

Summary
The Sir2 (silent information regulator 2) family of NAD-dependent deacetylases regulates aging and longevity across a wide variety of organisms, including yeast, worms, and flies. In mammals, the Sir2 ortholog Sirt1 promotes fat mobilization, fatty acid oxidation, glucose production, and insulin secretion in response to nutrient availability. We previously reported that an increased dosage of Sirt1 in pancreatic β cells enhances glucose-stimulated insulin secretion (GSIS) and improves glucose tolerance in beta cell-specific Sirt1-overexpressing (BESTO) transgenic mice at 3 and 8 months of age. Here, we report that as this same cohort of BESTO mice reaches 18–24 months of age, the GSIS regulated by Sirt1 through repression of Ucp2 is blunted. Increased body weight and hyperlipidemia alone, which are observed in aged males and also induced by a Western-style high-fat diet, are not enough to abolish the positive effects of Sirt1 on β cell function. Interestingly, plasma levels of nicotinamide mononucleotide (NMN), an important metabolite for the maintenance of normal NAD biosynthesis and GSIS in β cells, are significantly reduced in aged BESTO mice. Furthermore, NMN administration restores enhanced GSIS and improved glucose tolerance in the aged BESTO females, suggesting that Sirt1 activity decreases with advanced age due to a decline in systemic NAD biosynthesis. These findings provide insight into the age-dependent regulation of Sirt1 activity and suggest that enhancement of systemic NAD biosynthesis and Sirt1 activity in tissues such as β cells may be an effective therapeutic intervention for age-associated metabolic disorders such as type 2 diabetes.
Keywords: aging, BESTO mice, insulin secretion, pancreatic β cells, Sirt1, systemic NAD biosynthesis
Introduction
The silent information regulator 2 (Sir2) family of NAD-dependent deacetylases has been implicated in the regulation of aging and longevity in a wide variety of organisms, including yeast, worms, and flies (Blander & Guarente, 2004; Bordone & Guarente, 2005). An increased dosage of Sir2 proteins extends life span in each of these organisms, while deletion or mutation of Sir2 shortens life span (Kaeberlein et al., 1999; Tissenbaum & Guarente, 2001; Astrom et al., 2003; Howitz et al., 2003; Rogina & Helfand, 2004; Wood et al., 2004). In budding yeast, Sir2-mediated silencing at the rDNA loci is crucial for its ability to regulate life span because Sir2 suppresses homologous recombination and hence the formation and accumulation of toxic extrachromosomal rDNA circles, one of the primary causes of yeast aging (Gottlieb & Esposito, 1989; Sinclair & Guarente, 1997; Kaeberlein et al., 1999). In Caenorhabditis elegans, the Sir2 ortholog SIR-2.1 requires the forkhead transcription factor DAF-16 to promote life span extension (Tissenbaum & Guarente, 2001) and acts in a stress response pathway parallel to the insulin/IGF-1 signaling pathway that converges directly upon DAF-16 (Berdichevsky et al., 2006; Wang & Tissenbaum, 2006; Wang et al., 2006). In Drosophila, whole body and neuronal-specific overexpression of the Sir2 ortholog dSir2 extends life span (Rogina & Helfand, 2004). Finally, caloric restriction (CR), the single most consistent regimen to extend life span across a wide range of species, requires the Sir2 genes for CR-mediated life span extension in certain genetic backgrounds and conditions in these organisms (Lin et al., 2000, 2002, 2004; Anderson et al., 2003; Rogina & Helfand, 2004; Wang & Tissenbaum, 2006).
While it is not yet known whether the mammalian Sir2 ortholog Sirt1 similarly regulates aging and longevity in mammals, Sirt1 has been shown to regulate metabolic responses to changes in nutrient availability in multiple tissues (Bordone & Guarente, 2005; Moynihan & Imai, 2006). Sirt1 levels are increased in tissues such as muscle, brain, liver, and fat in response to fasting and CR in rodents (Al-Regaiey et al., 2004; Cohen et al., 2004; Nemoto et al., 2004; Rodgers et al., 2005). Up-regulation of Sirt1 in adipocytes in response to fasting promotes lipolysis and free fatty acid mobilization through repression of PPARγ, a nuclear hormone receptor that promotes adipogenesis (Picard et al., 2004). Sirt1 also promotes gluconeogenesis and represses glycolysis in hepatocytes in response to nutrient deprivation by interacting with and deacetylating PGC-1α, a key transcriptional regulator of glucose production in the liver (Rodgers et al., 2005). In skeletal muscle, Sirt1-mediated PGC-1α deacetylation is required to induce mitochondrial fatty acid oxidation genes in states of nutrient deprivation (Gerhart-Hines et al., 2007). Finally, we and others demonstrated that Sirt1 promotes insulin secretion in pancreatic β cells in response to glucose partly through repression of uncoupling protein 2 (Ucp2), a mitochondrial inner membrane protein that uncouples respiration from ATP production (Moynihan et al., 2005; Bordone et al., 2006).
Aging is one of the greatest risk factors for metabolic complications, such as obesity, glucose intolerance, and type 2 diabetes (Chang & Halter, 2003; Moller et al., 2003). It has been shown that a progressive decline in β cell function in the elderly is a major contributing factor to the pathophysiology of type 2 diabetes (Iozzo et al., 1999; Basu et al., 2003). A similar age-associated impairment of β cell function has also been demonstrated in rodents (Muzumdar et al., 2004). In our previous study, we reported that pancreatic beta cell-specific Sirt1-overexpressing (BESTO) transgenic mice exhibited enhanced glucose-stimulated insulin secretion (GSIS) and improved glucose tolerance compared to controls at both 3 and 8 months of age (Moynihan et al., 2005). BESTO islets had reduced levels of Ucp2 and correspondingly increased levels of ATP. Furthermore, BESTO pancreata and islets secreted more insulin compared to controls in response to the potent insulin secretagogue potassium chloride (KCl), which directly depolarizes β cells. Based on these findings, we hypothesized that increased Sirt1 dosage or activity in pancreatic β cells would provide life-long beneficial effects of enhanced β cell function on glucose homeostasis and prevent or delay the development of metabolic complications associated with aging.
To address this hypothesis, we followed the same cohorts of BESTO mice that we used in our previous study (Moynihan et al., 2005) and analyzed their β cell function at 18–24 months of age. Unexpectedly, we found that the improved glucose tolerance and enhanced GSIS were completely abolished as the BESTO mice reached these old ages. Furthermore, islets isolated from the aged BESTO mice also no longer showed Sirt1-mediated repression of Ucp2 expression or increased ATP levels compared to controls. Because Sirt1 expression remained high in the aged BESTO islets, our results suggest that either Sirt1 activity decreases with age, perhaps because of a decline in NAD biosynthesis, or the actions of some alternative age-associated factors, such as increased obesity and/or blood lipid levels, negate the Sirt1-mediated enhancement of GSIS. Recently, we have demonstrated that nicotinamide mononucleotide (NMN), a novel NAD biosynthetic metabolite circulating in plasma, plays a critical role in the regulation of NAD biosynthesis and GSIS in pancreatic β cells (Revollo et al., 2007b). Interestingly, we found that old BESTO mice have significantly reduced NMN levels in their plasma and that NMN administration restored the improved glucose tolerance and enhanced GSIS in the aged female BESTO mice. These findings strongly suggest that an age-associated decline in systemic NAD biosynthesis indeed accounts for the reduced activity of Sirt1 in aged pancreatic β cells. These surprising findings provide insight into the age-dependent regulation of Sirt1 activity and may open up new avenues for the treatment of age-associated metabolic complications such as impaired glucose tolerance and type 2 diabetes.
Results
Aged BESTO mice no longer show improved glucose tolerance or enhanced GSIS
We previously reported that an increased dosage of Sirt1 in pancreatic β cells results in enhanced GSIS and improved glucose tolerance in BESTO transgenic mice at 3 and 8 months of age (Moynihan et al., 2005). Based on these previous results, we were interested in examining whether these beneficial effects of improved β cell function would persist into old age in the BESTO mice. Surprisingly, the same cohort of BESTO male and female mice in two independent transgenic lines no longer exhibited such an improvement at 18–24 months of age (Fig. 1A and Supplementary Fig. S1A). Likewise, the enhancement of GSIS observed after glucose injection in the young BESTO mice (Moynihan et al., 2005) was also abolished in the aged BESTO male and female mice (Fig. 1B and Supplementary Fig. S1B). Consistent with these in vivo results, islets isolated from the aged BESTO mice and cultured in RPMI media for 18–20 h also no longer displayed the enhanced GSIS or increased ATP levels compared to controls (Fig. 1C,D) that was characteristic of their younger counterparts (Moynihan et al., 2005). Thus, contrary to our expectations, long-term overexpression of Sirt1 in pancreatic β cells does not result in life-long beneficial effects of improved β cell function in BESTO mice.
Fig. 1.
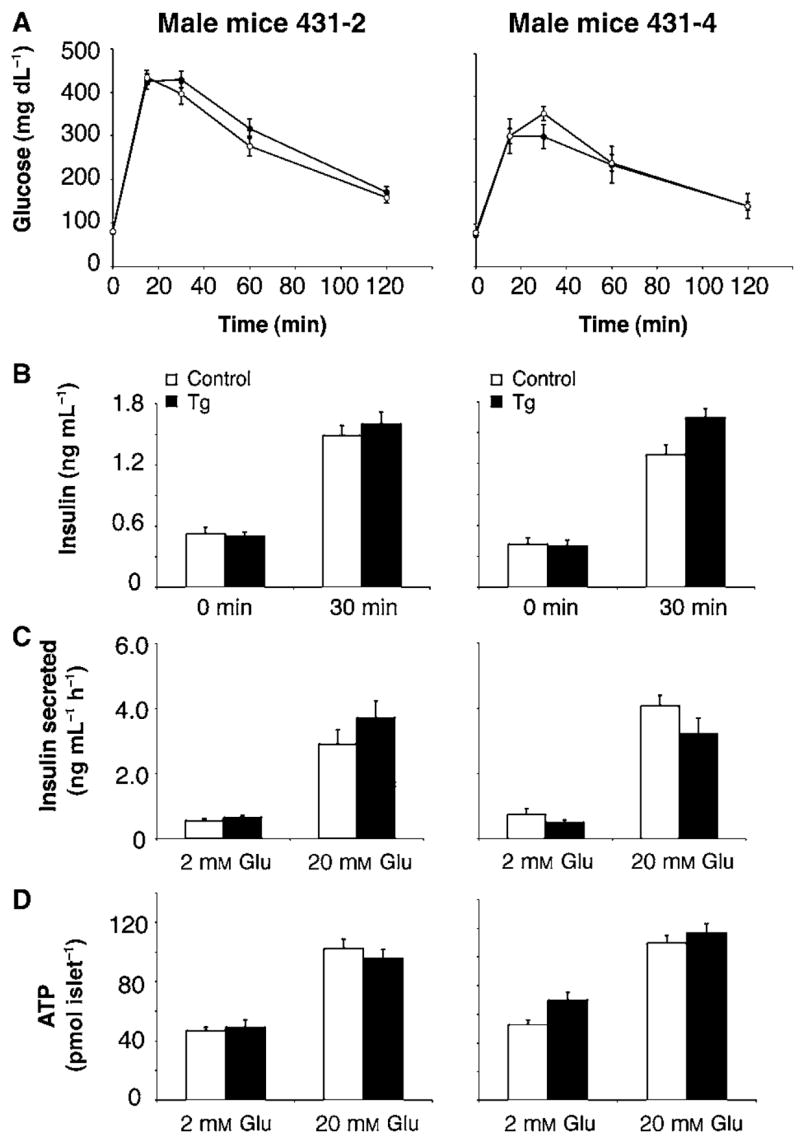
Intraperitoneal glucose tolerance tests (IPGTTs), plasma insulin levels, glucose-stimulated insulin secretion assays, and ATP levels in male BESTO and control mice and islets at 18–24 months of age. (A) Intraperitoneal glucose tolerance tests were conducted in male BESTO transgenic mice (closed circles) compared to controls (open circles) at 18–24 months of age in two independent transgenic lines. Following a 15 h fast, 2 g of 50% glucose (w/v) per kg body weight was injected intraperitoneally, and blood glucose values were assessed at 0, 15, 30, 60, and 120 min post-injection (line 431-2, n = 13–15; line 431-4, n = 6–7). (B) Plasma insulin levels in male BESTO mice (black bars) and controls (white bars) were measured at the 0 and 30 min time points during the IPGTTs (line 431-2, n = 13–15; line 431-4, n = 6–7). (C) Insulin secreted (ng mL−1 h−1) and (D) ATP levels (pmol islet−1) from transgenic (black bars) and control (white bars) islets were measured at the indicated glucose concentrations (line 431-2, n = 11; line 431-4, n = 9–12). Insulin secretion and ATP assays were performed on triplicate groups of 10 islets for each mouse. All results are expressed as mean ± standard error.
Sirt1-mediated repression of Ucp2 expression is lost in aged BESTO islets
One possible explanation for the loss of glucose-responsive phenotypes in the aged BESTO mice is that the Sirt1 transgene is no longer overexpressed. However, we found that Sirt1 protein levels were still high in islets isolated from aged BESTO mice compared to controls, comparable to the level of overexpression in the young BESTO islets (Fig. 2A, top panels).
Fig. 2.
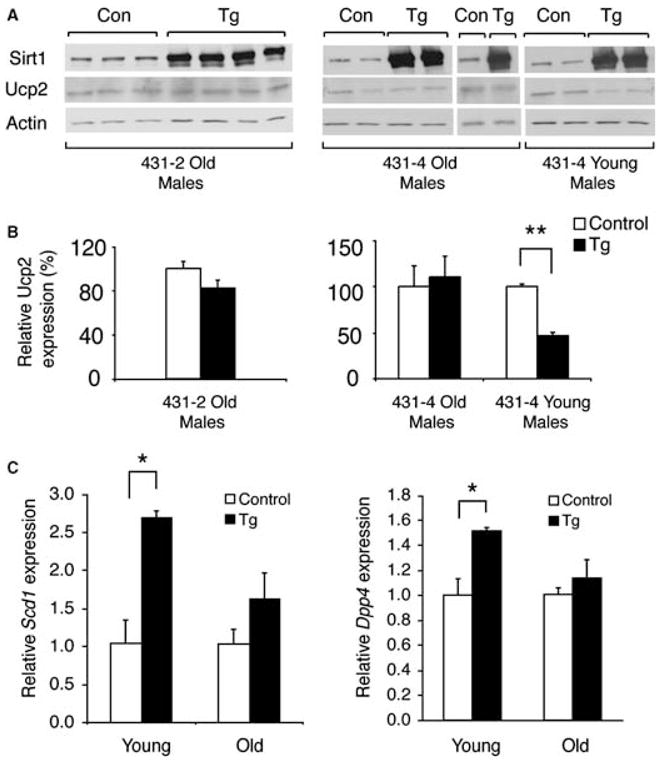
Sirt1 and uncoupling protein 2 (Ucp2) protein levels and stearoyl-CoA desaturase 1 (Scd1) and dipeptidyl peptidase IV (Dpp4) mRNA levels in islets isolated from aged BESTO and control mice. (A) Western blot analysis of Sirt1 and Ucp2 expression in transgenic (Tg) and control (Con) islet extracts from lines 431-2 and 431-4. Actin was used as a loading control for normalization. (B) Quantitation of the Ucp2 expression levels normalized to actin in BESTO transgenic islets (black bars) relative to control islets (white bars). (C) Quantitative real-time reverse transcriptase–polymerase chain reaction analysis of Scd1 and Dpp4 expression in BESTO transgenic islets (black bars) relative to control islets (white bars). The 3- to 5-month-old mice were considered young, while 18- to 19-month-old mice were considered old. All results are expressed as mean ± standard error. *P ≤ 0.05; **P ≤ 0.01.
We and others previously demonstrated that Sirt1 promotes GSIS partly through the repression of Ucp2 expression and the increase in ATP levels in pancreatic β cells (Moynihan et al., 2005; Bordone et al., 2006). In young BESTO islets, Ucp2 expression levels were significantly decreased compared to controls (Moynihan et al., 2005; see Fig. 2). However, despite maintaining high levels of Sirt1 overexpression, islets from aged BESTO mice no longer displayed reduced levels of Ucp2 (Fig. 2A,B), consistent with the lack of a difference in ATP content (Fig. 1D).
In order to determine whether the loss of Sirt1-dependent regulation with age was specific to Ucp2 or whether it was a more general phenomenon, we also examined the mRNA levels of stearoyl-CoA desaturase 1 (Scd1) and dipeptidyl peptidase IV (Dpp4), two genes found to be up-regulated by Sirt1 in microarray analyses comparing young BESTO and control islets (unpublished findings). While Sirt1 overexpression resulted in significant up-regulation of Scd1 (2.7-fold) and Dpp4 (1.5-fold) mRNA levels in young BESTO islets compared to controls, up-regulation of these two genes was reduced and no longer significantly different in old BESTO compared to control islets (Fig. 2C). Together, these results suggest either that Sirt1 activity decreases with advanced age or that the actions of some alternative age-associated factors negate the effect of Sirt1 in aged BESTO mice.
BESTO islets and mice still respond partially to other insulin secretagogues
In addition to being hyperresponsive to glucose, islets isolated from young BESTO mice also elicit a pronounced increase in insulin secretion in response to KCl, a potent insulin secretagogue, compared to controls (Moynihan et al., 2005). Because the enhancement of GSIS and the repression of Ucp2 were completely abolished in the aged BESTO mice and islets, we speculated that they also might have lost their ability to hyper-secrete insulin in response to other secretagogues, such as KCl. We found that islets isolated from BESTO mice at 18–24 months of age still maintained their ability to secrete higher levels of insulin in response to KCl (Fig. 3A). However, the extent of enhancement of insulin secretion in aged BESTO islets was less pronounced than it was in young BESTO islets (Moynihan et al., 2005). To confirm these results in vivo, we intraperitoneally injected arginine, which acts similarly to KCl in that it directly depolarizes the β cell membrane (Newsholme et al., 2005), to 20-month-old BESTO male mice and measured plasma insulin levels at 0, 2, and 5 min after arginine injection. Consistent with the in vitro results, the same aged BESTO mice that no longer showed enhanced GSIS maintained their ability to secrete more insulin in response to arginine than controls (Fig. 3B). Thus, interestingly, while Sirt1-stimulated GSIS was completely blunted as the BESTO mice reached old age, the Sirt1-mediated mechanism that acts downstream of β cell depolarization still remained partially intact in old BESTO islets.
Fig. 3.
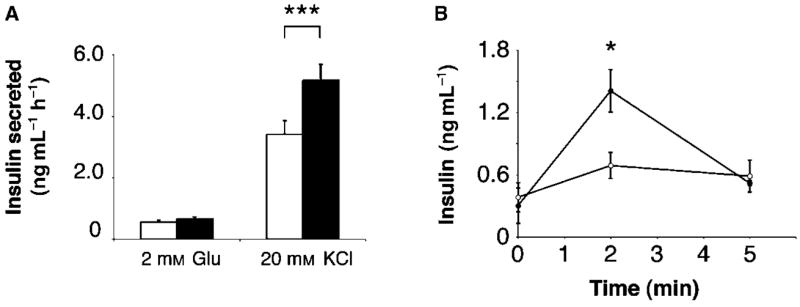
Insulin secretion in aged BESTO transgenic and control islets and mice in response to other insulin secretagogues. (A) Insulin secreted (ng mL−1 h−1) from transgenic (black bars) and control (white bars) islets was measured after stimulation with either 2 mM glucose or 20 mM KCl. Insulin secretion assays were performed on islets isolated from 18- to 24-month-old male BESTO and control mice from line 431-2 using triplicate groups of 10 islets for each mouse (n = 11). (B) Insulin secretion in response to arginine stimulation was measured in male BESTO mice (closed circles) compared to controls (open circles) from line 431-2 at 18–24 months of age. Following a 15 h fast, 1.5 g of arginine per kg body weight was injected intraperitoneally, and plasma insulin levels were assessed at 0, 2, and 5 min post-injection (n = 3–6). All results are expressed as mean ± standard error. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
BESTO mice maintain improved glucose tolerance and enhanced GSIS under a Western-style high-fat diet (HFD)
We observed that both aged BESTO and control male mice had increased body weights (Supplementary Fig. S2A) and statistically significant increases in lipid parameters such as cholesterol, triglycerides, and free fatty acids compared to the young BESTO and control male mice (Supplementary Fig. S3). Although similar differences in lipid profiles were not observed in female mice, we speculated that the increased body weights and blood lipid levels in the aged BESTO mice might contribute to their loss of glucose-responsive phenotypes.
To test this possibility, we placed BESTO and control mice at 2–4 months of age on a HFD containing 42% calories from fat for up to 30 weeks. After 12 weeks of this HFD treatment, BESTO and control mice both reached similar body weights to those of regular chow-fed mice aged 15–18 months (compare Supplementary Fig. S2A and B). While BESTO and control mice did not differ in the extent of the HFD-induced hyperglycemia, hyperinsulinemia, hypercholesterolemia, and hypertriglyceridemia over the course of the HFD treatment (Fig. 4A–E), both male and female BESTO mice still maintained significantly improved glucose tolerance and higher plasma insulin levels 15 min and 30 min post-glucose injection compared to controls after 12 weeks on the HFD (Fig. 5A,B). Even following 30 weeks of HFD treatment, the BESTO mice, which at that point were 9–11 months of age, still showed significantly improved glucose tolerance and enhanced GSIS compared to controls (Supplementary Fig. S4A,B). Additionally, islets from HFD-fed BESTO mice still displayed reduced Ucp2 expression compared to controls, similar to the level of repression observed in islets from young BESTO mice on a regular chow diet (Supplementary Figs S4C and 2). Therefore, although BESTO mice did not differ from controls in HFD-induced weight gain and lipidemia, Sirt1 still improved β cell function under this diabetogenic condition, suggesting that increased body weight and hyperlipidemia alone are not enough to abolish the positive effects of Sirt1 in β cell function.
Fig. 4.
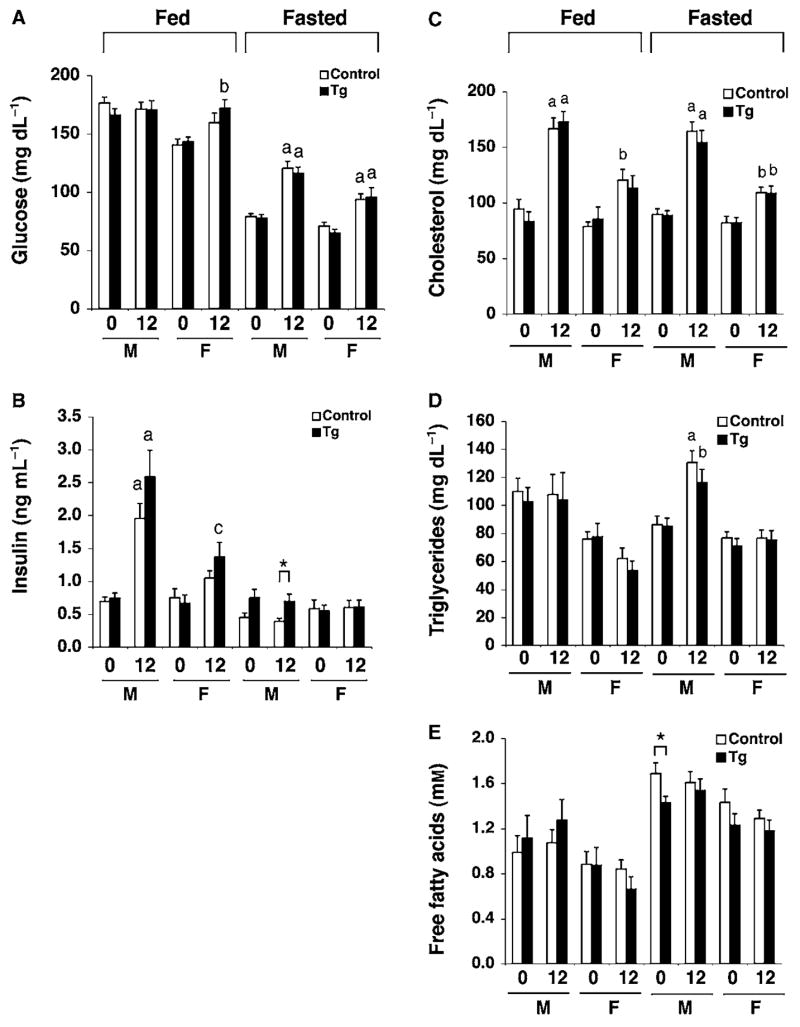
Plasma glucose, insulin, and lipid levels in BESTO transgenic and control mice fed a Western-style, high-fat diet (HFD). (A) Fed and fasted plasma glucose levels in male (M) and female (F) BESTO (black bars) and control (white bars) mice from line 431-4 before (0) and after (12 weeks) a HFD regimen. For fed glucose levels, n = 12–15. For fasted glucose levels, n = 5–15. (B) Fed and fasted plasma insulin levels in male (M) and female (F) BESTO (black bars) and control (white bars) mice before (0) and after (12) a HFD regimen. For fed insulin levels, n = 4–12. For fasted insulin levels, n = 5–15. a, P ≤ 0.001; b, P ≤ 0.01; c, P ≤ 0.05, compared to glucose or insulin levels before HFD feeding. (C–E) Fed and fasted plasma (C) cholesterol (D) triglycerides, and (E) total free fatty acid levels in male (M) and female (F) BESTO (black bars) and control (white bars) mice from line 431-4 before (0) and after (12 weeks) an HFD regimen (n = 6–15). a, P ≤ 0.001; b, P ≤ 0.01, compared to values before HFD feeding. All results are expressed as mean ± standard error. *P ≤ 0.05.
Fig. 5.
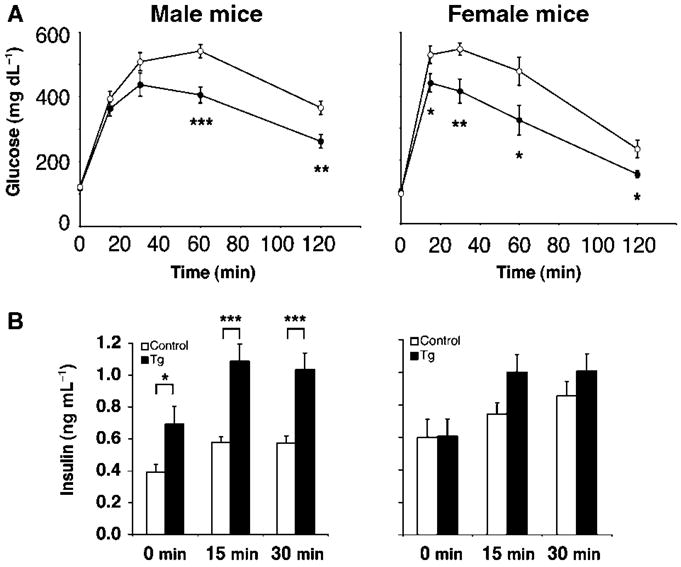
Intraperitoneal glucose tolerance tests (IPGTTs) in BESTO transgenic and control mice fed a Western-style high-fat diet (HFD). (A) IPGTTs were conducted in male and female BESTO transgenic (closed circles) and control (open circles) mice following 12 weeks on a HFD. Following a 15 h fast, 2 g of 50% glucose (w/v) per kg body weight was injected intraperitoneally, and blood glucose values were assessed at 0, 15, 30, 60, and 120 min post-injection (n = 9–14). (B) Plasma insulin levels in male and female transgenic mice (black bars) and controls (white bars) were measured at the 0, 15, and 30 min time points during the IPGTTs shown in (A). All results are expressed as mean ± standard error. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Administration of NMN restores the glucose-responsive phenotypes in aged BESTO female mice
An alternative explanation for the absence of glucose-responsive phenotypes in the aged BESTO mice is that Sirt1 activity decreases with advanced age. Because Sirt1 requires NAD for its enzymatic activity, a decline in NAD biosynthesis with age could result in reduced Sirt1 activity and subsequent loss of the glucose-responsive phenotypes in BESTO mice. To address this possibility, we initially assessed the NAD biosynthetic capability of pancreatic islets from young (5-month-old) and old (19-month-old) BESTO mice. Because the NAD biosynthetic machinery quickly adapts to culture conditions, we cultured isolated islets in RPMI media containing a physiological concentration (1 μM) of nicotinamide, a major precursor to NAD, for ~24 h prior to measuring the NAD content per 100 islets. No difference was detected in NAD content between young and old BESTO islets in vitro (Fig. 6A), suggesting that the NAD biosynthetic capability of the BESTO islets does not alter during aging.
Fig. 6.
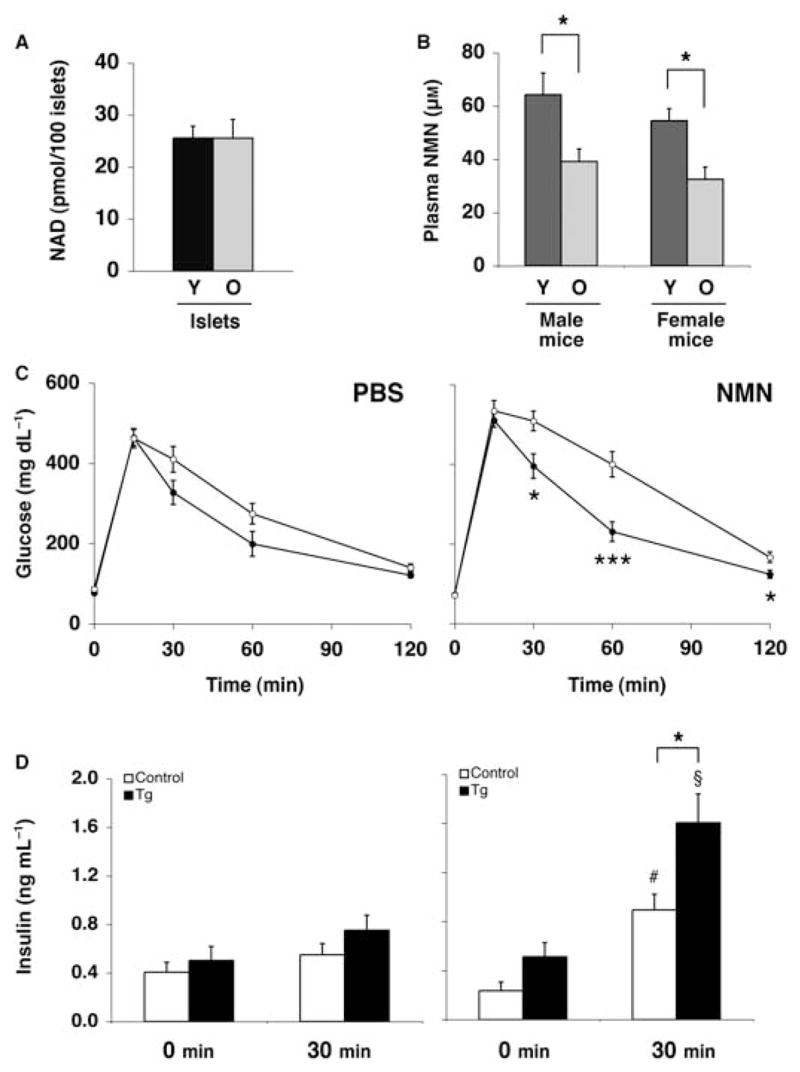
The effect of NMN administration on IPGTTs and plasma insulin levels in aged BESTO and control female mice. (A) NAD content in 100 cultured pancreatic islets isolated from young (Y, 5 months old) and old (O, 19 months old) BESTO mice. The measurements were conducted in duplicate or triplicate of primary islets pooled from three individual mice. (B) Plasma NMN levels in young (Y, 5 months old) and old (O, 19 months old) BESTO mice. The measurements were conducted with four individual mice for each sex and age. (C) IPGTTs were conducted in 20-month-old female BESTO transgenic mice (closed circles) and controls (open circles) from line 431-4 after mice were injected with PBS or NMN (500 mg kg−1 body weight) and fasted for 14 h (n = 10–15). (D) Plasma insulin levels were measured at 0 and 30 min post-glucose injection. #P ≤ 0.05; §P ≤ 0.01, compared to insulin levels 30 min after glucose injection in PBS-injected aged BESTO and control mice. All results are expressed as mean ± standard error. *P ≤ 0.05; ***P ≤ 0.001.
Of note, we recently demonstrated that NMN, an intermediate compound synthesized from nicotinamide in the NAD biosynthetic pathway (Revollo et al., 2007a), circulates systemically in mouse plasma and plays a critical role in the regulation of NAD biosynthesis and GSIS in pancreatic β cells (Revollo et al., 2007b). β cells depend on NMN in blood circulation for the maintenance of normal NAD and GSIS levels. Therefore, we next assessed the levels of NMN in plasma from young and old BESTO mice. Surprisingly, plasma NMN levels were significantly lower in both old male and female BESTO mice compared to young BESTO mice (Fig. 6B), suggesting that in vivo NAD levels must also be significantly reduced in the pancreatic β cells of the aged BESTO mice. In our recent study, we also demonstrated that administration of NMN ameliorates the defects in GSIS observed in mice and islets heterozygous for nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in the NAD biosynthetic pathway in mammals (Revollo et al., 2004). We therefore hypothesized that if the reduced plasma NMN levels in the aged BESTO mice contribute to their loss of phenotypes, we might be able to restore the improved glucose tolerance and enhanced GSIS in aged BESTO mice by administration of NMN.
We injected either phosphate-buffered saline (PBS) or NMN (500 mg kg−1 body weight) intraperitoneally to 20-month-old BESTO and control mice 14 h prior to performing IPGTTs. Glucose tolerance and GSIS did not differ significantly between BESTO and control mice following the PBS treatment (Fig. 6C,D, left panels). Interestingly, NMN-treated aged BESTO female mice resumed significantly improved glucose tolerance and enhanced GSIS compared to NMN-treated aged controls (Fig. 6C,D, right panels). While NMN administration slightly impaired glucose tolerance in both the BESTO and control mice compared to PBS-treated mice (Fig. 6C, right panel), it is important to note that NMN administration also augmented GSIS significantly in both BESTO and control mice (compare 30 min results in left and right panels in Fig. 6D). These phenomena appear to be sex specific, as we did not observe similar responses to the same dose of NMN in aged BESTO male mice (Supplementary Fig. S5). Nevertheless, NMN administration restored the positive effect of Sirt1 on glucose tolerance and GSIS, at least in the aged BESTO female mice. Taken together, these findings suggest that an age-associated decline in systemic NAD biosynthesis, and hence Sirt1 activity, accounts for the loss of the phenotypes in aged BESTO mice.
Discussion
In our previous study, we demonstrated that BESTO mice exhibit significantly enhanced GSIS and improved glucose tolerance at both 3 and 8 months of age (Moynihan et al., 2005). In BESTO islets, Sirt1 mediates the repression of Ucp2 and hence increases ATP levels in response to glucose stimulation, resulting in the enhancement of GSIS (Moynihan et al., 2005). Here, we speculated that increasing Sirt1 dosage or activity in pancreatic β cells would provide life-long beneficial effects of enhanced β cell function on glucose homeostasis in the process of aging. In this study, however, we made the unexpected finding that BESTO mice lose their glucose-responsive phenotypes with advanced age, despite maintaining high levels of Sirt1 over-expression in pancreatic β cells. First, both male and female BESTO mice from two independent lines no longer showed glucose-responsive phenotypes at 18–24 months of age (Fig. 1A,B and Supplementary Fig. S1). Second, islets isolated from these aged BESTO mice no longer showed enhanced GSIS in vitro or higher ATP levels compared to controls, consistent with the in vivo results (Fig. 1C,D). Third, aged BESTO islets no longer exhibited the Sirt1-mediated down-regulation of Ucp2 or the up-regulation of Scd1 and Dpp4 that was characteristic of their younger counterparts (Fig. 2). Together, these findings suggest that while Sirt1 overexpression in β cells enhances GSIS and improves glucose tolerance at younger ages, long-term overexpression of Sirt1 does not confer life-long beneficial effects of improved β cell function in mice.
These unexpected results raised an interesting question: Why do BESTO mice lose their glucose-responsive phenotypes with advanced age? Because Sirt1 expression remained high in the aged BESTO islets, two possible explanations were considered to account for the loss of the BESTO phenotypes: (i) Sirt1 activity decreases with advanced age or (ii) the actions of some alternative age-associated factors negate the Sirt1-mediated enhancement of GSIS. We first suspected that the increased body weight and circulating lipids might contribute to the loss of the BESTO phenotypes with age because these parameters were all significantly elevated in the old compared to young male mice (Supplementary Figs S2 and S3). However, following a Western-style HFD regimen for up to 30 weeks, the BESTO mice still maintained significantly improved glucose tolerance and enhanced GSIS compared to controls, and Ucp2 expression was still suppressed in islets from HFD-fed BESTO mice, as it was in islets from regular chow-fed young BESTO mice (Figs 4 and 5, and Supplementary Fig. S4). These results suggest that Sirt1 can still function to enhance GSIS and improve glucose tolerance even in the face of diabetogenic dietary conditions. Considering that Sirt1 mediates the repression of Ucp2 expression in β cells (Moynihan et al., 2005; Bordone et al., 2006), it is interesting to note that the results from HFD-fed BESTO mice are similar to those from HFD-fed Ucp2-deficient mice that also exhibit enhanced β cell glucose sensitivity compared to controls after HFD (Joseph et al., 2002). Therefore, our findings on HFD-fed BESTO mice reemphasize the notion that increasing Sirt1 dosage or activity in pancreatic β cells provides beneficial effects of enhanced β cell function on glucose homeostasis (Moynihan et al., 2005). These findings also suggest that common physiological changes observed in both aged and HFD-fed mice alone are not enough to abolish the positive effects of Sirt1 in β cells.
Because Sirt1 requires NAD for its enzymatic activity (Imai et al., 2000), it is also possible that a decline in NAD biosynthesis with age would result in a significant reduction of Sirt1 activity and subsequent loss of the glucose-responsive phenotypes. Although no difference was detected in the ability of young and old BESTO islets to synthesize NAD in culture conditions, we found that plasma levels of NMN, a critical component for β cells to maintain normal NAD biosynthesis and GSIS (Revollo et al., 2007b), were significantly reduced in old compared to young BESTO mice (Fig. 6A,B), indicating that in vivo NAD levels must also be reduced in old BESTO islets. Consistent with these findings, administration of NMN was able to restore improved glucose tolerance and enhanced GSIS compared to controls in aged BESTO female mice (Fig. 6C,D). While NMN leads to a slight impairment of glucose tolerance in both BESTO and control female mice (Fig. 6C), the levels of insulin secretion post-glucose injection in NMN-treated aged BESTO female mice were remarkably augmented compared to those in the PBS-treated counterparts (compare 30 min results in left and right panels, Fig. 6D), resulting in a pronounced difference in insulin secretion between aged BESTO and control mice (right panel, Fig. 6D). The restoration of increased GSIS and hence the improvement of glucose tolerance by NMN administration have also been observed in female Nampt-heterozygous mice, another mouse model in which plasma NMN levels are also reduced (Revollo et al., 2007b). Given that Nampt-mediated NAD biosynthesis has been shown to play an important role in the regulation of Sirt1 activity (Revollo et al., 2004), it is very likely that NMN administration restored the activity of over-expressed Sirt1 in β cells and thereby enhanced GSIS and improved glucose tolerance in aged BESTO female mice compared to old controls. Currently, why the aged BESTO male mice did not respond similarly to NMN administration remains unclear, and it should be noted that a similar gender disparity has also been observed in the case of the Nampt-heterozygous mice (Revollo et al., 2007b). Further investigation will be necessary to clarify this sex-dependent difference. Nonetheless, these findings strongly suggest that an age-dependent decline in plasma NMN levels contributes to decreased systemic NAD biosynthesis and reduced Sirt1 activity in aged animals (Fig. 7).
Fig. 7.
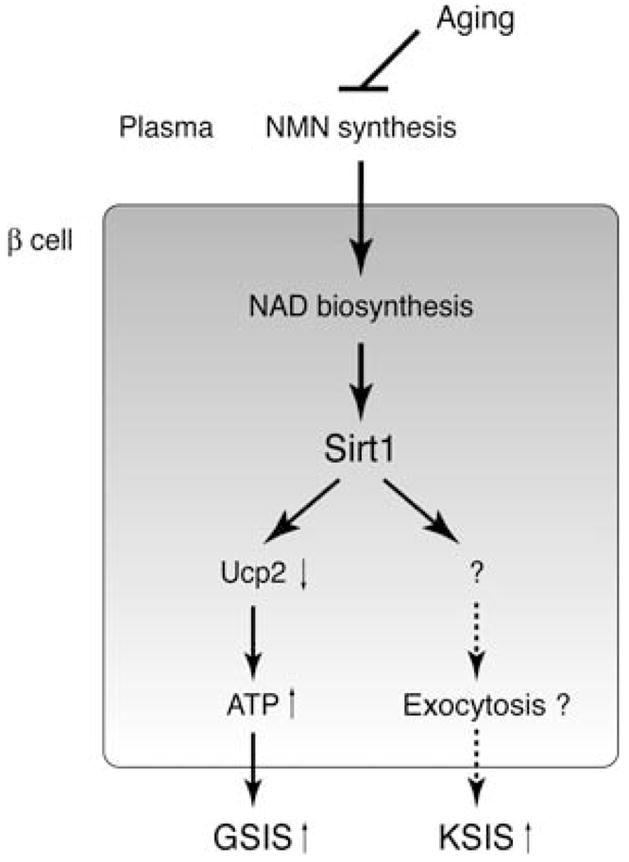
A model for the age-associated loss of the glucose-responsive phenotypes in aged BESTO mice. Sirt1 promotes insulin secretion through two independent mechanisms in pancreatic β cells. In one mechanism, Sirt1 mediates the repression of Ucp2 expression and thereby increases ATP content, resulting in the enhancement of GSIS. In the other mechanism, Sirt1 regulates the expression of an unidentified factor that acts downstream of β cell depolarization and possibly stimulates insulin granule exocytosis, resulting in the enhancement of (KCl)-stimulated insulin secretion (KSIS). Aging affects systemic NAD biosynthesis, perhaps at the level of biosynthesis of NMN in plasma, and decreases Sirt1 activity in pancreatic β cells of aged BESTO mice.
We previously proposed a model whereby Sirt1 regulates insulin secretion by at least two independent mechanisms, one of which acts upstream of depolarization through Ucp2, while the other acts downstream of depolarization (Moynihan et al., 2005). In this current study, we found that there is differential regulation of these two pathways with advanced age: while the Sirt1-promoted GSIS was completely blunted as the BESTO mice reached 18–24 months of age, the Sirt1-mediated mechanism that acts downstream of depolarization remained partially intact (Fig. 3). Although the molecular target of Sirt1 in the mechanism acting downstream of depolarization is still unclear, we currently suspect that Sirt1 might regulate the expression of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex proteins, based on our recent microarray analyses (unpublished findings). SNARE complex components are known to be critical for insulin granule exocytosis (Lam et al., 2005). Therefore, it will be interesting to examine whether Sirt1 indeed affects the function of SNAREs or SNARE-associated proteins in pancreatic β cells, resulting in the enhancement of KCl-stimulated insulin granule exocytosis (Fig. 7).
One of the greatest risk factors for the development of type 2 diabetes in humans is age (Wilson et al., 1986). Type 2 diabetes is characterized by a combination of defective insulin secretion and insulin resistance that results from a progressive age-associated decline in β cell function, increased visceral fat accumulation, and decreased physical activity, among other alterations (Iozzo et al., 1999; Basu et al., 2003; Moller et al., 2003). An age-associated decline in β cell function has also been reported in rat (Reaven et al., 1979, 1983, 1987; Muzumdar et al., 2004). Although the molecular mechanism for this age-dependent β cell defect is still unknown, the findings presented here suggest the possibility that a reduction in Sirt1 activity with age, likely caused by a decline in plasma NMN levels, contributes to this age-related impairment of β cell function (Fig. 7). If this is the case, it will be of great interest to examine whether administration of NMN can restore normal β cell function in the elderly. While further understanding of the precise molecular mechanisms by which Sirt1 activity is regulated with advanced age is necessary, our findings provide new insights into the age-associated pathophysiology of β cell function. Furthermore, our results suggest a novel approach for the development of an effective therapeutic strategy to activate Sirt1 in β cells by increasing systemic NAD biosynthesis in the treatment of age-associated metabolic disorders, such as impaired glucose tolerance and type 2 diabetes.
Experimental procedures
Animal experimentation
The BESTO transgenic mice have been described previously (Moynihan et al., 2005). BESTO mice (18- to 24-month-old) were used as the aged cohort of mice, while 3- to 5-month-old BESTO mice were considered the young cohort. Non-transgenic litter-mates were used as controls. The regular chow [PicoLab Rodent Diet 20 (Lab Diets, St Louis, MO, USA), 5053] contained protein (20%), fat (ether extract, 5.0%), far (acid hydrolysis, 5.6%), crude fiber (4.7%), nitrogen-free extract (52.9%), ash (6.1%), and vitamins, and 13.2% of total calories came from fat. The HFD consisted of chow containing 42% calories from fat (TD 88137; Harlan Teklad, Madison, WI, USA). The precise dietary composition of TD88137 is: casein, 195; DL-methionine, 3.0; sucrose, 341.46; cornstarch, 150.0; anhydrous milk fat, 210.0; cholesterol, 1.5; cellulose (fiber), 50.0; mineral mix (AIN-76), 35.0; calcium carbonate, 4.0; vitamin mix, 10.0; ethoxyquin (antioxidant), 0.04 (g kg−1). All animal procedures were approved by the Washington University Animal Studies Committee.
Fed and fasted glucose, insulin, and lipid measurements
Fed glucose, insulin, and lipid levels were measured between 09:00 and 10:00 hours, while fasted glucose, insulin, and lipid levels were measured after 15 h of overnight fasting. Glucose levels were determined using the Accu-Chek II glucometer (Roche Diagnostics, Indianapolis, IN, USA) with blood collected from the tail vein. For insulin and lipid measurements, blood was collected from the tail vein into chilled heparinized capillary tubes, plasma was separated by centrifugation, and samples were stored at −80 °C. Insulin levels were determined on 10 μL aliquots using rat insulin enzyme-linked immunosorbent assay (ELISA) kits with mouse insulin standards (Crystal Chem Inc., Downers Grove, IL, USA) at the Washington University Radio-immunoassay (RIA) Core facility. Cholesterol and triglycerides were measured by the Washington University Clinical Nutrition Research Unit (CNRU) Core facility using reagents from Thermo Electron Corporation (Waltham, MA, USA), while nonesterified free fatty acid levels were measured using reagents from Wako (Richmond, VA, USA).
IPGTTs and arginine stimulation
For the IPGTTs, mice were fasted for 15 h before being given an intraperitoneal injection of 50% dextrose (2 g kg−1 body weight). Blood was collected from the tail vein at 0, 15, 30, 60, and 120 min post-injection for determination of glucose values. Plasma for insulin measurements was also collected at 0, 15, and 30 min after glucose injection and stored at −80 °C. For the arginine stimulation experiments, mice were fasted for 15 h before being given an intraperitoneal injection of arginine (1.5 g kg−1 body weight). Blood was collected from the tail vein at 0, 2, and 5 min post-injection for both glucose and insulin measurements. For the NMN administration experiments, mice were injected with either PBS or NMN (500 mg kg−1 body weight) and fasted for 14 h prior to performing the IPGTTs as described earlier. All insulin samples were submitted to the Washington University RIA Core facility for ELISA analyses.
GSIS and ATP measurements from isolated islets
Islets were isolated by collagenase digestion as described previously (Moynihan et al., 2005). Briefly, after clamping off the pancreatic ducts prior to their entry site to the duodenum, pancreata were inflated with isolation buffer (10× Hank’s buffered salt solution, 10 mM 4-2-hydroxyethyl-1-piperazineethanesulfonic acid, 1 mM MgCl2, 5 mM glucose, pH 7.4) containing 0.375 mg mL−1 collagenase (Sigma, St Louis, MO, USA). The inflated pancreata were then removed, incubated at 37 °C for 15 min, and shaken vigorously. Islets were separated from acinar tissue after a series of washes and passages through a 70 μm nylon BD Falcon Cell Strainer (BD Biosciences, San Jose, CA, USA). Handpicked islets were cultured overnight in RPMI media containing 5 mM glucose, 2 mM L-glutamine, penicillin/streptomycin, and 10% fetal bovine serum (Gibco, Invitrogen, Carlsbad, CA, USA). The islets were then preincubated in Krebs–Ringer bicarbonate (KRB) buffer containing 2 mM glucose for 1 h at 37 °C. Islets of similar size were handpicked into groups of 10 islets in triplicate and incubated with 1 mL KRB buffer containing either 2 mM glucose, 20 mM glucose, or 20 mM KCl plus 2 mM glucose for 1 h at 37 °C. The supernatant was stored at −20 °C prior to insulin measurements. Islets were then washed two times with PBS, followed by extraction with extraction buffer (0.1 M NaOH, 0.5 mM ethylenediaminetetraacetic acid). After neutralizing the samples with 0.1 M HCl, ATP levels were measured using the ATP Bioluminescent Assay Kit (Sigma) according to the manufacturer’s instructions. Insulin levels of all samples were measured by radio-immunoassay at the Washington University RIA Core facility.
Western blotting
Protein extracts prepared from islets were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto Immobilon-P membranes (Millipore, Bedford, MA, USA). Membranes were blocked in Tris–buffered saline with 0.1% Tween-20 (TBS-T) and 5% dry milk (w/v). The primary antibodies used were affinity-purified polyclonal rabbit anti-mouse Sirt1 against an N-terminal fragment of mouse Sirt1 (1 : 5000), anti-Ucp2 [1 : 100, Santa Cruz Biotechnology (Santa Cruz, CA, USA) sc-6525], and anti-actin [1 : 5000, Calbiochem (La Jolla, CA, USA) #CP01]. Secondary antibodies included the horseradish peroxidase-conjugated anti-rabbit IgG (1 : 10 000, Amersham), anti-mouse IgM (1 : 2000, Calbiochem), and anti-goat IgG (1 : 10 000, Santa Cruz Biotechnology). Signals were visualized using the ECL Advance detection system (Amersham, Pittsburgh, PA, USA) and quantitated with the ImageQuant software (Molecular Dynamics, Sunnyvale, CA, USA).
Quantitative real-time reverse transcriptase–polymerase chain reaction (RT–PCR)
Total RNA samples were purified from isolated islets as described previously (Moynihan et al., 2005). cDNA synthesis and quantitative real-time RT–PCR were conducted as previously described (Revollo et al., 2004). Primer sequences are available upon request.
Islet NAD and plasma NMN measurements
The measurements of islet NAD and plasma NMN levels were conducted as described previously (Revollo et al., 2007b). Briefly, for NAD measurements in young and old BESTO islets, primary islets (~100 per sample) were isolated from 5- and 19-month-old BESTO mice, cultured in RPMI media containing 1 μM nicotinamide in 6 cm dishes, and harvested 24 h later in 800 μL of ice-cold PBS. Islets were lysed with 100 μL of 1 M perchloric acid on ice for 15 min. Lysates were cleared by centrifugation and neutralized by adding 33 μL of 3 M K2CO3 and incubating on ice for 10 min. After centrifugation, 100 μL of the supernatant was mixed with 300 μL of buffer A (50 mM K2PO4/KHPO4, pH 7.0) and loaded onto the column. The high performance liquid chromatography (HPLC) was run, and the amounts of NAD were quantitated based on the peak areas compared to a standard curve. The measurements were conducted in duplicate or triplicate of primary islets pooled from three individual mice. For NMN measurements, the HPLC was run at a flow rate of 0.7 mL min−1 in an isocratic condition. Then, 10 μL of freshly collected mouse plasma was extracted with 100 μL of 1 M perchloric acid, and the extracts were neutralized by adding 33 μL of 3 M K2CO3 and incubating on ice for 10 min. After clearing the extracts, 25 μL of the plasma extract was mixed with 275 μL of buffer and water. NMN levels were quantitated based on the peak areas compared to a standard curve. The measurements were conducted with four individual mice for each sex and age.
Statistical analysis
Unless otherwise indicated, all values are expressed as mean ± standard error, and statistical analyses were carried out using an unpaired Student’s t-test. Differences were considered to be statistically significant when P ≤ 0.05.
Supplementary Material
Fig. S1 IPGTTs and plasma insulin levels in female BESTO transgenic and control mice at 18–24 months of age. (A) IPGTTs were conducted in female BESTO transgenic mice (closed circles) compared to controls (open circles) at 18–24 months of age in two independent transgenic lines. Following a 15 h fast, 2 g of 50% glucose (w/v) per kg body weight was injected intraperitoneally, and blood glucose values were assessed at 0, 15, 30, 60, and 120 min post-injection. (B) Plasma insulin levels in female transgenic mice (black bars) and controls (white bars) were measured at the 0 and 30 min time points during the IPGTTs. Line 431-2, n = 10–13; line 431-4, n = 15–17. All results are expressed as mean ± standard error.
Fig. S2 Body weights of BESTO transgenic and control mice under a regular chow and a high-fat diet (HFD). Body weights of BESTO and control mice were measured during the indicated period of time under (A) a regular chow and (B) an HFD. (A) Because both 431-2 and 431-4 BESTO transgenic lines showed similar results, only the results from line 431-2 are shown. (B) Line 431-4 was used for this HFD experiment. All results are expressed as mean ± standard error.
Fig. S3 Fasted plasma lipid levels in young and old BESTO transgenic and control mice. Fasted plasma (A) cholesterol (B) triglycerides, and (C) total free fatty acid levels were measured in young and old BESTO (black bars) and control (white bars) male and female mice from line 431-4. Male mice, n = 6–10; female mice, n = 8–11. All results are expressed as mean ± standard error. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Fig. S4 IPGTTs and plasma insulin levels in BESTO transgenic and control mice and uncoupling protein 2 (Ucp2) expression levels in BESTO and control islets after 30-weeks of HFD feeding. (A) IPGTTs were conducted in male and female BESTO transgenic (closed circles) and control (open circles) mice from line 431-4 following 30 weeks on an HFD. Following a 15 h fast, 2 g of 50% glucose (w/v) per kg body weight was injected intraperitoneally, and blood glucose values were assessed at 0, 15, 30, 60, and 120 min post-injection (n = 6–7). (B) Plasma insulin levels in male and female transgenic mice (black bars) and controls (white bars) were measured at the 0 and 15 min time points during the IPGTTs shown in (A). (C) Western blot analysis of Ucp2 protein expression in transgenic (Tg) and control (Con) islet extracts from 431-4 mice fed a HFD for 30 weeks. Actin was used as a loading control for normalization. All results are expressed as mean ± standard error. *P ≤ 0.05; ***P ≤ 0.001.
Fig. S5 The effect of NMN administration on IPGTTs and plasma insulin levels in aged BESTO and control male mice. (A) IPGTTs were conducted in 20-month-old male BESTO transgenic mice (closed circles) and controls (open circles) from line 431-4 after mice were injected with PBS or NMN (500 mg kg−1 body weight) and fasted for 14 h (n = 13–17). (B) Plasma insulin levels were measured at 0 and 30 min post-glucose injection. #P ≤ 0.05; §P ≤ 0.01; ¶P ≤ 0.001, compared to insulin levels 30 min after glucose injection in PBS-injected aged BESTO and control mice. All results are expressed as mean ± standard error.
Acknowledgments
We would like to thank Jeffrey Gordon, Kerry Kornfeld, M. Alan Permutt, Kenneth Polonsky, Kelle Moley, and David Beebe for their helpful suggestions and discussions. We would also like to acknowledge the Diabetes Research and Training Center and the CNRU at Washington University for insulin and lipid measurements, respectively. Shin-ichiro Imai is an Ellison Medical Foundation Scholar in Aging, and is also supported by grants from the National Institute on Aging (AG024150), American Diabetes Association, Juvenile Diabetes Research Foundation, the Washington University CNRU (DK56341), and the National Center for Research Resources (C06RR015502).
Footnotes
This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/j.1474-9726.2007.00355.x (This link will take you to the article abstract).
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Al-Regaiey KA, Masternak MM, Bonkowski M, Sun L, Bartke A. Long-lived growth hormone receptor knockout mice: interaction of reduced IGF-1/insulin signaling and caloric restriction. Endocrinology. 2004;146:851–860. doi: 10.1210/en.2004-1120. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrom SU, Cline TW, Rine J. The Drosophila melanogaster sir2+ gene is nonessential and has only minor effects on position-effect variegation. Genetics. 2003;163:931–937. doi: 10.1093/genetics/163.3.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, Breda E, Oberg AL, Powell CC, Dalla Man C, Basu A, Vittone JL, Klee GG, Arora P, Jensen MD, Toffolo G, Cobelli C, Rizza RA. Mechanisms of the age-associated deterioration in glucose tolerance: contribution of alterations in insulin secretion, action, and clearance. Diabetes. 2003;52:1738–1748. doi: 10.2337/diabetes.52.7.1738. [DOI] [PubMed] [Google Scholar]
- Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell. 2006;125:1165–1177. doi: 10.1016/j.cell.2006.04.036. [DOI] [PubMed] [Google Scholar]
- Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A, Easlon EJ, Lin SJ, Guarente L. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AM, Halter JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab. 2003;284:E7–E12. doi: 10.1152/ajpendo.00366.2002. [DOI] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, De Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb S, Esposito RE. A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell. 1989;56:771–776. doi: 10.1016/0092-8674(89)90681-8. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Jarvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European group for the study of insulin resistance. J Clin Endocrinol Metab. 1999;84:863–868. doi: 10.1210/jcem.84.3.5542. [DOI] [PubMed] [Google Scholar]
- Joseph JW, Koshkin V, Zhang CY, Wang J, Lowell BB, Chan CB, Wheeler MB. Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes. 2002;51:3211–3219. doi: 10.2337/diabetes.51.11.3211. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam PP, Leung YM, Sheu L, Ellis J, Tsushima RG, Osborne LR, Gaisano HY. Transgenic mouse overexpressing syntaxin-1A as a diabetes model. Diabetes. 2005;54:2744–2754. doi: 10.2337/diabetes.54.9.2744. [DOI] [PubMed] [Google Scholar]
- Lin S-J, Defossez P-A, Guarente L. Life span extension by calorie restriction in S. cerevisiae requires NAD and Sir2. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- Lin S-J, Kaeberlein M, Andalis AA, Sturtz LA, Defossez P-A, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- Lin S-J, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller N, Gormsen L, Fuglsang J, Gjedsted J. Effects of ageing on insulin secretion and action. Horm Res. 2003;60:102–104. doi: 10.1159/000071233. [DOI] [PubMed] [Google Scholar]
- Moynihan KA, Imai S. Sirt1 as a key regulator orchestrating the response to caloric restriction. Drug Discov Today: Dis Mech. 2006;3:11–17. [Google Scholar]
- Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Muzumdar R, Ma X, Atzmon G, Vuguin P, Yang X, Barzilai N. Decrease in glucose-stimulated insulin secretion with aging is independent of insulin action. Diabetes. 2004;53:441–446. doi: 10.2337/diabetes.53.2.441. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- Newsholme P, Brennan L, Rubi B, Maechler P. New insights into amino acid metabolism, beta-cell function and diabetes. Clin Sci (Lond) 2005;108:185–194. doi: 10.1042/CS20040290. [DOI] [PubMed] [Google Scholar]
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Oliveira RM, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven EP, Gold G, Reaven GM. Effect of age on glucose-stimulated insulin release by the beta-cell of the rat. J Clin Invest. 1979;64:591–599. doi: 10.1172/JCI109498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven E, Curry D, Moore J, Reaven G. Effect of age and environmental factors on insulin release from the perfused pancreas of the rat. J Clin Invest. 1983;71:345–350. doi: 10.1172/JCI110775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven EP, Curry DL, Reaven GM. Effect of age and sex on rat endocrine pancreas. Diabetes. 1987;36:1397–1400. doi: 10.2337/diab.36.12.1397. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol. 2007a;23:164–170. doi: 10.1097/MOG.0b013e32801b3c8f. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Körner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W, Imai S. Nampt/PBEF/Visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007b;6:363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles – a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tissenbaum HA. Overlapping and distinct functions for a Caenorhabditis elegans SIR2 and DAF-16/FOXO. Mech Ageing Dev. 2006;127:48–56. doi: 10.1016/j.mad.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Wang Y, Oh SW, Deplancke B, Luo J, Walhout AJ, Tissenbaum HA. C. elegans 14-3-3 proteins regulate life span and interact with SIR-2.1 and DAF-16/FOXO. Mech Ageing Dev. 2006;127:741–747. doi: 10.1016/j.mad.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Wilson PW, Anderson KM, Kannel WB. Epidemiology of diabetes mellitus in the elderly. The Framingham study. Am J Med. 1986;80:3–9. doi: 10.1016/0002-9343(86)90532-2. [DOI] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 IPGTTs and plasma insulin levels in female BESTO transgenic and control mice at 18–24 months of age. (A) IPGTTs were conducted in female BESTO transgenic mice (closed circles) compared to controls (open circles) at 18–24 months of age in two independent transgenic lines. Following a 15 h fast, 2 g of 50% glucose (w/v) per kg body weight was injected intraperitoneally, and blood glucose values were assessed at 0, 15, 30, 60, and 120 min post-injection. (B) Plasma insulin levels in female transgenic mice (black bars) and controls (white bars) were measured at the 0 and 30 min time points during the IPGTTs. Line 431-2, n = 10–13; line 431-4, n = 15–17. All results are expressed as mean ± standard error.
Fig. S2 Body weights of BESTO transgenic and control mice under a regular chow and a high-fat diet (HFD). Body weights of BESTO and control mice were measured during the indicated period of time under (A) a regular chow and (B) an HFD. (A) Because both 431-2 and 431-4 BESTO transgenic lines showed similar results, only the results from line 431-2 are shown. (B) Line 431-4 was used for this HFD experiment. All results are expressed as mean ± standard error.
Fig. S3 Fasted plasma lipid levels in young and old BESTO transgenic and control mice. Fasted plasma (A) cholesterol (B) triglycerides, and (C) total free fatty acid levels were measured in young and old BESTO (black bars) and control (white bars) male and female mice from line 431-4. Male mice, n = 6–10; female mice, n = 8–11. All results are expressed as mean ± standard error. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Fig. S4 IPGTTs and plasma insulin levels in BESTO transgenic and control mice and uncoupling protein 2 (Ucp2) expression levels in BESTO and control islets after 30-weeks of HFD feeding. (A) IPGTTs were conducted in male and female BESTO transgenic (closed circles) and control (open circles) mice from line 431-4 following 30 weeks on an HFD. Following a 15 h fast, 2 g of 50% glucose (w/v) per kg body weight was injected intraperitoneally, and blood glucose values were assessed at 0, 15, 30, 60, and 120 min post-injection (n = 6–7). (B) Plasma insulin levels in male and female transgenic mice (black bars) and controls (white bars) were measured at the 0 and 15 min time points during the IPGTTs shown in (A). (C) Western blot analysis of Ucp2 protein expression in transgenic (Tg) and control (Con) islet extracts from 431-4 mice fed a HFD for 30 weeks. Actin was used as a loading control for normalization. All results are expressed as mean ± standard error. *P ≤ 0.05; ***P ≤ 0.001.
Fig. S5 The effect of NMN administration on IPGTTs and plasma insulin levels in aged BESTO and control male mice. (A) IPGTTs were conducted in 20-month-old male BESTO transgenic mice (closed circles) and controls (open circles) from line 431-4 after mice were injected with PBS or NMN (500 mg kg−1 body weight) and fasted for 14 h (n = 13–17). (B) Plasma insulin levels were measured at 0 and 30 min post-glucose injection. #P ≤ 0.05; §P ≤ 0.01; ¶P ≤ 0.001, compared to insulin levels 30 min after glucose injection in PBS-injected aged BESTO and control mice. All results are expressed as mean ± standard error.