30 s, and synthesis at 72°C for 30s, with a final extension at 72°C for 10 min.
Transactivation assays and cell culture
Plasmids containing cDNAs for 8 PXRs from 7 species (human, mouse, rat, rabbit, chicken [CXR], frog [BXRα and BXRβ], zebrafish), human vitamin D receptor (VDR; NR1I1), human Na+-taurocholate cotransporter (NTCP; SLC10A1), human organic anion transporting polypeptide (OATP; SLC21), as well as the reporter constructs tk-UAS-Luc and CYP3A4-PXRE-Luc, and ‘empty’ vectors pCDNA, PsG5, and PM2 were generously provided by SA Kliewer, JT Moore, and LB Moore (GlaxoSmithKline, Research Triangle Park, NC). The sea lamprey VDR clone (the ‘insertless’ full-length cDNA) was generously provided by G.K. Whitfield (University of Arizona College of Medicine, Tucson, AZ) (29). Mouse VDR (IMAGE clone 3710866) and pCMV-sport6 vectors were obtained from Invitrogen (Carlsbad, CA). The zebrafish RXRβ cDNA clone (IMAGE clone 5410111) was obtained from ATCC (Manassus, VA). The expression vectors were either full-length receptors (i.e., containing both a DBD and LBD; human, mouse, rat, rabbit, and chicken PXRs, as well as human, mouse, and sea lamprey VDRs) or GAL4/PXR chimeras that contain only the LBD of the PXR receptor (human SXR, BXRα, BXRβ, and zebrafish PXR) (7). For the full-length expression vectors, the reporter plasmid was CYP3A4-PXRE-Luc, a construct that contains a promoter element from CYP3A4 (recognized by PXR and VDR DBDs) driving luciferase expression. For the GAL4/LBD expression constructs, the reporter plasmid was tk-UAS-Luc, which contains GAL4 DNA binding elements driving luciferase expression. The sea lamprey VDR cDNA was co-transfected with zebrafish RXRβ (15 ng/well) for more robust expression.
It should be pointed out that cross-species differences in the DBDs of various PXRs could impact the ability of a particular PXR to activate the human CYP3A4-based promoter driving luciferase expression. However, this is unlikely to affect the pharmacology of the various ligands studied in this report, particularly as ligand activation of a particular receptor was normalized to a specific maximal activator. The most distantly related PXRs to the human PXR, zebrafish PXR and the frog BXRs, were studied using GAL4-LBD fusion constructs, so issues of cross-species differences in the DBD do not affect those receptors in this study. Also, the transactivation assays used will not detect compounds that bind to PXRs but do not transactivate (or transactivate very poorly). Other than for human PXR, suitable radioligands for other PXRs have not yet been developed.
The cell line used for the assays was HepG2 (human liver) cells stably expressing human NTCP, a transporter that can uptake conjugated bile acids. For experiments involving sulfated compounds, human OATP was co-transfected at 10 ng/well to facilitate uptake of sulfated steroids or bile salts. On day 1, 30,000 cells/well were seeded onto 96-well white opaque plates (Corning-Costar, Corning, NY, USA). On day 2, cells were transfected using the calcium phosphate precipitation method with expression vector or ‘empty’ control vector and luciferase reporter plasmid. On day 3, the cells were washed and then incubated with medium containing charcoal-dextran treated fetal bovine serum (Hyclone, Logan, UT, USA) and drugs or vehicle. On day 4, the cells were washed and the medium replaced with serum-free medium. Luciferase reagent with passive lysis buffer (Steady-Glo, Promega) was added. Two hours later, luminescence was measured in a 96-well luminometer (Analyst AD, Molecular Devices, Sunnyvale, CA, USA). An aliqot of lysed cells was taken to measure total protein content (Bio-Rad RC DC Protein Assay, Hercules, CA, USA). Experiments were performed in quadruplicate and repeated for a total of at least three times.
Activation of receptor by ligand was compared to receptor exposed to identical conditions without ligand (‘vehicle control’). In general, dimethyl sulfoxide (Sigma) was used as vehicle and was adjusted to be 1% (v/v) in all wells. A control was also run with transfection of ‘empty’ vector (i.e., lacking the receptor cDNA) and reporter vector to control for activation of reporter vector by endogenous receptor(s). In experiments with a variety of activators, activation by endogenous receptors was not seen. Concentration-response curves were fitted using Kaleidagraph software (Synergy Software, Reading, PA, USA). Data are presented throughout as mean ± S.E.M. In combining data from multiple experiments, the pooled variance was calculated by the formula , where there are N total data points among k groups, with n replicates in the ith group.
Determination of maximal activators for the various PXRs
Using previous comparative studies of vertebrate PXRs as a guide (6, 7), each PXR construct was tested with compounds previously shown to be robust activators of the respective receptors. Supplementary Table Ia shows the receptors tested, the type of reporter construct used, the amount of reporter construct and receptor cDNA transfected per well, and the activation of luciferase activity, relative to vehicle control, produced by a ‘maximal activator’. As can be seen, maximal induction of luciferase activity varies over almost 2 orders of magnitude. This is a result of differing levels of baseline (constitutive) activation and intrinsic differences in receptor properties. Mouse and chicken PXRs show high levels of constitutive activation and low maximal activation by ligand; in contrast, human VDR, mouse VDR, BXRα, BXRβ, and zebrafish PXR have low levels of constitutive activity and show greater than 10-fold maximal activation (6, 7, 20, 21, 37). This variability in activation makes it difficult to perform cross-species comparisons of differences in ligand efficacy.
To facilitate more reliable cross-species comparisons, complete concentration-response curves for ligands were determined in the same microplate as determination of response to a maximal activator. This allows for determination of ‘relative efficacy’, ε, defined as the maximal response to test ligand divided by the maximal response to a reference maximal activator (note than ε can exceed 1). Compounds with ε < 0.10 were designated ‘inactive.’ The maximal activators and concentrations eliciting maximal response are listed in Supplementary Table Ia. Fig. 3 shows concentration-response curves for human PXR, zebrafish PXR, and human VDR. The ordinate represents activation of the PXR or VDR, relative to vehicle control, and normalized to the maximal activator. For the human PXR data in Fig. 3A, rifampicin is the reference maximal activator (and hence has ε=1.0 by definition) while lithocholic acid has lower relative efficacy (approximately ε=0.15). Similarly, 5α-androstan-3α-ol and 1,25-(OH)2-vitamin D3 are the reference maximal compounds for zebrafish PXR and human VDR in Figs. 3B and 3C, respectively.
Plasma or serum concentrations of bile salts in vertebrates
Circulating total serum concentrations of deoxycholic acid, chenodeoxycholic acid, and cholic acid in humans are 0.23-0.89 μM, 0-1.61 μM, 0.08-0.91 μM, respectively (63). Total serum bile acids exceed 100 μM in patients with severe cholestasis (10). Bile acids are bound tightly to plasma proteins (approximately 70-95%) so the corresponding free plasma bile acid concentrations will be lower than the values above (64-66).
Total bile acid concentrations in Sprague-Dawly rat plasma are 8.26 ± 6.13 nmol/100 g body weight (67). With rats weighing approximately 400 g and assuming an estimated plasma volume of 5 mL/100 g body weight (68), this corresponds to 0.413 ± 0.31 μM total bile acid concentration in plasma. Concentration of total bile acids in mouse plasma is slightly higher than that in rats (52). Similarly, total bile acid concentrations in New Zealand rabbit plasma were 1.08 ± 0.44 nmol/100 g body weight (67). With rabbits weighing approximately 2000 g and assuming an estimated plasma volume of 20 mL/100 g body weight (69), this corresponds to 0.0027 ± 0.0011 μM. Total bile acid concentrations in hens are 0.075-0.15 μM (70). Concentrations of bile salts in fish or amphibian plasma or serum have not been reported.
Phylogenetic analysis
Sequences were aligned with Clustal X. Estimation of dN/dS (ω) ratios was carried out by maximum likelihood using a codon-based substitution model in PAML (Phylogenetic Analysis by Maximum Likelihood) version 3.13 (71). The input to PAML is a treefile of the phylogeny of the sequences to be studied and a file with aligned sequences. The phylogeny is based on the known phylogenetic relationships between the species to be studied, determined by a consensus of morphological and molecular data (44).
PAML can determine estimates of ω for models of varying complexity. The most commonly applied models are as follows (the PAML model numbers are shown in parentheses; the ‘sites’ refers to codons) (72, 73): model M0 (null model with a single ω ratio among all sites), M3 (“discrete” model, with 2 or more categories of sites with the ω ratio free to vary for each site), M7 (“Beta model”, ten categories of sites, with ten ω ratios in the range 0-1 taken from a discrete approximation of the beta distribution), and M8 (“Beta plus omega” model, ten categories of sites from a beta distribution as in M7 plus an additional category of sites with an ω ratio that is free to vary from 0 to greater than 1). PAML estimates the ω ratios that are allowed to vary under these models, as well as the proportion of sites (codons) with each ratio. We also utilized a combined branch-site model (‘model B’ of ref. (34)) in PAML to test for positive selection in specific lineages of the phylogeny. This model allows for a branch or branches of a phylogeny to be selected as ‘foreground’ and the remaining branches as ‘background.’ Model B is compared statistically to the simpler model of M3 with two site classes and no selection of specific lineages.
Of the PAML models listed above, only M3 and M8 can detect positive selection (i.e., ω > 1). Each PAML model generates a log-likelihood, indicating how well the models fit the input data. Some PAML models are “nested” within each other (e.g., M0 within M3, M7 within M8). In those cases, twice the log-likelihood difference between the two models is compared with a X2 distribution with degrees of freedom equal to the difference in degrees of freedom between the two models. P values for sites potentially under positive selection are obtained using a Bayesian approach in PAML (34). The accuracy and power of PAML models increases with more sequences and longer length sequences (74). Simpler PAML models are preferred unless a more complex model fits the observed data significantly better. Analysis was performed on publicly available PXR and VDR coding sequences, either from cDNA sequences or predicted from genomic DNA sequences. VDR was chosen for comparison as the gene most closely related to PXR for which sequence data is available in mammals and other vertebrates (CAR is only found in mammals). Separate PAML analyses were performed on datasets of full-length sequence and restricted to either DBD or LBD, the two major domains of NRs, and to datasets consisting of all available vertebrate species and restricted to mammals only. Mammalian and fish PXRs have an approximately 40 amino acid ‘insertion domain’ between LBD helices 1 and 3 (termed ‘H1-H3 insert’) although there is little sequence similarity between mammalian and fish sequences (7, 53). The H1-H3 insert is absent in Xenopus laevis BXRα and BXRβ (21) and only partially present in chicken PXR (20). The H1-H3 insert sequence was excluded in PAML analyses that include both mammalian and non-mammalian PXR genes due to uncertainties in alignment and lack of sequence for some PXRs.
For PXR, full-length sequence data is available for 11 genes from 10 species (accession numbers in parentheses; unless otherwise specified accession numbers are from GenBank): fugu (SINFRUG76306; http://genome.jgi-psi.org/fugu3/fugu3), zebrafish (AF454673 and AF502918), Xenopus laevis BXRα (BC041187) and BXRβ (AF305201), chicken CXR (AF276753), mouse (AF031814), rat (AF151377), rabbit (AF188476), rhesus monkey (AF454671), chimpanzee (Nov. 2003 chimpanzee Arachne assembly, NCBI Build 1 version 1, UCSC: panTro1), and human (NM_003889). PAML analyses of full-length sequence and DBD are of the above 11 sequences (5 mammals). Partial sequence essentially containing only the LBD is available for dog (AF454670) and pig (AF454672); this allows for PAML analysis of the LBD of PXRs to include 13 sequences total (7 mammals).
For VDR, full-length sequence data is available for 11 species: sea lamprey (AY249863), fugu (SINFRUG63876; http://genome.jgi-psi.org/fugu3/fugu3), zebrafish (NM_130919), bastard halibut (AB037674), Xenopus laevis (U91846), chicken (AF011356), Japanese quail (U12641), mouse (NM_009504), rat (NM_017058), cotton-top tamarin monkey (AF354232), and human (NM_000376). PAML analyses of full-length sequence and DBD are of the above 11 sequences (4 mammals). Partial sequence covering the DBD is available for the chimpanzee (Nov. 2003 chimpanzee Arachne assembly, NCBI Build 1 version 1, UCSC: panTro1); this allows for PAML analysis of the DBD of VDRs to include 12 sequences total (5 mammals).
Supplementary Material
Acknowledgments
M.D.K. thanks Dr. Anna Di Rienzo (University of Chicago, Department of Human Genetics) for hosting him in her laboratory and for helpful discussions. The authors thank Dr. Alan F. Hofmann for critical reading of the manuscript. This work was supported by an internal grant from the University of Chicago Department of Pathology (Vinay Kumar, MD, chairman) and a Robert Priest Fellowship to M.D.K. L.R.H. was supported by funding from the Zoological Society of San Diego, CA. E.G.S. was supported by NIH grants GM60346 and P30 CA21765 Cancer Center Support grant, by the NIH/NIGMS Pharmacogenetics Research Network and Database (U01 GM61374, http://pharmgkb.org) under grant U01 GM61393, and by American Lebanese Syrian Associated Charities (ALSAC). The authors also gratefully acknowledge supply of plasmids by SA Kliewer, JT Moore, and LB Moore (GlaxoSmithKline, Research Triangle Park, NC), supply of the sea lamprey VDR clone by G.K. Whitfield (University of Arizona College of Medicine, Tucson, AZ), and the excellent technical assistance of C. Kline (St. Jude Children’s Research Hospital).
References
- 1.Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, Ohlsson R, Postlind H, Blomquist P, Berkenstram A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci U S A. 1998;95:12208–13. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–23. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dussault I, Yoo H-D, Lin M, Fan M, Batta AK, Salen G, Erickson SK, Forman BM. Identification of an endogenous ligand that activates pregnane X receptor-mediated sterol clearance. Proc Natl Acad Sci U S A. 2003;100:833–8. doi: 10.1073/pnas.0336235100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodwin B, Gauthier KC, Umetani M, Watson MA, Lochansky MI, Collins JL, Leitersdorf E, Mangelsdorf DJ, Kliewer SA, Repa JJ. Identification of bile acid precursors as endogenous ligands for the nuclear xenobiotic pregnane X receptor. Proc Natl Acad Sci U S A. 2003;100:223–8. doi: 10.1073/pnas.0237082100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NCO, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- 6.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, Goodwin B, Liddle C, Blanchard SG, Willson TM, Collins JL, Kliewer SA. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275:15122–7. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 7.Moore LB, Maglich JM, McKee DD, Wisely B, Willson TM, Kliewer SA, Lambert MH, Moore JT. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol Endocrinol. 2002;16:977–86. doi: 10.1210/mend.16.5.0828. [DOI] [PubMed] [Google Scholar]
- 8.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–74. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blumberg B, Sabbagh W, Juguilon H, Bolado J, van Meter CM, Ong ES, Evans RM. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998;12:3195–205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer S, Beuers U, Spengler U, Zwiebel FM, Koebe H-G. Hepatic levels of bile acids in end-stage chronic cholestatic liver disease. Clin Chim Acta. 1996;251:173–86. doi: 10.1016/0009-8981(96)06305-x. [DOI] [PubMed] [Google Scholar]
- 11.Schuetz EG, Strom S, Yasuda K, Lecureur V, Assem M, Brimer C, Lamba J, Kim RB, Ramachandran V, Komoroski BJ, Venkataramanan R, Cai H, Sinal CJ, Gonzalez FJ, Schuetz JD. Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J Biol Chem. 2001;276:39411–8. doi: 10.1074/jbc.M106340200. [DOI] [PubMed] [Google Scholar]
- 12.Maglich JM, Stoltz CM, Goodwin B, Hawkins-brown D, Moore JT, Kliewer SA. Nuclear pregnane X receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic distribution. Mol Pharmacol. 2002;62:638–46. doi: 10.1124/mol.62.3.638. [DOI] [PubMed] [Google Scholar]
- 13.Hartley DP, Dai X, He YD, Carlini EJ, Wang B, Huskey SW, Ulrich RG, Rushmore TH, Evers R, Evans DC. Activators of the rat pregnane X receptor differentially modulate hepatic and intestinal gene expression. Mol Pharmacol. 2004;65:1159–71. doi: 10.1124/mol.65.5.1159. [DOI] [PubMed] [Google Scholar]
- 14.Teng S, Jekerle V, Piquette-Miller M. Induction of ABCC3 (MRP3) by pregnane X receptor. Drug Metab Dispos. 2003;31:1296–9. doi: 10.1124/dmd.31.11.1296. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J Biol Chem. 2004;279:49517–22. doi: 10.1074/jbc.M409041200. [DOI] [PubMed] [Google Scholar]
- 16.Staudinger J, Liu Y, Mada A, Habeebu S, Klaassen CD. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos. 2001;29:1467–72. [PubMed] [Google Scholar]
- 17.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A. 2001;98:3375–80. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Kliewer SA, Gonzalez FJ, Sinal CJ. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem. 2003;278:45062–71. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- 19.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 20.Handschin C, Podvinec M, Meyer UA. CXR, a chicken xenobiotic-sensing orphan nuclear receptor, is related to both mammalian pregnane X receptor (PXR) and constitutive androstane receptor (CAR) Proc Natl Acad Sci U S A. 2000;97:10769–74. doi: 10.1073/pnas.97.20.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blumberg B, Kang H, Bolado J, Chen H, Craig AG, Moreno TA, Umesano K, Perlmann T, De Robertis EM, Evans RM. BXR, an embryonic orphan nuclear receptor activated by a novel class of endogenous benzoate metabolites. Genes Dev. 1998;12:1269–77. doi: 10.1101/gad.12.9.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Burch PE, Cooney AJ, Lanz RB, Pereira FA, Wu J, Gibbs RA, Weinstock G, Wheeler DA. Genomic analysis of the nuclear receptor family: new insights into structure, regulation, and evolution from the rat genome. Genome Res. 2004;14:580–90. doi: 10.1101/gr.2160004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagey LR. Bile acid biodiversity in vertebrates: chemistry and evolutionary implications Physiology and Pharmacology. University of California, San Diego; San Diego: 1992. [Google Scholar]
- 25.Hofmann AF, Schteingart CD, Hagey LR. Species differences in bile acid metabolism. In: Paumgartner G, Beuers U, editors. International Falk Symposiumml: Bile acids in liver diseases. Kluwer Academic Publishers; Dordrecht: 1995. pp. 3–30. [Google Scholar]
- 26.Une M, Hoshita T. Natural occurrence and chemical synthesis of bile alcohols, higher bile acids, and short side chain bile acids. Hiroshima J Med Sci. 1994;43:37–67. [PubMed] [Google Scholar]
- 27.Goto T, Holzinger F, Hagey LR, Cerrè C, Ton-Nu H-T, Schteingart CD, Steinbach JH, Shneider BL, Hofmann AF. Physicochemical and physiological properties of 5α-cyprinol sulfate, the toxic bile salt of cyprinid fish. J Lipid Res. 2003;44:1643–51. doi: 10.1194/jlr.M300155-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–6. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 29.Whitfield GK, Dang HTL, Schluter SF, Bernstein RM, Bunag T, Manzon LA, Hsieh G, Dominguez CE, Youson JH, Haussler MR, Marchalonis JJ. Cloning of a functional vitamin D receptor from the lamprey (Petromyzon marinus), an ancient vertebrate lacking a calcified skeleton and teeth. Endocrinology. 2003;144:2704–16. doi: 10.1210/en.2002-221101. [DOI] [PubMed] [Google Scholar]
- 30.Hofmann AF, Mosbach EH, Sweeley CC. Bile acid composition of bile from germ-free rabbits. Biochim Biophys Acta. 1969;176:204–7. doi: 10.1016/0005-2760(69)90092-7. [DOI] [PubMed] [Google Scholar]
- 31.Cali JJ, Hsieh C-L, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991;266:7779–83. [PMC free article] [PubMed] [Google Scholar]
- 32.Axelson M, Ellis E, Mörk B, Garmark K, Abrahamsson A, Björkhem I, Ericzon B-G, Einarsson C. Bile acid synthesis in culture human hepatocytes: support for an alternative biosynthetic pathway to cholic acid. Hepatology. 2000;31:1305–12. doi: 10.1053/jhep.2000.7877. [DOI] [PubMed] [Google Scholar]
- 33.Clark AG, Glanowski S, Nielsen R, Thomas PD, Kejariwal A, Todd MA, Tanenbaum DM, Civello D, Lu F, Murphy B, Ferriera S, Wang G, Zheng X, White TJ, Sninsky JJ, Adams MD, Cargill M. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science. 2003;302:1960–3. doi: 10.1126/science.1088821. [DOI] [PubMed] [Google Scholar]
- 34.Yang Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–73. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- 35.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–33. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 36.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. 2.1 Å crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry. 2003;42:1430–8. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 37.Grün F, Venkatesan RN, Tabb MM, Zhou C, Cao J, Hemmati D, Blumberg B. Benzoate X receptors α and β are pharmacologically distinct and do not function as xenobiotic receptors. J Biol Chem. 2002;277:43691–7. doi: 10.1074/jbc.M206553200. [DOI] [PubMed] [Google Scholar]
- 38.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Ann Rev Biochem. 2003;72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 39.Thornton JW. Evolution of vertebrate steroid receptors from an ancestral estrogen receptor by ligand exploitation and serial genome expansions. Proc Natl Acad Sci U S A. 2001;98:5671–6. doi: 10.1073/pnas.091553298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thornton JW, Need E, Crews D. Resurrecting the ancestral steroid receptor: ancient origin of estrogen signaling. Science. 2003;301:1714–7. doi: 10.1126/science.1086185. [DOI] [PubMed] [Google Scholar]
- 41.Bentley PJ. Comparative vertebrate endocrinology. Third ed. Cambridge University Press; Cambridge (UK): 1998. [Google Scholar]
- 42.Haslewood GAD. Comparative studies of bile salts. Myxinol disulphate, the principal bile salt of hagfish (Myxinidae) Biochem J. 1966;100:233–7. doi: 10.1042/bj1000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haslewood GAD, Tökés L. Comparative studies of bile salts: bile salts of the lamprey Petromyzon marinus L. Biochem J. 1969;114:179–84. doi: 10.1042/bj1140179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carroll RL. Vertebrate paleontology and evolution. Freeman; New York: 1988. [Google Scholar]
- 45.Nakagawa M, Une M, Takenaka S, Kuramoto T, Abukawa D, Iinuma K. Urinary bile alcohol profile in infants with intrahepatic cholestasis: identification of 5β-cholestane-3α,7α,24,25-tetrol. Acta Paediatrica. 1999;88:1078–82. doi: 10.1080/08035259950168126. [DOI] [PubMed] [Google Scholar]
- 46.Yousef IM, Perwalz S, Lamireau T, Tuchweber B. Urinary bile acid profile in children with inborn errors of bile acid metabolism and chronic cholestasis; Screening technique using Electrospray tandem mass-spectrometry (ES/MS/MS) Med Sci Monit. 2003;9:MT21–MT31. [PubMed] [Google Scholar]
- 47.Ichimiya H, Yanagisawa J, Nakayama F. Significance of bile alcohol in urine of a patient with cholestasis: identification of 5β-cholestane-3α,7α,12α,26,27-pentol (5β-cyprinol) and 5β-cholestane-3α,7α,12α,26-tetrol (27-deoxy-5β-cyprinol) Chem Pharm Bull. 1984;32:2874–7. doi: 10.1248/cpb.32.2874. [DOI] [PubMed] [Google Scholar]
- 48.Nakagawa M, Une M, Takenaka S, Tazawa Y, Nozaki S, Imanaka T, Kuramoto T. Urinary bile acid profiles in healthy and cholestatic children. Clin Chim Acta. 2001;314:101–6. doi: 10.1016/s0009-8981(01)00636-2. [DOI] [PubMed] [Google Scholar]
- 49.Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36:703–22. doi: 10.1081/dmr-200033475. [DOI] [PubMed] [Google Scholar]
- 50.Yeh TH, Wang DY, Deng JF, Chen SK, Hwang DF. Short-term toxicity of grass carp bile powder, 5α-cyprinol and 5α-cyprinol sulfate in rats. Comp Biochem Physiol Part C. 2002;131:1–8. doi: 10.1016/s1532-0456(01)00247-2. [DOI] [PubMed] [Google Scholar]
- 51.Xuan BH, Thi TX, Nguyen ST, Goldfarb DS, Stokes MB, Rabenou RA. Ichthyotoxic ARF after fish gallbladder ingestion: a large case series from Vietnam. Am J Kidney Dis. 2003;41:220–4. doi: 10.1053/ajkd.2003.50008. [DOI] [PubMed] [Google Scholar]
- 52.Meerman L, Koopen NR, Bloks V, Van Goor H, Havinga R, Wolthers BG, Kramer W, Stengelin S, Müller M, Kuipers F, Jansen PLM. Biliary fibrosis associated with altered bile composition in a mouse model of erythropoietic protoporphyria. Gastroenterology. 1999;117:696–705. doi: 10.1016/s0016-5085(99)70464-6. [DOI] [PubMed] [Google Scholar]
- 53.Maglich JM, Caravella JA, Lambert MH, Willson TM, Moore JT, Ramamurthy L. The first completed genome sequence from a telost fish (Fugu rubripes) adds significant diversity to the nuclear receptor superfamily. Nucleic Acids Res. 2003;31:4051–8. doi: 10.1093/nar/gkg444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forman BM, Tzameli I, Choi H-S, Chen J, Simha D, Seol W, Evans RM, Moore DD. Androstane metabolites bind to and deactivate the nuclear receptor CAR-β. Nature. 1998;395:612–5. doi: 10.1038/26996. [DOI] [PubMed] [Google Scholar]
- 55.Handschin C, Blaettler S, Roth A, Looser R, Oscarson M, Kaufmann MR, Podvinec M, Gnerre C, Meyer UA. The evolution of drug-activated nuclear receptors: one ancestral gene diverged into two xenosensor genes in mammals. Nuclear Receptor. 2004;2:7. doi: 10.1186/1478-1336-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dehal P, Satou Y, Campbell RK, et al. The draft genome of Ciona intestinalis: insights into vertebrate origins. Science. 2002;298:2157–67. doi: 10.1126/science.1080049. [DOI] [PubMed] [Google Scholar]
- 57.Bertrand S, Brunet F, Escriva H, Parmentier G, Laudet V, Robinson-Rechavi M. Evolutionary genomics of nuclear receptors: from twenty-five ancestral genes to derived endocrine systems. Mol Biol Evol. 2004;21:1923–37. doi: 10.1093/molbev/msh200. [DOI] [PubMed] [Google Scholar]
- 58.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–28. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 59.Watkins RE, Noble SM, Redinbo MR. Structural insights in the promiscuity and function of the human pregnane X receptor. Curr Opin Drug Discov Devel. 2002;5:150–8. [PubMed] [Google Scholar]
- 60.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature. 1997;395:137–43. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 61.Zhang J, Kuehl P, Green ED, et al. The human pregnane X receptor: genomic structure and identification and functional characterization of natural allelic variants. Pharmacogenetics. 2001;11:555–72. doi: 10.1097/00008571-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 62.Hagenbuch B, Dawson P. The sodium bile salt cotransporter family SLC10. Pflugers Arch. 2004;447:566–70. doi: 10.1007/s00424-003-1130-z. [DOI] [PubMed] [Google Scholar]
- 63.Burtis CA, Ashwood ER, editors. Tietz Textbook of Clinical Chemistry. Third ed. W.B. Saunders Company; Philadelphia: 1999. [Google Scholar]
- 64.Ceryak S, Bouscarel B, Fromm H. Comparative binding of bile acids to serum lipoproteins and albumin. J Lipid Res. 1993;34:1661–74. [PubMed] [Google Scholar]
- 65.Cowen AE, Korman MG, Hofmann AF, Thomas PJ. Metabolism of lithocholate in healthy man. III. Plasma disappearance of radioactivity after intravenous injection of labeled lithocholate and its derivatives. Gastroenterology. 1975;69:77–82. [PubMed] [Google Scholar]
- 66.Rudman D, Kendall FE. Bile acid content of human serum. II. The binding of cholanic acids by human plasma proteins. J Clin Invest. 1957;36:538–42. doi: 10.1172/JCI103451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kasbo J, Saleem M, Perwaiz S, Mignault D, Lamireau T, Tuchweber B, Yousef I. Biliary, fecal and plasma deoxycholic acid in rabbit, hamster, guinea pig, and rat: comparative study and implication in colon cancer. Biol Pharm Bull. 2002;25:1381–4. doi: 10.1248/bpb.25.1381. [DOI] [PubMed] [Google Scholar]
- 68.Stahl AM, Gillen CM, Takamata A, Nadel ER, Mack GW. Reduced blood-to-tissue albumin movement after plasmapheresis. Shock. 2003;19:440–7. doi: 10.1097/01.shk.0000051757.08171.58. [DOI] [PubMed] [Google Scholar]
- 69.Lurie F, Jahr JS, Driessen B. Changes in circulating blood and plasma volume after hemoglobin-based oxygen carrier infusion and additional infusion of colloid solutions. Am J Ther. 2002;9:425–30. doi: 10.1097/00045391-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 70.Bromidge ES, Wells JW, Wight PAL. Elevated bile acids in the plasma of laying hens fed rapeseed meal. Res Vet Sci. 1985;39:378–28. [PubMed] [Google Scholar]
- 71.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–6. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 72.Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–17. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- 73.Yang Z, Nielsen R, Goldman N, Pedersen AK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–49. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anisimova M, Bielawski JP, Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 2001;18:1585–92. doi: 10.1093/oxfordjournals.molbev.a003945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.