

Abstract
This review deals with the most recent findings on the antimalarial, antimycobacterial, and antifungal properties of fatty acids, with particular emphasis on novel marine fatty acids. The first section deals with the most recent and some background literature on what has been the latest developments with respect to fatty acids as antimalarial agents and the importance of enzyme inhibition, in particular the inhibition of the enoyl-ACP-reductase (Fab I) of P. falciparum, the principal agent responsible for malaria. This section of the review also emphasizes the latest antimalarial research with the very long-chain Δ5,9 fatty acids from sponges. The second section of the review deals with the recent literature on the antimycobacterial activity of fatty acids and the importance of enzyme inhibition, in particular the inhibition of the enoyl-ACP-reductase (InhA) of M. tuberculosis for antimycobacterial activity. The inhibitory activities of the Δ5,9 fatty acids against InhA as well as that of the α-methoxylated fatty acids are also discussed. The importance of Δ5,9 fatty acids as topoisomerase I inhibitors and its connection to cancer is also reviewed. The last part of the review, the antifungal section, also emphasizes the most recent research with antifungal fatty acids and the importance of enzyme inhibition, in particular N-myristoyltransferase (NMT) inhibition, for antifungal activity. This last section of the review emphasizes the latest research with the α-methoxylated fatty acids but the importance of acetylenic fatty acids is also considered.
1. Introduction
Fatty acids are ubiquitous in nature but marine organisms, in particular sponges, have provided some of the most interesting structural varieties. Many of these marine fatty acids originate from unusual biosynthetic pathways and excellent reviews have appeared in recent years as to the fatty acid structural types present in these organisms, their possible role in membranes, and their biogenesis [1–4]. However, little is known, or has been reviewed, as to the biomedical potential of these unusual sponge fatty acids; in particular as to what differences exist in their bioactivity as compared to what has been reported for the more common fatty acids. Due to recent research activity, we are now beginning to learn more as to the potential of these marine compounds to combat infectious diseases such as malaria, tuberculosis, and fungal infections. In particular, the sponge very long-chain Δ5,9 fatty acids and the α-methoxylated fatty acids are beginning to unveil some interesting structure-activity relationships with respect to the biomedical potential of these lipids. The aim of this review is to present the progress as to what is known of the bioactivity of these unusual marine fatty acids and what future research directions we could envisage.
2. Malaria
2.1. Introduction to the disease
Malaria is one of the most important tropical parasitic diseases [5]. It is estimated by the World Health Organization (WHO) that there are around 300–500 million acute clinical malarial cases every year and around 1 million deaths do occur every year [5]. Malaria is mainly encountered within the poorest populations in the World and it is widely spread in Africa, Asia, and in several South American countries. Malaria is mainly caused by four species of Plasmodium, but Plasmodium falciparum is responsible for the most severe and deadly form of the disease and is to blame for 90% of malaria-related deaths occurring in Africa [5].
Treatment of malaria is of high priority to the National Institutes of Health (NIH) and the magnitude of the problem calls for multiple approaches to tackle this world-wide problem. At present the therapeutic efforts are concentrating in three main areas: (a) vaccine development, (b) drug development, and (c) pathogenesis. Within drug development there is a constant need to develop new drugs to improve existing ones for the treatment of malarial infections due to the unending problem of the growing resistance to known drugs. There is urgency to identify and characterize unique parasite biochemical pathways that may serve as targets for new drugs, to determine the mode of action of existing and potential new drugs, and to elucidate possible mechanisms of resistance to existing drugs. At present available drugs respond to three classes of compounds: (a) aryl aminoalcohol compounds such as quinine, (b) antifolates-dihydrofolate reductase inhibitors such as pyrimethamine, and (c) artemisinin derivatives. Artemisinin was first isolated in 1970 by Chinese scientists from Artemisia annua [6–7]. However, treatment with only one drug is not sufficient and now there is a general agreement between scientists that combinations of two drugs probably offer the best option for treatment and reduces the risk of resistance. Examples of drug combinations are the artemisinin-amodiaquine pair and the artemether-lumefantine combination (Coartem) [8]. More recent developments call for fixed dose formulations which are becoming popular within the pharmaceutical industry since they improve patient compliance by reducing the daily dose, e.g., less tablets per day, they are less expensive, and the formulation reduces the risk of patients taking only one of the active drugs in a combinatorial treatment, which can contribute to the development of resistance [5].
2.2. Fatty acid biosynthesis as a target in antimalarial chemotherapy
Malaria chemotherapy is an area that is in continuous growth and revision due to the limited number of drugs presently available, the severe side effects of available drugs, and the continuous development of resistance developed by the parasite to some of these drugs [9]. Plasmodium falciparum, the most deadly malarial parasite of the phylum Apicomplexa, has been found to contain an apicoplast, an organelle that originally arised from a cyanobacterium through a secondary endosymbiotic process and thus possesses two membranes [10]. The apicoplast is indispensable for the malarial parasite since several vital metabolic processes for the parasite do occur at this site. Among these processes isoprene biosynthesis, haem biosynthesis, and fatty acid biosynthesis take place. However, the fatty acid biosynthesis that does occur at the apicoplast is different from the fatty acid biosynthesis normally found in humans or in other higher eukaryotes. Higher eukaryotes normally use a type I fatty acid synthase (FAS I) system, where each fatty acid biosynthetic step is catalyzed by a single protein with multiple domains. On the other hand, in the apicoplast a type II fatty acid synthase (FAS II) system is operative, where each fatty acid biosynthetic pathway is carried out by a discrete enzyme encoded by a different gene [11]. This type II FAS system is absent in humans since we are eukaryotic in nature but is common in bacteria and algae [12]. If we were to interfere with the plasmodial type II FAS system we can then destroy the parasite without harming the human host. Fatty acid biosynthesis is critical for the parasite inasmuch as fatty acids are needed for the construction of cell membranes, they are an important source of energy, they play a key role in signal transduction as well as in protein acylation, and they are needed for the growth, differentiation and homeostasis in P. falciparum. It is also known that lipid biosynthesis is elevated during the erythrocytic phases of the parasite [12]. When the parasite is invading a host it needs to protect itself by creating a so-called parasitophorous vacuole, in part as a protection from the immune system of the host. In this process the parasite needs to make its own fatty acids de novo so as to form and expand its membrane. In P. falciparum the principal membrane fatty acids are decanoic acid (10:0), lauric acid (12:0), and myristic acid (14:0).
There are several enzymes responsible for the biosynthesis of fatty acids in P. falciparum as well as in a typical type II fatty acid biosynthetic scheme (Scheme 1). This biosynthetic pathway incorporates several enzymes that can be inhibited by drugs. Some interesting drug examples are isoniazid (which inhibits Fab I), triclosan (which also inhibits Fab I), and thiolactomycin and derivatives (that inhibit Fab B and Fab H) [13–14]. Among these enzymes, the enoyl-ACP reductase (PfFabI enzyme) has been particularly favored for inhibition and chemical intervention. The active site of the PfFabI enzyme is known and a crystal structure of the enzyme is available incorporating the triclosan inhibitor and the cofactor NADH in the active site [15].
Scheme 1.
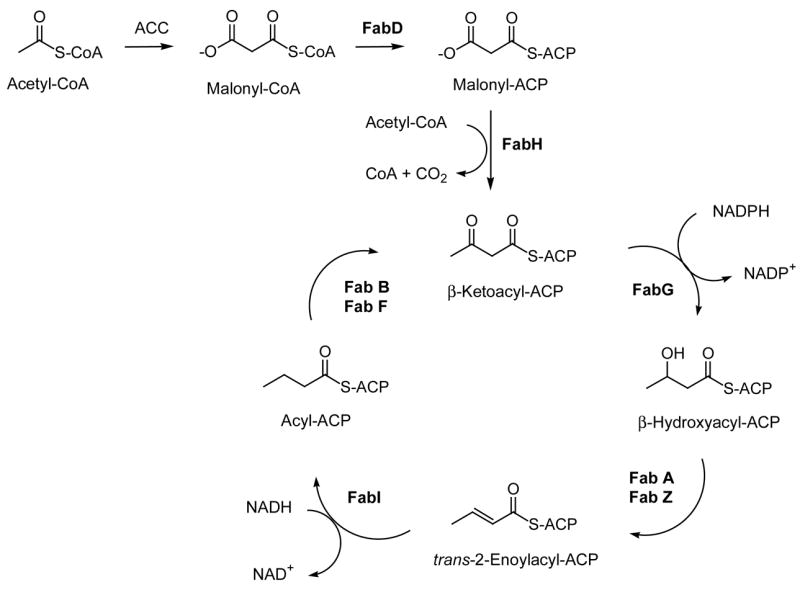
Type II fatty acid biosynthesis and the key enzymes in the process. ACC: acetyl-CoA carboxylase, ACP: acyl carrier protein, FabD: ACP transacylase, FabH: β-ketoacyl-ACP synthase III, FabG: β-ketoacyl-ACP reductase, FabA: β-hydroxydecanoyl–ACP dehydratase/isomerase, FabZ: β-hydroxyacyl-ACP dehydratase FabI: enoyl-ACP reductase, FabB: β-ketoacyl-ACP synthase I, FabF: β-ketoacyl-ACP synthase II.
The PfFabI enzyme has a greater sequence similarity with the corresponding plant enzyme than with the enoyl reductase (ENR) of bacterial origin. Among the many potential PfFabI inhibitors tested so far triclosan (Fig. 1) remains the most potent with an IC50 = 14 ng/ml (50 nM) but the compound is not suitable for therapeutic use because of human health and environmental risks [16]. Triclosan binding traps the PfFab I enzyme in the nonproductive NAD+ cofactor state and prevents the binding of NADH [17]. Therefore, it is evident that the plasmodial Fab I enzyme is a good target for treating malaria.
Fig. 1.
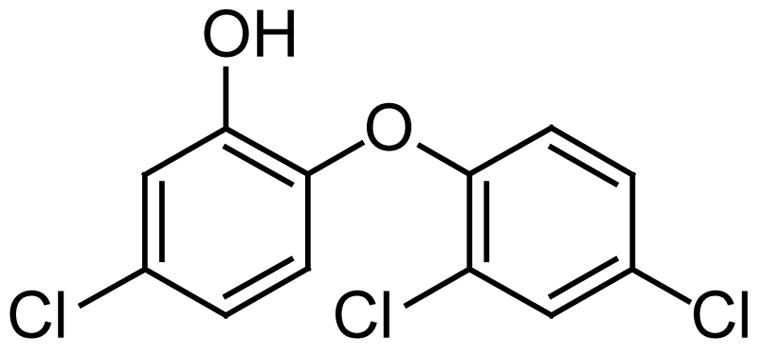
The structure of Triclosan.
2.3. Fatty acids as antimalarial agents
The antimalarial effect of fatty acids has received some consideration in the past but the realization that fatty acids themselves might inhibit the fatty acid biosynthetic machinery of the parasite P. falciparum has only been recently contemplated as a likely strategy to combat the parasite. Earlier work in 1992 by Kumaratilake and collaborators reports on the antimalarial properties of n-3 and n-6 polyunsaturated fatty acids, where acids such as 22:6 (n-3) and 20:5 (n-3) were the best in the studied series for the in vitro killing of intraerythrocytic forms of P. falciparum [18]. These investigators found that the degree of unsaturation was critical for the antiplasmodial effect of the fatty acids towards the parasite, inasmuch as the toxicity was reported to follow the order 22:6 (n-3) > 20:5 (n-3) > 20:4 (n-6) > 18:1 (n-9) > 22:0, where docosahexaenoic acid [22:6 (n-3)] was the best in the studied series of fatty acids in killing P. falciparum (>90% death) at concentrations of 20–40 μg/ml [18]. The methyl esters of the fatty acids were reported to be as potent as the free acids in killing the parasite. The authors also pointed out in their study that these fatty acids were not toxic to either normal red blood cells (RBC) or parasitized red blood cells (PRBC) cells and did not induce hemolysis. The binding of the fatty acids to albumin in vivo was also discussed as unlikely to inhibit the antimalarial effect of the polyunsaturated fatty acids [18]. These investigators also suggested that lipid peroxidation was the most likely mechanism responsible for the antiplasmodial activity displayed by the polyunsaturated fatty acids [18].
Later work in 1995 by Krugliak and collaborators reported on the antiplasmodial effect of a series of C18 fatty acids against the FCR3 strain of P. falciparum, and these fatty acids displayed some inhibitory activity (≤ 200 μg/ml) against both the intact infected cells and the free parasites [19]. In this particular work oleic acid (9–18:1) was the most inhibitory fatty acid with an IC50 of 23 μg/ml, linoleic acid (9,12–18:2) displayed an IC50 of 76 μg/ml, but linolenic acid (9,12,15–18:3) only displayed an IC50 of 92 μg/ml. In contrast to what was reported before by Kumaratilake [18], polyunsaturation was postulated as not been critical for the inhibitory effect displayed by the C18 fatty acids. In addition, monodiglycerides of oleic acid did not cause any inhibition (> 3 mM) of the parasite, thus implying that the free fatty acid was critical for the antiplasmodial activity. The authors were not able to present a reasonable antiplasmodial mechanism that could account for their antiplasmodial observations with these fatty acids. However, these investigators concluded that the C18 fatty acids do not inhibit parasitic growth by inducing lipid peroxidation, the acids do not uncouple mitochondria and do not promote oxidative damage to the infected membrane cells. These investigators did not consider the possibility of the inhibition of the P. falciparum fatty acid biosynthesis as a possible explanation for the observed results with the C18 fatty acids.
In 2005, a naturally occurring C18 fatty acid, named scleropyric acid (Fig. 2), was isolated from the twigs of Scleropyrum wallichianum, and also shown to display good antiplasmodial activity (IC50 = 7.2 μg/mL) against a K1 multidrug-resistant strain of P. falciparum [20]. These results are important, because the authors do underline once more the potential of C18 fatty acids as antimalarial agents and that acetylenic fatty acids also merit further exploration as antiplasmodial compounds [20].
Fig. 2.
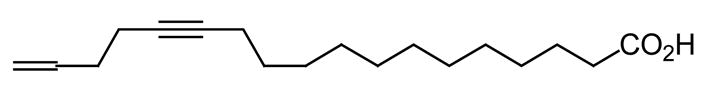
The structure of scleropyric acid.
Dr. Deniz Tasdemir of the School of Pharmacy of The University of London has developed the first type II FAS-target-based antimalarial screening approach employed in the field of natural products research [12]. This screening protocol was applied to the most unusual sponge marine fatty acids from the sponge Agelas oroides [21]. The sponge was extracted with methanol and partitioned between hexane, chloroform, and aqueous methanol (Kupchan partition) and the fatty acid composition of the hexane fractions from the sponge were tested. First, the in vitro antiplasmodial activity of the crude fatty acid extracts, and fractions of purified mixed fatty acid mixtures (by HPLC), were tested on the bloodstage forms of the multidrug-resistant K1 strain of P. falciparum as shown in Table 1. The bioassay measured the incorporation of radiolabelled 3H-hypoxanthine, which is taken up by the parasite for purine salvage and DNA synthesis, and this was an indication of parasite growth [22]. In this antiplasmodial bioassay the fraction containing the very long-chain all cis C23–C26 Δ5,9 fatty acids (Fig. 3) displayed considerable antiprotozoal activity (IC50’s = 12–16 μg/ml), and they were more cytotoxic than linoleic acid (IC50 > 20 μg/ml).
Table 1.
The FabI inhibitory and antiprotozoal activity of mixtures of Δ5,9 fatty acids from Agelas oroides (IC50 values are in μg/ml, atriclosan, bartemisinin, cphodophyllotoxin)
| Fatty Acid | PfFabI inhibition | Plasmodium falciparum | L6 cells |
|---|---|---|---|
| Standard | 0.014a | 0.0455 b | 0.009c |
| 23:2 Δ5,9
24:2 Δ5,9 |
0.35 | 12.1 | 83.6 |
| 24:2 Δ5,9
25:2 Δ5,9 26:2 Δ5.9 |
0.35 |
16.3 |
84.7 |
| 18:2 Δ9,12 | > 100 | >20 | 76.3 |
Fig. 3.
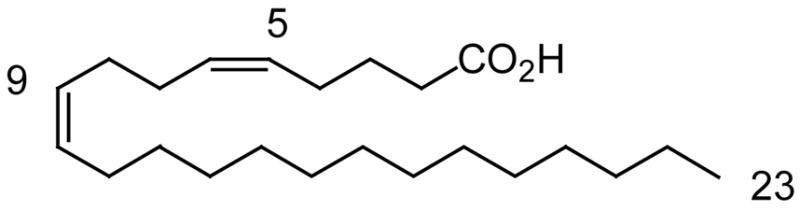
The structure of a Δ5,9 very-long chain fatty acid.
These same Δ5,9 fatty acids were also tested simultaneously for enzyme inhibitory activity on the recombinant FabI enzyme of P. falciparum (PfFab I), in order to determine if the antiplasmodial activity, at least in part, was due to FabI inhibition. The enzyme was heterologously expressed in E. coli and purified as described by Perozzo [15]. The enzyme assay employs crotonyl-CoA as the substrate and in the presence of NADH, Fab I reduces the conjugated double bond to yield butyrylCoA (Fig. 4). The transformation of NADH to NAD+ is determined by an absorption decrease at 340 nm. In the presence of a FabI inhibitor the oxidation of NADH to NAD+ will be affected. This enzymatic transformation can be followed spectrophotometrically for 1 minute and the IC50 values are determined from dose-response curves. As we can see from Table 1 the marine C23–C26 Δ5,9 fatty acids were good inhibitors (IC50’s = 0.35 μg/ml) of the P. falciparum enoyl-ACP reductase (FabI) enzyme that catalyses the final reduction step of the fatty acid chain elongation cycle in P. falciparum. Linoleic acid was not as good an inhibitor of the PfFabI enzyme (IC50 >100 μg/ml) (Table 1). This finding demonstrated, for the first time, that the marine Δ5,9 fatty acids can inhibit the FabI enzyme of P. falciparum, which seems to be the potential intracellular target of the fatty acids. It is clear that we need to better understand the mechanism of inhibition and the mode of binding of these interesting marine Δ5,9 fatty acid inhibitors to the PfFab I enzyme to pave the way for the development of other more potent lipid inhibitors. However, it seems clear that the very long chains might be a prerequisite for the good enzyme inhibition observed.
Fig. 4.
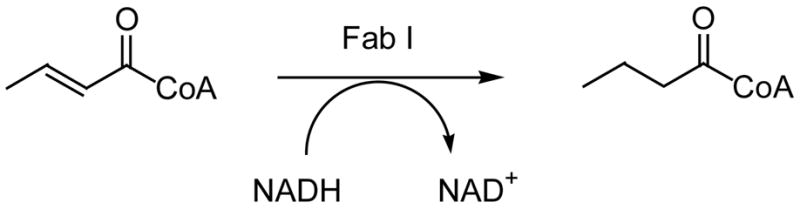
The FabI enzyme assay (340 nm, 1min).
It is important to mention that in addition to the antiplasmodial and PfFab I inhibitory studies described above it was also essential to study the cytotoxicity of the Δ5,9 fatty acids on mammalian cells. For this purpose, rat skeletal myoblasts L6 cells were used and good was the fact that the marine C23–C26 Δ5,9 fatty acids had almost no cytotoxicity on mammalian L6 cells as can also be seen from Table 1 [21]. Therefore, these results indicate that the Δ5,9 fatty acids may be of use against the parasite without damage to the host.
3. Tuberculosis
3.1 Introduction to the disease
Tuberculosis (TB) continues to be a serious health concern since the emergence of multidrug-resistant tuberculosis (MDR-TB) in the 1980s and the more recent cases of extensively drug-resistant tuberculosis (XDR-TB) [23–24]. The mycobacteria become resistant since their slow growth phase increases its ability to adapt and acquire new resistance mechanisms. Presently the first line therapy against TB includes the use of isoniazid (inhibits the synthesis of mycolic acids in the mycobacteria cell wall) (Fig. 5) and rifampin (inhibits RNA synthesis), which are probably the most effective mycobacterial drugs available today. If the mycobacteria become resistant (the so-called MDR-TB strains) to these two drugs then the second-line agents available today are a series of fluoroquinolones (inhibit DNA gyrase) and some aminoglycosides (inhibit protein synthesis) including the injectable second-line drugs capreomycin, kanamycin, and amikacin which target protein synthesis. It is estimated that around 2% of the culture-confirmed TB cases in the US were resistant to both isoniazid and rifampin. On the other hand, if the mycobacterium also became resistant to these second-line agents it would also be extensively drug-resistant (the so-called XDR-TB strains) and then there is a very small chance to treat the disease since other newer drugs are seriously limited by gastrointestinal, renal, and/or neurological toxicities. At this stage the disease could be considered basically “untreatable” and then it becomes a scary public-health issue. Therefore, the search for new ways to treat TB is of primary importance and urgency.
Fig. 5.
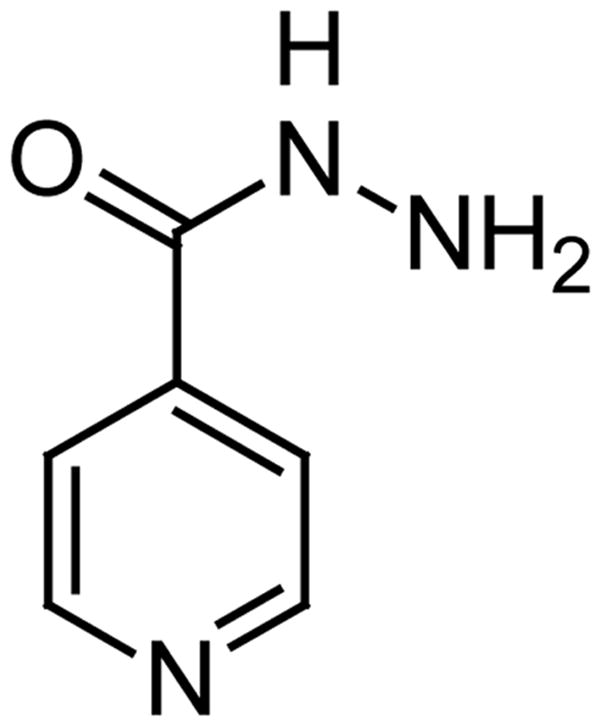
The structure of Isoniazid
3.2. Fatty acid biosynthesis as a target in antimycobacterial chemotherapy
Similar to what we have discussed before for P. falciparum, mycobacteria also display a dual mechanism for the synthesis of fatty acids, i.e., an eukaryotic FASI system and a prokaryotic FASII system [25–26]. The FASI synthetase makes fatty acids between 16 and 24 carbons, while the FASII system elongates the C16 acids to longer chains, which are the precursors of the mycolic acids [25]. Chemical intervention with the FASII synthetase of mycobacteria is also an attractive target for the development of new antimycobacterial fatty acids. For example, 2-alkynoic fatty acids such as the 2-hexadecynoic acid (Fig. 6), 2-octadecynoic acid, and 2-nonadecynoic acid have been synthesized and they are toxic to mycobacteria, such as Mycobacterium tuberculosis H37Rv, with minimum inhibitory concentrations (MICs) of 20–25 μM [27]. These 2-alkynoic fatty acids have been shown to inhibit InhA, the enoyl-ACP reductase (the analog of PfFabI) that catalyzes the NADH-specific reduction of 2-trans-enoyl-ACP [27]. The crystal structure of the M. tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate is also known [28]. In addition to InhA inhibition, recent work has also shown that the 2-alkynoic fatty acids are also metabolized into 3-keto acids which block fatty acid biosynthesis (in the case of 2-hexadecynoic acid the 3-keto acid is the 3-ketohexadecanoic acid), and into 3-alkynoic fatty acids which inhibit β-oxidation in mycobacteria (in the case of 2-hexadecynoic acid the 3-alkynoic acid is the 3-hexadecynoic acid). This dual inhibition of fatty acid biosynthesis and fatty acid degradation, in addition to InhA inhibition, are mechanisms responsible for the antimycobacterial effect of the 2-alkynoic fatty acids. However, it is important to point out that the effect of the 2-alkynoic acids on eukaryotes is different from that observed in mycobacteria also due to the different localization of the fatty acid synthesis and degradation enzymes in these systems, which suggests that the 2-alkynoic fatty acids can be specifically used against mycobacteria without toxicity to the host [27].
Fig. 6.
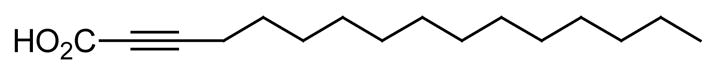
The structure of 2-hexadecynoic acid.
3.3. Other antimycobacterial fatty acids
The mycobactericidal effect of fatty acids has been known for some time and excellent reviews exist, but we will limit our survey here to the most recent findings and some previous work so as to get a better idea of the fatty acid structural characteristics known to be effective against TB [29–32]. Earlier studies by Kondo and Kanai established that among a series of linear-chain saturated fatty acids (C2-C20) myristic acid displayed the strongest bactericidal activity at 0.04 mM against a highly virulent strain (Ravenel) of Mycobacterium bovis and an avirulent strain (H37Ra) of M. tuberculosis [30]. Later, Saito and collaborators studied the cytotoxicity of a selected number of fatty acids against seventy-one strains of 15 species of rapidly growing mycobacteria using the agar dilution method at pH 7.0 [31]. Among a series of linear chain saturated fatty acids between 2 and 20 carbons, lauric acid (C12:0) was the most active (MIC’s 6.25–25 μg/ml) and capric acid (C10:0) was the second most active with MIC’s in the range of 50–100 μg/ml [31]. Other saturated fatty acids between C2–C20 were essentially nontoxic to all the studied mycobacteria. Unsaturated fatty acids with 16–20 carbons were also highly toxic to the group IV mycobacteria, being palmitoleic acid a highly toxic acid with MIC’s in the range of 3.2–6.25 μg/ml, while oleic acid also showed a similar profile. A possible antibacterial mechanism was postulated to proceed via disruption of the bacterial cell membrane resulting in a change in membrane permeability [31].
Marine sponges are the primary source of α-methoxylated fatty acids [3]. Recently, the antimycobacterial effect of a selected number of these compounds was studied. For this purpose, a series of saturated even-chain 2-methoxylated fatty acids between 8 and 14 carbons were synthesized [32]. The 2-methoxylated C10–C14 acids were prepared from the corresponding 2-hydroxylated fatty acids, while the 2-methoxyoctanoic acid was synthesized starting with heptaldehyde [32]. All of the methoxylated fatty acids displayed some degree of inhibition (between 2–99%) of Mycobacterium tuberculosis H37Rv at 6.25 μg/ml. The most effective fatty acid was the (±)-2-methoxydecanoic acid (Fig. 7) with a minimum inhibitory concentration (MIC) of 200–239 μM against M. tuberculosis H37Rv as determined by both the microplate alamar blue assay (MABA) and the green fluorescent protein assay (GFP). In this bioassay capric acid also displayed a MIC of 200 μM, and with an IC50 > 300 μM both compounds were not toxic to mammalian Vero cells (African green monkey kidney cells) [32]. However, the antimycobacterial activity displayed by the (±)-2-methoxydecanoic acid was not better than the one found for capric acid, thus implying that α-methoxylation does not help much in increasing the antimycobacterial activity of fatty acids [32].
Fig. 7.
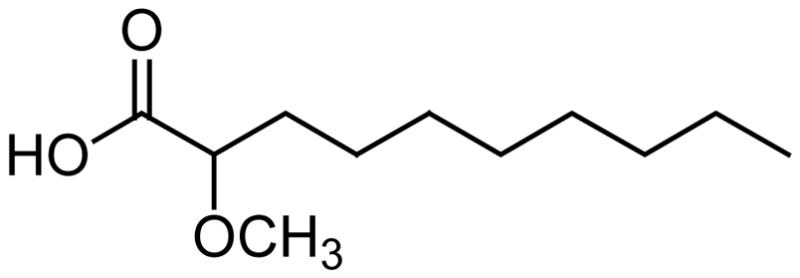
The structure of 2-methoxydecanoic acid.
In a separate study the marine fatty acids 23:2Δ5,9 and 24:2Δ5,9 were tested against the mycobacterial enoyl reductase FabI (also called InhA) from M. tuberculosis and the bacterial Fab I (EcFabI) from Escherichia coli. The inhibitory activity of the 23:2Δ5,9 and 24:2Δ5,9 fatty acids on InhA yielded an IC50 of 9.4 μg/ml, but the Δ5,9 fatty acids were quite potent against EcFab I with an IC50 of 0.50 μg/ml [21]. Typical C16–C18 fatty acids were inactive against EcFab I at the concentrations tested (IC50s > 50–100 μg/ml). These results indicated that while the very long-chain Δ5,9 fatty acids are moderate inhibitors of InhA, they are more promising inhibitors of the FabI of E. coli [21]. However, the antibacterial activity of Δ5,9 fatty acids against either M. tuberculosis or E. coli has not been tested so far.
The general conclusion that can be drawn from the recent literature in this area is that most saturated fatty acids are inactive towards mycobacteria (with the exception of the C10 and C12 fatty acids), whereas unsaturated fatty acids do show activity that depends on the degree of unsaturation, chain length, and the bacterial species tested [29–34]. Inhibition of fatty acid biosynthesis seems to be the preferred mechanism by which alkynoic fatty acids are toxic to mycobacteria, thus making InhA also an attractive target for further studies. The best fatty acid candidates for this type of inhibition, therefore, seem to be the 2-alkynoic fatty acids, since the Δ5,9 fatty acids, as well as the α-methoxylated fatty acids, do not look promising for further antimycobacterial studies. However, the Δ5,9 fatty acids merit further studies in other antibacterial bioassays, in particular against E. coli.
4. The Δ5,9 fatty acids as topoisomerase I inhibitors
In addition to the findings that the very long-chain Δ5,9 fatty acids are effective against the FabI enzyme of P. falciparum and somewhat effective against the InhA of M. tuberculosis additional research has been done on their interaction with another important enzyme, namely the topoisomerase I. There are several literature reports describing the inhibitory effects of fatty acids towards topoisomerase I [35–43]. Topoisomerases I is a key enzyme in the breaking and repair of DNA strands and is involved in making the necessary topological changes to DNA for key cellular processes such as replication, transcription, and recombination [36]. Topoisomerases have also evolved as key cellular targets for the development of effective anticancer drugs.
Fatty acids are known inhibitors of topoisomerase I [35–42]. Suzuki and collaborators reported that while saturated fatty acids (C6–C22) display no inhibition of the topo-I enzyme (even at concentrations of the acid as high as 2000 μM), cis-monounsaturated fatty acids (C14–C22) do exhibit inhibition of the enzyme [35]. The geometry and position of the double bonds as well as the carbon chain lengths were found to be critical for the inhibitory process [35]. For example, while oleic acid (cis-18:1 Δ9) inhibits topoisomerase I with an IC50 of 31 μM, the trans-isomer, namely elaidic acid (trans-18:1 Δ9) does not inhibit the enzyme (IC50 > 1000 μM) [35]. Other interesting topo-I lipid inhibitors include conjugated C18 and eicosapentaenoic fatty acids [36,38], very-long chain (C26-C30) Δ5,9 fatty acids [39,40], and phospholipids containing unsaturated fatty acids [41]. However, among the very long-chain Δ5,9 fatty acids the (5Z,9Z)-5,9-heptacosadienoic acid (Fig. 8) effectively inhibits human topoisomerase I with an IC50 of 0.9 μM, while other similar Δ5,9 fatty acids inhibit the enzyme with IC50’s in the range of 1–3 μM [39,40]. This shows that chain length is critical for good topo-I inhibition since we have shown that the short-chain (5Z,9Z)-hexadecadienoic acid only inhibits topo-I at the high concentrations of 800 μM [43].
Fig. 8.
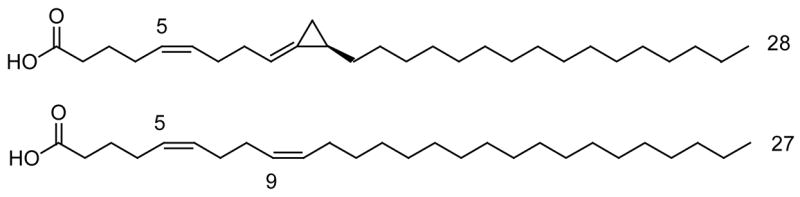
The two most potent Topo-I Δ5,9 fatty acids inhibitors reported to date.
Some Δ5,9 fatty acids are also cytotoxic to cancer cells. For example, a mixture of (5Z,9Z)-23-methyl-5,9-tetracosadienoic acid and (5Z,9Z)-22-methyl-5,9-tetracosadienoic acid display cytotoxicity towards mouse Ehrlich carcinoma cells with ED50 of 1.8 μg/mL and show a hemolytic effect on mouse erythrocytes [44]. Interesting to also mention here is that the (5E,9Z)-6-bromo-5,9-heptacosadienoic acid, isolated in 1995 by Kobayashi and collaborators from the Okinawan sponge Xestospongia sp., is also cytotoxic to L1210 murine leukaemia cells (IC50 = 11 μg/mL) and to KB human epidermoid cancer cells (IC50 = 18.5 μg/mL) [45]. Surprisingly, palmitic acid has also been reported to be cytotoxic to several leukemic cell lines (Molt-4, HL-60, K-562) with IC50’s in the range of 10–15 μg/ml [37]. However, palmitic acid is not cytotoxic to normal human dermal fibroblasts since an IC50 of 75 μg/ml was reported [37]. We should mention that in contrast to what was reported by Suzuki, Harada and collaborators found that palmitic acid does inhibit the topoisomerase I enzyme with an IC50 of 3–10 μM, but not the topo-II enzyme [37]. We should mention that our research group has also found that palmitic acid does inhibit the human topoisomerase I enzyme, but at concentrations of 400 μM (N. Carballeira, unpublished results). All of these findings suggest that the cytotoxicity of fatty acids to cancer cell lines might be due, at least in part, to the inhibition of topoisomerase I [37].
While most of the fatty acid topoisomerase I inhibitory work has been performed with normal-chain fatty acids, there is just one report dealing with the topoisomerase I inhibitory activity of methyl-branched iso and anteiso fatty acids [42]. J.H. Jung and collaborators reported on the topoisomerase I inhibitory activity of compounds from a Streptomyces sp. strain KM86-9B isolated from a marine sponge, and observed that the compounds responsible for the activity were a series of saturated iso and anteiso C15–C17 fatty acids [42]. The best topo-I inhibitions were reported at concentrations of 100 μg/ml for these methyl-branched fatty acids [42].
Based on this short survey of the fatty acid literature we can easily see that the most promising fatty acid topoisomerase I inhibitors to date are the marine very long-chain Δ5,9 fatty acids, in particular the (5Z,9Z)-5,9-heptacosadienoic acid (Fig. 8), and further studies in this direction could certainly yield important drug leads.
5. Fungal Infections
5.1 Introduction to the disease
Fungal infections can result in a series of severe diseases [46–49]. Candida albicans, for example, is common in the oral cavity, in the lower gastrointestinal tract, and in the female genital tract [46–48]. However, in a weakened immune system C. albicans can overgrow and invade surrounding tissues leading to a condition known as candidiasis. Candidiasis is one of the early symptoms of patients infected with the HIV virus, but can also occur in infants and patients with diabetes [49]. Another fungal organism, Aspergillus niger, is a recognized cause of pulmonary aspergillosis, an infection of the lungs caused by inhalation of airborne Aspergillus spores. A. niger can also lead to otomycosis, a common fungal infection of the ear in the tropics [47–48].
Fungal infections were considered curable in the 1970s since they were mainly superficial, but with the emergence of invasive fungal infections, fungal sepsis has increased three-fold in the US between 1979 and 2004 [50]. Around 70–90% of all invasive mycoses are from Candida spp. and around 10–20% arise from Aspergillus spp. In addition, the increasing emergence of fungal resistance to currently available antimycotic agents presents a serious health-problem. The many resistant strains may arise from the use of immunomodulatory agents in the precention of rejection in bone marrow and organ transplants, the use of antineoplastic agents, and the indiscriminate use of antibiotics [51].
Among the fungal antimycotic agents available today, the azoles are quite popular (Fig. 9). The azoles are imidazole and triazole derivatives that interfere with the synthesis of ergosterol, an important constituent of fungal membranes [52]. Among the azoles we should mention fluconazole and voriconazole (Fig. 9). Voriconazole has a broader activity and better bioavailability than fluconazole [52]. Other antifungal agents are the polyenes, such as Amphotericin B, that binds primarily to ergosterol in the fungal membranes altering membrane permeability thus resulting in leakage of cellular components and eventually cell death. Another set of known antifungal agents are the allyl amines, such as terbinafine (Fig. 10), which interfere with ergosterol biosynthesis by targeting the enzyme squalene oxidase [53].
Fig. 9.
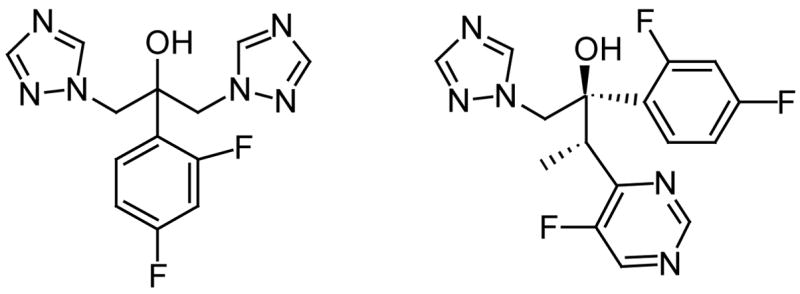
The structures of fluconazole (left) and voriconazole (right).
Fig. 10.
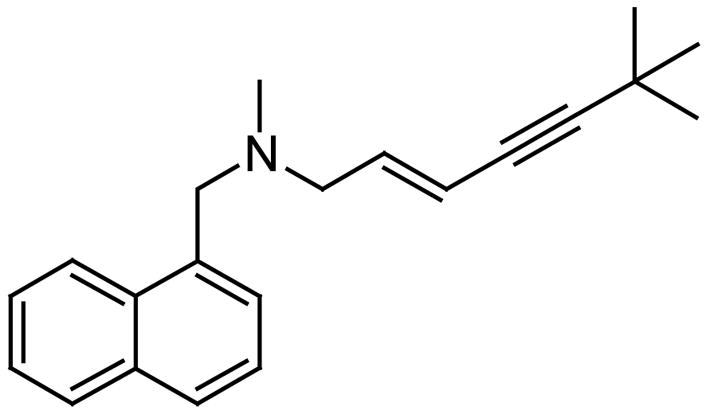
The structure of terbinafine.
5.2. Antifungal fatty acids
The antifungal properties of many fatty acids and possible mechanisms of fungitoxicity have been previously studied [29,54]. A study of the cytotoxicity of several short-chain fatty acids on three strains of C. albicans revealed that capric acid (10:0) caused the greatest reduction of the infectivity titers (≥ 6.75 log 10CFU) at 37°C as compared to the control at 10 mM [54]. Transmission electron microscopy (TEM) studies on C. albicans showed disorganization of the cytoplasm due to a disrupted or disintegrated plasma membrane caused by a hydrostatic turgor pressure within the cell. A similar mechanism of action was proposed for (Z)-9-heptadecenoic acid (17:1), which inhibits the growth and germination of the fungi Phytophthora infestans and Idriella bolleyi [55]. The mechanism of inhibition of the (Z)-9-heptadecenoic acid (17:1) was explained in terms of membrane disruption resulting in the release of intracellular electrolytes and proteins and eventually cytoplasmic disintegration of mycelia [55].
Acetylenic fatty acids, in particular the 2-alkynoic fatty acids (FA) have also been known to be fungitoxic [56–57]. The fungal activity of these compounds depends on the FA chain length and pH of the medium [56–57]. The optimal chain lengths between 8 and 16 carbons have been established for the 2-alkynoic FA to exert maximum fungistatic effects [56]. However, the 2-hexadecynoic acid has received the most attention for its antifungal, antimicrobial, and cytotoxic properties [58–60]. The cytotoxicity of the 2-hexadecynoic acid has been ascribed to its ability to inhibit the elongation of saturated and unsaturated FA as well as its potential to inhibit the fatty acid acylation process, particularly triacylglycerol synthesis [59]. Therefore, fatty acid biosynthesis inhibition is also a mechanism responsible for the antifungal activity of fatty acids.
More recently, a novel acetylenic fatty acid, namely the 6-nonadecynoic acid, was isolated from an ethanol extract of the roots of Pentagonia gigantifolia [61]. This acetylenic fatty acid was reported to be fungistatic and specific to fluconazole-susceptible and resistant C. albicans strains with MIC values (0.52 μg/ml) comparable to those of amphotericin B (0.52 μg/ml) and fluconazole (0.29 μg/ml) [61]. The antifungal mechanism was suggested to be due to the interference of the compound with fungal sphingolipid biosynthesis [61]. However, later this fatty acid was synthesized and found to be fungitoxic to C. neoformans ATCC 66031 but inactive towards C. albicans ATCC 14053 and C. albicans ATCC 60193 [62]. This discrepancy might be due to strain variability.
In an effort to combine both acetylenic functionalities (i.e, a triple bond at both C-2 and C-6) in one fatty acid the novel fatty acids 2,6-hexadecadiynoic acid, 2,6-nonadecadiynoic acid, and 2,9-hexadecadiynoic acid were recently synthesized in two steps and in 11–18% overall yields starting from either 1,5-hexadiyne or 1,8-nonadiyne [63]. Among all the diynoic compounds the 2,6-hexadecadiynoic acid (Fig. 11) displayed the best overall antifungal activity against both the fluconazole-resistant C. albicans strains ATCC 14053 and ATCC 60193 (MIC = 11 μM) and against C. neoformans ATCC 66031 (MIC < 5.7 μM). The 2,9-hexadecadiynoic acid did not display any significant cytotoxicity against the fluconazole resistant C. albicans strains, but it showed fungitoxicity against C. neoformans ATCC 66031 with a MIC value of <5.8 μM. Other fatty acids, such as 2-hexadecynoic acid, 5-hexadecynoic acid, 9-hexadecynoic acid, and 6-nonadecynoic acid were also synthesized and their antifungal activities compared [63]. The 2-hexadecynoic acid, a known antifungal fatty acid, exhibited the best antifungal activity (MIC = 9.4 μM) against the fluconazole resistant C. albicans ATCC 14053 strain, but it showed a MIC value of only 100 μM against C. albicans ATCC 60193. The fatty acids 2,6-hexadecadiynoic acid and 2-hexadecynoic acid also displayed a MIC of 140–145 μM towards M. tuberculosis H37Rv in Middlebrook 7H12 medium. This study concluded that the 2,6-hexadecadiynoic acid exhibited the best fungitoxicity profile compared to the other acetylenic analogues. Its advantage over the 2-hexadecynoic acid relied on the fact that it was less toxic to mammalian cells. For example, while the 2-hexadecynoic acid displayed an IC50 of 14.4 μg/mL against Vero cells, the 2,6-hexadecadiynoic acid displayed an IC50 of 46.3 μg/mL [63]. Therefore, the 2,6-hexadecadiynoic acid fatty acid has the potential to be further evaluated for use in topical antifungal formulations [63].
Fig. 11.
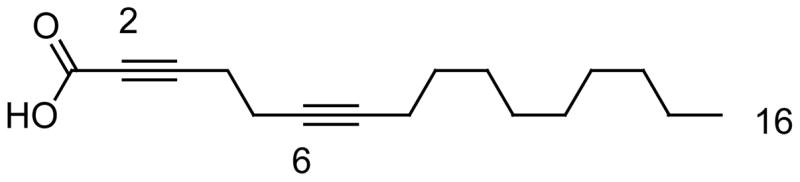
The structure of the 2,6-hexadecadiynoic acid.
Cyclopropane fatty acids also possess antifungal activity, although less studied. Majusculoic acid (Fig. 12) is a recent example of a natural cyclopropane fatty acid with proven antifungal activity against C. albicans ATCC 14503 (MIC = 8 μM) and Candida glabrata (MIC = 19.3 μM) [64]. The accumulation of a C17 cyclopropane fatty acid in a Pseudomonas spp. has been ascribed as one of the agents causing the antagonistic effect of the pseudomonad on the fungi Pythium ultimum [65]. However, interest in cyclopropane fatty acids has also grown with the discovery that the pathogenicity of M. tuberculosis is highly dependent on the presence of cyclopropyl moieties in their membrane lipids [66]. Therefore, cyclopropane fatty acids merit further investigation as antifungals since they could either inhibit key enzymes responsible for the biosynthesis of fungal fatty acids or cause serious disruptions in fungal membranes. Nevertheless, we should mention that cyclopropene fatty acids have been known for some time to be toxic due to the inhibition of the stearoyl-CoA desaturase in humans [67–68].
Fig. 12.
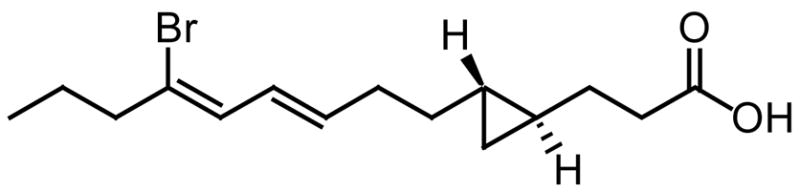
Majusculoic acid.
Several tetradecanoic acid (14:0) analogues have also been studied in vitro for their ability to inhibit the enzyme N-myristoyltransferase (NMT) [69]. Many fungal species synthesize a variety of N-myristoyl proteins using NMT as catalyst [69]. Incorporation of these myristic acid analogues into the fungi competes with 14:0 for binding to NMT and myristoylation of fungi proteins. This event perturbs fungal protein function such as protein folding, resulting in fungal growth inhibition. An example of a NMT inhibitor is the (±)-2-bromotetradecanoic acid with MICs between 0.01–0.04 mM against C. albicans, C. neoformans, Saccharomyces cerevisiae and A. niger [69]. Another interesting tetradecanoic acid analogue is the 4-oxatetradecanoic acid, which is fungicidal to C. neoformans, producing a 10,000-fold reduction in viable cell number 1 h after administration [70]. Metabolic labeling studies confirmed that the 4-oxatetradecanoic acid is a substrate for C. neoformans N-myristoyltransferase, despite the fact that its antifungal effect could also be due to a perturbation of the fungal lipid metabolism. Interestingly, the 4-oxatetradecanoic acid was not as effective against C. albicans as it was against C. neoformans [70].
Very few marine sponge fatty acids have been screened as antifungal agents. Despite the fact that the antifungal activity of the sponge Δ5,9 fatty acids have not been studied, another class of sponge fatty acids, namely the α-methoxylated fatty acids (Fig. 7) have received considerable attention [71]. In particular, the antifungal activity of the (±)-2-methoxytetradecanoic acid (2-OMe-14:0), and several synthetic analogs, has received particular emphasis since they are myristic acid analogs and potential NMT inhibitors [71–72]. The (±)-2-methoxytetradecanoic acid was initially isolated form the Caribbean sponge Callyspongia fallax [73] and it has been synthesized [71]. Antifungal studies demonstrated that it displays activity against C. albicans (ATCC 14053) in RPMI medium and Aspergillus niger (ATCC 16404) and Cryptococcus neoformans (ATCC 66031) in SDB medium at the minimum inhibitory concentration (MIC) of 100 μM, which compares to the fungitoxicity of a 2-iodotetradecanoic acid against the same fungi [67]. One possible mechanism of fungitoxicity was postulated as inhibition of the fungal N-myristoyltransferase (NMT). However, it was also demonstrated that the 2-OMe-14:0 acid was not synergistic with Amphotericin B against the tested fungi [71].
The inhibitory effect of the 2-OMe-14:0 acid against the isolated NMT enzyme has not been tested. However, molecular modeling calculations (performed by Dr. Amiram Goldblum of the Hebrew University of Jerusalem) on a partial model of the Saccharomyces cerevisiae-NMT (Sc-NMT) enzyme using the consistent valence force field (cvff force field) parameterized using peptide and protein structures revealed that the CoA derivative of the (S)-2-OMe-14:0 binds much stronger to the enzyme than the (R)-2-OMe-14:0 (Fig. 13). In fact, the S-enantiomer displays hydrogen bonding to the NH of a threonine 205 in the active site, while the R-enantiomer collides with the backbone according to this model (Fig. 13). However, optically pure 2-methoxytetradecanoic acids were in need to be synthesized in order to test these predictions.
Fig. 13.

Representation of the S (left) and the R (right) enantiomers of the CoA of the 2-OMe:14:0 inside the active site of a partial model of the enzyme Sc-NMT. The methoxy oxygen of the S-enantiomer hydrogen bonds to a Threonine 205 (left). (Picture courtesy of Prof. Amiram Goldblum, Hebrew University of Jerusalem).
In such an effort, the unprecedented (±)-2-methoxy-4-oxatetradecanoic acid (Fig. 14) and the optically pure (S)-2-methoxy-4-oxatetradecanoic acid were synthesized in six steps and in 11–14% overall yields [72]. In this case, the S-enantiomer of the 4-oxa-2-OMe-14:0 was also calculated to be a better inhibitor of the enzyme than the R-enantiomer, but not as good as the (S)-2-methoxytetradecanoic acid (A. Goldblum, unpublished results). When experimentally tested, the methoxylated 4-oxa compounds displayed selective fungitoxicity in the range of 0.08–0.22 mM against C. neoformans ATCC 66031 and A. niger ATCC 16404, but no significant activity against C. albicans ATCC 14053 and ATCC 60193 (> 2.6 mM) [72]. Interesting was the finding that the 4-oxa functionality decreased the antifungal effect of the parent 2-OMe-14:0 acid against C. albicans, while retaining the activity against C. neoformans and A. niger [72]. Albeit being reasonable substrates for N-myristoyltransferases (NMTs), the racemic and the S-enantiomer of the oxygenated 2-methoxylated compounds showed no significant difference in antifungal activity. This finding suggested an alternative mechanism of fungitoxicity other than NMT inhibition for these α-methoxylated fatty acids [72]. However, in contrast to what has been found with M. tuberculosis, it is evident that α-methoxylation increases the antifungal potential of myristic acid analogues. Nevertheless, the acetylenic fatty acids continue to be better antifungal compounds, although probably more toxic to mammalian cells, than the α-methoxylated fatty acids studied to date.
Fig. 14.
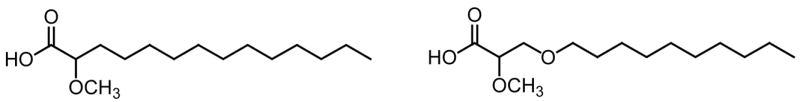
Structures of racemic 2-methoxytetradecanoic acid (left) and its 4-oxa analog (right).
6. Summary
From what we have reviewed herein it is clear that marine fatty acids still hold promise to treat infectious diseases. The Δ5,9 fatty acids are probably the best fatty acid inhibitors of the P. falciparum Fab I enzyme known to date and further study in this direction should yield interesting results. On the other hand, the Δ5,9 fatty acids are not as good inhibitors of the M. tuberculosis InhA enzyme but hold promise as inhibitors of the E. coli Fab I enzyme. The 2-alkynoic fatty acids display good antimycobacterial activity but toxicity to mammalian cells continues to remain a problem. It is evident that α-methoxylation does not increase the antimycobacterial activity of fatty acids. As topoisomerase I inhibitors the marine Δ5,9 fatty acids continue to be the most potent fatty acid topo I inhibitors reported to date and chain length seems to play an important role in their inhibition. Topo I inhibition seems to be partially responsible for the toxicity displayed by some of these Δ5,9 fatty acids towards cancer cell lines. Finally, acetylenic fatty acids are the best antifungal fatty acids reported so far, but again, toxicity remains a serious issue for their general acceptance. It is evident that α-methoxylation increases the antifungal properties of fatty acids probably due to better solubility or increased acidity. However, NMT inhibition does not seem to be the only mechanism by which these myristic acid analogues display their toxicity.
Acknowledgments
We are indebted to Prof. Keykavous Parang (College of Pharmacy, University of Rhode Island) for the antifungal testing of some of the compounds presented here, to Prof. Deniz Tasdemir (College of Pharmacy, University of London) for the antimalarial and Fab I bioassays of the very long chain Δ5,9 fatty acids, and to Prof. Scott Franzblau (College of Pharmacy, University of Illinois at Chicago) for the antimycobacterial testing of some of the compounds presented in this review. The financial support of the National Institutes of Health (grant no. S06GM08102), which made possible the synthesis and the biotesting of many of the compounds presented here, is greatly appreciated.
Abbreviations
- ATCC
American type culture collection
- ACP
Acyl carrier protein
- Fab I
Enoyl-ACP reductase
- FAS
Fatty acid synthase
- HPLC
High performance liquid chromatography
- MIC
Minimum inhibitory concentration
- NMT
N-myristoyltransferase
- MDR-TB
Multidrug-resistant tuberculosis
- Sc-NMT
Saccharomyces cerevisiae-NMT
- Topo-I
Topoisomerase I
- XDR-TB
Extensively drug-resistant tuberculosis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that app that apply to the journal pertain.
References
- 1.Djerassi C, Lam WK. Sponge phospholipids. Acc Chem Res. 1991;24:69–75. [Google Scholar]
- 2.Dembitsky VM, Rezanka T, Srebnik M. Lipid compounds of freshwater sponges: Family Spongillidae, class Demospongiae. Chem Phys Lipids. 2003;123:117–155. doi: 10.1016/s0009-3084(03)00020-3. [DOI] [PubMed] [Google Scholar]
- 3.Carballeira NM. New advances in the chemistry of methoxylated lipids. Prog Lipid Res. 2002;41:437–456. doi: 10.1016/s0163-7827(02)00005-x. [DOI] [PubMed] [Google Scholar]
- 4.Berge JP, Barnathan G. Fatty acids from lipids of marine organisms: Molecular biodiversity, roles as biomarkers, biologically active compounds, and economical aspects. Adv Biochem Eng Biotechnol. 2005;96:49–125. doi: 10.1007/b135782. [DOI] [PubMed] [Google Scholar]
- 5.Thayer AM. Fighting Malaria. Chem Eng News. 2005;83:69–82. [Google Scholar]
- 6.Klayman DL. Qinghaosu (Artemisinin): an antimalarial drug from China. Science. 1985;228:1049–1055. doi: 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- 7.Acton N, Klayman DL. Artemisinin, a new sesquiterpene lactone endoperoxide from Artemisia annua. Planta Medica. 1985;47:442–445. doi: 10.1055/s-2007-969543. [DOI] [PubMed] [Google Scholar]
- 8.Piola P, Fogg C, Bajunirwe F, Biraro S, Grandesso F, Ruzagira E, Babigumira J, Kigozi I, Kiguli J, Kyomuhendo J, Ferradini L, Taylor W, Checchi F, Guthmann JP. Supervised versus unsupervised intake of six-dose artemether-lumefantrine for treatment of acute, uncomplicated Plasmodium falciparum malaria in Mbarara, Uganda: a randomised trial. The Lancet. 2005;365:1467–1473. doi: 10.1016/S0140-6736(05)66416-1. [DOI] [PubMed] [Google Scholar]
- 9.Thayer AM. Preventing Malaria. Chem Eng News. 2005;83:85–95. [Google Scholar]
- 10.McFadden GI, Reith M, Munholland J, Lang-Unnasch N. Plastid in human parasites. Nature. 1996;381:482. doi: 10.1038/381482a0. [DOI] [PubMed] [Google Scholar]
- 11.Waller RF, Keeling PJ, Donald RG, Striepen B, Handman E, Lang-Unnasch N, Cowman AF, Besra GS, Roos DS, McFadden GI. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc Natl Acad Sci USA. 1998;95:12352–12357. doi: 10.1073/pnas.95.21.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tasdemir D. Type II fatty acid biosynthesis, a new approach in antimalarial natural product discovery. Phytochemistry Reviews. 2006;5:99–108. [Google Scholar]
- 13.Heath RJ, White SW, Rock CO. Lipid biosynthesis as a target for antibacterial agents. Prog Lipid Res. 2001;40:467–497. doi: 10.1016/s0163-7827(01)00012-1. [DOI] [PubMed] [Google Scholar]
- 14.Jones SM, Urch JE, Kaiser M, Brun R, Harwood JL, Berry C, Gilbert IH. Analogues of thiolactomycin as potential antimalarial agents. J Med Chem. 2005;48:5932–5941. doi: 10.1021/jm049067d. [DOI] [PubMed] [Google Scholar]
- 15.Perozzo R, Kuo M, Sidhu AbS, Valiyaveettil JT, Bittman R, Jacobs WR, Fidock DA, Sacchettini JC. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J Biol Chem. 2002;277:13106–13114. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 16.Kapoor M, Reddy CC, Krishnasastry MV, Surolia N, Surolia A. Slow-tight-binding inhibition of enoyl-acyl carrier protein reductase from Plasmodium falciparum by triclosan. Biochem J. 2004;381:719–724. doi: 10.1042/BJ20031821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu JZ, Lee PJ, Waters NC, Prigge ST. Fatty acid synthesis as a target for antimalarial discovery. Comb Chem High Throughput Screen. 2005;8:15–26. doi: 10.2174/1386207053328192. [DOI] [PubMed] [Google Scholar]
- 18.Kumaratilake LM, Robinson BS, Ferrante A, Poulos A. Antimalarial properties of n-3 and n-6 polyunsaturated fatty acids: In vitro effects on Plasmodium falciparum and in vivo effects on P. berghei. J Clin Invest. 1992;89:961–967. doi: 10.1172/JCI115678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krugliak M, Deharo E, Shalmiev G, Sauvain M, Moretti C, Ginsburg H. Antimalarial effects of C18 fatty acids on Plasmodium falciparum in culture and on Plasmodium vinckei petteri and Plasmodium yoelii nigeriensis in vivo. Exp Parasitol. 1995;81:97–105. doi: 10.1006/expr.1995.1097. [DOI] [PubMed] [Google Scholar]
- 20.Suksamrarn A, Buaprom M, Udtip S, Nuntawong N, Haritakun R, Kanokmedhakul S. Antimycobacterial and antiplasmodial unsaturated carboxylic acid from the twigs of Scleropyrum wallichianum. Chem Pharm Bull. 2005;53:1327–1329. doi: 10.1248/cpb.53.1327. [DOI] [PubMed] [Google Scholar]
- 21.Tasdemir D, Topaloglu B, Perozzo R, Brun R, O’Neill R, Carballeira NM, Zhang X, Tonge PJ, Linden A, Rüedi P. Marine natural products from the Turkish sponge Agelas oroides that inhibit the enoyl reductase from Plasmodium falciparum, Mycobacterium tuberculosis and Escherichia coli. Bioorg Med Chem. 2007;15:6834–6845. doi: 10.1016/j.bmc.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 22.Sperandeo NR, Brun R. Synthesis and biological evaluation of pyrazolylnaphthoquinones as new potential antiprotozoal and cytotoxic agents. Chem Bio Chem. 2003;4:69–72. doi: 10.1002/cbic.200390016. [DOI] [PubMed] [Google Scholar]
- 23.Rouhi AM. Tuberculosis: a tough adversary. Chem Eng News. 1999;77(20):52–70. [Google Scholar]
- 24.Nachega JB, Chaisson RE. Tuberculosis drug resistance: a global threat. Clin Infect Dis. 2003;36:S24–30. doi: 10.1086/344657. [DOI] [PubMed] [Google Scholar]
- 25.Marrakchi H, Laneelle G, Quemard A. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiol. 2000;146:289–296. doi: 10.1099/00221287-146-2-289. [DOI] [PubMed] [Google Scholar]
- 26.Oliveir JS, Vasconcelos IB, Moreira IS, Santos DS, Basso LA. Enoyl reductases as targets for the development of anti-tubercular and anti-malarial agents. Curr Drug Targets. 2007;8:399–411. doi: 10.2174/138945007780058942. [DOI] [PubMed] [Google Scholar]
- 27.Morbidoni HR, Vilcheze C, Kremer L, Bittman R, Sacchettini JC, Jacobs WR., Jr Dual inhibition of mycobacterial fatty acid biosynthesis and degradation by 2-alkynoic acids. Chem Biol. 2006;13:297–307. doi: 10.1016/j.chembiol.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Rozwarski DA, Vilcheze C, Sugantino M, Bittman R, Sacchettini JC. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J Biol Chem. 1999;274:15582–15589. doi: 10.1074/jbc.274.22.15582. [DOI] [PubMed] [Google Scholar]
- 29.Kabara JJ, Swieczkowski DM, Conley AJ, Truant JP. Fatty acids and derivatives as antimicrobial agents. Antimicrob Agents Chemother. 1972;2:23–28. doi: 10.1128/aac.2.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kondo E, Kanai K. The relationship between the chemical structure of fatty acids and their mycobactericidal activity. Japan J Med Sci Biol. 1977;30:171–178. doi: 10.7883/yoken1952.30.171. [DOI] [PubMed] [Google Scholar]
- 31.Saito H, Tomioka H, Yoneyama T. Growth of group IV mycobacteria on medium containing various saturated and unsaturated fatty acids. Antimicrob Agents Chemother. 1984;26:164–169. doi: 10.1128/aac.26.2.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carballeira NM, Cruz H, Kwong CD, Wan B, Franzblau S. 2-Methoxylated fatty acids in marine sponges: Defense mechanism against mycobacteria? Lipids. 2004;39:675–680. doi: 10.1007/s11745-004-1281-8. [DOI] [PubMed] [Google Scholar]
- 33.Seidel V, Taylor PW. In vitro activity of extracts and constituents of Pelagonium against rapidly growing mycobacteria. Int J Antimicrob Agents. 2004;23:613–619. doi: 10.1016/j.ijantimicag.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Stavri M, Schneider R, O’Donnell G, Lechner D, Bucar F, Gibbons S. The antimycobacterial components of hops (Humulus lupulus) and their dereplication. Phytother Res. 2004;18:774–776. doi: 10.1002/ptr.1527. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki K, Shono F, Kai H, Uno T, Uyeda M. Inhibition of topoisomerases by fatty acids. J Enzyme Inhibition. 2000;15:357–366. doi: 10.1080/14756360009040693. [DOI] [PubMed] [Google Scholar]
- 36.Mizushina Y, Tsuzuki T, Eitsuka T, Miyazawa T, Kobayashi K, Ikawa H, Kuriyama I, Yonezawa Y, Takemura M, Yoshida H, Sakaguchi K. Inhibitory action of conjugated C18-fatty acids on DNA polymerases and DNA topoisomerases. Lipids. 2004;39:977–983. doi: 10.1007/s11745-004-1319-y. [DOI] [PubMed] [Google Scholar]
- 37.Harada H, Yamashita U, Kurihara H, Fukushi E, Kawabata J, Kamei Y. Antitumor activity of palmitic acid found as a selective cytotoxic substance in a marine alga. Anticancer Res. 2002;22:2587–2590. [PubMed] [Google Scholar]
- 38.Yonezawa Y, Tsuzuki T, Eitsuka T, Miyazawa T, Hada T, Uryu K, Murakami-Nakai C, Ikawa H, Kuriyama I, Takemura M, Oshige M, Yoshida H, Sakaguchi K, Mizushina Y. Inhibitory effect of conjugated eicosapentaenoic acid on human DNA topoisomerases I and II. Arch Biochem Biophys. 2005;435:197–206. doi: 10.1016/j.abb.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 39.Nemoto T, Ojika M, Sakagami Y. Amphimic acids, novel unsaturated C28 fatty acids as DNA topoisomerase I inhibitors from an Australian sponge Amphimedon sp. Tetrahedron Lett. 1997;38:5667–5670. [Google Scholar]
- 40.Nemoto T, Yoshino G, Ojika M, Sakagami Y. Amphimic acids and related long-chain fatty acids as DNA topoisomerase I inhibitors from an Australian sponge, Amphimedon sp: Isolation, structure, synthesis, and biological evaluation. Tetrahedron. 1997;53:16699–16710. [Google Scholar]
- 41.Mizushima T, Natori S, Sekimizu K. Inhibition of Escherichia coli DNA topoisomerase I activity by phospholipids. Biochem J. 1992;285:503–506. doi: 10.1042/bj2850503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee HK, Lee DS, Kim J, Kim JS, Im KS, Jung JH. Topoisomerase I inhibitors from the Streptomyces sp. strain KM86-9B isolated from a marine sponge. Arch Pharm Res. 1998;21:729–733. doi: 10.1007/BF02976766. [DOI] [PubMed] [Google Scholar]
- 43.Carballeira NM, Betancourt JE, Orellano EA, González FA. Total synthesis and biological evaluation of (5Z,9Z)-5,9-hexadecadienoic acid, an inhibitor of human topoisomerase I. J Nat Prod. 2002;65:1715–1718. doi: 10.1021/np0202576. [DOI] [PubMed] [Google Scholar]
- 44.Makarieva TN, Santalova EA, Gorshkova IA, Dmitrenok AS, Guzii AG, Gorbach VI, Svetashev VI, Stonik VA. A new cytotoxic fatty acid (5Z,9Z)-22-methyl-5,9-tetracosadienoic acid and the sterols from the far eastern Sponge Geodinella robusta. Lipids. 2002;37:75–80. doi: 10.1007/s11745-002-0866-6. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Ishibashi M, Sasaki T, Kobayashi J. New bromine-containing unsaturated fatty acid derivatives from the Okinawan marine sponge Xestospongia sp. J Chem Research (M) 1995:901–923. [Google Scholar]
- 46.Akpan A, Morgan R. Oral candidiasis. Postgrad Med J. 2002;78:455–459. doi: 10.1136/pmj.78.922.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Araiza J, Canseco P, Bonifaz A. Otomycosis: clinical and mycological study of 97 cases. Rev Laryngol Otol Rhinol (Bord) 2006;127:251–254. [PubMed] [Google Scholar]
- 48.Mugliston T, O’Donoghue G. Otomycosis-a continuing problem. J Laryngol Otol. 1985;99:327–333. doi: 10.1017/s002221510009678x. [DOI] [PubMed] [Google Scholar]
- 49.Fidel PL., Jr Distinct protective host defenses against oral and vaginal candidiasis. Med Mycol. 2002;40:359–375. [PubMed] [Google Scholar]
- 50.Chakrabarti A. Microbiology of systemic fungal infections. J Postgrad Med. 2005;51:16–20. [PubMed] [Google Scholar]
- 51.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 52.Cha R, Sobel JD. Fluconazole for the treatment of candidiasis: 15 years experience. Expert Rev Anti Infect Ther. 2004;2:357–366. doi: 10.1586/14787210.2.3.357. [DOI] [PubMed] [Google Scholar]
- 53.Loo DS. Systemic antifungal agents: an update of established and new therapies. Adv Dermatol. 2006;22:101–124. doi: 10.1016/j.yadr.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 54.Bergsson G, Arnfinnsson J, Steingrimsson O, Thormar H. In vitro killing of Candida albicans by fatty acids and monoglycerides. Antimicrob Agents Chemother. 2001;45:3209–3212. doi: 10.1128/AAC.45.11.3209-3212.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Avis TJ, Bélanger RR. Specificity and mode of action of the antifungal fatty acid cis-9-heptadecenoic acid produced by Pseudozyma flocculosa. Appl Environ Microbiol. 2001;67:956–960. doi: 10.1128/AEM.67.2.956-960.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gershon H, Shanks L. Antifungal properties of 2-alkynoic acids and their methyl esters. Can J Microbiol. 1978;24:593–597. doi: 10.1139/m78-096. [DOI] [PubMed] [Google Scholar]
- 57.Gershon H, Shanks L. Antifungal activity of fatty acids and derivatives: Structure activity relationships. In: Kabara JJ, editor. The pharmacological effect of lipids. American Oil Chemical Society; Champaign, Illinois: 1978. pp. 51–62. [Google Scholar]
- 58.Konthikamee W, Gilbertson JR, Langkamp H, Gershon H. Effect of 2-alkynoic acids on in vitro growth of bacterial and mammalian cells. Antimicrob Agents Chemother. 1982;22:805–809. doi: 10.1128/aac.22.5.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wood R, Lee T. Metabolism of 2-hexadecynoate and inhibition of fatty acid elongation. J Biol Chem. 1981;256:12379–12386. [PubMed] [Google Scholar]
- 60.Upreti GC, Matocha M, Wood R. Effect 2-hexadecynoic acid on cultured 7288C hepatoma cells. Lipids. 1981;16:315–322. doi: 10.1007/BF02534955. [DOI] [PubMed] [Google Scholar]
- 61.Li XC, Jacob MR, ElSohly HN, Nagle DG, Smillie TJ, Walker LA, Clark AM. Acetylenic acids inhibiting azole-resistant Candida albicans from Pentagonia gigantifolia. J Nat Prod. 2003;66:1132–1135. doi: 10.1021/np030196r. [DOI] [PubMed] [Google Scholar]
- 62.Carballeira NM, Sanabria D, Parang K. Total synthesis and further scrutiny of the in vitro antifungal activity of 6-nonadecynoic acid. Arch Pharm Chem Life Sci. 2005;338:441–443. doi: 10.1002/ardp.200500102. [DOI] [PubMed] [Google Scholar]
- 63.Carballeira NM, Sanabria D, Cruz C, Parang K, Wan B, Franzblau S. 2,6-Hexadecadiynoic acid and 2,6-nonadecadiynoic acid: Novel synthesized acetylenic fatty acids as potent antifungal agents. Lipids. 2006;41:507–511. doi: 10.1007/s11745-006-5124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacMillan JB, Molinski TF. Majusculoic Acid, a brominated cyclopropyl fatty acid from a marine cyanobacterial mat assemblage. J Nat Prod. 2005;68:604–606. doi: 10.1021/np049596k. [DOI] [PubMed] [Google Scholar]
- 65.Ellis RJ, Timms-Wilson TM, Bailey MJ. Identification of conserved traits in fluorescent pseudomonads with antifungal activity. Environ Microbiol. 2000;2:274–284. doi: 10.1046/j.1462-2920.2000.00102.x. [DOI] [PubMed] [Google Scholar]
- 66.Glickman MS, Cox JS, Jacobs WR. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–727. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- 67.Raju PK, Reiser R. Hepatic stearoyl-CoA desaturase activity in mice as affected by early postnatal dietary cyclopropene fatty acids. J Nutr. 1973;103:904–907. doi: 10.1093/jn/103.6.904. [DOI] [PubMed] [Google Scholar]
- 68.Raju PK, Reiser R. Inhibition of stearoyl coenzyme A desaturase by sterculate in mouse liver microsomes. J Biol Chem. 1972;247:3700–3701. [PubMed] [Google Scholar]
- 69.Parang K, Knaus EE, Wiebe LI, Sardari S, Daneshtalab M, Csizmadia F. Synthesis and antifungal activities of myristic acid analogs. Arch Pharm Pharm Med Chem. 1996;329:475–482. doi: 10.1002/ardp.19963291102. [DOI] [PubMed] [Google Scholar]
- 70.Langer CA, Lodge JA, Travis SJ, Caldwell JE, Lu T, Li Q, Bryant ML, Devadas B, Gokel GW, Kobayashi GS, Gordon JI. 4-Oxatetradecanoic acid is fungicidal for Cryptococcus neoformans and inhibits replication of human immunodeficiency virus I. J Biol Chem. 1992;267:17159–17169. [PubMed] [Google Scholar]
- 71.Carballeira NM, Ortiz D, Parang K, Sardari S. Total synthesis and in vitro-antifungal activity of (±)-2-methoxytetradecanoic acid. Arch Pharm Pharm Med Chem. 2004;337:152–155. doi: 10.1002/ardp.200300824. [DOI] [PubMed] [Google Scholar]
- 72.Carballeira NM, O’Neill R, Parang K. Racemic and optically active 2-methoxy-4-oxatetradecanoic acids: Novel synthetic fatty acids with selective antifungal properties. Chem Phys Lipids. 2005;136:47–54. doi: 10.1016/j.chemphyslip.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 73.Carballeira NM, Pagán M. New methoxylated fatty acids from the Caribbean sponge Callyspongia fallax. J Nat Prod. 2001;64:620–627. doi: 10.1021/np000537q. [DOI] [PubMed] [Google Scholar]