

Abstract
Patricia Goldman-Rakic played a groundbreaking role in investigating the cognitive functions subserved by dorsolateral prefrontal cortex and the key role of dopamine in that. The work discussed here builds on that including: 1) Studies of children predicted to have lower levels of prefrontal dopamine but otherwise basically normal brains (children treated for phenylketonuria [PKU]). Those studies changed medical guidelines, improving the children’s lives. 2) Studies of visual impairments (in contrast sensitivity and motion perception) in PKU children due to reduced retinal dopamine and due to excessive phenylalanine during the first postnatal weeks. Those studies, too, changed medical guidelines. 3) Studies of working memory and inhibitory control differences in typically developing children due to differences in catechol-O-methyltransferase (COMT) genotype, which selectively affect prefrontal dopamine levels. 4) Studies of gender differences in the effect of COMT genotype on cognitive performance in older adults. 5) A hypothesis about fundamental differences between attention deficit hyperactivity disorder (ADHD) that includes hyperactivity and ADHD of the inattentive type. Those disorders are hypothesized to differ in the affected neural system, underlying genetics, responsiveness to medication, comorbidities, and cognitive and behavioral profiles. These sound quite disparate but they all grew systematically out the base laid down by Patricia Goldman-Rakic.
Keywords: attention deficit hyperactivity disorder, cognitive control, catechol-o-methyltransferase, DAT1, DRD4, executive function, inhibition, phenylalanine hydroxylase, phenylketonuria, visual contrast sensitivity, working memory
Introduction
This paper is for a special issue in honor of Patricia Goldman-Rakic. Therefore, I will briefly trace the history leading up to the recent findings to be discussed here to honor Pat’s contribution them.
Historical Background
Before I ever met Pat, I hypothesized for my doctoral dissertation that some of the cognitive changes seen in human infants between 7 and 12 months of age were made possible in part by maturational changes in dorsolateral prefrontal cortex. One of the principal reasons to hypothesize that was the striking similarity between Piaget’s A-not-B task (Piaget 1937), on which infants improve between 7 and 12 months of age, and the delayed-response task that Jacobsen had introduced to study the functions of prefrontal cortex in rhesus monkeys (Jacobsen 1935). Indeed, Pat had been at the forefront of neuroscientists showing in precise, elegant studies that success on delayed response depends specifically on dorsolateral prefrontal cortex (e.g., Alexander and Goldman 1978; and Rosvold 1970).
Wanting to get converging evidence from a very different behavioral paradigm, I devised a transparent barrier detour task (called “object retrieval”) based on the work of Moll and Kuypers (1977). Whereas A-not-B and delayed response were hiding tasks that imposed brief delays between hiding and retrieval, in the object retrieval task nothing was hidden and there was no delay. Yet both A-not-B/delayed response and object retrieval required holding information in mind and inhibiting a strong action tendency (i.e., they required working memory plus inhibition). For the former, subjects had to hold in mind where the reward had been hidden (updating that on each trial) and they had to inhibit the tendency to repeat a rewarded response when the location of hiding changed. For the latter (object retrieval), subjects had to inhibit the very strong pull to try to reach directly for their visible goal through a closed, transparent side of the box, substituting a detour reach around to the one open side. They also had to hold in mind the route they had looked through when they had looked in the box opening now that they were looking through a closed side of the box, and had to mentally coordinate reaching along the route through the opening with the very different route present to their eyes now that they were looking through a closed side of the box.
For the dissertation I documented that improvement on these tasks occurred over the same age period in infants and that although there were very large individual differences in when landmarks on the tasks were reached, there was remarkable consistency across these tasks in when given infants reached those landmarks (Diamond 1991a, 1991b). Typically, it happened at the very same age and never more than 2 weeks apart. However, I had no data specifically linking my findings in infants to the brain. I contacted Patricia Goldman-Rakic: “Can I test your monkeys on my tasks (monkeys with lesions to dorsolateral prefrontal cortex, monkeys with control lesions, and normal infant monkeys)?” Pat only wondered why it had taken me so long to ask. As a postdoctoral fellow in Pat’s lab I embarked on a systematic program of research to chart the developmental progression of infant rhesus monkeys on the 3 tasks, the effect of lesions of dorsolateral prefrontal cortex and of posterior parietal cortex on adult rhesus monkeys‘ performance of those tasks, and the effect of lesions of dorsolateral prefrontal cortex on infant rhesus monkeys’ performance of the tasks (Diamond and Goldman-Rakic 1985, 1986, 1989). This was the first time that the contribution of a specific neural region to an aspect of cognitive development had been demonstrated. It encouraged people that rigorous experimental work addressing brain–behavior relations was possible even in infants and that work in neuroscience might inform work in developmental psychology and vice versa.
Much work remained to be done, however. Even if I were right that prefrontal maturation helped make possible the cognitive advances evidenced by improved A-not-B, delayed response, and object retrieval performance, what about prefrontal cortex was changing that made these cognitive advances possible? Again, I turned to the pioneering work of Patricia Goldman-Rakic and colleagues. They had shown that depleting dorsolateral prefrontal cortex of dopamine impairs delayed-response performance as severely as does removing dorsolateral prefrontal cortex altogether (Brozoski et al. 1979; Sawaguchi and Goldman-Rakic 1991). They had also shown that levels of dopamine are increasing in the rhesus brain during the period that infant rhesus monkeys are improving on my tasks (Brown and Goldman 1977; Brown et al. 1979).
Thus, I hypothesized that perhaps one of the changes that was improving the functioning of prefrontal cortex during development was increasing levels of dopamine in prefrontal cortex. As an initial way of looking at the role of dopamine in modulating cognitive functions dependent on prefrontal cortex early in life in humans, I set out to study a group of children who, there was reason to believe, had lower levels of dopamine in prefrontal cortex, but otherwise basically normal brains. Those were children treated early and continuously for the genetic disorder, phenylketonuria (PKU).
Typically in PKU there is mutation in the phenylalanine hydroxylase gene (located in the q22–q24.1 region of chromosome 12) such that the phenylalanine hydroxylase enzyme is absent, inactive, or markedly less active (e.g., Güttler et al. 1987). Without that enzyme to metabolize phenylalanine into tyrosine, phenylalanine levels in the blood skyrocket and levels of tyrosine drop. (Some tyrosine is still available directly through diet but the other main route to tyrosine [through hydroxylating phenylalanine] is inoperative.) Without treatment, the extremely high levels of phenylalanine wreak havoc on the brain causing widespread, severe brain damage, and severe mental retardation. Treating PKU by reducing dietary intake of phenylalanine has to be a compromise between the need for protein and the need to minimize phenylalanine intake. That compromise is needed because no food contains phenylalanine in isolation; it is a component of protein; the only way to restrict its intake is to restrict the intake of protein. For many years, medical guidelines had maintained that if phenylalanine levels in blood did not go over 600 μmol/L (10 mg/dL), children with PKU were under adequate treatment. Certainly their IQs were in the normal range and they showed no signs of gross brain damage. However, there were more and more reports that these children were still showing some cognitive deficits, deficits very much like those found with prefrontal cortex damage or dysfunction (Pennington et al. 1985; Faust et al. 1986; Welsh et al. 1990). Those reports, though, were not affecting medical practice. In part, no one could imagine a mechanism that would produce what the researchers were reporting—a selective deficit limited to prefrontal cortex cognitive functions.
Ah, but none of the physicians and researchers in inborn errors of metabolism working on PKU had been in the laboratory of Patricia Goldman-Rakic in Yale’s Stirling Hall of Medicine, where just one floor below was the laboratory of Robert Roth. Had they spent time in Stirling they might have realized that Bob Roth’s work in neuropharmacology provided exactly the mechanism needed to account for the selective prefrontal cognitive deficits researchers were reporting in treated PKU children. Restricting the intake of phenylalanine reduces the imbalance between the levels of phenylalanine and tyrosine in the bloodstream, but does not normalize them. Phenylalanine levels are still somewhat too high and tyrosine levels are still too low; it’s just that the imbalance is now moderate instead of enormous. Phenylalanine and tyrosine compete to cross the blood–brain barrier and the protein carriers have a higher affinity for phenylalanine than for tyrosine (Oldendorf 1973; Pardridge 1977; Pardridge and Oldendorf 1977; Miller et al. 1985). If the ratio of phenylalanine to tyrosine is modestly elevated in the bloodstream, the upshot will be a modest decrease in the amount of tyrosine reaching the brain. Most brain regions, such as the striatum, are insensitive to small decreases in the amount of the raw material (tyrosine) from which dopamine is made (Cooper et al. 2002). However, the dopamine projection to prefrontal cortex is unusual. The dopamine neurons that project to prefrontal cortex have a higher baseline rate of firing and a higher rate of dopamine turnover, making the prefrontal dopamine system exquisitely sensitive to small changes in the level of available tyrosine, changes too small to affect the dopamine system elsewhere, such as in the striatum (Thierry et al. 1976; Bannon et al. 1981; Bradberry et al. 1989; Tam et al. 1990). These special properties of the dopamine neurons that project to prefrontal cortex provide a mechanism by which children treated for PKU might show deficits limited to prefrontal cortex. (The dopamine neurons innervating most of the striatum, such as the caudate and putamen, originate in the substantia nigra; Cooper et al. 2002. The dopamine neurons that innervate prefrontal cortex are located in the ventral tegmental area [VTA]. Dopamine neurons from the VTA also innervate the nucleus accumbens, but even in the VTA there is some segregation between the prefrontal dopamine innervation and the accumbens dopamine innervation (Sesack and Carr 2002). Thus, although both the striatum and prefrontal cortex receive dopaminergic innervation, that input comes from different neurons that have different properties).
Colleagues and I confirmed the selective reduction in dopamine and dopamine metabolites in prefrontal cortex in 2 animal models of PKU, demonstrating that those reductions produced deficits in cognitive abilities dependent on prefrontal cortex, and that those cognitive deficits were closely linked to the degree of dopamine reduction in prefrontal cortex (Diamond et al. 1994). The same was not true for norepinephrine or serotonin or for other brain regions such as the caudate-putamen or the nucleus accumbens. We also studied children with PKU, testing infants every month, toddlers every 3 months, and young children every 6 months, for 4 years, along with their siblings, matched controls, and children from the general population (Diamond et al. 1997; Diamond 2001). PKU children whose phenylalanine levels were 360–600 μmol/L (6–10 mg/dL) were impaired on all 6 tasks that required working memory plus inhibition dependent on dorsolateral prefrontal cortex. That was evident in every age range (infants, toddlers, and young children) and regardless of who we compared them with (other children with PKU with lower phenylalanine levels, their own siblings, matched controls, or children from the general population). At each age range, they performed worse than each of the comparison groups on the tasks that required working memory plus inhibition (see Fig. 1 for examples). There was also a direct, inverse relation between their plasma phenylalanine levels and their performance on these working memory + inhibition tasks; the higher their current phenylalanine levels, the worse their performance on each of the 6 tasks. These same children performed fine on all 10 control tasks, most of which required the functions of posterior parietal cortex or the medial temporal lobe. Thus, their cognitive deficits were limited to the functions dependent on prefrontal cortex, though the entire brain was receiving a modest reduction in tyrosine. This work, building on that of others, changed the national medical guidelines for the treatment of PKU throughout North America and most of Europe. The guidelines now are that phenylalanine levels should be kept between 120 and 360 μmol/L (2–6 mg/dL). Subsequent research has shown that this change has improved children’s lives (Stemerdink et al. 1999; Huijbregts et al. 2002).
Figure 1.
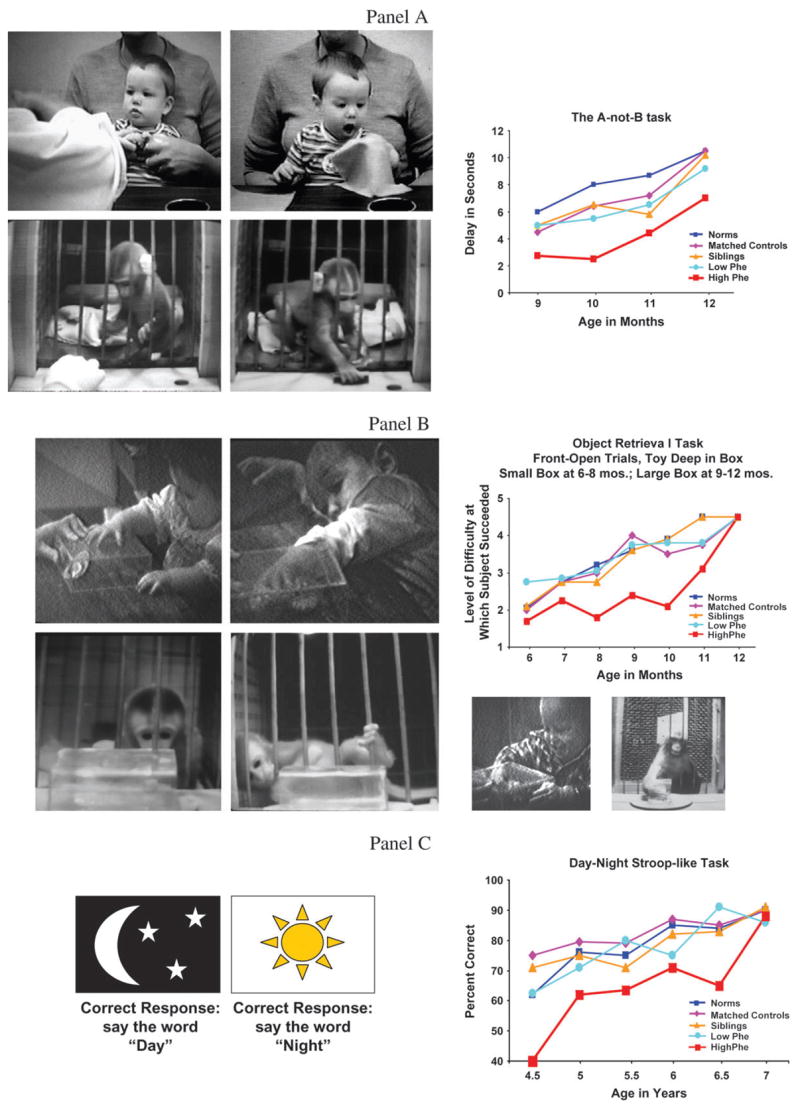
Examples of tasks requiring working memory and inhibition, dependent on prefrontal cortex, on which children with PKU whose phenylalanine levels are 360–600 μmol/L show deficits. Top Panel (Panel A): The A-not-B task. When the side on which the reward is hidden changes, human infants, infant monkeys, and monkeys with lesions of dorsolateral prefrontal cortex (not shown) often err by reaching back to where they were previously rewarded (Column 2), even though they had been watching the reward being placed in the new location only moments earlier (Column 1). Infants with PKU with “higher” Phe levels (360–600 μmol/L) perform significantly worse than all other groups—other PKU children with lower Phe levels, their own siblings, matched controls, or children from the general population. Middle Panel (Panel B): The object retrieval task. The youngest human and rhesus infants try to reach directly for the visible reward through the side of the box they happen to be looking (Column 1). They try persistently, but only reach through the side they are looking. Rhesus infants, slightly older human infants (and monkeys with lesions of dorsolateral prefrontal cortex, not shown here) often take elaborate steps to move themselves or the transparent box to change which side they are looking through, but they still only reach through the side they are looking (Column 2). Older human infants or monkeys can readily reach through the open side of the box while looking through a closed side (see photos below graph). Infants with PKU with higher Phe levels performed significantly worse than all other groups—other PKU children with lower Phe levels, their own siblings, matched controls, or children from the general population. Although the graphs may make it look like the children with higher Phe levels eventually catch up, that appearance is only an artifact of using the same task over a large age range. When tasks for the next age range are introduced, the children with higher Phe levels once again perform worse than the other groups on those tasks requiring working memory and inhibition, dependent on prefrontal cortex. Bottom Panel (Panel C): The Day–Night Stroop-like task (Gerstadt et al. 1994; Diamond et al. 2002). This task requires holding 2 rules in mind and inhibiting saying what the stimuli really represent. Indeed, the opposite of what they represent (that represented by the other stimulus) is the correct response. This task is appropriate for children 4–7 years age. Again, children with PKU with higher Phe levels (though much lower than they would be without treatment and considered at the time to be adequately treated) performed significantly worse than all other groups—other PKU children with lower Phe levels, their own siblings, matched controls, or children from the general population.
Visual Function in Children Treated Early and Consistently for PKU
It so happens that there is another set of dopamine neurons that share all the same properties as the dopamine neurons that project to prefrontal cortex. Those are the dopamine neurons in the retina. They, too, have rapid baseline firing and dopamine turnover rates, and are unusually sensitive to the level of available tyrosine (Fernstrom and Fernstrom 1988; Iuvone et al. 1989). Moreover, the competition between phenylalanine and tyrosine at the blood-retinal barrier is comparable to that at the blood–brain barrier (Rapoport 1976; Tornquist and Alm 1986). To be consistent, I had to predict that visual functions sensitive to the level of dopamine in the retina should be impaired in PKU children with plasma phenylalanine levels of 360–600 μmol/L (6–10 mg/dL). Retinal dopamine appears to be important for contrast sensitivity (the ability to detect differences in luminance or brightness of adjacent regions in a visual display). Persons with poorer contrast sensitivity require a greater difference in luminance (e.g., lines of darker gray against a background of fainter gray) if they are to detect the lines than people with better contrast sensitivity. Certainly, patients with Parkinson’s disease, who have greatly reduced dopamine levels, have impaired sensitivity to contrast (Bodis-Skrandies and Gottlob 1986; Bodis-Wollner et al. 1987; Bodis-Wollner 1990). We predicted that children with PKU children whose plasma phenylalanine levels were 360–600 μmol/L (6–10 mg/dL) would also be impaired. Indeed, they were, and at every level of contrast and every spatial frequency (Diamond and Herzberg 1996).
We had found converging evidence from 2 very different domains, vision and cognition, in support of our hypothesis about the mechanism causing cognitive deficits in PKU children when their phenylalanine levels were maintained at what had been thought to be safe levels (3–5 times normal; 360–600 μmol/L). We should have been opening the champagne. One discrepancy troubled me, however. The prefrontal cognitive deficits were closely related to children’s current levels of phenylalanine. The visual deficits were not. The deficit in contrast sensitivity was closely related to what the children’s phenylalanine levels had been during the first month of life. By the time we studied contrast sensitivity, we knew what range of phenylalanine levels produced a deficit and so we only sampled from that range. Having a far more truncated range of current phenylalanine levels could easily have accounted for the failure to find a relation between contrast sensitivity and current phenylalanine levels. However, a child born with PKU is usually not started on treatment for the disorder until about 10–14 days of age. The visual system is maturing very rapidly during the days right after birth (e.g., Slater and Johnson 1998; Cavallini et al. 2002). Perhaps the excessively high levels of phenylalanine reaching the brain during those first days after birth impair the visual system. To test that hypothesis we brought in pairs of siblings, both of whom had PKU, as well as children from the general population. The importance of the sibling pairs was that although PKU in the first child born with the disorder in a family was not detected until the heel prick test after birth, amniocentesis was performed for all later-born children and so it was known if any of those children had PKU before birth. The first-born children started dietary treatment on average at 11 days of age, whereas the later-born children started dietary treatment on average at 3 days of age. Although contrast sensitivity generally improves with age, and by definition all of our first-born PKU children were older then their sibling with PKU, the first PKU sibling in a family had poorer contrast sensitivity at low levels of contrast than did his or her younger PKU sibling and than did children from the general population. The children whose brains were exposed to massive levels of phenylalanine for the first 10 days of life showed no deficits in visual acuity or in determining form from texture, but they were impaired in contrast sensitivity at very low levels of contrast and in determining form from motion (Amso et al. in preparation). These deficits were evident over 10 years later when we tested the children. This is in striking parallel to the findings of Daphne Maurer and Terri Lewis who have tested children who received very degraded visual input for only the first few weeks after birth (children born with congenital cataracts who had surgery to correct their eyesight within the first month of life; Maurer and Lewis 2001). Based on a verbal report of our PKU sibling results, though the paper is not yet out, the National Institutes of Health Consensus Conference on PKU recommended that treatment for PKU should be initiated earlier than was currently done, and no later than 7–10 days after birth. Evidently, there is still some role for current phenylalanine levels in the contrast sensitivity deficit of PKU children, as high phenylalanine levels during the first 10 days of age were related to deficits only at very low contrast. Both neonatal and current phenylalanine levels appear to matter.
The COMT Gene in Children and the Elderly, and Gender Differences
Scientific results are rarely perfectly neat and clean. The summary in the Historical Background section above on cognitive deficits in children treated early and continuously for PKU was accurate as far as it went, but it omitted a finding for which we had no explanation at the time. In addition to the cognitive tasks mentioned in that section, we also administered 3 tasks that tax working memory and are dependent on dorsolateral prefrontal cortex. Children with PKU who had phenylalanine levels of 360–600 μmol/L were not impaired on those tasks (2 self-ordered pointing tasks and one temporal order memory task). We had expected that performance on all tasks dependent on dorsolateral prefrontal cortex would be impaired in those children. Converging evidence from patients with frontal lobe damage (Petrides and Milner 1982), neuroimaging of healthy subjects performing these tasks (Petrides et al. 1993), and animal models (Petrides 1995) convincingly showed that dorsolateral prefrontal cortex is critical for success on these tasks, yet we had found no deficit. Shortly thereafter, Trevor Robbins, Angela Roberts, and colleagues (Collins et al. 1998) showed in monkeys that although lesions of prefrontal cortex impair performance on self-ordered pointing, depleting prefrontal cortex of dopamine does not. That was consistent with our PKU finding. We predicted prefrontal cognitive deficits produced by reduced dopamine in prefrontal cortex due to a small reduction in available tyrosine. For some reason, not fully understood even to this day, although self-ordering pointing appears to recruit prefrontal cortex, it is not sensitive to reductions in dopamine levels in prefrontal cortex. Further exploration of this took us into molecular genetics.
The dopamine system in prefrontal cortex differs from the dopamine system in other brain regions in yet another away (besides the higher baseline firing and dopamine turnover rates mentioned earlier), in that prefrontal cortex contains significantly less dopamine transporter protein than other brain regions such as the striatum (Sesack et al. 1998 [rat]; Sanchez-Gonzalez and Cavada 2003 [monkey]). Dopamine transporter provides the best mechanism for clearing released dopamine from extracellular space. Its relative dearth in prefrontal cortex makes prefrontal cortex more dependent on secondary mechanisms, such as the catechol-O-methyltransferase (COMT) enzyme, for terminating the action of released dopamine. The COMT enzyme accounts for more than 60% of the dopamine degradation in prefrontal cortex, but for less than 15% of the dopamine degradation in the striatum (Karoum et al. 1994). Administering Tolcapone (a selective COMT inhibitor) to Parkinson’s patients, improves their performance on tasks that require working memory plus inhibition dependent on prefrontal cortex (Gasparini et al. 1997), but does not improve Parkinsonian motor symptoms associated with striatal function (Chong et al. 2000).
Because prefrontal cortex is more dependent on the COMT enzyme than other neural regions, variations in the COMT gene (located within the q11 band of human chromosome 22), which codes for the COMT enzyme, should disproportionately affect prefrontal cortex, leaving other brain regions (such as the striatum) relatively unaffected. A common variation in the COMT gene (a single base pair substitution of guanine for adenine [CGTG for CATG]) translates into a substitution of methionine for valine at one location on the gene (codon 108/158: AGVKD vs. AGMKD; Lachman et al. 1996). The methionine variant results in a more sluggish COMT enzyme; it methylates dopamine at only about 25% the rate of the COMT enzyme containing valine (Lotta et al. 1995; Lachman et al. 1996). Thus, the methionine variant of the COMT gene leaves dopamine around longer in prefrontal cortex. It has been shown in adults to result in better performance on prefrontal cognitive tasks requiring working memory plus inhibition (Egan et al. 2001; Malhotra et al. 2002) and to result in more efficient prefrontal functioning when cognitive performance is held constant (Egan et al. 2001).
We predicted that because the COMT enzyme affects dopamine levels in prefrontal cortex, variations in the COMT gene should affect performance on the kinds of cognitive tasks on which PKU children with phenylalanine levels of 360–600 μmol/L were impaired (tasks that tax both working memory and inhibition) but should not affect performance on tasks on which PKU children did fine (such as self-ordered pointing) even though self-ordered pointing requires prefrontal cortex. We predicted that based on of our hypothesis that PKU children with phenylalanine levels of 360–600 μmol/L have selective reductions in dopamine in prefrontal cortex, a prediction fortified by the findings of Collins et al. (1998) that depleting prefrontal cortex of dopamine impairs monkeys’ performance on tasks requiring working memory and inhibition (the delayed-response task) but does not impair performance on self-ordered pointing (exactly the pattern we had found in treated PKU children). Therefore, we predicted that variations in the level of dopamine in prefrontal cortex caused by variations in the COMT enzyme should similarly not affect self-ordered pointing performance, but should affect performance on tasks requiring working memory plus inhibition.
We conducted the first study of the effect of variations in the COMT enzyme on children’s performance (Diamond et al. 2004). Sure enough, children homozygous for the methionine version of the COMT gene performed better on our Dots-Mixed tasks (Davidson et al. 2006), which requires working memory plus inhibition, but no better on self-ordered pointing, recall memory (dependent on the medial temporal lobe), or mental rotation (dependent on posterior parietal cortex; see Fig. 2).
Figure 2.
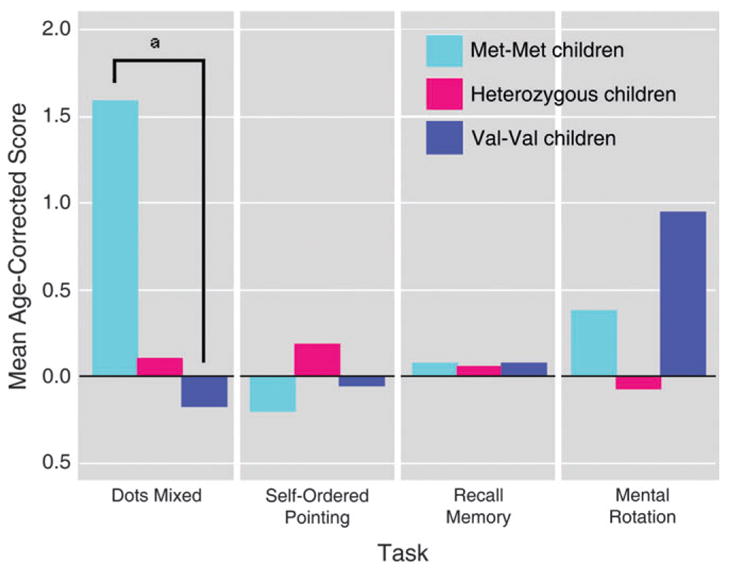
Performance by COMT genotype of healthy normal children on cognitive tasks. Children homozygous for the methionine variant of the COMT gene performed significantly better than children homozygous for the valine version of the gene on the dots-mixed task, which requires holding 2 higher-order rules in mind and switching between inhibiting a prepotent response and making it, and which depends on prefrontal cortex. All groups performed comparably on self-ordered pointing, which also depends on prefrontal cortex, but which is not sensitive to the level of dopamine in prefrontal cortex. No difference between Met–Met and Val–Val children was found on the 6-, 8-, or 12-item self-ordered pointing trials or on all combined. All groups also performed comparably on the 2 control tasks: recall memory and mental rotation. Standard deviations were as follows for the Met–Met, Val–Met, and Val–Val genotypes, respectively: dots-mixed task—2.01, 3.37, and 2.74; self-ordered pointing task—0.99, 1.06, and 1.05; recall memory task—0.07, 0.15, and 0.13; mental rotation—2.98, 2.44, and 1.92. Reprinted from Diamond et al. (2004), with permission.
Never one to leave well enough alone, I asked, “How come Dan Weinberger’s group studying adults, and my group studying children, did not find larger effects of variations in the COMT gene? Sure, our effects were significant, but why weren’t they larger?” The optimum amount of dopamine in prefrontal cortex is an intermediate amount; too much or too little dopamine results in poorer performance on cognitive tasks dependent on prefrontal cortex (Zahrt et al. 1997; Mattay et al. 2003). I reasoned that perhaps in some children and young adults having the methionine version of the COMT gene might result in pushing prefrontal dopamine levels past the optimum point; perhaps it resulted in too much dopamine in prefrontal cortex for some people. That would explain why the results at a group level were significant but not larger.
Levels of dopamine decrease in the brain with aging (e.g., Suhara et al. 1991; Volkow et al. 1996, 1998; Wong et al. 1997). Therefore, I predicted that the methionine variant of the COMT gene would be more universally beneficial among older adults, as it would be more likely to help a larger percentage of them to reach the optimal point on the dopamine curve, rather than going past it. I hypothesized that the effect of COMT genotype on prefrontally-dependent cognitive functions should be much larger among the elderly than among children or young adults. I asked Art Kramer, an expert on cognitive aging, to collaborate on the study with me.
When Art’s postdoctoral fellow, Paige Scaile, reported the results to me, I was dumbfounded. No effect of COMT genotype was evident in the results of elderly adults. Amy Arnsten reported that that was consistent with her results with aged monkeys; moderately increasing dopamine levels helped young monkeys but not the old monkeys (personal commun.). How was one to make sense of this?
The percentage of females in the population greatly increases with advancing age. Indeed, all of Arnsten’s aged monkeys were female. There is also considerable evidence that males perform better, or no worse, if slightly stressed but females perform worse under stress, even slight stress (Shors and Miesegaes 2002; Shors and Leuner 2003; Shansky et al. 2004). We know that the dopamine system in prefrontal cortex is acutely sensitive to stress; stress increases prefrontal dopamine levels (Thierry et al. 1976; Reinhard et al. 1982; Roth et al. 1988; Deutch and Roth 1990; Arnsten and Goldman-Rakic 1998; Arnsten 1999, 2000). Might it be possible that females have higher (more nearly optimal) baseline levels of dopamine whereas males have slightly too low baseline levels of dopamine? Hence, males’ performance improves when dopamine levels are slightly increased but females’ performance does not. This would be consistent with males being more susceptible to disorders of too little dopamine (e.g., attention deficit hyperactivity disorder [ADHD]) and females being more susceptible to disorders of too much dopamine (e.g., depression). Certainly, there is evidence that COMT activity is reduced epigenetically by estrogen and that COMT enzyme activity is roughly 30% lower in females than males (Cohn and Axelrod 1971; Boudikova et al. 1990). If this line of reasoning were correct, then having a still more sluggish COMT enzyme would not be beneficial to women. Indeed, whereas the more sluggish methionine-containing COMT enzyme might be more beneficial to men; the faster-acting valine version of the COMT enzyme might be more beneficial to women. I contacted Art and Paige, “Quick. Analyze your results separately for men and women.” Everything fell into place. Males homozygous for the methionine version of the COMT gene performed much better on the Wisconsin Card Sort; whereas females homozygous for the valine version of the COMT gene showed superior performance on the task (Scaile et al., in preparation). We would predict that this gender difference in the effects of COMT genotype on prefrontally-dependent cognitive functions should be even greater in young adults, because young women have higher circulating levels of estrogen, and estrogen facilitates dopamine tone and downregulates COMT enzymatic activity (Ho et al. 2006).
The methionine and valine versions of the COMT gene occur in almost equal frequencies in Europe and North America (Floderus and Wetterberg 1981; Spielman and Weinshilboum 1981; Siervogel et al. 1984; Palmatier et al. 1999). Therefore, both versions of the gene must confer certain advantages. The methionine version of COMT gene is associated with better executive function (better performance on cognitive skills that rely on prefrontal cortex) at least in males, but it is also associated with more sensitively to stress, higher anxiety, and higher reactivity to lower levels of stress (Zubieta et al. 2003). If females have slightly higher baseline levels of dopamine, then they might be expected to have a slight advantage in executive functions but they might also be expected to be somewhat more reactive to stress.
Two other Genes that Affect Dopamine, DAT1 and DRD4, and Different Variants of ADHD
As noted above, prefrontal cortex differs from the striatum in that prefrontal cortex has relatively little dopamine transporter, whereas the striatum has a rich supply. Given that, one would expect polymorphisms in the gene that codes for the dopamine transporter, the DAT1 gene, to have a greater effect on the striatum than on prefrontal cortex and, indeed, they do (Durston et al. 2005). Yet another difference between the dopamine systems in prefrontal cortex and the striatum is that the dopamine receptor subtype, DRD4, is present in humans in prefrontal cortex but not in the striatum (Meador-Woodruff et al. 1996). Hence one would expect that polymorphisms in the DRD4 gene would have a greater effect on prefrontal cortex than on the striatum, and indeed they do (Durston et al. 2005).
There is much evidence of abnormalities in the striatum in ADHD that includes hyperactivity (e.g., ADHD of the combined type; Teicher et al. 1996; Filipek et al. 1997; Vaidya et al. 1998; Schrimsher et al. 2002; Durston et al. 2003). For example, functional neuroimaging studies report less striatal activity in children with ADHD that includes hyperactivity while they are performing response-inhibition tasks compared with age-matched controls (Vaidya et al. 1998, 2005). I have hypothesized that although ADHD that includes hyperactivity is caused by a primary problem in the striatum and in the prefrontal–striatal system more generally, that ADHD of the inattentive type (where there is truly no hyperactivity) is a primary problem in prefrontal cortex and in the prefrontal–parietal circuit more generally (Diamond 2005). It follows, if I am correct, that polymorphisms in the DAT1 gene should be more closely associated with ADHD that includes hyperactivity and that polymorphisms of the DRD4 gene should be more closely associated with inattention. Indeed, although levels of hyperactive-impulsive symptoms are correlated with the number of DAT1 high-risk alleles, levels of inattentive symptoms are not (Waldman et al. 1998). DAT binding specifically in the striatum has been found to be related to motor hyperactivity but not to inattentive symptoms (Jucaite et al. 2005). On the other hand, sustained attention and other executive functions have been found to be worse in children with the 7-repeat-allele polymorphism of the DRD4 gene (Auerbach et al. 2001).
A role for polymorphisms of the DAT1 gene in ADHD that includes hyperactivity is consistent with the efficacy of methylphenidate in treating those forms of ADHD, as methylphenidate acts directly on DAT function (Shenker 1992; Seeman and Madras 1998; Volkow et al. 1998; Dresel et al. 2000). Indeed, most children (perhaps as many as 90%) with ADHD that includes hyperactivity respond positively to methylphenidate (Ritalin) and over two-thirds respond positively to methylphenidate in moderate to high doses (Barkley et al. 1991; Barkley 2001; Milich et al. 2001; Weiss et al. 2003).
On the other hand, a significant percentage of children with ADHD without hyperactivity are not helped by methylphenidate and those who are helped often do best at low doses (Barkley et al. 1991; Barkley 2001; Milich et al. 2001; Weiss et al. 2003). Many of the latter individuals are helped by amphetamines, such as Adderall. There is considerable overlap in the mechanisms of action of methylphenidate and amphetamines, but there is a significant difference. Although both inhibit reuptake of dopamine and norepinephrine, only amphetamines also promotes release of those neurotransmitters. Recent research suggests that low doses of methylphenidate (the dosages likely to be efficacious in treating ADHD without hyperactivity) may also release norepinephrine (Ishimatsu et al. 2002). Although ADHD children with hyperactivity appear to have a deficit in inhibitory control and often cannot sit still, ADHD children without hyperactivity appear to more “under-powered” and hypoactive. They do better when a risk or challenge gets their adrenaline pumping and although they, too, appear distractible, it may be more that as their interest wanes in what they are supposed to be doing they go looking for distraction, rather than that they are unable to inhibit responses to distraction if properly motivated (Diamond 2005).
There is also some evidence for differential responsivity to nicotine. There are marked similarities in the neurobiological and psychological effects of nicotine and methylphenidate (e.g., Pomerleau 1997). In particular, nicotine may act directly on the dopamine transporter in the same way as does methylphenidate (Krause et al. 2000, 2002, 2003). It has been hypothesized that individuals with ADHD that includes hyperactivity who are not taking stimulant medication may try to self-medicate by smoking. Certainly, unmedicated adolescents with ADHD that includes hyperactivity smoke far more than do their medicated ADHD peers and their non-ADHD peers (Whalen et al. 2003). Krause et al. (2003) report that individuals with ADHD that includes hyperactivity are far more likely to smoke than are individuals with ADHD of the inattentive type: “It was striking how many of the 20–40 years old patients in our group, who had shown symptoms of hyperactivity and impulsivity in childhood, were smokers: 9 smoked and only 3 were nonsmokers. The opposite was shown in the patients with only inattentive symptoms throughout their whole life: only 2 smoked, 7 were nonsmokers” (p. 610–611). (It should be noted, however, that Tercyak et al. (2002) report the opposite.)
A role for polymorphisms of the DAT1 gene in ADHD that includes hyperactivity is consistent with the centrality of the striatum in ADHD that includes hyperactivity as DAT plays a particularly important role in the striatum and is consistent with the efficacy of methylphenidate in treating those forms of ADHD (and perhaps with nicotine as a self-medication) because methylphenidate and nicotine act directly on DAT function. The predictions outlined here concerning ADHD with and without hyperactivity, as well as additional predictions, are summarized in Table 1 and laid out in Diamond (2005).
Table 1.
Comparison of the Characteristics of ADHD (Attention Deficity Hyperactivity Disorder) and ADD (Attention Deficit Disorder)
| ADHD that includes hyperactivity (i.e., children with ADHD-combined-type and ADHD-hyperactive) | ADD (i.e., children with ADHD-inattentive excluding those with significant hyperactivity even if they fail to meet criterion on 7 hyperactivity items [those children are really ADHD-combined-type]) |
|---|---|
| • Hyperactive, always on the go, impulsive | • A significant subset are hypoactive, sluggish, with quite slow response speeds |
| • Primary deficit in response inhibition | • Primary deficit in working memory—especially prominent in auditory processing because of the demands it places on working memory |
| • Often insufficiently self-conscious | • Tend to be overly self-conscious |
| • Social problems because too assertive and impulsive—butt in, take things belonging to others, fail to wait their turn, & act without first considering the feelings of others | • Social problems because too passive, shy, or withdrawn. |
| • Tend to be extroverted | • More likely to be introverted |
| • Externalizing behaviors, such as conduct disorder, aggressivity, disruptive behavior, and even oppositional defiant disorder are far more commonly comorbid with ADHD than with ADD | • Internalizing disorders, such as anxiety or depression, are somewhat more common in children with ADD than those with ADHD. ADD children tend to socially isolated or withdrawn.
Reading & language deficits, & problems with mental mathematical calculations are more commonly comorbid with ADD than with ADHD |
| • Respond positively to methylphenidate (Ritalin) | • A significant percentage are not helped by methylphenidate |
| • Most respond positively to methylphenidate in moderate to high doses | • Those who are helped by methylphenidate often do best at low doses |
| • There are marked similarities in the neurobiological and psychological effects of nicotine and methylphenidate. Those with ADHD are more likely to smoke than are those with ADD. | |
| • Methylphenidate reduces catecholamine reuptake. Addressing reuptake appears to be sufficient to help most individuals with ADHD. | • A significant subset are helped by amphetamines rather than methylphenidate. Amphetamines both reduce reuptake and increase release of catecholamines. A marked deficit in the release of dopamine (DA) and norepinephrine (NE) might cause sluggishness and under-arousal. |
| • People with ADD are not so much easily distracted as easily bored. Their problem lies more in motivation (under-arousal) than it does in inhibition. | |
| • Challenge or risk, something to literally get their adrenaline pumping, can be key to keeping their attention and to eliciting optimum performance. Individuals with ADD, though typically shy, may engage in risk-taking and thrill-seeking activities as ways to experience a level of engagement they have difficulty sustaining in their daily lives. | |
| • Converging evidence for a primary disturbance in the striatum. | • A primary disturbance in prefrontal cortex is implicated. |
| • The primary neural circuit affected may be a frontal-striatal one. | • The primary neural circuit affected may be a frontal-parietal one. |
| • Polymorphisms in the DAT1 gene are associated ADHD. This is consistent with the centrality of the striatum in ADHD because DAT plays a particularly important role there. It is also consistent with the efficacy of methylphenidate because DAT is the primary target for the clinical action of methylphenidate. | • The 7-repeat allele polymorphism of the DRD4 gene is more strongly linked to ADD then to ADHD. That is consistent with the centrality of prefrontal cortex in ADD because the DRD4 receptor is present in prefrontal cortex but not in the striatum in humans. |
Note: DRD4, D4 dopamine receptor.
It was Patricia Goldman-Rakic’s groundbreaking work on prefrontal cortex, on the cognitive functions it subserves and on the role of dopamine in prefrontal cortex, that paved the way for all the work discussed above.
Acknowledgments
The author gratefully acknowledges grant support from the National Institute on Drug Abuse (NIDA R01 #DA19685) and from the Canada Research Chairs Program during the writing of this paper, and the support of the National Science Foundation (NSF Doctoral Dissertation Grant #BNS 8013-4471978 and NSF Graduate Fellowship), Danforth Foundation (a graduate Fellowship), Sloan Foundation (a postdoctoral fellowship), March of Dimes (Social and Behavioral Sciences Research Grants #12-253 & #12-0554), National Institute of Mental Health (NIMH Postdoctoral Fellowship #F32 MH09007 & NIMH R01 #MH41842), and National Institute of Child Health and Human Development (NICHD R01 #HD34346) for funds that made possible the research reported here.
Funding to pay the Open Access publication charges for this article was provided by NIH R01 grant #DA19685-16A2.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.0/uk/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Conflict of Interest: None declared.
References
- Alexander GE, Goldman PS. Functional development of the dorsolateral prefrontal cortex: an analysis utilizing reversible cryogenic depression. Brain Res. 1978;143:233–249. doi: 10.1016/0006-8993(78)90566-8. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Development of the cerebral cortex: XIV. Stress impairs prefrontal cortical function. J Am Acad Child Adolesc Psychiatry. 1999;38:220–222. doi: 10.1097/00004583-199902000-00024. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT. Stress impairs prefrontal cortical function in rats and monkeys: role of dopamine D1 and norepinephrine a-1 receptor mechanisms. Prog Brain Res. 2000;126:183–192. doi: 10.1016/S0079-6123(00)26014-7. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Goldman-Rakic PS. Noise stress impairs prefrontal cortical cognitive function in monkeys: evidence for a hyperdopaminergic mechanism. Arch Gen Psychiatry. 1998;55:362–368. doi: 10.1001/archpsyc.55.4.362. [DOI] [PubMed] [Google Scholar]
- Auerbach JG, Benjamin J, Faroy M, Geller V, Ebstein R. DRD4 related to infant attention and information processing: a developmental link to ADHD? Psychiatr Genet. 2001;11:31–35. doi: 10.1097/00041444-200103000-00006. [DOI] [PubMed] [Google Scholar]
- Bannon MJ, Bunney EB, Roth RH. Mesocortical dopamine neurons: rapid transmitter turnover compared to other brain catecholamine systems. Brain Res. 1981;218:376–382. [PubMed] [Google Scholar]
- Barkley RA. The inattentive type of ADHD as a distinct disorder: what remains to be done. Clin Psychol Sci Pract. 2001;8:489–493. [Google Scholar]
- Barkley RA, DuPaul GJ, McMurray MB. Attention deficit disorder with and without hyperactivity: clinical response to three dose levels of methylphenidate. Pediatrics. 1991;87:519–531. [PubMed] [Google Scholar]
- Bodis-Wollner I. Visual deficits related to dopamine deficiency in experimental animals and Parkinson’s disease patients. Trends Neural Sci. 1990;13:296–302. doi: 10.1016/0166-2236(90)90113-o. [DOI] [PubMed] [Google Scholar]
- Bodis-Wollner I, Marx MS, Mitra S, Bobak P, Mylin L, Yahr M. Visual dysfunction in Parkinson’s disease. Loss in spatiotemporal contrast sensitivity. Brain. 1987;110:1675–1698. doi: 10.1093/brain/110.6.1675. [DOI] [PubMed] [Google Scholar]
- Boudikova B, Szumlanski C, Maidak B, Weinshilboum R. Human liver catechol-O-methyltransferase pharmacogenetics. Clin Pharmacol Ther. 1990;48:381–389. doi: 10.1038/clpt.1990.166. [DOI] [PubMed] [Google Scholar]
- Bradberry CW, Karasic DH, Deutsch AY, Roth RH. Regionally-specific alterations in mesotelencephalic dopamine synthesis in diabetic rats: associations with precursor tyrosine. J Neural Transm. 1989;78:221–229. doi: 10.1007/BF01249231. [DOI] [PubMed] [Google Scholar]
- Brown RM, Crane AM, Goldman PS. Regional distribution of monoamines in the cerebral cortex and subcortical structures of the rhesus monkey: concentrations and in vivo synthesis rates. Brain Res. 1979;168:133–150. doi: 10.1016/0006-8993(79)90132-x. [DOI] [PubMed] [Google Scholar]
- Brown RM, Goldman PS. Catecholamines in neocortex of rhesus monkeys: regional distribution and ontogenetic development. Brain Res. 1977;124:576–580. doi: 10.1016/0006-8993(77)90960-x. [DOI] [PubMed] [Google Scholar]
- Brozoski TJ, Brown RM, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science. 1979;205:929–932. doi: 10.1126/science.112679. [DOI] [PubMed] [Google Scholar]
- Cavallini A, Fazzi E, Viviani V, Astori MG, Zaviero S, Bianchi PE, Lanzi G. Visual acuity in the first two years of life in healthy term newborns: an experience with the Teller Acuity Cards. Funct Neurol. 2002;17:87–92. [PubMed] [Google Scholar]
- Chong DJ, Suchowersky O, Szumlanski C, Weinshilboum RM, Brant R, Campbell NR. The relationship between COMT genotype and the clinical effectiveness of tolcapone, a COMT inhibitor, in patients with Parkinson’s disease. Clin Neuropharmacol. 2000;23:143–148. doi: 10.1097/00002826-200005000-00003. [DOI] [PubMed] [Google Scholar]
- Cohn CK, Axelrod J. The effect of estradiol on catechol-O-methyltransferase activity in rat liver. J Life Sci. 1971;10:1351–1354. doi: 10.1016/0024-3205(71)90335-3. [DOI] [PubMed] [Google Scholar]
- Collins P, Roberts AC, Dias R, Everitt BJ, Robbins TW. Perseveration and strategy in a novel spatial self-ordered task for nonhuman primates: effect of excitotoxic lesions and dopamine depletions of the prefrontal cortex. J Cogn Neurosci. 1998;10:332–354. doi: 10.1162/089892998562771. [DOI] [PubMed] [Google Scholar]
- Cooper JR, Roth RH, Bloom FE. The biochemical basis of neuropharmacology. New York: Oxford University Press; 2002. [Google Scholar]
- Davidson MC, Amso D, Anderson LC, Diamond A. Development of cognitive control and executive functions from 4-13 years: evidence from manipulations of memory, inhibition, and task switching. Neuropsychologia. 2006;44:2037–2078. doi: 10.1016/j.neuropsychologia.2006.02.006. (Mar 29; [Epub ahead of print]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutch AY, Roth RH. The determinants of stress-induced activation of the prefrontal cortical dopamine system. Prog Brain Res. 1990;85:367–403. doi: 10.1016/s0079-6123(08)62691-6. [DOI] [PubMed] [Google Scholar]
- Diamond A. Frontal lobe involvement in cognitive changes during the first year of life. In: Gibson KR, Petersen AC, editors. Brain maturation and cognitive development: comparative and cross-cultural perspectives. New York: Aldine de Gruyter; 1991a. pp. 127–180. [Google Scholar]
- Diamond A. Neuropsychological insights into the meaning of object concept development. In: Carey S, Gelman R, editors. The epigenesis of mind: essays on biology and knowledge. Hillsdale (NJ): Lawrence Erlbaum Associates; 1991b. pp. 67–110. [Google Scholar]
- Diamond A. A model system for studying the role of dopamine in prefrontal cortex during early development in humans. In: Nelson C, Luciana M, editors. Handbook of developmental cognitive neuroscience. Cambridge (MA): MIT Press; 2001. pp. 433–472. [Google Scholar]
- Diamond A. ADD (ADHD without hyperactivity), a neurobiologically and behaviorally distinct disorder from ADHD (with hyperactivity) Dev Psychopathol. 2005;17:807–825. doi: 10.1017/S0954579405050388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond A, Briand L, Fossella J, Gehlbach L. Genetic and neurochemical modulation of prefrontal cognitive functions in children. Am J Psychiatry. 2004;161:125–132. doi: 10.1176/appi.ajp.161.1.125. [DOI] [PubMed] [Google Scholar]
- Diamond A, Ciaramitaro V, Donner E, Djali S, Robinson MB. An animal model of early-treated PKU. J Neurosci. 1994;14:3072–3082. doi: 10.1523/JNEUROSCI.14-05-03072.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond A, Goldman-Rakic PS. Evidence for involvement of prefrontal cortex in cognitive changes during the first year of life: comparison of performance of human infant and rhesus monkeys on a detour task with transparent barrier. Abstr Soc Neurosci (Part II) 1985;11:832. [Google Scholar]
- Diamond A, Goldman-Rakic PS. Comparative development in human infants and infant rhesus monkeys of cognitive functions that depend on prefrontal cortex. Abstr Soc Neurosci. 1986;12:742. [Google Scholar]
- Diamond A, Goldman-Rakic PS. Comparison of human infants and rhesus monkeys on Piaget’s A-not-B task: evidence for dependence on dorsolateral prefrontal cortex. Exp Brain Res. 1989;74:24–40. doi: 10.1007/BF00248277. [DOI] [PubMed] [Google Scholar]
- Diamond A, Herzberg C. Impaired sensitivity to visual contrast in children treated early and continuously for PKU. Brain. 1996;119:523–538. doi: 10.1093/brain/119.2.523. [DOI] [PubMed] [Google Scholar]
- Diamond A, Kirkham NZ, Amso D. Conditions under which young children can hold two rules in mind and inhibit a prepotent response. Dev Psychol. 2002;38:352–362. [PubMed] [Google Scholar]
- Diamond A, Prevor M, Callender G, Druin DP. Prefrontal cortex cognitive deficits in children treated early and continuously for PKU. Monogr Soc Res Child Dev. 1997;62:1–207. [PubMed] [Google Scholar]
- Dresel S, Krause J, Krause KH, LaFougere C, Brinkbaumer K, Kung HF, Hahn K, Tatsch K. Attention deficit hyperactivity disorder: binding of [99mTc]TRODAT-1 to the dopamine transporter before and after methylphenidate treatment. Eur J Nucl Med. 2000;27:1518–1524. doi: 10.1007/s002590000330. [DOI] [PubMed] [Google Scholar]
- Durston S, Fossella JA, Casey BJ, Hulshoff Pol HE, Galvan A, Schnack HG, Steenhuis MP, Minderaa RB, Buitelaar JK, Kahn RS, et al. Differential effects of DRD4 and DAT1 genotype on fronto-striatal gray matter volumes in a sample of subjects with attention deficit hyperactivity disorder, their unaffected siblings, and controls. Mol Psychiatry. 2005;10:678–685. doi: 10.1038/sj.mp.4001649. [DOI] [PubMed] [Google Scholar]
- Durston S, Tottenham NT, Thomas KM, Davidson MC, Eigsti IM, Yang Y, Ulug AM, Casey BJ. Differential patterns of striatal activation in young children with and without ADHD. Biol Psychiatry. 2003;53:871–878. doi: 10.1016/s0006-3223(02)01904-2. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, Goldman D, Weinberger DR. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci USA. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust D, Libon D, Pueschel S. Neuropsychological functioning in treated phenylketonuria. Int J Psychiatry Med. 1986;16:169–177. doi: 10.2190/2209-39kw-bekl-w80j. [DOI] [PubMed] [Google Scholar]
- Fernstrom JD, Fernstrom MH. Tyrosine availability and dopamine synthesis in the retina. In: Bodis-Wollner I, Piccolino M, editors. Dopaminergic mechanisms in vision. New York: Alan Liss; 1988. pp. 59–70. [Google Scholar]
- Filipek PA, Semrud-Clikeman M, Steingard RJU, Renshaw PF, Kennedy DN, Biederman J. Volumetric MRI analysis comparing subjects having attention-deficit hyperactivity disorder with normal controls. Neurology. 1997;48:589–601. doi: 10.1212/wnl.48.3.589. [DOI] [PubMed] [Google Scholar]
- Floderus Y, Wetterberg L. The inheritance of human erythrocyte catechol-O-methyltransferase activity. Clin Genet. 1981;19:392–395. doi: 10.1111/j.1399-0004.1981.tb00732.x. [DOI] [PubMed] [Google Scholar]
- Gasparini M, Fabrizio E, Bonifati V, Meco G. Cognitive improvement during Tolcapone treatment in Parkinson’s disease. J Neural Transm. 1997;104:887–894. doi: 10.1007/BF01285556. [DOI] [PubMed] [Google Scholar]
- Gerstadt C, Hong Y, Diamond A. The relationship between cognition and action: performance of 3 1/2-7 year old children on a Stroop-like day-night test. Cognition. 1994;53:129–153. doi: 10.1016/0010-0277(94)90068-x. [DOI] [PubMed] [Google Scholar]
- Goldman PS, Rosvold HE. Localization of function within the dorsolateral prefrontal cortex of the rhesus monkey. Exp Neurol. 1970;27:291–304. doi: 10.1016/0014-4886(70)90222-0. [DOI] [PubMed] [Google Scholar]
- Güttler F, Ledley FD, Lidsky AS, DiLella AG, Sullivan SE, Woo SLC. Correlation between polymorphic DNA haplotypes at the phenylalanine hydroxylase locus and clinical phenotypes of phenylketonuria. J Pediatr. 1987;110:68–71. doi: 10.1016/s0022-3476(87)80290-1. [DOI] [PubMed] [Google Scholar]
- Ho JY-P, Chen M-J, Sheu WH-H, Yi Y-C, Tsai AC-W, Guu H-F, Ho ES-C. Differential effects of oral conjugated equine estrogen and transdermal estrogen on atherosclerotic vascular disease risk markers and endothelial function in healthy postmenopausal women. Hum Reprod. 2006;21:2715–2720. doi: 10.1093/humrep/del245. [DOI] [PubMed] [Google Scholar]
- Huijbregts SCJ, de Sonneville LMJ, Licht R, van Spronsen FJ, Sergeant JA. Short-term dietary interventions in children and adolescents with treated phenylketonuria: effects on neuropsychological outcome of a well-controlled population. J Inherit Metab Disord. 2002;25:419–430. doi: 10.1023/a:1021205713674. [DOI] [PubMed] [Google Scholar]
- Ishimatsu M, Kidani Y, Tsuda A, Akasu T. Effects of methylphenidate on the membrane potential and current in neurons of the rat locus coeruleus. J Neurophysiol. 2002;87:1206–1212. doi: 10.1152/jn.00463.2001. [DOI] [PubMed] [Google Scholar]
- Iuvone PM, Tigges M, Fernandes A, Tigges J. Dopamine synthesis and metabolism in rhesus monkey retina: development, aging, and the effects of monocular visual deprivation. Vis Neurosci. 1989;2:465–471. doi: 10.1017/s0952523800012360. [DOI] [PubMed] [Google Scholar]
- Jacobsen CF. Functions of the frontal association areas in primates. Arch Neurol Psychiatry. 1935;33:558–560. [Google Scholar]
- Jucaite A, Fernell E, Halldin C, Forssberg H, Farde L. Reduced midbrain dopamine transporter binding in male adolescents with attention-deficit/hyperactivity disorder: association between striatal dopamine markers and motor hyperactivity. Biol Psychiatry. 2005;57:229–238. doi: 10.1016/j.biopsych.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Karoum F, Chrapusta SJ, Egan MF. 3-Methoxytryramine is the major metabolite of released dopamine in the rat frontal cortex: reassessment of the effects of antipsychotics on the dynamics of dopamine release and metabolism in the frontal cortex, nucleus accumbens, and striatum by a simple two pool model. J Neurochem. 1994;63:972–979. doi: 10.1046/j.1471-4159.1994.63030972.x. [DOI] [PubMed] [Google Scholar]
- Krause KH, Dresel SH, Krause J, Kung HF, Tatsch K. Increased striatal dopamine transporter in adult patients with attention deficit hyperactivity disorder: effects of methylphenidate as measured by single photon emission computed tomography. Neurosci Lett. 2000;285:107–110. doi: 10.1016/s0304-3940(00)01040-5. [DOI] [PubMed] [Google Scholar]
- Krause KH, Dresel SH, Krause J, Kung HF, Tatsch K, Ackenheil M. Stimulant-like action of nicotine on striatal dopamine transporter in the brain of adults with attention deficit hyperactivity disorder. Int J Neuropsychopharmacol. 2002;5:111–113. doi: 10.1017/S1461145702002821. [DOI] [PubMed] [Google Scholar]
- Krause KH, Dresel SH, Krause J, LaFougere C, Ackenheil M. The dopamine transporter and neuroimaging in attention deficit hyper-activity disorder. Neurosci Biobehav Rev. 2003;27:605–613. doi: 10.1016/j.neubiorev.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Lachman HM, Papolos DF, Saito T, Yu YM, Szumlanski CL, Weinshilboum RM. Human catechol O-methyltransferase pharmacogenetics: description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics. 1996;6:243–250. doi: 10.1097/00008571-199606000-00007. [DOI] [PubMed] [Google Scholar]
- Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melen K, Julkunen I, Taskinen J. Kinetics of human soluble and membrane-bound catechol-O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry. 1995;34:4202–4210. doi: 10.1021/bi00013a008. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Kestler LJ, Mazzanti C, Bates JA, Goldberg T, Goldman D. A functional polymorphism in the COMT gene and performance on a test of prefrontal cognition. Am J Psychiatry. 2002;159:652–654. doi: 10.1176/appi.ajp.159.4.652. [DOI] [PubMed] [Google Scholar]
- Mattay VS, Goldberg TE, Fera F, Hariri AR, Tessitore A, Egan MF, Kolachana B, Callicott JH, Weinberger DR. Catechol O-methyltransferase val158-met genotype and individual variation in the brain response to amphetamine. Proc Natl Acad Sci USA. 2003;100:6186–6191. doi: 10.1073/pnas.0931309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer D, Lewis TL. Visual acuity: the role of visual input in inducing postnatal change. Clin Neurosci Res. 2001;1:239–247. [Google Scholar]
- Meador-Woodruff JH, Damask SP, Wang J, Haroutunian V, Davis KL, Watson SJ. Dopamine receptor mRNA expression in human striatum and neocortex. Neuropsychopharmacology. 1996;15:17–29. doi: 10.1016/0893-133X(95)00150-C. [DOI] [PubMed] [Google Scholar]
- Milich R, Balentine AC, Lynam DR. ADHD combined type and ADHD predominantly inattentive type are distinct and unrelated disorders. Clin Psychol Sci Pract. 2001;8:463–488. [Google Scholar]
- Miller L, Braun LD, Pardridge WM, Oldendorf WH. Kinetic constants for blood-brain, barrier amino acid transport in conscious rats. J Neurochem. 1985;45:1427–1432. doi: 10.1111/j.1471-4159.1985.tb07209.x. [DOI] [PubMed] [Google Scholar]
- Moll L, Kuypers HGJM. Premotor cortical ablations in monkeys: contralateral changes in visually guided reaching behavior. Science. 1977;198:317–319. doi: 10.1126/science.410103. [DOI] [PubMed] [Google Scholar]
- Palmatier MA, Kang AM, Kidd KK. Global variation in the frequencies of functionally different catechol-O-methyltransferase alleles. Biol Psychiatry. 1999;46:557–567. doi: 10.1016/s0006-3223(99)00098-0. [DOI] [PubMed] [Google Scholar]
- Pardridge W. Regulation of amino acid availability to the brain. In: Wurtman RJ, Wurtman JJ, editors. Nutrition and the brain. NY: Raven Press; 1977. pp. 141–204. [Google Scholar]
- Pardridge WM, Oldendorf WH. Transport of metabolic substrates through the blood-brain barrier. J Neurochem. 1977;28:5–12. doi: 10.1111/j.1471-4159.1977.tb07702.x. [DOI] [PubMed] [Google Scholar]
- Pennington BF, VanDoornick WJ, McCabe LL, McCabe ERB. Neuropsychological deficits in early treated phenylketonuric children. Am J Ment Defic. 1985;89:467–474. [PubMed] [Google Scholar]
- Petrides M. Impairments on nonspatial self-ordered and externally ordered working memory tasks after lesions of the mid-dorsal part of the lateral frontal cortex in the monkey. J Neurosci. 1995;15:359–375. doi: 10.1523/JNEUROSCI.15-01-00359.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrides M, Alivisatos B, Meyer E, Evans AC. Functional activation of the human frontal cortex during performance of verbal working memory tasks. Proc Natl Acad Sci USA. 1993;90:878–882. doi: 10.1073/pnas.90.3.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrides M, Milner B. Deficits on subject-ordered tasks after frontal- and temporal-lobe lesions in man. Neuropsychologia. 1982;20:249–262. doi: 10.1016/0028-3932(82)90100-2. [DOI] [PubMed] [Google Scholar]
- Piaget J. La naissance de l‘intelligence chez l’enfant. In: Cook M, editor. 1952: The origins of intelligence in children. New York: Basic Books; 1937. [Google Scholar]
- Pomerleau CS. Co-factors for smoking and evolutionary psychobiology. Addiction. 1997;92:397–408. [PubMed] [Google Scholar]
- Oldendorf WH. Stereospecificity of blood brain barrier permeability to amino acids. Am J Physiol. 1973;224:967–969. doi: 10.1152/ajplegacy.1973.224.4.967. [DOI] [PubMed] [Google Scholar]
- Rapoport SI. Sites and functions of the blood-aqueous and blood-vitreous barriers of the eye. In: Rapoport SI, editor. Blood-brain barrier in physiology and medicine. New York: Raven Press; 1976. pp. 207–232. [Google Scholar]
- Reinhard JF, Bannon MJ, Jr, Roth RH. Acceleration by stress of dopamine synthesis and metabolism in prefrontal cortex: antagonism by diazepam. Nauyn-Schmiedeberg’s Arch Pharmacol. 1982;318:374–377. doi: 10.1007/BF00501182. [DOI] [PubMed] [Google Scholar]
- Roth RH, Tam SY, Ida Y, Yang JX, Deutch AY. Stress and the mesocorticolimbic dopamine systems. Ann N Y Acad Sci. 1988;537:138–147. doi: 10.1111/j.1749-6632.1988.tb42102.x. [DOI] [PubMed] [Google Scholar]
- Sanchez-Gonzalez MA, Cavada C. Dopamine transporter expression in the primate brain. Society for Neuroscience, 33rd Annual Meeting, Abstract Viewer/Itinerary Planner.2003. [Google Scholar]
- Sawaguchi T, Goldman-Rakic PS. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science. 1991;251:947–950. doi: 10.1126/science.1825731. [DOI] [PubMed] [Google Scholar]
- Schrimsher GW, Billingsley RL, Jackson EF, Moore BD., 3rd Caudate nucleus volume asymmetry predicts attention-deficit hyperactivity disorder (ADHD) symptomatology in children. J Child Neurol. 2002;17:877–884. doi: 10.1177/08830738020170122001. [DOI] [PubMed] [Google Scholar]
- Seeman P, Madras BK. Anti-hyperactivity medication: methylphenidate and amphetamine. Mol Psychiatry. 1998;3:386–396. doi: 10.1038/sj.mp.4000421. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Carr DB. Selective prefrontal cortex inputs to depamine cells: implications for schizophrenia. Physiology & Behavior. 2002;77:513–517. doi: 10.1016/s0031-9384(02)00931-9. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Hawrylak VA, Matus C, Guido MA, Levey AI. Dopamine axon varicosities in the prelimbic division of the rat prefrontal cortex exhibit sparse immunoreactivity for the dopamine transporter. J Neurosci. 1998;18:2697–2708. doi: 10.1523/JNEUROSCI.18-07-02697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shansky RM, Glavis-Bloom C, Lerman D, McRae P, Benson C, Miller K, Cosand L, Horvath TL, Arnsten AF. Estrogen mediates sex differences in stress-induced-prefrontal cortex dysfunction. Mol Psychiatry. 2004;9:531–538. doi: 10.1038/sj.mp.4001435. [DOI] [PubMed] [Google Scholar]
- Shenker A. The mechanism of action of drugs used to treat attention-deficit hyperactivity disorder: focus on catecholamine receptor pharmacology. Adv Pediatr. 1992;39:337–382. [PubMed] [Google Scholar]
- Shors TJ, Leuner B. Estrogen-mediated effects on depression and memory formation in females. J Affect Disord. 2003;74:85–96. doi: 10.1016/s0165-0327(02)00428-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G. Testosterone in utero and at birth dictates how stressful experience will affect learning in adulthood. Proc Natl Acad Sci USA. 2002;99:13955–13960. doi: 10.1073/pnas.202199999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siervogel RM, Weinshilboum R, Wilson AF, Elston RC. Major gene model for the inheritance of catechol-O-methyltransferase activity in five large families. Am J Med Genet. 1984;19:315–323. doi: 10.1002/ajmg.1320190214. [DOI] [PubMed] [Google Scholar]
- Skrandies W, Gottlob I. Alterations of visual contrast sensitivity in Parkinson’s disease. Hum Neurobiol. 1986;5:255–259. [PubMed] [Google Scholar]
- Slater A, Johnson SP. Visual sensory and perceptual abilities of the newborn: beyond the blooming, buzzing confusion. In: Simion F, Butterworth G, editors. The development of sensory, motor and cognitive capacities in early infancy: from perception to cognition. Hove, UK: Psychology Press Ltd; 1998. pp. 121–141. [Google Scholar]
- Spielman RS, Weinshilboum RM. Genetics of red cell COMT activity: analysis of thermal stability and family data. Am J Med Genet. 1981;10:279–290. doi: 10.1002/ajmg.1320100311. [DOI] [PubMed] [Google Scholar]
- Stemerdink NBA, van der Molen MW, Kalverboer AF, van der Meere JJ, Huisman J, Jong LW, Slijper FME, Verkerk PH, van Spronsen FJ. Prefrontal dysfunction in early and continuously treated phenylketonuria. Dev Neuropsychol. 1999;16:29–57. [Google Scholar]
- Suhara T, Fukuda H, Inoue O, Itoh T, Suzuki K, Yamasaki T, Tateno Y. Age-related changes in human D1 dopamine receptors measured by positron emission tomography. Psychopharmacology. 1991;103:41–45. doi: 10.1007/BF02244071. [DOI] [PubMed] [Google Scholar]
- Tam SY, Elsworth JD, Bradberry CW, Roth RH. Mesocortical dopamine neurons: high basal firing frequency predicts tyrosine dependence of dopamine synthesis. J Neural Transm. 1990;81:97–110. doi: 10.1007/BF01245830. [DOI] [PubMed] [Google Scholar]
- Teicher MH, Ito Y, Glod CA, Barber NI. Objective measurement of hyperactivity and attentional problems in ADHD. J Am Acad Child Adolesc Psychiatry. 1996;35:334–342. doi: 10.1097/00004583-199603000-00015. [DOI] [PubMed] [Google Scholar]
- Tercyak KP, Lerman C, Audrain J. Association of attention-deficit hyperactivity disorder symptoms with levels of cigarette smoking in a community sample of adolescents. J Am Acad Child Adolesc Psychiatry. 2002;41:799–805. doi: 10.1097/00004583-200207000-00011. [DOI] [PubMed] [Google Scholar]
- Thierry AM, Tassin JP, Blanc G, Glowinski J. Selective activation of the mesocortical DA system by stress. Nature. 1976;263:242–244. doi: 10.1038/263242a0. [DOI] [PubMed] [Google Scholar]
- Tornquist P, Alm A. Carrier-mediated transport of amino acids through the blood-retinal and blood-brain barriers. Graefes Arch Clin Exp Ophthalmol. 1986;224:21–25. doi: 10.1007/BF02144127. [DOI] [PubMed] [Google Scholar]
- Vaidya CJ, Austin G, Kirkorian G, Ridlehuber HW, Desmond JE, Glover GH, Gabrieli JDE. Selective effects of methylphenidate in attention deficit hyperactivity disorder: a functional magnetic resonance study. Proc Natl Acad Sci USA. 1998;95:14494–14499. doi: 10.1073/pnas.95.24.14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya CJ, Bunge SA, Dudukovic NM, Zalecki CA, Elliott GR, Gabrieli JDE. Altered neural substrates of cognitive control in childhood ADHD: evidence from functional magnetic resonance imaging. Am J Psychiatry. 2005;162:1605–1613. doi: 10.1176/appi.ajp.162.9.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Ding YS, Fowler JS, Wang G, Logan J, Gately SJ, Hitzemann R, Smith G, Fields D, Gur R. Dopamine transporters decrease with age in healthy subjects. J Nucl Med. 1996;37:554–558. [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Ding YS, Gur RC, Gatley J, Logan J, Moberg PJ, Hitzemann R, Smith G, et al. Parallel loss of presynaptic and postsynaptic dopamine markers in normal aging. Ann Neurol. 1998;44:143–147. doi: 10.1002/ana.410440125. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Gatley SJ, Logan J, Ding YS, Hitzemann R, Pappas N. Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am J Psychiatry. 1998;155:1325–1331. doi: 10.1176/ajp.155.10.1325. [DOI] [PubMed] [Google Scholar]
- Waldman ID, Rowe DC, Abramowitz A, Kozel ST, Mohr JH, Sherman SL, Cleveland HH, Sanders ML, Gard JM, Stever C. Association and linkage of the dopamine transporter gene and attention-deficit hyperactivity disorder in children: heterogeneity owing to diagnostic subtype and severity. Am J Hum Genet. 1998;63:1767–1776. doi: 10.1086/302132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M, Worling D, Wasdell M. A chart review study of the inattentive and combined types of ADHD. J Atten Disord. 2003;7:1–9. doi: 10.1177/108705470300700101. [DOI] [PubMed] [Google Scholar]
- Welsh MC, Pennington BF, Ozonoff S, Rouse B, McCabe ERB. Neuropsychology of early-treated phenylketonuria: specific executive function deficits. Child Dev. 1990;61:1697–1713. [PubMed] [Google Scholar]
- Whalen CK, Jamner LD, Henker B, Gehricke JG, King PS. Is there a link between adolescent cigarette smoking and pharmacotherapy for ADHD? Psychol Addict Behav. 2003;17:332–335. doi: 10.1037/0893-164X.17.4.332. [DOI] [PubMed] [Google Scholar]
- Wong DF, Young D, Wilson PD, Meltzer CC, Gjedde A. Quantification of neuroreceptors in the living human brain: III. D2-like dopamine receptors: theory, validation, and changes during normal aging. J Cereb Blood Flow Metab. 1997;17:316–330. doi: 10.1097/00004647-199703000-00009. [DOI] [PubMed] [Google Scholar]
- Zahrt J, Taylor JR, Mathew RG, Arnsten AFT. Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci. 1997;17:8528–8535. doi: 10.1523/JNEUROSCI.17-21-08528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta JK, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, Koeppe RA, Stohler CS, Goldman D. COMT val158met genotype affects muopioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–1243. doi: 10.1126/science.1078546. [DOI] [PubMed] [Google Scholar]