

Abstract
Objective
Graft-versus-host disease (GVHD) is the major cause of morbidity and mortality following allogeneic hematopoietic stem cell transplantation. Models of immunodeficient mice that consistently and efficiently reconstitute with xenoreactive human T cells would be a valuable tool for the in vivo study of GVHD, as well as other human immune responses.
Materials and Methods
We developed a consistent and sensitive model of human GVHD by retro-orbitally injecting purified human T cells into sublethally irradiated NOD/SCID-β2mnull recipients. In addition, we characterized for the first time the trafficking patterns and expansion profiles of xenoreactive human T cells in NOD/SCID-β2mnull recipients using in vivo bioluminescence imaging.
Results
All NOD/SCID-β2mnull mice conditioned with 300 cGy of total body irradiation and injected with 1 × 107 human T cells exhibited human T cell engraftment, activation, and expansion, with infiltration of multiple target tissues and a subsequent greater than 20% loss of pretransplant body weight. Importantly, histological examination of the GVHD target tissues revealed changes consistent with human GVHD. Furthermore, we also showed by in vivo bioluminescence imaging that the development of lethal GVHD in the NOD/SCID-β2mnull recipients was dependent upon the initial retention and early expansion of human T cells in the retroorbital sinus cavity.
Conclusion
Our NOD/SCID-β2mnull mouse model provides a system to study the pathophysiology of acute GVHD induced by human T cells and aids in the development of more effective therapies for human GVHD.
Introduction
Graft-versus-host disease (GVHD)1 remains a major cause of morbidity and mortality following allogeneic hematopoietic stem cell transplantation (HSCT) and donor lymphocyte infusion [1]. Many of the therapeutic strategies that are being developed to control GVHD while maintaining the beneficial graft-versus-leukemia and/or graft-versus-infection effects provided by donor lymphocyte infusion after allogeneic HSCT require ex vivo T cell stimulation and expansion. Unfortunately, multiple preclinical and clinical studies have demonstrated that these ex vivo expanded T cells exhibit decreased survival and function in vivo, including reduced alloreactivity and GVHD potential [2–6]. Recently, others and we described an ex vivo method for expansion of human T (huT) cells, using anti-CD3 and anti-CD28 monoclonal antibodies that are covalently attached to superparamagnetic microbeads (CD3/CD28 beads) [7,8]. Autologous T cells expanded from patients with HIV infections and a variety of cancers using this CD3/CD28 bead activation technology have been well-tolerated; infusion of these activated T cells demonstrated preliminary evidence of therapeutic effects in initial clinical trials [9–13]. However, it remains unknown whether allogeneic huT cells polyclonally expanded with CD3/CD28 beads promote engraftment, induce GVHD and provide a graft-versus-leukemia effect.
Models of immunodeficient mice that consistently and efficiently reconstitute with xenoreactive huT cells would be a valuable tool for the development of more effective therapies for GVHD and to study the pathophysiology of the disease. In most xenograft models, huT cells are deleted or, if present, are tolerant or anergic due, presumably, to their continuous stimulation by mouse xenoantigens [14–16]. Initial experiments using SCID mice, which lack functional T and B-cells, demonstrated that anywhere from 1% to 20% of unconditioned SCID mice could develop xenogeneic GVHD (X-GVHD) following intraperitoneal (i.p.) injection of extremely high numbers (50–100 × 106) of human peripheral blood mononuclear cells (huPBMCs) [14,17–23]. Nonobese diabetic (NOD)/SCID mice exhibit reduced NK activity, macrophage function and serum hemolytic complement activity in addition to the deficit in mature T and B cells [24,25]. Although increased huT cell engraftment levels and X-GVHD have been reported for NOD/SCID mice, these mice are still constrained by the presence of residual NK cell activity and poor human CD4+ T cell engraftment [23,24,26–30]. Recently, this residual NK cell activity was virtually eliminated by backcrossing the β2-microglobulin-null (β2mnull) allele onto the NOD/SCID background [31]. Compared to NOD/SCID controls, unconditioned NOD/SCID-β2mnull mice injected i.p. with huPBMCs exhibit similar [30], or increased [31], huT cell engraftment with 6- to 7-fold higher numbers of CD4+ T cells [31]. However, <20% of unconditioned NOD/SCID-β2mnull recipients injected i.p. with huPBMCs develop X-GVHD [30,31].
Multiple strategies have been explored to increase huT cell engraftment and the incidence of lethal X-GVHD in immunodeficient mice, including: (i.) pretransplant conditioning with sublethal irradiation [19,23,26,32–40], (ii.) targeted reduction of murine natural killer (NK) cell activity with antibodies directed towards specific cell membrane markers (anti-asialo-GM1 [19,33–35] or anti-CD122 [23,36,39]), (iii.) targeted reduction of murine macrophages with clodronate–containing liposomes [37], (iv.) using newborn mice which lack a fully developed innate immune system [32], (v.) administration of interleukin-15 (IL-15) [38,40], and/or (vi.) generating new mouse strains with additional defects in the innate immune system [37]. In general, these protocols have significantly improved huT cell engraftment and thereby, increased the incidence of lethal X-GVHD to anywhere from 50% to 100%. In the present report, we developed a consistent and sensitive model of human GVHD that does not require the use of depleting antibodies, clodronate–containing liposomes, or the administration of human cytokines. We show that all NOD/SCID-β2-m−/− mice conditioned with 300 cGy of total body irradiation (TBI) developed lethal X-GVHD following retroorbital (r.o.) administration of purified huT cells. Consistent with X-GVHD, these mice exhibited huT cell engraftment, activation, and expansion, with infiltration of multiple target tissues and a subsequent >20% loss of pretransplant body weight. Importantly, histological examination of the GVHD target tissues revealed changes consistent with human GVHD.
Materials and methods
Mice and conditioning regimen
NOD/SCID-β2mnull [31,41,42] were obtained from Taconic Farms (Germantown, NY, USA) or from The Jackson Laboratory (Bar Harbor, ME, USA). Animals were fed autoclaved food and water, and all manipulations were performed on a laminar flow bench. Animal used were 8 to 14 weeks old. Animal care and euthanasia were approved by the Washington University Medical School Animal Studies Committee. Mice received 250 or 300 cGy of total body irradiation (TBI), using a Shepard Mark IV Cesium 137 irradiator, between 4 to 24 hours prior to the human cell injection. Control mice were irradiated but did not receive human cells.
Human cell isolation and transplantation
Heparinized whole blood samples were obtained from healthy volunteers (n=5). PBMCs were isolated by density centrifugation on Histopaque-1077 (Sigma-Aldrich, St. Louis, MO, USA), washed twice, counted and resuspended in PBS/0.1% human serum albumin. HuT cells were isolated from PBMCs by depletion of non-T cells (negative selection) using the human Pan T Cell Isolation Kit II (Miltenyi Biotech, Auburn, CA, USA) in combination with an AutoMACS cell separation device (Miltenyi). Cell suspensions containing 5 × 106 or 10 × 106 huT cells in 0.2 mL of PBS were injected intravenously via the tail vein or into the retro-orbital (r.o.) space [43] in the irradiated mice.
Construction of CBRluc-egfp Retroviral Vector
Click Beetle Red luciferase (CBRluc) was modified from pCBR (Promega Corporation, Madison, WI, USA) by PCR to add ΔU3 vector sequence and a Kozak sequence to the 5′ end (Forward, 5′-GGCTAGCGTAGACGGCATCGCAGCTTGGATACACGCCGCCCACGTGAAGGCT GCCGACCCCGGGGTGGACCATCCTCTAGACTGCCATGGTAAAGCGTGAG-3′). The 3′end of pCBR was modified to eliminate the stop codon of CBRluc and introduce a gly-gly-ser linker and a BamHI restriction site (Reverse, 5′-CGGTGGATCCGATCCTCCTCCACCGCCGGCCTTCAC-3′). The modified pCBR product was then digested with Acc1 and BamH1 and the insert was ligated to the pEGFP-N1 (Clontech, Palo Alto, CA, USA) vector to generate pCBR-eGFP-N1. To construct retroviral vector, the ΔU3ΔCD34-tk75 vector [44] was digested with Acc1 and Not1 to remove ΔCD34-tk75 and ligated to a Acc1/Not1 fragment of pCBR-eGFP-N1 to generate ΔU3CBRluc-enhanced green fluorescent protein (EGFP). Vesicular stomatitis virus G envelope protein pseudotyped viruses were prepared by packaging the ΔU3CBRluc-EGFP retroviral vectors in 293GPG cells as previously described. [44]
Ex-vivo activation, transduction, and expansion of human T cells
PBMCs were washed twice in HBSS and inoculated into 300 ml Lifecell tissue culture bags (Baxter, Deerfield, IL, USA) at a concentration of 1×106 cells per ml in serum-free Stemline T cell medium (Sigma-Aldrich) supplemented with L-Glu. CD3/CD28 antibody-coated magnetic beads (Xcyte™Dynabeads®, Xcyte Therapies, Inc., Seattle, WA, USA) [8] were washed once in HBSS and added at a ratio of 3 beads per one cell. After 24 hours at 37 °C, 50 U/ml IL-2 (Sigma) was added. Culture bags were incubated for 3 to 7 days without a change of medium. On day 4 or 8, cells were completely removed from the culture bags and magnetic beads were removed by bead immobilization using a strong magnetic field. A 96% to 99% bead reduction could be achieved routinely. Human T cells were genetically modified with CBRluc/egfp by adding ΔU3CBRluc-EGFP retrovirus 48 hours after CD3/CD28 bead stimulation.
GVHD evaluation
Mice were labeled as having developed lethal GVHD only if they had severe weight loss (>20%), hunched posture, ruffled fur, reduced mobility, tachypnea, high engraftment of huT cells in blood, huT cell infiltration in target tissues (fluorescence-activated cell sorter (FACS), histology and immunohistochemistry), and high serum levels of human cytokines. Mice were bled from the r.o. sinus under anesthesia two days after the human cell injection, and later once per week. Blood was collected in EDTA-coated tubes and analyzed for huT cell subsets using FACS. Plasma was isolated from the remaining blood and stored at −80°C for later determination of human cytokines. At the time of sacrifice, tissues (bone marrow, spleen, liver, lung and kidney) were harvested and divided into two parts. For histology and immunohistochemistry staining the organs were fixed immediately in 10% buffered formalin and then embedded in paraffin. For flow cytometry analysis, single cell suspensions were prepared as we previously described. [45] Briefly, the spleen, liver, lung and kidney were pressed through nylon mesh with the blunt end of a syringe and erythrocytes were removed by hypotonic lysis with 154 mmol/L ammonium chloride, 10 mmol/L potassium bicarbonate, and 0.1 mmol/L ethylenediaminetetraacetic acid (EDTA). Cell suspensions were centrifuged at 500 × g for 15 minutes, filtered through a 70-μm cell strainer, and resuspended in PBS containing 0.5% bovine serum albumin and 1 mmol/L EDTA. The bone marrow was harvested by flushing the femurs and tibias with PBS. The resulting cell pellet was then depleted of erythrocytes, filtered, and resuspended as described above.
Flow cytometry analysis for the detection of engraftment
Single-cell suspensions from the peripheral blood, spleen, bone marrow, liver, lung, and kidney were incubated with fluorescently labeled monoclonal antibodies, then 5-color FACS analysis of human antigens was performed on an FC-500 flow cytometer (Becton Dickinson, San Jose, CA, USA) and data were analyzed using FLOWJO software (Tree Star, San Carlos, CA, USA). All samples were blocked first with purified anti-mouse CD16/CD32 (FcR III/II Receptor) and human FcR-Blocking. The mouse antibodies used were biotin anti-mouse CD45 (LCA, Ly-5, 30-F11) (pan-murine leukocytes) and streptavidin-PE-Texas Red. The human antibodies used were CD45-allophycocyanin (APC)(pan-human leukocytes), CD3-cyanin 7 (Cy7)(pan-T cells), CD4-phycoerythrin (PE)(T-helper), CD8-fluorescein isothiocyanate (FITC)(T-cytotoxic), CD25-PE (IL-2R), CD30- (FITC)(Ki-1), and CD69-FITC. All antibodies were purchased from BD Pharmingen (San Diego, CA, USA). The proportion of human cells was calculated as follows: % huCD45+ = [huCD45+ / (huCD45+ + mCD45+)] × 100%.
Histology and immunohistochemistry
Paraffin-embedded sections (5 μm) were cut and stained with hematoxylin-eosin for histology. Selected sections were also immunohistochemically stained with anti-human CD3, CD8 and CD45 antibodies purchased from Dako (Carpinteria, CA, USA) and anti-human CD4 antibody purchased from BioGenex (San Ramon, CA, USA) using DAKO EnVision™ system. Slides were coded and read by a pathologist expert in human GVHD, using a blinded technique. A semiquantitative scoring system was used to assess abnormalities associated with GVHD or allograft rejection. [46]
Bioluminescence Imaging of Animals
Human T cell trafficking in NOD/SCID-β2mnull recipients was assessed non-invasively by bioluminescence with an IVIS 100 CCD camera (Xenogen, Alameda, CA, USA) as described. [47,48] Briefly, mice were injected i.p. with D-luciferin (150 μg/g in PBS) and imaged 10 min later with the IVIS (1–60 sec exposure, binning 8, f-stop 1, FOV 15 cm). Anesthesia was induced and maintained during imaging by vaporizer delivery of 2–2.5% isofluorane. Total photon flux (photons/sec) was quantified on images using a rectangular region of interest encompassing the entire abdomen and thorax. Specific identification of tissues and organs was based on location and imaging characteristics of each tissue type. Due to the desire to repetitively image the animals over time, we did not dissect tissues for specific verification in each animal. However, based on extensive experience and other dissection protocols, we are reasonably confident that these foci indeed represent lymph nodes or other tissues as noted.
Measurement of human cytokines
Mouse serum was analyzed for human IFN-γ TNF-α IL-2, IL-4, IL-6, and IL-10 with a cytometric bead array combined with flow cytometry assay (Human Th1/Th2 Cytokine CBA-II, Becton Dickinson).
Statistical analysis
Values are expressed as mean ± standard deviation (SD). Comparison between groups was performed using unpaired Student’s t test, two tailed, 95% confidence intervals, or analysis of variance (ANOVA). Kaplan-Meier survival curves were compared using the log-rank test. P values < 0.05 were considered as statistically significant. All statistical analyses were performed using StatView software (Abacus Concepts, Berkeley, CA, USA).
Results
Development of a xenogeneic model of human GVHD
To evaluate whether we could induce lethal X-GVHD in NOD/SCID-β2mnull mice, we sublethally irradiated animals with 250 cGy of TBI and injected them intravenously, either through the lateral tail vein (i.v.) or retro-orbitally (r.o.), the following day, with 5 × 106 or 10 × 106 huT cells. Following injection, we assessed huT cell engraftment and the development of X-GVHD by performing weekly FACS analyses of the peripheral blood and recording body weights every 3 to 4 days. NOD/SCID-β2mnull mice that were injected with huT through the lateral tail vein exhibited only small and transient huT cell engraftment and failed to develop lethal X-GVHD, irrespective of the huT cell dose (Fig. 1A,B). We found a similar failure of the huT cells to engraft and induce X-GVHD when 5 × 106 huT cells were injected r.o. In contrast, we observed significantly increased huT cell engraftment (P < 0.0007 compared to other conditions; Fig. 1A) and lethal X-GVHD (P < 0.0002 compared to other conditions; Fig. 1B) when NOD/SCID-β2mnull mice were injected r.o. with 10 × 106 huT cells. Overall, 20 of 34 NOD/SCID-β2mnull mice conditioned with 250 cGy of TBI and injected with 10 × 106 huT cells r.o. developed lethal X-GVHD (Fig. 1B), with a median survival time of 22 days.
Figure 1.

Human T cell engraftment and development of lethal X-GVHD in NOD/SCID-β2mnull mice. NOD/SCID-β2mnull mice (n = 59) were sublethally irradiated with 250 cGy of TBI and injected through the lateral tail vein (i.v.) or retro-orbital (r.o.) venous plexus with 5 × 106 or 10 × 106 naive purified huT cells the following day. (A) Human T cell engraftment. Peripheral blood samples collected from mice that developed (GVHD; open circles) or did not develop (No GVHD; closed circles) lethal GVHD were labeled with mAbs specific for human and murine CD45 and analyzed by flow cytometry. The proportion of human cells in the peripheral blood was calculated as follows: % huCD45+ = [huCD45+ / (huCD45+ + mCD45+)] × 100%. Maximum huT cell engraftment is shown for each mouse and typically occurred between 2 and 3 weeks after huT injection. (B) Kaplan-Meier survival analysis. Lethal X-GVHD was only observed when 10 × 106 huT cell were injected r.o. Results are pooled from four separate experiments with different donors.
We found several significant differences between the NOD/SCID-β2mnull mice that developed lethal X-GVHD and those that survived long term without evidence of GVHD. Compared to animals that failed to develop lethal X-GVHD, NOD/SCID-β2mnull mice with lethal X-GVHD exhibited significantly increased: i.) weight loss (P < 0.05 from day 4 onward; Fig. 2A), ii.) huT engraftment in the peripheral blood (P = 0.0004 at day 24 post-transplantation; Fig. 2B), iii.) expansion of human CD4+ and human CD8+ T cells in the peripheral blood (Fig. 2C), iv.) up-regulation of the T cell activation markers CD25 and CD30 on huT in the peripheral blood (P < 0.01 at days 10 and 20 post-transplantation; Fig. 2D), and v.) human IFN-γ serum levels (P < 0.05 at days 10 and 20 post-transplantation; Fig. 2E). In addition to these differences, NOD/SCID-β2mnull mice with lethal X-GVHD also displayed several clinical signs of GVHD including hunched posture, ruffled fur, reduced activity, tachypnea, and anemia. These symptoms were not evident in mice that failed to develop GVHD (data not shown). Furthermore, we detected no significant quantities of human IL-2, IL-4, IL-6, IL-10, or TNFα in the serum of any of the NOD/SCID-β2mnull mice examined (data not shown). In summary, we found that 59% of NOD/SCID-β2mnull mice developed lethal X-GVHD if they were conditioned with 250 cGy and retro-orbitally injected the following day with 10 × 106 huT cells.
Figure 2.

Characterization of mice that developed or did not develop lethal X-GVHD. NOD/SCID-β2mnull mice (n=34) were sublethally irradiated with 250cGy of TBI and injected r.o. with 10 × 106 naive purified huT cells. Mice that developed lethal X-GVHD (n = 20; open bars) were then compared to animals that failed to develop lethal X-GVHD (n=14; closed bars). (A) Percent change in pretransplant body weight. (B) Kinetics of huT cell engraftment in the peripheral blood. The proportion of human cells in the peripheral blood was determined by flow cytometry as described in the legend to Fig. 1. (C) CD4+/CD8+ huT cell ratio. The percentage of human CD45+CD3+ T cells in the peripheral blood that expressed CD4 or CD8 was determined by flow cytometry. (D) Expression of T cell activation markers. The percentage of human CD45+CD3+ T cells in the peripheral blood that expressed CD25, CD30, or CD69 at 10 or 20 days after huT cell injection was determined by flow cytometry. Normal human PBMCs were used as controls in all analyses to establish consistent gating criteria for CD25, CD30, and CD69. (E) Human IFN-γ serum levels. Serum was obtained from peripheral blood samples collected at the indicated times after huT cell injection and human IFN-γ levels were determined using a cytometric bead assay (BD Biosciences). Values of p indicate statistically significant differences between the two groups. NS indicates differences were not statistically significant.
In vivo bioluminescence imaging of human T cell trafficking in NOD/SCID-β2mnull mice
To evaluate why the route of huT cell administration affected huT cell engraftment and the development of lethal X-GVHD in NOD/SCID-β2mnull mice, we performed in vivo bioluminescence imaging (BLI) studies using huT cells that were genetically modified with a click beetle red luciferase/enhanced green fluorescent protein (CBRluc/egfp) dual function reporter gene (huTCBRluc/EGFP). In these studies, we transduced CD3/CD28 bead activated PBMCs with a CBRluc/egfp retrovirus (30% huT cell transduction efficiency; data not shown) and injected 10 × 106 huTCBRluc/EGFP cells either through the lateral tail vein (i.v.) or r.o. into sublethally (250 cGy) irradiated NOD/SCID-β2mnull mice. Similar to the results we obtained with naïve (unmanipulated) huT cells (Fig. 1B), NOD/SCID-β2mnull mice injected with huTCBRluc/EGFP cells through the lateral tail vein failed to develop lethal X-GVHD, whereas 3 of 5 mice that were injected with huTCBRluc/EGFP cells r.o. developed lethal X-GVHD (Fig. 3A). Furthermore, as before, mice injected r.o. exhibited significantly increased weight loss (Fig. 3B; p < 0.02) and huT cell engraftment in the peripheral blood (Fig. 3C; p<0.04 and 0.02 on days 7 and 14) compared to mice that were injected i.v. with huTCBRluc/EGFP cells. It is important to note that the standard deviations presented in Fig. 3B and C are small because one of the two mice that received huT cells r.o. but did not develop lethal X-GVHD had significant weight loss (~ 10%) and huT cell engraftment (14% at day 14).
Figure 3.

In vivo BLI of huT cells following i.v. or r.o. administration. NOD/SCID-β2mnull mice were sublethally irradiated with 250 cGy of TBI and injected through the lateral tail vein (i.v.; n = 5) or retro-orbital (r.o.; n = 5) venous plexus with 10 × 106 CBRluc/egfp transduced huT (huTCBRluc/EGFP) cells the following day. (A) Kaplan-Meier survival analysis. Lethal X-GVHD was only observed when huTCBRluc/EGFP cells were injected r.o. (B) Percent change in pretransplant body weight. (C) Kinetics of huT cell engraftment in the peripheral blood. The proportion of human cells in the peripheral blood was determined by flow cytometry as described in the legend to Fig. 1. (D–G) BLI of luciferase activity. The trafficking and expansion of huTCBRluc/EGFP cells following i.v. or r.o. injection was assessed in the dorsal (D) and ventral positions (E) by BLI (1h: 1 hour, 1d: 1 day, 4d: 4 days, 7d: 7 days, 14d: 14 days, 21d: 21 days post huT injection). One representative animal for each group is shown over time. Photon flux is indicated in the color scale bar. Overall quantifications of emitted photons in the dorsal (F) and ventral (G) positions are shown graphically.
Serial whole body BLI revealed very different trafficking patterns and expansion profiles between the i.v. and r.o. routes of huT cell administration. As shown in Figure 3D–G, huTCBRluc/EGFP cells injected through the lateral tail vein immediately trafficked to the lungs and failed to expand during the first two weeks after infusion. In contrast, we observed that a significant portion of the r.o.-injected cells remained in the retroorbital sinus cavity and trafficked to secondary lymphoid organs during the first 7 days after huTCBRluc/EGFP cell infusion (Fig. 3D). This altered trafficking of the r.o.-injected cells was associated with a greater than 10-fold increase in the CBRluc/EGFP BLI signal during the first 2 weeks after huT cell infusion (Fig. 3F–G), with the huTCBRluc/EGFP cells accumulating in the skin, lymph nodes and gut. This homing pattern and strong BLI signal of the r.o.-injected huTCBRluc/EGFP cells remained until death from GVHD. Surprisingly, the CBRluc/EGFP BLI signal of the i.v.-injected huT cells increased greater than 1 order of magnitude between days 14 and 28 after huTCBRluc/EGFP cells infusion. However, this late expansion of the huTCBRluc/EGFP cells was not associated with the development of X-GVHD (n=0/5). Interestingly, we observed that the 2 r.o.-injected animals that failed to develop lethal X-GVHD exhibited huTCBRluc/EGFP cell trafficking patterns and expansion profiles that were identical to the i.v.-injected huTCBRluc/EGFP cells (data not shown). This observation suggests that lethal X-GVHD will not develop when r.o.-injected huTCBRluc/EGFP cells arrive directly into the blood stream.
Organ distribution and phenotype of huT in NOD/SCID-β2mnull mice that developed lethal X-GVHD
To more thoroughly characterize the X-GVHD induced by huT cells following r.o. injection of 10 × 106 huT cells into 250 cGy conditioned NOD/SCID-β2mnull mice, we used flow cytometry to analyze the presence and phenotype of huT cells in organs obtained from animals that developed lethal GVHD. In all flow cytometry analyses, we used normal human PBMCs as controls to establish consistent gating criteria (Fig. 4A). We detected significant amounts of both CD4+ and CD8+ huT cells in the blood, spleen, liver, lung, kidney, and bone marrow (BM) of NOD/SCID-β2mnull mice that developed lethal X-GVHD (Fig. 4B). The percentage of hematopoietic cells that were huT cells (either CD4+ or CD8+ T cells) ranged from 10% in the BM to approximately 60% in the liver, lung, and kidney (Fig. 4B). Additionally, although both CD4+ and CD8+ huT cells were present in the organs of animals that developed lethal X-GVHD, we observed a skewing of the CD4+ to CD8+ T cell ratio towards CD4+ T cells in the tissues, particularly the lung and kidney.
Figure 4.

Human T cell infiltration and phenotype in tissues of NOD/SCID-β2mnull mice that develop lethal GVHD. NOD/SCID-β2mnull mice (n=34) were sublethally irradiated with 250cGy of TBI and injected r.o. with 10 × 106 naive purified huT cells. When possible, moribund animals with X-GVHD were euthanized and portions of blood, spleen, liver, lung, kidney and bone marrow (BM) were analyzed by flow cytometry. (A) Representative FACS analysis of blood, spleen, and liver harvested from a NOD/SCID-β2mnull mouse that developed lethal X-GVHD. In all FACS analyses, normal human PBMCs were used as controls to establish consistent gating criteria. (B) Human T cell subset composition. The percentage of human CD45+CD3+ T cells in the indicated tissues that expressed CD4 or CD8 was determined by flow cytometry. (C) Expression of T cell activation markers. The percentage of human CD45+CD3+ T cells in the indicated tissues that expressed CD25, CD30, or CD69 was determined by flow cytometry
In addition to evaluating the CD4+ and CD8+ T cell subset composition in the organs of NOD/SCID-β2mnull mice with lethal X-GVHD, we also analyzed the expression of the T cell activation markers CD25, CD30, and CD69. Similar to huT cells that were detected in the blood of mice that developed lethal X-GVHD (Fig 2D), we observed increased expression of all three activation markers on huT cells present in the various organs tested (Fig 4C). On average, anywhere from 5% to >35% of the huT cells present in the various organs displayed an activated phenotype. Taken together, these flow cytometry analyses clearly demonstrate that activated CD4+ and CD8+ huT cells infiltrate multiple organs in NOD/SCID-β2mnull mice that developed lethal X-GVHD.
Increased lethal X-GVHD in NOD/SCID-β2mnull mice conditioned with 300 cGy TBI
Since only 59% of the NOD/SCID-β2mnull mice developed lethal X-GVHD when they were conditioned with 250 cGy and injected r.o. the following day with 10 × 106 huT cells, we evaluated whether increasing the irradiation dose to 300 cGy TBI and reducing the time between conditioning and huT cell injection from 24 hours to less than 6 hours would increase the incidence of lethal X-GVHD. All (8/8) NOD/SCID-β2mnull mice that were conditioned with 300 cGy TBI and injected with 10 × 106 huT cells developed lethal X-GVHD, with a median survival time of 15 days (Fig. 5A). Consistent with the X-GVHD observed in animals conditioned with 250 cGy of TBI, NOD/SCID-β2mnull mice conditioned with 300 cGy of TBI exhibited weight loss (Fig. 5B), huT engraftment in the peripheral blood (Fig. 5C), a preserved CD4+/CD8+ ratio (Fig. 5D,6B), increased human IFN-γ serum levels (Fig. 5E), and up-regulation of CD25 (Fig. 5F,6C), CD30 (Fig. 6D), and CD69 (data not shown) on a significant percentage of huT cells in the blood, spleen, liver, lung, kidney, and BM.
Figure 5.

Retention of X-GVHD-inducing potential following ex vivo activation of human T cells. NOD/SCID-β2mnull mice were sublethally irradiated with 300 cGy of TBI and left untreated (control; n = 4) or injected r.o. with 10 × 106 naive (n = 8; open bars) 4-day activated (n = 9; closed bars), or 8-day activated (n = 5; hatched bars) huT cells. Activated T cells were prepared by incubating human PBMCs with CD3/CD28 beads at a ratio of 3 beads per cell in the presence of IL-2 (50 U/mL) for 4 or 8 days. (A) Kaplan-Meier survival analysis. Mice injected with naive or activated huT cells displayed similar overall survivals that were significantly decreased compared to the untreated control. (B) Kinetics of huT cell engraftment in the peripheral blood. The proportion of huT cells in the peripheral blood was determined by flow cytometry as described in the legend to Fig. 1. (C) Percent change in pretransplant body weight. (D) CD4+/CD8+ huT cell ratio. The percentage of human CD45+CD3+ T cells in the peripheral blood that expressed CD4 or CD8 was determined by flow cytometry. (E) Human IFN-γ serum levels. Serum was obtained from peripheral blood samples collected at the indicated times after huT cell injection and human IFN-γ levels were determined using a cytometric bead assay (BD Biosciences). (F) Kinetics of CD25 expression on naive and activated huT cells. The percentage of human CD45+CD3+ T cells in the peripheral blood that expressed CD25 was determined by flow cytometry. Normal human PBMCs were used as controls in all analyses to establish consistent gating criteria for CD25. Values of p in (C–F) indicate statistically significant differences between the naive and activated huT cell groups. NS indicates differences were not statistically significant.
Figure 6.

Tissue infiltration of naive and activated human T cells in NOD/SCID-β2mnull mice. NOD/SCID-β2mnull mice were sublethally irradiated with 300 cGy of TBI and left untreated (control; n = 4) or injected r.o. with 10 × 106 naive (n = 8; open bars) 4-day activated (n = 9; closed bars), or 8-day activated (n = 5; hatched bars) huT cells. Activated T cells were prepared by incubating human PBMCs with CD3/CD28 beads at a ratio of 3 beads per cell in the presence of IL-2 (50 U/mL) for 4 or 8 days. Moribund animals with X-GVHD were euthanized and portions of blood, spleen, liver, lung, and kidney were analyzed by flow cytometry. (A) Human T cell infiltration of different tissues. The percentage of human CD45+CD3+ T cells in the indicated tissues was determined by flow cytometry. (B) CD4+/CD8+ huT cell ratio. The percentage of human CD45+CD3+ T cells in the peripheral blood that expressed CD4 or CD8 was determined by flow cytometry. (C,D) Expression of T cell activation markers. The percentage of human CD45+CD3+ T cells in the indicated tissues that expressed CD25 and CD30 was determined by flow cytometry. Normal human PBMCs were used as controls in all analyses to establish consistent gating criteria for CD25 and CD30. Values of p in indicate statistically significant differences between the naive and activated huT cell groups. NS indicates differences were not statistically significant.
Retention of X-GVHD-inducing potential following ex vivo activation of huT cells
In addition to evaluating the effect of the irradiation dose and timing of huT administration on the development of lethal X-GVHD, we also examined, in the same set of experiments, the consequence of ex vivo activation on the X-GVHD-inducing potential of huT cells in NOD/SCID-β2mnull mice. To prepare activated huT cells, we cultured human PBMCs for 4- or 8-days in media supplemented with 50 U/ml IL-2 and CD3/CD28 beads. After 4- or 8-days, the CD3/CD28 bead activated huT cells expanded 5-fold and 12-fold, respectively. Furthermore, the CD4+ to CD8+ T cell ratio changed from 1.6 ± 0.5 at the start of the activation protocol to 1.7 ± 0.4 at 4-days post-activation and 1.1 ± 0.2 at 8-days post-activation (average from 3 experiments with different donors; p = NS).
To evaluate the X-GVHD-inducing potential of the CD3/CD28-bead-activated huT cells, we conditioned NOD/SCID-β2mnull mice with 300 cGy of TBI and injected them r.o., within 6 hours, with 10 × 106 naive or activated huT cells. Similar to animals that received naive huT cells, NOD/SCID-β2mnull mice that received activated huT cells developed lethal X-GVHD with a median survival (Fig. 5A) and weight loss (Fig. 5B) that were not significantly different than animals that received naive huT cells. Interestingly, NOD/SCID-β2mnull mice injected with 4-day activated huT cells exhibited percentages of huT cells in the blood, spleen, liver, and lung (Fig. 5C, 6A), as well as human IFN-γ serum levels (Fig. 5E) that were all significantly increased compared to animals that received either naive or 8-day activated huT cells.
In addition to evaluating huT cell engraftment and organ infiltration, we also compared the activation status of the naive and activated huT cells present in the peripheral blood and tissues of the NOD/SCID-β2mnull mice. On the day of injection, <2% of the naive huT cells expressed CD25, CD30, or CD69. However, all 3 of these T cell activation markers were up-regulated following injection of the naive huT cells into the NOD/SCID-β2mnull mice. Overall, the percentage of huT cells in the peripheral blood that expressed CD25, CD30, and CD69 increased from <2% at day 3 to 28% ± 20%, 32% ± 21%, and 39% ± 14%, respectively, by day 16 after injection of naive huT cells (Fig. 5F and data not shown). At the time of sacrifice, anywhere from 10% to >80% of the injected naive huT cells present in the spleen, liver, lung, kidney, and BM expressed CD25, CD30, and CD69 (Fig. 6C,D; data not shown for CD69). These data further demonstrate that naive huT cells become activated and infiltrate multiple organs following injection into NOD/SCID-β2mnull mice.
In contrast to the naive huT cells, 95%, 43%, and 75% of the 4-day activated and 100%, 9%, and 78% of the 8-day activated huT cells expressed CD25, CD30, and CD69, respectively, at the time of injection (data not shown). Within 3 days after injection, however, 6.3 ± 3.1% of the 4-day activated and 50.7 ± 9.4% of the 8-day activated huT cells expressed CD25 in the peripheral blood, and 3.9 ± 2% and 7.4 ±2.9, respectively, expressed CD30. From day 3 onwards, the percentage of 4-day activated huT cells in the peripheral blood that expressed CD25, CD30, or CD69 increased at a rate that was similar to mice that received naive huT cells. In contrast, the percentage of 8-day activated huT cells in the peripheral blood that expressed CD25, CD30, or CD69 remained at a level that was similar to mice that received 4-day activated huT cells. Furthermore, similar to mice that received naive huT cells, we observed increased expression of all three T cell activation markers on huT cells present in the spleen, liver, lung, and kidney of mice injected with activated huT cells (Fig. 6C,D). In summary, although we observed several differences in the phenotype and engraftment of the activated cells as compared to naive huT cells after injection into NOD/SCID-β2mnull mice, these studies clearly demonstrated that huT cells retain their X-GVHD-inducing potential following ex vivo activation and expansion with CD3/CD28 beads and low levels of IL-2.
Histopathology of NOD/SCID-β2mnull mouse X-GVHD
Histopathologic and immunohistochemical analyses were performed on the liver, skin, gut, lung, kidney and salivary glands obtained from NOD/SCID-β2mnull mice that developed lethal GVHD induced by naive or activated huT cells. The histologic features of acute GVHD observed in the liver, skin and gut were very similar to those seen in human GVHD disease (Fig. 7). In the liver, the characteristic findings included portal lymphocytic infiltration (Fig. 7B), endotheliitis and hepatocyte apoptosis (Fig. 7C). A variable degree of bile duct damage with intraepithelial lymphocytic infiltration was also noted. This was in marked contrast to mice that did not develop GVHD, in which none of these histologic findings in the liver was evident (Fig. 7A). In the skin, acute GVHD manifested as apoptosis and basal vacuolar damage of the keratinocytes in the epidermis and hair follicles (Fig. 7E). Lymphocytic infiltration was noted in both epidermis and superficial dermis. The injury secondary to GVHD in the gut was relatively milder than that seen in other organs, and was evidenced by the presence of apoptotic enterocytes in the mucosa (Fig. 7F). Ulceration and significant crypt dropout were not observed.
Figure 7.
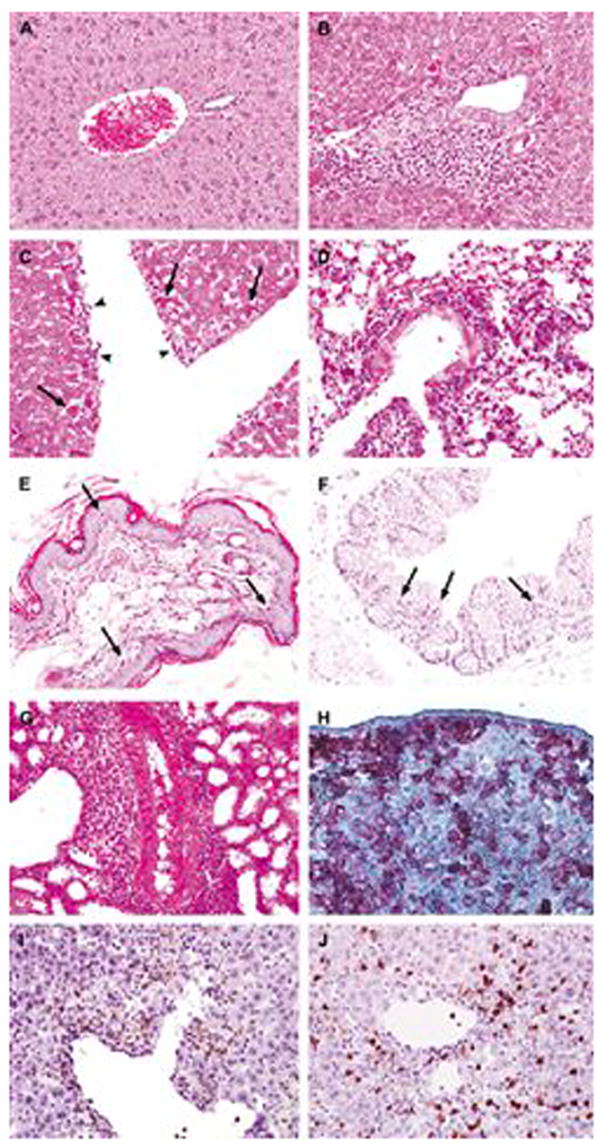
Histopathologic and immunohistochemical analysis of X-GVHD in NOD/SCID-β2mnull mice. NOD/SCID-β2mnull mice were sublethally irradiated with 300 cGy of TBI and injected r.o. with 10 × 106 naive or 10 × 106 activated huT cells. Moribund animals with X-GVHD were euthanized and portions of skin, liver, small intestine, colon, lung, kidney, salivary gland and spleen were saved for histopathologic and immunohistochemical analyses. (A) Liver from a mouse that did not show clinical or histological signs of X-GVHD following injection of huT. This liver shows an unremarkable portal tract and healthy hepatocytes. (B,C) X-GVHD in the liver characterized by (B) dense lymphocytic infiltrates in the portal tract and hepatocyte apoptosis, and (C) endotheliitis (head arrows). (D) X-GVHD in the lung showing perivascular and interstitial lymphocytic infiltration. (E) X-GVHD in the skin showing apoptosis and basal vacuolar damage of the keratinocytes. (F) X-GVHD in the colon showing apoptosis in the mucosa. (G) X-GVHD in the kidney showing prominent perivascular lymphocytic infiltration. (H) An example of immunohistochemical stains showing huT cells in X-GVHD expressing human CD45 in the spleen, (I,J) portal lymphocytes in liver X-GVHD consisting of huT lymphocytes expressing either human CD4 (I) or human CD8 (J). Slides in panels B–G were obtained from mice that received activated huT cells. Slides in panels A and H–J were obtained from mice that received naive huT cells. There were no significant differences in the pathologic damage originated by naive or activated huT cells. Black arrows indicate apoptotic cells.
The major histologic findings in the lung were perivascular, peribronchiolar and interstitial lymphocytic infiltration (Fig. 7D), accompanied by endotheliitis and bronchial epithelial apoptosis. Alveolar damage with intraalveolar edema was observed in some of the mice. Perivascular and interstitial lymphocytic infiltration was also prominent in the kidney (Fig. 7G), with a variable degree of tubulitis, epithelial apoptosis and endotheliitis. Although acute GVHD has not been well described in human lung and kidney, the histologic observations in the NOD/SCID-β2mnull mice were believed to represent acute GVHD because they recapitulated those seen in acute lung and kidney allograft rejection in humans. It has been well established that acute hepatic GVHD in humans shares major histologic features with acute allograft rejection. In addition, acute GVHD was noted in the salivary gland, which was also characterized by perivascular and interstitial lymphocytic infiltration, endotheliitis and apoptosis. Necrotizing vasculitis with thrombosis was present in some of the mice (data not shown). Immunohistochemical stains demonstrated that the infiltrative lymphocytes in various organs were huT cells that coexpressed CD45 and CD3 (Fig. 7H). They were composed of both CD4 (Fig. 7I) and CD8 (Fig. 7J) positive cells.
We developed a new scale to characterize X-GVHD in this model, following the same alterations observed in different target tissues in human GVHD. We evaluated different tissues from mice that developed lethal GVHD, and we showed that there is no significant difference in the damage observed originated by naive (n=8) or 4-day activated (n=9) huT cells (Fig. 8). Interestingly, however, we found significantly less damage to the liver, lung, skin, and kidney of mice that received 8-day activated huT cells compared to animals that received either naïve or 4-day activated huT cells. As controls, a group of mice were used that received a sub-lethal conditioning regimen and later were injected with huT cells, but did not develop lethal GVHD (n=8). This control group did not present the typical features observed in lethal GVHD (p<0.005).
Figure 8.

X-GVHD score. NOD/SCID-β2mnull mice were sublethally irradiated with 300 cGy of TBI and injected r.o. with 10 × 106 naive (n = 8; open bars) 4-day activated (n = 9; closed bars), or 8-day activated (n = 5; hatched bars) huT cells. Moribund animals with X-GVHD were euthanized and portions of skin, liver, small intestine, colon, lung, kidney, and salivary gland were saved for histopathologic analysis. Tissues were scored for pathologic damage as described in the Materials and Methods. NOD/SCID-β2mnull mice that received 250 cGy of TBI and were injected r.o. with 10 × 106 naive huT cells but did not develop lethal X-GVHD served as no X-GVHD controls. Data represent mean ± SD.
Discussion
Human acute GVHD is believed to occur in 3 phases: (phase 1) recipient conditioning, (phase 2) donor T cell activation, and (phase 3) effector cells mediating GVHD [49]. In this study, we found evidence that all three of these phases were replicated in the development of lethal X-GVHD in NOD/SCID-β2mnull mice injected with huT cells.
Studies in both humans and animals have demonstrated that acute GVHD is closely associated with the intensity and toxicity of the recipient conditioning regimen [49]. In this study, we found that TBI (phase 1) was essential to consistently induce lethal acute X-GVHD in NOD/SCID-β2mnull recipients injected with huT cells. Although we did not evaluate the development of X-GVHD in the absence of pre-transplant irradiation, others have reported no [31] or little [30] lethal X-GVHD following injection of huT into unconditioned NOD/SCID-β2mnull mice. In contrast, lethal X-GVHD is observed following administration of huT and human IL-15 to nonirradiated SCID recipients pre-treated with a monoclonal antibody to murine CD122 (to deplete residual host NK cells) [38]. In this study, we found that 20 of 34 (59%) NOD/SCID-β2mnull recipients developed lethal X-GVHD following pre-transplant conditioning with 250 cGy of TBI and r.o. injection of 10 × 106 purified huT the following day. Importantly, we increased the incidence of lethal X-GVHD in these mice to 100% (8/8) by increasing the TBI dose to 300 cGy and by decreasing the time between irradiation and huT cell injection to < 6 h. Therefore, our studies indicate that 300 cGy of TBI and retroorbital injection of huT cells within 6 h is required to attain consistent lethal X-GHVD in NOD/SCID-β2mnull recipients.
In addition to the requirement for pretransplant irradiation, we also found that the development of lethal X-GVHD in NOD/SCID-β2mnull recipients was dependent upon the route of human T cell administration. When 107 human T cells were injected via the lateral tail vein into 250 cGy NOD/SCID-β2mnull mice no lethal X-GVHD developed. However, when we administered the same cells through the retroorbital venous plexus they induced lethal X-GVHD in 59% of the similarly conditioned recipients and in all recipients irradiated with 300 cGy of TBI. Previously, Price et. al. [50] reported no biologically significant differences in the organ distribution patterns and blood decay profiles between the lateral tail vein and retroorbital routes of i.v. administration when radiolabeled chemicals (sodium pertechnetate), antibodies, or cells (thymocytes or melanoma cells) were injected into unconditioned mice. Since it was unclear why there was such a dramatic difference in the incidence of lethal X-GVHD in our studies, we used in vivo BLI to compare the trafficking patterns and expansion profiles of luciferase-modified human T cells following lateral tail vein or retroorbital injection. These studies clearly revealed that the development of lethal X-GVHD in NOD/SCID-β2mnull recipients was dependent upon the initial retention and early expansion of huT in the retroorbital sinus cavity and local regional draining lymph nodes. This profile of engraftment and early expansion was observed in all mice that were r.o.-injected with huT and developed lethal X-GVHD. In contrast, all recipients that received huT through the lateral tail vein, as well as r.o.-injected mice that failed to develop lethal X-GVHD, showed initial huT migration to the lungs and no huT expansion during the first two weeks after infusion. Interestingly, although these recipients failed to develop lethal X-GVHD, we did observe a dramatic expansion of the transferred huT cells between two and three weeks after infusion. This late huT cell expansion without the development of lethal X-GVHD may be attributed to the absence of irradiation-induced inflammatory changes that contribute to GVHD pathophysiology. [49] In murine allogeneic transplant models, others and we have shown that the severity of GVHD is reduced or abolished following delayed infusion of alloreactive donor T cells. [45,51–54]
The mechanism/s by which pre-transplant irradiation of NOD/SCID-β2mnull recipients increases the incidence of lethal X-GVHD induced by huT cells remains unknown. One possibility is that TBI provides additional immunosuppression, thereby permitting increased huT cell engraftment and subsequent development of lethal X-GVHD [23]. Consistent with this hypothesis, we found significantly increased huT cell engraftment in the peripheral blood of NOD/SCID-β2mnull mice after increasing the TBI dose from 250 cGy to 300 cGy and decreasing the time between irradiation and huT cell injection from 24 h to <6 h. A second possibility is that the pre-transplant irradiation prompts massive cytokine induction, also known as the “cytokine storm”, which promotes huT cell engraftment, activation and development of lethal X-GVHD [49]. In allogeneic transplantation, the TBI-induced release of TNF-α, IL-1, IL-6, and lipopolysaccharide (LPS) promotes the development of acute GVHD through the activation of host antigen presenting cells (APC) and up-regulation of MHC antigens [55–57]. Furthermore, it is interesting to note that in human/murine mixed lymphocyte cultures performed in the absence of human APCs, only activated, but not resting, murine APCs can stimulate resting huT cells [58]. This inability of huT cells to interact effectively with resting murine APCs may explain why TBI, and the subsequent activation of murine APCs, is required to induce lethal X-GVHD in NOD/SCID-β2mnull recipients. One final factor likely contributing to the development of lethal X-GVHD in irradiated NOD/SCID-β2mnull mice is the probable increased trafficking of xenoreactive huT cells to GVHD target tissues following total body irradiation. Indeed, Chakraverty et al [59] recently showed in a murine allogeneic transplant model that irradiation-induced tissue inflammation is a prerequisite for the migration of alloreactive T cells to GVHD target tissues and the induction of GVHD.
During the second phase of human acute GVHD, allogeneic donor T cells become activated by encountering residual host APCs or donor-derived APCs, which cross present host alloantigens [49]. Although this allo-response is believed to be mediated primarily by dendritic cells (DCs), other nonprofessional APCs, including B cells, macrophages and non-hematopoietic cells, may play a role. We report here that both human CD4+ and CD8+ T cells become activated following r.o. injection into irradiated NOD/SCID-β2mnull mice. The up-regulation of the early T cell activation markers CD25, CD30, and CD69 and the secretion of large quantities of human IFN-γ characterize the huT cell activation. Interestingly, both CD25 and CD30 up-regulation, as well as serum levels of human IFN-γ, served as useful predictors for which mice will develop lethal X-GVHD following the injection of huT cells. Future experiments will determine whether therapies targeting these markers will mitigate X-GVHD induced by huT cells in the NOD/SCID-β2mnull mice.
Because we injected cell populations that were >99% CD3+, the xenoreactive human CD4+ and CD8+ T cells were most likely stimulated by murine APCs. However, it remains unknown which specific murine APC population mediated this activation in the NOD/SCID-β2mnull mice. In fact, the abundance and function of DCs, as well as other APC subsets, in NOD/SCID-β2mnull mice and most other immunodeficient mouse strains have not been evaluated. Determining the lineage and function of APCs that mediate the graft-versus-host reaction in the NOD/SCID-β2mnull X-GVHD model should help elucidate the pathophysiology of human acute GVHD at the cellular and molecular levels.
Another interesting question that arises from our studies is the mechanism by which human CD8+ T cells become activated in the irradiated NOD/SCID-β2mnull recipients. Previous studies by others have established that human CD4+ T cells can recognize and become activated by xenoantigens presented via murine MHC class II molecules [60,61]. Since NOD/SCID-β2mnull mice have normal MHC class II function, they should retain their ability to present xenoantigens to human CD4+ T cells. However, because of the lack of β2m, which is important for the assembly of stable MHC I molecules, NOD/SCID-β2mnull mice are considered to be MHC class I deficient and therefore, residual APCs in these mice should be poor stimulators of human CD8+ T cells. One possible explanation for our detection of activated human CD8+ T cells in irradiated NOD/SCID-β2mnull mice may reside in the fact that functional class I molecules can assemble in the absence of β2m [62,63]. Alternatively, it may be that the human CD8+ T cells are displaying an activated phenotype because of homeostatic proliferation or by the recognition of xenoantigens being cross-presented by activated human CD4+ cells present in the graft.
During the final phase of human acute GVHD, effector donor T cells mediate tissue injury in various organs through direct cytotoxic activity or the production of inflammatory cytokines [49]. In this study, we demonstrated high numbers of activated huT cells in the spleen, liver, lung, kidney and bone marrow of NOD/SCID-β2mnull mice that developed lethal X-GVHD by flow cytometry and immunohistochemistry. However, the most significant finding was that the histopathology of X-GVHD induced by huT cells in the NOD/SCID-β2mnull mice closely resembled what is observed in human acute GVHD.
Data from both mouse and human allogeneic studies have shown that donor T cells exhibit decreased allo-reactivity and GVHD potential following ex vivo activation and expansion [2–6]. This loss of allo-reactivity is extremely undesirable because both the conversion to full donor chimerism and the anti-leukemia effect after allogeneic HSCT are mediated, at least in part, by alloreactive donor T cells. We chose to evaluate the CD3/CD28 bead method of T cell stimulation because several recent studies by others have demonstrated that huT cells activated and expanded by CD3/CD28 beads maintain a broad T cell repertoire with retention of antigen specific CD4 and CD8 cells, are capable of reactivation upon re-stimulation, and survive long term following infusion into patients with HIV infections or a variety of cancers [9–12]. Unique to our protocol, however, is the use of low concentrations of IL-2 (50 U/ml) added only 24 hours after culture initiation. Using this activation protocol, we can report that CD3/CD28 bead-activated huT cells retain the capacity to recognize antigen, proliferate, and induce lethal X-GVHD in NOD/SCID-β2mnull recipients. In fact, compared to mice injected with naive huT cells, NOD/SCID-β2mnull recipients injected with 4-day CD3/CD28 bead activated T cells displayed increased engraftment and serum IFN-γ levels, with similar weight loss, histological GVHD scores, and induction of lethal X-GVHD. Interestingly, although the X-GVHD-inducing potential of 4- and 8-day activated huT cells were similar, we found that the engraftment, in-vivo expansion, and tissue infiltration of 8-day activated huT cells was significantly decreased compared to cells that were cultured for only 4 days. This decreased huT cell engraftment and expansion following prolonged ex-vivo activation may explain why Bondaza et al [39] observed decreased lethal X-GVHD following the i.p. injection of 12-day CD3/CD28 bead activated huT into anti-CD122 treated and irradiated NOD/SCID recipients (58% lethal X-GVHD for 12-day activated huT versus 100% for non-activated huT). In summary, our findings further illustrate the preservation of huT cell function following CD3/CD28 bead stimulation and will allow us to evaluate the efficacy of GVHD prevention strategies, such as suicide gene therapy [39], requiring ex vivo T cell activation and expansion.
The present study describes the conditions that allow the induction of acute X-GVHD in NOD/SCID-β2mnull recipient mice following injection of purified huT cells. Importantly, the pathogenesis of this acute X-GVHD was very similar to human allogeneic acute GVHD. The xenoreactive huT cells initiated a rapidly progressing illness characterized by tissue injury in various organs, including the gut, liver and skin. Therefore, this NOD/SCID-β2mnull model provides a system to study the pathophysiology of acute GVHD induced by huT cells and aids in the development of therapeutic strategies for GVHD.
Table 1.
X-GVHD histopathologic scoring system.
| Organ | Histologic findings | Organ | Histologic findings |
|---|---|---|---|
| Small bowel | Villous blunting
Luminal sloughing cellular debris Crypt cell apoptosis Crypt regeneration Outright crypt destruction (loss) Lamina propria lymphocytic infiltrate Mucosal ulceration |
Colon | Surface colonocyte vacuolation
Surface colonocyte attentuation Crypt cell apoptosis Crypt regeneration Outright crypt destruction (loss) Lamina propria lymphocytic infiltrate Mucosal ulceration |
| Lung | Perivascular lymphocytic infiltrate
Interstitial lymphocytic infiltrate Peribronchiolar lymphocytic infiltrate Endotheliitis Bronchial epithelial apoptosis Bronchial epithelial detachment Alveolar edema Alveolar debris Alveolar damage |
Kidney | Perivascular lymphocytic infiltrate
Interstitial lymphocytic infiltrate Tubulitis Vasculitis Glomerulitis Interstitial fibrosis Tubular atrophy Glomerular sclerosis Apoptosis |
| Salivary gland | Perivascular lymphocytic infiltrate
Interstitial lymphocytic infiltrate Vasculitis Epithelial apoptosis Regeneration Luminal debris Loss of duct or acinus Necrosis |
Liver | Portal lymphocytic infiltrate
Bile duct lymphocytic infiltrate Bile duct epithelial apoptosis Bile duct epithelial sloughing Endotheliitis Hepatocyte apoptosis Microabscess Hepatocyte mitosis Cholestasis Steatosis |
| Skin | Basal vacuolar damage
Epidermal lymphocytic infiltrate Apoptosis in epidermis/follicle Dermal lymphocytic infiltrate Cleft and microvesicle formation Separation epidermis from dermis |
Spleen | Splenomegaly
Structural damage Fibrosis Vasculitis |
A semiquantitative scoring system was used to assess abnormalities associated with GVHD or allograft rejection. The scoring system designated 0 as normal, 0.5 as focal and rare, 1.0 as focal and mild, 2.0 as diffuse and mild, 3.0 as diffuse and moderate, and 4.0 as diffuse and severe. Scores were added to provide a total score for each specimen.
Acknowledgments
This research was supported by National Institutes of Health grants RO1 CA83845 (JFD), R21 CA110489 (JFD), and P50 CA94056(DP-W). We thank Dave Hess (Washington University School of Medicine) for technical assistance.
Footnotes
Abbreviations used in this paper: GVHD, graft vs host disease; HSCT, hematopoietic stem cell transplantation; huT, human T cells; X-GVHD, xenogeneic GVHD; huPBMCs, human peripheral blood mononuclear cells; β2mnull, β2-microglobulin-null; TBI, total body irradiation; r.o., retro-orbital; CBRluc, Click Beetle Red luciferase; EGFP, enhanced green fluorescent protein; BLI, bioluminescence imaging; EDTA, ethylenediaminetetraacetic acid.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Appelbaum FR. The current status of hematopoietic cell transplantation. Annu Rev Med. 2003;54:491–512. doi: 10.1146/annurev.med.54.101601.152456. [DOI] [PubMed] [Google Scholar]
- 2.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 3.Contassot E, Murphy W, Angonin R, Pavy JJ, Bittencourt MC, Robinet E, Reynolds CW, Cahn JY, Herve P, Tiberghien P. In vivo alloreactive potential of ex vivo-expanded primary T lymphocytes. Transplantation. 1998;65:1365–1370. doi: 10.1097/00007890-199805270-00014. [DOI] [PubMed] [Google Scholar]
- 4.Drobyski WR, Majewski D, Ozker K, Hanson G. Ex vivo anti-CD3 antibody-activated donor T cells have a reduced ability to cause lethal murine graft-versus-host disease but retain their ability to facilitate alloengraftment. J Immunol. 1998;161:2610–2619. [PubMed] [Google Scholar]
- 5.Tiberghien P. Use of suicide gene-expressing donor T-cells to control alloreactivity after haematopoietic stem cell transplantation. J Intern Med. 2001;249:369–377. doi: 10.1046/j.1365-2796.2001.00809.x. [DOI] [PubMed] [Google Scholar]
- 6.Weijtens M, van Spronsen A, Hagenbeek A, Braakman E, Martens A. Reduced graft-versus-host disease-inducing capacity of T cells after activation, culturing, and magnetic cell sorting selection in an allogeneic bone marrow transplantation model in rats. Hum Gene Ther. 2002;13:187–198. doi: 10.1089/10430340252769725. [DOI] [PubMed] [Google Scholar]
- 7.Trickett A, Kwan YL. T cell stimulation and expansion using anti-CD3/CD28 beads. J Immunol Methods. 2003;275:251–255. doi: 10.1016/s0022-1759(03)00010-3. [DOI] [PubMed] [Google Scholar]
- 8.Kalamasz D, Long SA, Taniguchi R, Buckner JH, Berenson RJ, Bonyhadi M. Optimization of human T-cell expansion ex vivo using magnetic beads conjugated with anti-CD3 and Anti-CD28 antibodies. J Immunother. 2004;27:405–418. doi: 10.1097/00002371-200409000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Levine BL, Bernstein WB, Aronson NE, Schlienger K, Cotte J, Perfetto S, Humphries MJ, Ratto-Kim S, Birx DL, Steffens C, Landay A, Carroll RG, June CH. Adoptive transfer of costimulated CD4+ T cells induces expansion of peripheral T cells and decreased CCR5 expression in HIV infection. Nat Med. 2002;8:47–53. doi: 10.1038/nm0102-47. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein WB, Cox JH, Aronson NE, Tracy L, Schlienger K, Ratto-Kim S, Garner R, Cotte J, Zheng Z, Winestone L, Liebig C, Galley LM, Connors M, Birx DL, Carroll RG, Levine BL. Immune reconstitution following autologous transfers of CD3/CD28 stimulated CD4(+) T cells to HIV-infected persons. Clin Immunol. 2004;111:262–274. doi: 10.1016/j.clim.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Thompson JA, Figlin RA, Sifri-Steele C, Berenson RJ, Frohlich MW. A phase I trial of CD3/CD28-activated T cells (Xcellerated T cells) and interleukin-2 in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2003;9:3562–3570. [PubMed] [Google Scholar]
- 12.Laport GG, Levine BL, Stadtmauer EA, Schuster SJ, Luger SM, Grupp S, Bunin N, Strobl FJ, Cotte J, Zheng Z, Gregson B, Rivers P, Vonderheide RH, Liebowitz DN, Porter DL, June CH. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood. 2003;102:2004–2013. doi: 10.1182/blood-2003-01-0095. [DOI] [PubMed] [Google Scholar]
- 13.Bonyhadi M, Frohlich M, Rasmussen A, Ferrand C, Grosmaire L, Robinet E, Leis J, Maziarz RT, Tiberghien P, Berenson RJ. In vitro engagement of CD3 and CD28 corrects T cell defects in chronic lymphocytic leukemia. J Immunol. 2005;174:2366–2375. doi: 10.4049/jimmunol.174.4.2366. [DOI] [PubMed] [Google Scholar]
- 14.Tary-Lehmann M, Saxon A. Human mature T cells that are anergic in vivo prevail in SCID mice reconstituted with human peripheral blood. J Exp Med. 1992;175:503–516. doi: 10.1084/jem.175.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tary-Lehmann M, Lehmann PV, Schols D, Roncarolo MG, Saxon A. Anti-SCID mouse reactivity shapes the human CD4+ T cell repertoire in hu-PBL-SCID chimeras. J Exp Med. 1994;180:1817–1827. doi: 10.1084/jem.180.5.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tary-Lehmann M, Saxon A, Lehmann PV. The human immune system in hu-PBL-SCID mice. Immunol Today. 1995;16:529–533. doi: 10.1016/0167-5699(95)80046-8. [DOI] [PubMed] [Google Scholar]
- 17.Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature. 1988;335:256–259. doi: 10.1038/335256a0. [DOI] [PubMed] [Google Scholar]
- 18.Hesselton RM, Koup RA, Cromwell MA, Graham BS, Johns M, Sullivan JL. Human peripheral blood xenografts in the SCID mouse: characterization of immunologic reconstitution. J Infect Dis. 1993;168:630–640. doi: 10.1093/infdis/168.3.630. [DOI] [PubMed] [Google Scholar]
- 19.Murphy WJ, Bennett M, Anver MR, Baseler M, Longo DL. Human-mouse lymphoid chimeras: host-vs.-graft and graft-vs.-host reactions. Eur J Immunol. 1992;22:1421–1427. doi: 10.1002/eji.1830220614. [DOI] [PubMed] [Google Scholar]
- 20.Hoffmann-Fezer G, Gall C, Zengerle U, Kranz B, Thierfelder S. Immunohistology and immunocytology of human T-cell chimerism and graft-versus-host disease in SCID mice. Blood. 1993;81:3440–3448. [PubMed] [Google Scholar]
- 21.Martino G, Anastasi J, Feng J, Mc Shan C, DeGroot L, Quintans J, Grimaldi LM. The fate of human peripheral blood lymphocytes after transplantation into SCID mice. Eur J Immunol. 1993;23:1023–1028. doi: 10.1002/eji.1830230506. [DOI] [PubMed] [Google Scholar]
- 22.Garcia S, Dadaglio G, Gougeon ML. Limits of the human-PBL-SCID mice model: severe restriction of the V beta T-cell repertoire of engrafted human T cells. Blood. 1997;89:329–336. [PubMed] [Google Scholar]
- 23.Tournoy KG, Depraetere S, Pauwels RA, Leroux-Roels GG. Mouse strain and conditioning regimen determine survival and function of human leucocytes in immunodeficient mice. Clin Exp Immunol. 2000;119:231–239. doi: 10.1046/j.1365-2249.2000.01099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, McKenna S, Mobraaten L, Rajan TV, Greiner DL, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154:180–191. [PubMed] [Google Scholar]
- 25.Prochazka M, Gaskins HR, Shultz LD, Leiter EH. The nonobese diabetic scid mouse: model for spontaneous thymomagenesis associated with immunodeficiency. Proc Natl Acad Sci U S A. 1992;89:3290–3294. doi: 10.1073/pnas.89.8.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorin NC, Piantadosi S, Stull M, Bonte H, Wingard JR, Civin C. Increased risk of lethal graft-versus-host disease-like syndrome after transplantation into NOD/SCID mice of human mobilized peripheral blood stem cells, as compared to bone marrow or cord blood. J Hematother Stem Cell Res. 2002;11:277–292. doi: 10.1089/152581602753658466. [DOI] [PubMed] [Google Scholar]
- 27.Hesselton RM, Greiner DL, Mordes JP, Rajan TV, Sullivan JL, Shultz LD. High levels of human peripheral blood mononuclear cell engraftment and enhanced susceptibility to human immunodeficiency virus type 1 infection in NOD/LtSz-scid/scid mice. J Infect Dis. 1995;172:974–982. doi: 10.1093/infdis/172.4.974. [DOI] [PubMed] [Google Scholar]
- 28.Kerre TC, De Smet G, De Smedt M, Zippelius A, Pittet MJ, Langerak AW, De Bosscher J, Offner F, Vandekerckhove B, Plum J. Adapted NOD/SCID model supports development of phenotypically and functionally mature T cells from human umbilical cord blood CD34(+) cells. Blood. 2002;99:1620–1626. doi: 10.1182/blood.v99.5.1620. [DOI] [PubMed] [Google Scholar]
- 29.Greiner DL, Shultz LD, Yates J, Appel MC, Perdrizet G, Hesselton RM, Schweitzer I, Beamer WG, Shultz KL, Pelsue SC, et al. Improved engraftment of human spleen cells in NOD/LtSz-scid/scid mice as compared with C.B-17-scid/scid mice. Am J Pathol. 1995;146:888–902. [PMC free article] [PubMed] [Google Scholar]
- 30.Berney T, Molano RD, Pileggi A, Cattan P, Li H, Ricordi C, Inverardi L. Patterns of engraftment in different strains of immunodeficient mice reconstituted with human peripheral blood lymphocytes. Transplantation. 2001;72:133–140. doi: 10.1097/00007890-200107150-00026. [DOI] [PubMed] [Google Scholar]
- 31.Christianson SW, Greiner DL, Hesselton RA, Leif JH, Wagar EJ, Schweitzer IB, Rajan TV, Gott B, Roopenian DC, Shultz LD. Enhanced human CD4+ T cell engraftment in beta2-microglobulin-deficient NOD-scid mice. J Immunol. 1997;158:3578–3586. [PubMed] [Google Scholar]
- 32.Pflumio F, Lapidot T, Murdoch B, Patterson B, Dick JE. Engraftment of human lymphoid cells into newborn SCID mice leads to graft-versus-host disease. Int Immunol. 1993;5:1509–1522. doi: 10.1093/intimm/5.12.1509. [DOI] [PubMed] [Google Scholar]
- 33.Shpitz B, Chambers CA, Singhal AB, Hozumi N, Fernandes BJ, Roifman CM, Weiner LM, Roder JC, Gallinger S. High level functional engraftment of severe combined immunodeficient mice with human peripheral blood lymphocytes following pretreatment with radiation and anti-asialo GM1. J Immunol Methods. 1994;169:1–15. doi: 10.1016/0022-1759(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 34.Sandhu JS, Gorczynski R, Shpitz B, Gallinger S, Nguyen HP, Hozumi N. A human model of xenogeneic graft-versus-host disease in SCID mice engrafted with human peripheral blood lymphocytes. Transplantation. 1995;60:179–184. [PubMed] [Google Scholar]
- 35.Tsuchida M, Brown SA, Tutt LM, Tan J, Seehafer DL, Harris JP, Xun CQ, Thompson JS. A model of human anti-T-cell monoclonal antibody therapy in SCID mice engrafted with human peripheral blood lymphocytes. Clin Transplant. 1997;11:522–528. [PubMed] [Google Scholar]
- 36.Cao T, Leroux-Roels G. Antigen-specific T cell responses in human peripheral blood leucocyte (hu-PBL)-mouse chimera conditioned with radiation and an antibody directed against the mouse IL-2 receptor beta-chain. Clin Exp Immunol. 2000;122:117–123. doi: 10.1046/j.1365-2249.2000.01340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Rijn RS, Simonetti ER, Hagenbeek A, Hogenes MC, de Weger RA, Canninga-van Dijk MR, Weijer K, Spits H, Storm G, van Bloois L, Rijkers G, Martens AC, Ebeling SB. A new xenograft model for graft-versus-host disease by intravenous transfer of human peripheral blood mononuclear cells in RAG2−/− gammac−/− double-mutant mice. Blood. 2003;102:2522–2531. doi: 10.1182/blood-2002-10-3241. [DOI] [PubMed] [Google Scholar]
- 38.Roychowdhury S, Blaser BW, Freud AG, Katz K, Bhatt D, Ferketich AK, Bergdall V, Kusewitt D, Baiocchi RA, Caligiuri MA. IL-15 but not IL-2 rapidly induces lethal xenogeneic graft-versus-host disease. Blood. 2005;106:2433–2435. doi: 10.1182/blood-2005-04-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bondanza A, Valtolina V, Magnani Z, Ponzoni M, Fleischhauer K, Bonyhadi M, Traversari C, Sanvito F, Toma S, Radrizzani M, La Seta-Catamancio S, Ciceri F, Bordignon C, Bonini C. Suicide gene therapy of graft-versus-host disease induced by central memory human T lymphocytes. Blood. 2006;107:1828–1836. doi: 10.1182/blood-2005-09-3716. [DOI] [PubMed] [Google Scholar]
- 40.Sun A, Wei H, Sun R, Xiao W, Yang Y, Tian Z. Human interleukin-15 improves engraftment of human T cells in NOD-SCID mice. Clin Vaccine Immunol. 2006;13:227–234. doi: 10.1128/CVI.13.2.227-234.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kollet O, Peled A, Byk T, Ben-Hur H, Greiner D, Shultz L, Lapidot T. beta2 microglobulin-deficient (B2m(null)) NOD/SCID mice are excellent recipients for studying human stem cell function. Blood. 2000;95:3102–3105. [PubMed] [Google Scholar]
- 42.Angelopoulou MK, Rinder H, Wang C, Burtness B, Cooper DL, Krause DS. A preclinical xenotransplantation animal model to assess human hematopoietic stem cell engraftment. Transfusion. 2004;44:555–566. doi: 10.1111/j.1537-2995.2004.03285.x. [DOI] [PubMed] [Google Scholar]
- 43.Pinkerton W, Webber M. A Method of Injecting Small Laboratory Animals by the Ophthalmic Plexus Route. Proc Soc Exp Biol Med. 1964;116:959–961. doi: 10.3181/00379727-116-29420. [DOI] [PubMed] [Google Scholar]
- 44.Rettig MP, Ritchey JK, Meyerrose TE, Haug JS, DiPersio JF. Transduction and selection of human T cells with novel CD34/thymidine kinase chimeric suicide genes for the treatment of graft-versus-host disease. Mol Ther. 2003;8:29–41. doi: 10.1016/s1525-0016(03)00142-4. [DOI] [PubMed] [Google Scholar]
- 45.Rettig MP, Ritchey JK, Prior JL, Haug JS, Piwnica-Worms D, DiPersio JF. Kinetics of in vivo elimination of suicide gene-expressing T cells affects engraftment, graft-versus-host disease, and graft-versus-leukemia after allogeneic bone marrow transplantation. J Immunol. 2004;173:3620–3630. doi: 10.4049/jimmunol.173.6.3620. [DOI] [PubMed] [Google Scholar]
- 46.Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J, Jr, Ferrara JL. Tumor necrosis factor- alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102:1882–1891. doi: 10.1172/JCI4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luker GD, Bardill JP, Prior JL, Pica CM, Piwnica-Worms D, Leib DA. Noninvasive bioluminescence imaging of herpes simplex virus type 1 infection and therapy in living mice. J Virol. 2002;76:12149–12161. doi: 10.1128/JVI.76.23.12149-12161.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luker GD, Pica CM, Song J, Luker KE, Piwnica-Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat Med. 2003;9:969–973. doi: 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- 49.Reddy P. Pathophysiology of acute graft-versus-host disease. Hematol Oncol. 2003;21:149–161. doi: 10.1002/hon.716. [DOI] [PubMed] [Google Scholar]
- 50.Price JE, Barth RF, Johnson CW, Staubus AE. Injection of cells and monoclonal antibodies into mice: comparison of tail vein and retroorbital routes. Proc Soc Exp Biol Med. 1984;177:347–353. doi: 10.3181/00379727-177-41955. [DOI] [PubMed] [Google Scholar]
- 51.Sykes M, Sheard MA, Sachs DH. Graft-versus-host-related immunosuppression is induced in mixed chimeras by alloresponses against either host or donor lymphohematopoietic cells. J Exp Med. 1988;168:2391–2396. doi: 10.1084/jem.168.6.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson BD, Truitt RL. Delayed infusion of immunocompetent donor cells after bone marrow transplantation breaks graft-host tolerance allows for persistent antileukemic reactivity without severe graft-versus-host disease. Blood. 1995;85:3302–3312. [PubMed] [Google Scholar]
- 53.Blazar BR, Lees CJ, Martin PJ, Noelle RJ, Kwon B, Murphy W, Taylor PA. Host T cells resist graft-versus-host disease mediated by donor leukocyte infusions. J Immunol. 2000;165:4901–4909. doi: 10.4049/jimmunol.165.9.4901. [DOI] [PubMed] [Google Scholar]
- 54.Billiau AD, Fevery S, Rutgeerts O, Landuyt W, Waer M. Crucial role of timing of donor lymphocyte infusion in generating dissociated graft-versus-host and graft-versus-leukemia responses in mice receiving allogeneic bone marrow transplants. Blood. 2002;100:1894–1902. doi: 10.1182/blood-2002-02-0419. [DOI] [PubMed] [Google Scholar]
- 55.Xun CQ, Thompson JS, Jennings CD, Brown SA, Widmer MB. Effect of total body irradiation, busulfan-cyclophosphamide, or cyclophosphamide conditioning on inflammatory cytokine release and development of acute and chronic graft-versus-host disease in H-2-incompatible transplanted SCID mice. Blood. 1994;83:2360–2367. [PubMed] [Google Scholar]
- 56.Chang RJ, Lee SH. Effects of interferon-gamma and tumor necrosis factor-alpha on the expression of an Ia antigen on a murine macrophage cell line. J Immunol. 1986;137:2853–2856. [PubMed] [Google Scholar]
- 57.Pober JS, Orosz CG, Rose ML, Savage CO. Can graft endothelial cells initiate a host anti-graft immune response. Transplantation. 1996;61:343–349. doi: 10.1097/00007890-199602150-00001. [DOI] [PubMed] [Google Scholar]
- 58.Lucas PJ, Bare CV, Gress RE. The human anti-murine xenogeneic cytotoxic response. II. Activated murine antigen-presenting cells directly stimulate human T helper cells. J Immunol. 1995;154:3761–3770. [PubMed] [Google Scholar]
- 59.Chakraverty R, Cote D, Buchli J, Cotter P, Hsu R, Zhao G, Sachs T, Pitsillides CM, Bronson R, Means T, Lin C, Sykes M. An inflammatory checkpoint regulates recruitment of graft-versus-host reactive T cells to peripheral tissues. J Exp Med. 2006;203:2021–2031. doi: 10.1084/jem.20060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lucas PJ, Shearer GM, Neudorf S, Gress RE. The human antimurine xenogeneic cytotoxic response. I. Dependence on responder antigen-presenting cells. J Immunol. 1990;144:4548–4554. [PubMed] [Google Scholar]
- 61.Duchosal MA, Mauray S, Ruegg M, Trouillet P, Vallet V, Aarden L, Tissot JD, Schapira M. Human peripheral blood leukocyte engraftment into SCID mice: critical role of CD4(+) T cells. Cell Immunol. 2001;211:8–20. doi: 10.1006/cimm.2001.1822. [DOI] [PubMed] [Google Scholar]
- 62.Bix M, Raulet D. Functionally conformed free class I heavy chains exist on the surface of beta 2 microglobulin negative cells. J Exp Med. 1992;176:829–834. doi: 10.1084/jem.176.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Glas R, Franksson L, Ohlen C, Hoglund P, Koller B, Ljunggren HG, Karre K. Major histocompatibility complex class I-specific and -restricted killing of beta 2-microglobulin-deficient cells by CD8+ cytotoxic T lymphocytes. Proc Natl Acad Sci U S A. 1992;89:11381–11385. doi: 10.1073/pnas.89.23.11381. [DOI] [PMC free article] [PubMed] [Google Scholar]