

Abstract
CATERPILLER is a mammalian gene family with signature NBD and LRR domains. Several members of this family are positive regulators of inflammatory responses. Others, however, exert negative effects on proinflammatory responses. These data are particularly convincing when shRNA/siRNA are used. This review focuses on the Monarch-1/PYPAF7 gene with brief discussions of CLR16.2/NOD3, PYPAF2/PAN1/NALP2, and PYPAF3.
Keywords: CLR, NOD, NLR, NALP, Plant R genes, PYPAF, PAN
The CATERPILLER (CLR) gene family
The discovery of the CLR gene family by our group was prompted by our long-term interest in the master transcriptional regulator of MHC-II, the class II transactivator (CIITA) [1]. Analyses of domain structures of CIITA by us and others have led to the general depiction of this protein as containing an N-terminal acidic domain that is critical for transcription activation, a mid-region nucleotide-binding domain (NBD) and a C-terminal leucine rich region (LRR) [2, 3]. Mutations of these three regions are detrimental to the capacity of CIITA to activate MHC-II genes. By comparing the domain architecture of CIITA to published gene sequences, it became clear to us that other genes encode proteins with the same structure, including the then just-discovered NOD1 protein [3]. Most remarkably, a subgroup of plant disease-resistance genes encodes proteins with a similar nucleotide-binding domain, and LRR motifs [4]. Thus we noted over five years ago that the NBD motifs and LRR of these plant proteins are similar to CIITA with respect to both spacing and size, and suggested a divergent family of genes with a similar domain structure which protects against infectious agents in both mammals and plants. In a separate paper, the Nunez group, proposed a similar concept based on their studies of NOD1 and NOD2, as well as other proteins with similar domains [5].
The simultaneous publication of the initial drafts of the human genome by Celera and a public consortium in early 2001 allowed a rapid acceleration of our own effort to complete the search of the CLR gene family [6]. Based on bioinformatics data culled from the Celera human genome scaffold data and the National Center for Biotechnology Information (NCBI), we described in detail the CLR family, which in humans is comprised of over 20+ family members. The genomic organization of all the family members is remarkably similar with large NBD exons (≃1500 nt) and LRR exons of either ≃76 nt which we referred to as a singlet LRR, or a doublet of 174 nt. The N-terminal end containing either a CARD or pyrin domain is typically ≃300 nt long. We also noted and compared the relatedness of this family to the NAIP and IPAF/CARD12, and to its more distant cousin, the APAF-1 gene. From a historic perspective, it is rather remarkable that the entire family and its close relatives were identified in one felt swoop, testifying to the impact of the human genome project. Soon thereafter, another group reported on a nearly-identical gene family and named it the nucleotide-oligomerization domain (NOD) family after its founding members NOD1 and NOD2 [7, 8]. A more recent proposal suggests the name NACHT-LRR or NLR [9]. Several others reported on the pyrin-containing subgroup or members of this subgroup, which were given various names including PAN, PYPAF and NALP [10–12].
A seminal step forward in understanding the biologic function of CLR genes was the association of certain members of this family with human diseases. Notable examples are the genetic association of CIAS-1 to three autoinflammatory diseases and of NOD2 to Crohn’s disease and Blau’s syndrome. Other notable progress includes the findings that NOD1, NOD2 and CIAS-1 are all required for cellular responses to degradation products of bacterial peptidoglycans. This has led to the concept that these CLR proteins function as “sensors” of pathogenic products. Finally, another fundamental step forward was the finding that some CLR proteins are important components of the “inflammasome” [13]. The formation of this multi-protein complex leads to the activation of caspase-1 and subsequent production of mature IL-1β. These topics have been discussed in several recent reviews and will not be a focus here [12, 14]. Instead, this review will focus on a pyrin-containing CLR protein which we refer to as Monarch-1. This protein is found to be a negative regulator of NF-κB activation and subsequent cytokine and chemokine production. Additional examples of negative regulators have also emerged from other studies, and will be briefly mentioned.
The Monarch-1/PYAPF7 gene
The 3′ fragment of the gene encoding Monarch-1 was first identified as a gene whose expression is induced by nitric oxide in the leukemic line, HL60 [15]. The full length sequence was first cloned and named PYPAF7 [16]. Simultaneously, we also cloned the full length sequence and named it CLR19.3 or Monarch-1 [17]. The gene is located on human chromosome 19q13.4, among a cluster of nine pyrin-containing CLR genes. The full-length cDNA has a 3189-bp open reading frame (accession number AY116204) encoded by 10 exons. The protein consists of a predicted N-terminal pyrin domain; a central nucleotide-binding domain, that belongs to the NACHT subfamily of NTPases containing seven signature motifs; and a leucine rich region. In addition, we found that there are at least four Monarch-1 splice forms resulting from differential splicing of the LRR (accession numbers AY116205, AY116206, and AY116207) (Fig. 1). Preliminary evidence indicates that these splice forms exhibit different functions, suggesting that varying the length of the LRR may serve to alter the ultimate biologic activity of the gene product (J. Lich, unpublished observations).
Figure 1. Monarch-1 isoforms.
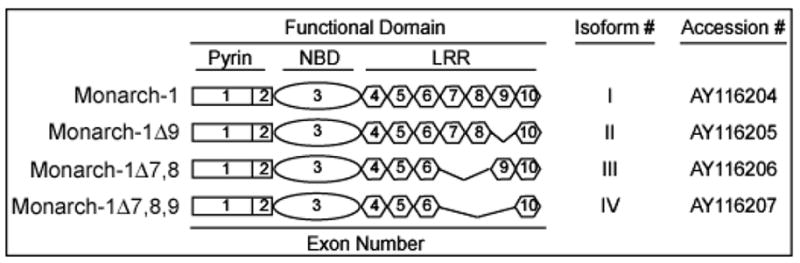
Four different isoforms encoding Monarch-1 have been identified. These isoforms differ due to mRNA splicing of exons encoding the LRR domain. Listed above is the common name followed by an exon diagram for each isoform. In addition, the isoform number (I–IV) and accession number are shown.
Monarch-1 expression
Monarch-1 is found to be expressed in monocytes and granulocytes with notably high expression in eosinophils, while a panel of over 70 tissues/cells shows little or no expression [16, 17]. This myeloid restricted pattern of expression provided the initial clue that the protein may be a regulator of the immune response, inflammation and host response to pathogens. In examining the mRNA expression, we found that Monarch-1 expression is reduced by greater than 65–80% after an 1 h treatment with purified E. coli LPS, which activates TLR4, or peptidoglycan/Pam3Cys, which activates TLR2 [18]. Similarly, live bacteria including M. tuberculosis and P. gingivalis all resulted in reduced Monarch-1. This reduced expression is found in the THP-1 cell line and, more convincingly, in human peripheral blood granulocytes and monocytes. Confirmation of this reduction in Monarch-1 was also verified at the protein level. This response in Monarch-1 expression is not limited to TLRs. Indeed, expression is also down-regulated upon exposure of PBMC to TNFα or IFNγ [18]. These results, coupled with the functional studies to be reviewed below, have led to hypothesis that Monarch-1 functions as a negative regulator of inflammatory responses induced by TLRs and cytokines.
In contrast to the observations that Monarch-1 levels decline rapidly under inflammatory conditions, another group has shown that nitric oxide increases the expression of Monarch-1 [15]. This has been confirmed by us and others [17]. Thus an interesting proposition is that nitric oxide may serve as a negative feedback loop to induce Monarch-1 expression, which then attenuates an ongoing inflammatory response by inhibiting NF-κB activation as well as proinflammatory cytokine and chemokine expression.
Monarch-1 generally exhibits a cytoplasmic pattern of expression. Bertin and colleagues found that when expressed together with ASC (Apoptotic Speck containing protein with a CARD), the two proteins co-localize to punctate cytoplasmic structures. However, attempts to co-precipitate Monarch-1 and ASC have not been successful ([16] and J. Lich, unpublished observations). This parallels the interaction between CIAS-1 and ASC, which can be detected in a yeast two-hybrid system, but not by co-precipitation of overexpressed proteins in mammalian cells. Interestingly, however, co-precipitation with disease-associated CIAS-1 mutants has been demonstrated, indicating that disease-associated mutation resulted in enhanced interaction with ASC. The confirmation of the physical interaction of Monarch-1 with ASC awaits studies analyzing endogenous proteins.
Monarch-1 function
Bertin et al. found that while Monarch-1 alone does not have much demonstrated function in 293T cells, simultaneous overexpression of Monarch-1 and ASC in these cells is found to result in NF-kB and caspase-1 activation. However our findings with siRNA directed at Monarch-1 in the monocytic cell line, THP-1, showed very different findings. In THP-1 cells, silencing Monarch-1 causes a dramatic enhancement of NF-κB activation upon TLR stimulation, suggesting that endogenous Monarch-1 functions as a negative regulator of the NF-κB response in monocytes. Furthermore, the production of the proinflammatory cytokines IL-6 and IL-1β is elevated in Monarch-1 silenced THP-1 monocytes in response to TLR4 agonists (LPS), TLR2 agonists (Pam3Cys) or whole bacteria (M. tuberculosis) [18]. This negative regulatory role was confirmed in reciprocal experiments utilizing THP-1 cells stably expressing elevated levels of Monarch-1. In these cells, greatly reduced levels of proinflammatory cytokines are produced in response to TLR stimulation (our unpublished observations). Together with the siRNA results, these finding demonstrate that Monarch-1 performs a negative regulatory role in controlling the inflammatory response.
Toward identifying the molecular mechanisms of Monarch-1 activity
Monarch-1 functions as a negative regulator of inflammation by suppressing the production of proinflammatory cytokines and chemokines [18]. Based on reporter gene assays, Monarch-1 exerts this anti-inflammatory function by inhibiting NF-κB activation. Inhibition of NF-κB is not unique to Monarch-1. Indeed, this phenotype is shared among several CLR proteins such as PYPAF2 and CLR16.2 [19, 20]. Unfortunately, little is known regarding the molecular mechanisms by which Monarch-1 or these other CLR proteins exert this anti-inflammatory activity.
Monarch-1 suppresses TLR-induced expression of NF-κB responsive cytokines in monocytes. Therefore, to determine how Monarch-1 suppresses NF-κB, we focused our studies on the role of Monarch-1 in TLR signaling. In this pathway, TLRs associate with cytoplasmic adaptor proteins, such as MyD88, that recruit the serine/threonine kinases IRAK1 and IRAK4. IRAK4 phosphorylates IRAK1, which activates the autophosphorylation activity of IRAK1. This leads to the accumulation of a hyperphosphorylated species of IRAK1 [21]. This critical step is required for activation of TRAF6 and downstream MAP3 kinases. These MAP3 kinases activate the IKK complex (composed of IKKα, β, and γ), which promotes the activation of RelA/p50 NF-κB dimers. This “canonical” pathway proceeds rapidly and leads to the production of inflammatory cytokines and chemokines including IL-6, TNF-α and IL-8 [22].
Our studies have led to the finding that Monarch-1 intersects the TLR signaling pathway through its association with IRAK1 [18]. The association with IRAK1 is mediated through the NBD of Monarch-1 and is induced following TLR stimulation. Importantly, this association results in a sharp reduction in cellular levels of hyperphosphorylated IRAK1. It is not clear whether this reduction results from an inability to phosphorylate IRAK1 or is due to a rapid turnover of hyperphosphorylated IRAK1 in the presence of Monarch-1. Nevertheless, the loss of hyperphosphorylated IRAK1 would, presumably, hinder the activation of downstream signaling molecules including NF-κB. This supports our findings from reporter gene assays, where Monarch-1 expression inhibits IRAK1-mediated NF-κB activation [18]. Furthermore, it provides a possible explanation for the enhanced level of NF-κB activation following TLR stimulation of monocytes in which Monarch-1 expression has been silenced.
In addition to suppressing canonical NF-κB activation, our studies have found that Monarch-1 also inhibits “noncanonical” NF-κB activation. This alternative pathway occurs subsequent to the canonical pathway and is activated downstream of TLRs as well as certain TNF family receptors [23–25]. Unlike the canonical pathway, which can be activated by many different upstream kinases, the noncanonical pathway is strictly dependent upon the MAP3 kinase NIK [26]. Upon activation, NIK recruits IKKα and NF-κB2/p100, which leads to the processing of p100 to its active form p52. Activation of this noncanonical pathway augments the initial proinflammatory response by regulating the transcriptional activation of chemokines such as CXCL12, CXCL13 and CCL5 [27] (Fig. 2).
Figure 2. Monarch-1 suppresses both canonical and noncanonical NF-κB activation.
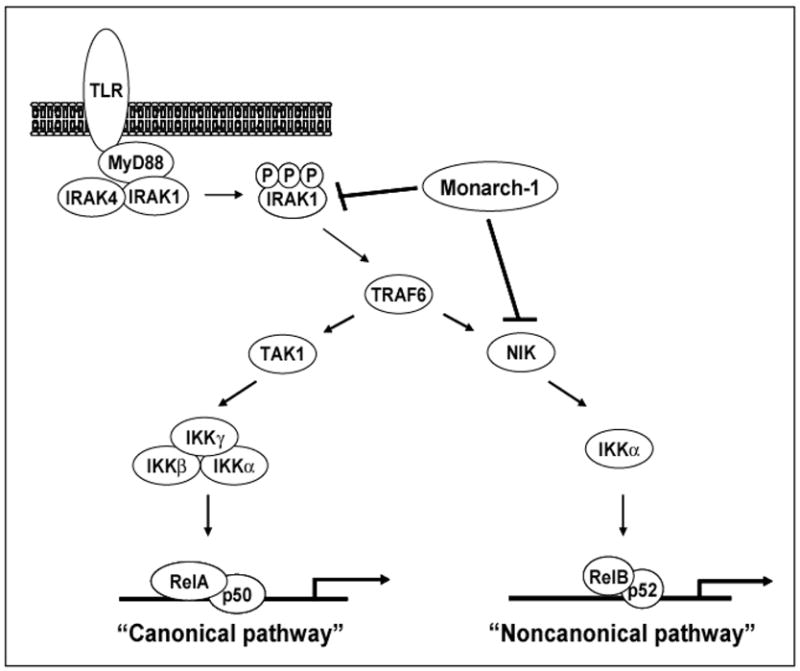
Upon TLR stimulation, the adaptor protein MyD88 is recruited to the receptor complex. MyD88 then recruits IRAK4 and IRAK1 which results in the accumulation of hyperphosphorylated IRAK1. This leads to the activation of TRAF6 which triggers NF-κB activation via MAP3 kinases such as TAK1 and NIK. While the canonical pathway can be activated by a number of upstream kinases, the noncanonical pathway is strictly dependent upon NIK. Monarch-1 is capable of blocking both pathways through its interactions with IRAK1 and NIK. Monarch-1 suppresses canonical NF-κB activation by blocking the accumulation of hyperphosphorylated IRAK1. The noncanonical pathway is also inhibited as the association of Monarch-1 with NIK leads to rapid proteasome dependent degradation of the kinase.
Analogous to our findings concerning Monarch-1 and IRAK1, Monarch-1 associates with NIK following activation of the kinase and inhibits NIK-induced NF-κB activation (J. Lich et al. submitted). This was observed both in reporter gene assays and in THP-1 monocytes where elevated levels of Monarch-1 inhibited p100 processing. Surprisingly, Monarch-1 mediated inhibition of NIK stems, at least in part, from the ability of Monarch-1 to induce rapid proteasome mediated degradation of the kinase. To our knowledge, this is the first report of a CLR protein that controls inflammatory signaling pathways through a proteasome mediated process.
Based on these studies, a mechanism is emerging where Monarch-1 functions to negatively regulate innate immune responses by associating with and suppressing key signaling molecules. In the case of IRAK1, a significant reduction in hyperphosphorylated IRAK1 is observed in TLR stimulated monocytes expressing elevated levels of Monarch-1 [18]. As stated earlier, the mechanism by which Monarch-1 reduces hyperphosphorylated IRAK1 is not clear. However, the results from our NIK studies have led to the hypothesis that hyperphosphorylated IRAK1 is a target for Monarch-1-induced proteasome mediated degradation. Indeed, in HEK293 cells expressing both IRAK1 and Monarch-1, the addition of proteasome inhibitors rescues hyperphosphorylated IRAK1. This indicates that this species of IRAK1 can form in the presence of Monarch-1; however, its accumulation is limited by proteasome mediated degradation (J. Lich, unpublished observations). Current studies are underway to determine if Monarch-1 induces proteasome mediated degradation of hyperphosphorylated IRAK1 in TLR stimulated monocytes.
The degradation of signaling molecules following cellular activation provides a potent mechanism for controlling inflammatory signaling pathways. Our results support a model where activation leads to the association of Monarch-1 with the active forms of the signaling proteins IRAK1 and NIK. This interaction leads to rapid degradation of these proteins, thereby attenuating the inflammatory signaling. Other CLR proteins have been shown to associate with signaling proteins and suppress their ability to activate NF-κB. For instance, NOD2 associates with TAK1 and inhibits the ability of TAK1 to activate NF-κB [28]. Similarly, PYPAF2 associates with ASC and the IKK complex and prevents NF-κB activation downstream of a number of activators [19]. It remains to be seen if these CLR proteins mediate NF-κB suppression through proteasome-mediated pathways.
Other negative regulatory CLR proteins
Although this review is primarily focused on Monarch-1, several CLRs exhibit negative regulatory functions including the CLR16.2/NOD3, PYPAF2/PAN1/NALP2, and PYPAF3. CLR16.2 is expressed primarily in T cells, and similar to Monarch-1, its expression declines rapidly following cell stimulation [20]. When present, CLR16.2 inhibits NF-κB, AP-1 and NFAT transcriptional activation in Jurkat T cells downstream of CD3/CD28 stimulation or treatment with PMA/ionomycin. This suggests that CLR16.2 functions as a novel suppressor of T cell activation.
PYPAF2 is expressed more broadly than CLR16.2 or Monarch-1 and mRNA can be detected in many different cell types and tissues, including non-hematopoietic cells [19, 29]. In addition, distinct from Monarch-1 and CLR16.2, PYPAF2 expression is induced following treatment of cells with LPS or proinflammatory cytokines. PYPAF2 inhibits NF-κB activation downstream of many different upstream activators. This was shown directly in reporter gene assays and indirectly in THP-1 monocytes, where siRNA mediated silencing of PYPAF2 results in increased expression of ICAM1. It appears that PYPAF2 performs this function through its association with the IKK complex [19]. In addition to the IKK complex, PYPAF2 also associates with ASC. However, in this case, PYPAF2 acts as a positive regulator by promoting ASC-mediated caspase-1 activation and IL-1β release. This may result from the ability of PYPAF2 to form an inflammasome complex. Indeed, another report subsequently found that PYPAF2 forms a large protein complex consisting of ASC, pro-caspase-1, and CARDINAL/CARD8 [13].
Similar to PYAF2, PYPAF3 is broadly expressed and induced by LPS or proinflammatory cytokines [29]. However, in contrast to PYPAF2, PYPAF3 has no measured effect on NF-κB activation. Instead, PYPAF3 suppresses ASC-mediated IL-1β release by inhibiting the processing and activation of pro-caspase-1 and pro-IL-1β. This was confirmed in THP-1 monocytes expressing elevated levels of PYPAF3. How PYPAF3 performs this function has not been elucidated. However, PYPAF3 associates with pro-caspase-1 and pro-IL-1β in co-precipitations suggesting that it forms a large protein complex similar to that formed by PYPAF2. As mentioned above, PYPAF2 also associates with pro-caspase-1, but different from PYPAF3, functions to promote IL-1b release. Thus, even though PYPAF2 and PYPAF3 form similar protein complexes, they have different functional outcomes.
Current challenges and future directions
Ectopic expression of CLR proteins in model cell lines provides a convenient and powerful system to explore the molecular mechanisms influenced by these newly discovered genes. However there is some concern surrounding such overexpression systems, as genes of this family have exhibited both positive and negative functions, depending on the dosage used. For instance, Bruey et al. found that PYAPF2 enhances ASC-mediated IL-1β release at low doses [19]. This effect was lost as increasing amounts of PYPAF2 were expressed. Furthermore, in our laboratory, we have observed that higher levels of CIITA are actually detrimental to MHC class II expression. Even in gene deletion mice, three different strains of NOD2−/−mice exhibit different positive and negative responses to pathogenic stimulants [14]. Hence while awaiting the availability of gene deletion mice, the utilization of shRNA or siRNA in combination with ectopic expression would be most informative.
While much has been learned concerning how CLR genes function in inflammation and immunity, there is still much to be explored. For instance, NOD1, NOD2, and CIAS1 respond to components of peptidoglycan. However, it remains unknown whether or not other CLR family members, such as Monarch-1, also respond to microbial derived molecules. In addition, very little is known concerning the molecular mechanisms by which the majority of CLR proteins function. Several CLR proteins have been shown to form large protein complexes collectively termed ‘inflammasomes” [30]. These complexes have been implicated in processing IL-1β and IL-18. PYPAF2 associates with these complexes [13]. However, it is not known how the formation of these large multi-protein complexes relates to the NF-κB suppressive activity of PYPAF2. Most importantly, with regards to disease associated CLRs, only after these molecular mechanisms have been elucidated will we be able to understand how certain mutations lead to autoinflammatory conditions in humans.
Acknowledgments
J.L. is supported by the American Cancer Society. J. P-Y.T. is a Sandler’s Awardee and supported through the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ting JP, Davis BK. Annu Rev Immunol. 2004;19:19. [Google Scholar]
- 2.Riley JL, Westerheide SD, Price JA, Brown JA, Boss JM. Immunity. 1995;2:533–543. doi: 10.1016/1074-7613(95)90033-0. [DOI] [PubMed] [Google Scholar]
- 3.Harton JA, Ting JP. Mol Cell Biol. 2000;20:6185–6194. doi: 10.1128/mcb.20.17.6185-6194.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ausubel FM. Nat Immunol. 2005;6:973–979. doi: 10.1038/ni1253. [DOI] [PubMed] [Google Scholar]
- 5.Inohara N, Nunez G. Oncogene. 2001;20:6473–6481. doi: 10.1038/sj.onc.1204787. [DOI] [PubMed] [Google Scholar]
- 6.Harton JA, Linhoff MW, Zhang J, Ting JP. J Immunol. 2002;169:4088–4093. doi: 10.4049/jimmunol.169.8.4088. [DOI] [PubMed] [Google Scholar]
- 7.Inohara Chamaillard, McDonald C, Nunez G. Annu Rev Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 8.Inohara N, Nunez G. Nat Rev Immunol. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 9.Martinon F, Tschopp J. Trends Immunol. 2005;26:447–454. doi: 10.1016/j.it.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Fiorentino L, Stehlik C, Oliveira V, Ariza ME, Godzik A, Reed JC. J Biol Chem. 2002;277:35333–35340. doi: 10.1074/jbc.M200446200. Epub 32002 Jul 35331. [DOI] [PubMed] [Google Scholar]
- 11.Grenier JM, Wang L, Manji GA, Huang WJ, Al-Garawi A, Kelly R, Carlson A, Merriam S, Lora JM, Briskin M, DiStefano PS, Bertin J. FEBS Lett. 2002;530:73–78. doi: 10.1016/s0014-5793(02)03416-6. [DOI] [PubMed] [Google Scholar]
- 12.Tschopp J, Martinon F, Burns K. Nat Rev Mol Cell Biol. 2003;4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- 13.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 14.Eckmann L, Karin M. Immunity. 2005;22:661–667. doi: 10.1016/j.immuni.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Shami PJ, Kanai N, Wang LY, Vreeke TM, Parker CH. Br J Haematol. 2001;112:138–147. doi: 10.1046/j.1365-2141.2001.02491.x. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, Geddes BJ, Briskin M, DiStefano PS, Bertin J. J Biol Chem. 2002;277:29874–29880. doi: 10.1074/jbc.M203915200. Epub 22002 May 29817. [DOI] [PubMed] [Google Scholar]
- 17.Williams KL, Taxman DJ, Linhoff MW, Reed W, Ting JP. J Immunol. 2003;170:5354–5358. doi: 10.4049/jimmunol.170.11.5354. [DOI] [PubMed] [Google Scholar]
- 18.Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, Kurtz S, Coffield VM, Accavitti-Loper MA, Su L, Vogel SN, Braunstein M, Ting JP. J Biol Chem. 2005;3:3. [Google Scholar]
- 19.Bruey JM, Bruey-Sedano N, Newman R, Chandler S, Stehlik C, Reed JC. J Biol Chem. 2004;279:51897–51907. doi: 10.1074/jbc.M406741200. Epub 52004 Sep 51828. [DOI] [PubMed] [Google Scholar]
- 20.Conti BJ, Davis BK, Zhang J, O'Connor W, Jr, Williams KL, Ting JP. J Biol Chem. 2005;280:18375–18385. doi: 10.1074/jbc.M413169200. Epub 12005 Feb 18310. [DOI] [PubMed] [Google Scholar]
- 21.Li S, Strelow A, Fontana EJ, Wesche H. Proc Natl Acad Sci U S A. 2002;99:5567–5572. doi: 10.1073/pnas.082100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonizzi G, Karin M. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Muller JR, Siebenlist U. J Biol Chem. 2003;278:12006–12012. doi: 10.1074/jbc.M210768200. Epub 12003 Jan 12028. [DOI] [PubMed] [Google Scholar]
- 24.Mordmuller B, Krappmann D, Esen M, Wegener E, Scheidereit C. EMBO Rep. 2003;4:82–87. doi: 10.1038/sj.embor.embor710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, Klaus GG, Johnston LH, Ley SC. Embo J. 2002;21:5375–5385. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao G, Harhaj EW, Sun SC. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 27.Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M. Embo J. 2004;23:4202–4210. doi: 10.1038/sj.emboj.7600391. Epub 2004 Oct 4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen CM, Gong Y, Zhang M, Chen JJ. J Biol Chem. 2004;9:9. doi: 10.1074/jbc.M400682200. [DOI] [PubMed] [Google Scholar]
- 29.Kinoshita T, Wang Y, Hasegawa M, Imamura R, Suda T. J Biol Chem. 2005;280:21720–21725. doi: 10.1074/jbc.M410057200. Epub 22005 Apr 21726. [DOI] [PubMed] [Google Scholar]
- 30.Petrilli V, Papin S, Tschopp J. Curr Biol. 2005;15:R581. doi: 10.1016/j.cub.2005.07.049. [DOI] [PubMed] [Google Scholar]