

Abstract
In spite of great advances in cancer therapy, there is considerable current interest in developing anticancer agents with a new mode of action because of the development of resistance by cancer cells towards current anticancer drugs. A growing number of studies have shown that some of the cationic antimicrobial peptides (AMPs), which are toxic to bacteria but not to normal mammalian cells, exhibit a broad spectrum of cytotoxic activity against cancer cells. Such studies have considerably enhanced the significance of AMPs, both synthetic and from natural sources, which have been of importance both for an increased understanding of the immune system and for their potential as clinical antibiotics. The electrostatic attraction between the negatively charged components of bacterial and cancer cells and the positively charged AMPs is believed to play a major role in the strong binding and selective disruption of bacterial and cancer cell membranes, respectively. However, it is unclear why some host defense peptides are able to kill cancer cells when others do not. In addition, it is not clear whether the molecular mechanism(s) underlying the antibacterial and anticancer activities of AMPs are the same or different. In this article, we review various studies on different AMPs that exhibit cytotoxic activity against cancer cells. The suitability of cancer cell-targeting AMPs as cancer therapeutics is also discussed.
1. Introduction
Despite recent advances in treatment modalities, cancer remains a major source of morbidity and mortality throughout the world. In the United States, cancer is the leading cause of death for individuals less than 85 years of age [1]. Moreover, the incidence of many cancers, including cancers of the skin, prostate, breast, and kidney, continues to increase [2]. “Cancer” is, in fact, a general term that refers to over 100 distinct diseases affecting many different tissues and cell types. However, all forms of cancer are characterized by abnormal cell growth resulting from a relatively small number of inherited or environmentally-induced genetic mutations [3]. Hanahan and Weinberg [4] have argued that in order for a cell to become cancerous, it must acquire six unique traits as a result of altered cell physiology. These defining traits of cancer cells are: 1) the ability to generate their own growth signals or respond to weak growth signals that are ignored by healthy cells; 2) insensitivity to antiproliferative signals; 3) resistance to cellular suicide mechanisms that normally cause aberrant cells to die by apoptosis; 4) the capacity for limitless replication; 5) the ability to stimulate new blood vessel development in order to allow for tumour growth; and 6) the capacity to invade tissues, at first locally, and later to spread or metastasize throughout the body. Although localized cancers can often be successfully treated by surgery and/or radiation therapy, chemotherapy remains the usual treatment of choice for advanced or metastatic disease [5]. However, the use of conventional chemotherapeutic agents that typically target rapidly dividing cancer cells is often associated with deleterious side-effects caused by inadvertent drug-induced damage to healthy cells and tissues [6, 7]. Moreover, cancer cells that are quiescent or slowly proliferating are refractory to the cytotoxic effect of chemotherapeutic drugs that act at the level of DNA synthesis [8]. Cancer cells also frequently become resistant to chemotherapy as a consequence of cellular changes that include increased expression of drug-detoxifying enzymes and drug transporters, altered interactions between the drug and its target, an increased ability to repair DNA damage, and defects in the cellular machinery that mediate apoptosis [9]. The development of a new class of anticancer drugs that lack the toxicity of conventional chemotherapeutic agents and are unaffected by common mechanisms of chemoresistance would be a major advance in cancer treatment. In this article, we review the recent studies on naturally occurring, as well as some selected hybrid and synthetic cationic antimicrobial peptides (AMPs) that exhibit anticancer activities. The biochemical properties and anticancer activities of some of these AMPs are summarized in Table 1.
2. Antimicrobial Peptides
Naturally occurring AMPs probably represent one of the first evolved and successful forms of chemical defense of eukaryotic cells against bacteria, protozoa, fungi, and viruses [10]. AMPs have been found in every species that has been tested, including bacteria, fungi, plants, and animals. A very large number of AMPs isolated from nature and thousands of synthetic variants have been found to have a broad spectrum of antimicrobial activities. For a regularly updated list of plant and animal AMPs, see the websites: http://www.bbcm.univ.trieste.it/~tossi/antimic.html and http://aps.unmc.edu/AP/main.php. Most AMPs kill both Gram-positive and Gram-negative bacteria, while a significant number of these bactericidal peptides have been shown to have anticancer and antiviral activities [10, 11]. Some of these peptides have been found to have lipopolysaccharide (LPS) neutralizing ability and the capacity to recruit the adaptive immune response [12]. The growing problem of resistance to conventional antibiotics is a global public health problem and the need for new antibiotics has stimulated interest in the development of AMPs as human therapeutics [13]. Several AMPs have already entered pre-clinical and clinical trials to promote wound healing and for the treatment of cystic fibrosis, catheter site infections, acne, and patients undergoing stem cell transplantation [14-16].
In addition to antibacterial activities of AMPs, recent studies on their anticancer activities are exciting and are most relevant for this review [17]. While not all AMPs are able to kill cancer cells, those that do can be placed into two broad categories: (a) AMPs that are highly potent against bacteria and cancer cells but not against normal mammalian cells and (b) AMPs that are cytotoxic for bacteria, cancer cells, and normal mammalian cells. The structural and functional properties of AMPs are briefly outlined below.
Although the amino acid sequences of different AMPs are highly heterogeneous and a great variation in their secondary structures has been reported. AMPs are generally cationic (i.e., the net charge at neutral pH varies from +2 to +9) and amphipathic, which enables the peptides to interact with and disrupt lipid membranes. Most AMPs are very short in length, containing 5 to 40 amino acid residues, while a few of them contain more than 40 residues. Positively charged residues such as Lys and Arg and substantial hydrophobic residues (~30% or more) are commonly found in these peptides. An important set of AMPs, such as tritrpticin, lactoferricins and indolicidin, are rich in Trp and Arg residues [18]. Studies have shown that all-D-amino-acid analogs have identical activity but opposite chirality relative to natural all-L-peptides; this property has been utilized in designing AMPs that can resist proteolytic degradation. Unlike currently available conventional antibiotics, which typically interact with a specific target protein, most of these cationic AMPs target the cell membrane of invading microorganisms, leading to cell lysis and death [10, 18, 19, 20]. Thus, AMPs offer the possibility of a new class of therapeutics agents, which are complementary to existing antibiotics, and to which bacteria may not be able to develop resistance.
Most of the linear AMPs are unstructured in solution (an exception is LL37, a human antimicrobial peptide [20, 21]), while the cyclic peptides due to the presence of one or more Cys-Cys disulfide bonds form β-sheets [22]. The amphipathicity of AMPs is enhanced upon the induction of specific secondary structures, such as α-helices, β-sheets, or extended polyproline-like helices; this amphipathicity is thought to play a key role in their antimicrobial mechanism of action. The mechanism by which an AMP executes its function depends on a number of physicochemical properties: the amino acid sequence, net charge, amphipathicity, hydrophobicity, structural folding (includes secondary structure, dynamics and orientation) in membranes, oligomerization, peptide concentration, and membrane composition [19]. There are several mechanisms of membrane-disruption proposed to explain the activity of AMPs (Figure 1). Some of the models used to identify the membrane-disrupting process are carpet model [23], barrel-stave model [19], toroidal-pore wormhole model [24], and detergent-type membrane lytic mechanism [25, 26]. In the carpet model, AMPs assemble on the surface of the membrane to disrupt the membrane via barrel-stave, toroidal-pore, or detergent-type mechanisms. In the barrel-stave model [19, 27-31], AMPs insert with a transmembrane orientation in membranes and aggregate to form a traditional ion-channel pore. In the toroidal-pore model [24, 32], the peptides are located closer to the head group region with an initial orientation parallel to the lipid bilayer surface. In this orientation, the hydrophilic side of the helix is exposed to the hydrophilic lipid head groups and the water phase outside bilayers, while the hydrophobic face of the helix is buried in the hydrophobic core of the membrane to minimize the net free energy of folding process. Aggregation of peptides to a sufficient local concentration increases the curvature strain on the membrane surface to extent that toroidal-pores form. In the detergent-type mechanism [26], the peptides first carpet the surface of the lipid bilayer like the beginning stages of the toroidal-pore model. Following this step, the peptide aggregation leads to a sufficient high local concentration where the amphipathic nature of the peptide allows them to behave like detergents and break the lipid membrane into small fragments. These fragments can be like bicelles or micelles [20]. There are other models such as the sinking raft model [33] or the molecular electroporation [34], which have not received much attention in the field, but could be useful to explain the antimicrobial activity of certain AMPs. In the sinking raft model, AMPs bound to the cell membrane introduce a large membrane curvature due to a mass imbalance, which make AMPs sink and generate transient pores in membranes. On the other hand, in the molecular electroporation model, AMPs create a difference in the electrical potential across the membrane leading to pore formation through electroporation.
Figure 1. Models of AMP-induced membrane permeabilization.

Cationic linear antimicrobial peptides are initially unstructured monomers (most AMPs) or helical and aggregated (for example, LL-37) in aqueous solution. They bind to negatively charged membrane surface by electrostatic interactions. In the carpet model, peptides bind to phospholipid head groups and align themselves parallel to the membrane surface in a carpet-like fashion until a critical threshold concentration is reached. In the barrel stave model, peptides self aggregate in the membrane once a critical threshold concentration of peptide is reached, resulting in the formation of a transmembrane pore lined by peptide oriented such that the hydrophilic face forms the inner channel while the hydrophobic face is on the outside. Since an AMP has to span the entire thickness of the lipid bilayer in this model, a minimum of ~20 residues for α-helical peptide and ~8 residues for a β-sheet peptide is required to function via the barrel stave mechanism. The toroidal pore model is an extension of the barrel stave model and postulates that at some critical concentration of peptide curvature strain induces the membranes to curve inward, resulting in the formation of a pore that is lined by both peptide and lipid headgroups. Repulsive interactions between the positively charged residues of the peptide are minimized due to the presence of the negatively charged phospholipids in the pore-lining. While the formation of these pores depend on the lipid:peptide ration and the ionic selectivity depend on the membrane composition, the lifetime of these pores seem to vary. The detergent-like model proposes that peptides intercalate in between the phospholipid head groups in a cone-like fashion causing curvature strain and micellization at local regions of high peptide density or when preformed peptide aggregates interact with lipid membranes. Recent NMR studies suggested that after sufficient time (may be a month or longer) AMPs fragment the lipid bilayers (even those containing toroidal pores) to form bicelle or micelle-like structures. Other membrane-disruptive and non-membrane disruptive mechanisms of some AMPs are discussed in the text.
Bacterial membranes are negatively charged with lipids such as phosphatidylglycerol (PG), cardiolipin (CL), or phosphatidylserine (PS). Electrostatic interaction between these negatively charged lipids and the positively charged AMPs enable the cationic peptides to bind bacterial membranes. The outer membrane of a Gram-negative bacteria is also negatively charged as it contains anionic lipopolysaccharides (LPS); LPS is normally stabilized by the divalent cations like Ca2+ and Mg2+ but AMPs diplaces them to interact with the outer membrane. On the other hand, mammalian cell membranes consist largely of zwitterionic phospholipids (neutral in net charge) such as phosphatidylethanolamine (PE), phosphatidylcholine (PC), or sphingomyelin (SM) and are therefore less attractive to cationic AMPs. In addition, cholesterol present in mammalian membranes makes it harder for AMPs to disrupt lipid bilayer structures. Therefore, the AMPs are selectively toxic to bacteria.
Solid-state NMR approaches have been developed and successfully utilized to determine the secondary structure and location of the peptide in lipid bilayers, and also to determine the structure of the lipid bilayers. For example, solid-state NMR studies have determined the structure and bilayer-surface orientation of several linear AMPs including MSI-78 [32, 35-37], MSI-594 [35], magainin2 [38], MSI-843 [39], PGLa [40-43], subtilosin A [44], KIAGAKI [45], fragments of granulysin [46], and other peptides [47-50]. These studies support the carpet mechanism and rule out the barrel-stave mechanism as the mode of action for these AMPs. Thus, these peptides have been shown to induce positive curvature strain that leads to toroidal-type membrane disruption [24, 32], which is consistent with other biophysical studies on magainin peptides. These peptides also induce significant disorder in the hydrophobic core of lipid bilayers. On the other hand, peptides like pardaxin [27] and alamethicin [29-31] prefer a transmembrane orientation and disrupt membranes via the barrel-stave mechanism. Interestingly, pardaxin changes its orientation depending on the membrane composition, which is consistent with its multiple biological activities. A recent solid-state NMR study has reported a change in the orientation of PGLa when the concentration of the peptide was increased [41-43]. Similar studies have also been reported for non-linear AMPs. Solid-state NMR studies on non-helical peptides like protegrin have revealed their mechanism of action as well [51].
While the above-mentioned mechanisms explain the bactericidal activity of AMPs due to the disruption of the membrane integrity that lead to leakage of ions and metabolites and depolarization, there are AMPs that act on putative key intracellular targets in bacteria without disrupting the cell membrane [Hwang and Vogel, 1998]. These AMPs inhibit the synthesis of protein or cell-wall, inhibit the activities of certain enzymes, interfere with the metabolic processes of microbes, or interact with DNA or RNA.
Certain AMPs have been shown to have antiviral activities: they inhibit the replication of enveloped viruses such as influenza A virus [52], vesicular stomatitis virus (VSV) and human immunodeficiency virus (HIV-1) [53, 54]. The generally accepted antiviral mechanism of action is a direct interaction of these AMPs with the envelope of the virus, leading to permeation of the envelope and, eventually, lysis of the virus particle, analogous to the pore-formation mechanism mentioned above for antibacterial activity of AMPs. However, other mechanisms for antiviral activities have also been proposed: T22 ([(Tyr5,12, Lys7)-polyphemusin II]) a 18 amino acids peptide analogue of polyphemusin II, an AMP isolated from the hemocyte debris of American horseshoe crabs (Limulus polyphemus), was found to specifically inhibit the ability of T cell line-tropic HIV-1 to induce cell fusion [55]. On the other hand, lactoferricin, which is an N-terminal fragment of lactoferrin, inhibits the binding and uptake of human papilloma virus, human cytomegalovirus, and herpes simplex virus into human cells [56-58].
3. Membrane Differences that Contribute to the Selectivity of Antimicrobial Peptides for Cancer Cells
Changes in the membrane of a cell have important implications in the progression of cancer, as they play a key role in the cell’s response to its environment. The cell membrane of a malignant tumor cell may influence its ability to grow even without the signals that would normally inspire growth, as well as to attach and respond to neighboring cells differently. The cell membrane may also affect a cancer cell’s motility, aiding in invasion and metastasis. Therefore, it is important to look at the differences between normal cells, non-malignant tumor cells, and malignant tumor cells, as well as how these differences arise during the progression from a normal cell to a tumor cell. These differences likely give insight into possible treatments and prevention. The cell membrane is a site of many such differences, and will be further examined at the molecular level.
Fundamental differences exist between the cell membranes of malignant cells and normal cells that likely account for the ability of certain AMPs to kill cancer cells (hereafter referred to as anticancer peptides; ACPs) while sparing healthy cells. In this regard, electrostatic interactions between cationic ACPs and anionic cell membrane components are believed to be a major factor in the selective killing of cancer cells by ACPs. Cancer cell membranes typically carry a net negative charge due to a higher than normal expression of anionic molecules such as PS (<9% of the total phospholipids of membranes) [59, 60] and O-glycosylated mucins [61, 62]. In addition, the negative membrane potential of cancer cells may also contribute to the selective cytotoxic activity of ACPs [63]. In contrast to neoplastic cells, electrostatic interactions between ACPs and untransformed cells are not favored because of the overall neutral charge conferred on healthy cells by the zwitterionic nature of their major membrane components, e.g., SM, PE, and PC [64]. Therefore, it is believed that the ACPs could kill cancer cells via one of the aforementioned mechanisms to disrupt the cell membrane. Another possibility is the induction of apoptosis in cancer cells via mitochondrial membrane disruption following ACP uptake into the cytoplasm. Other factors that contribute to the preferential killing of cancer cells by ACPs include membrane fluidity and cell-surface area. The membrane fluidity of cancer cells is greater than that of untransformed cells [65, 66], which may enhance the lytic activity of ACPs by facilitating membrane destabilization. Leuschner and Hansel [67] have suggested that cholesterol, which is a major component of eukaryotic cell membranes [68], may protect eukaryotic cells from the cytolytic effect of antimicrobial peptides by altering membrane fluidity and thereby interfering with the membrane insertion of lytic peptides. In this regard, the rate of membrane insertion by the AMP cecropin and its analogues is reduced when the cholesterol content of the membrane is increased [69, 70]. Interestingly, some breast and prostate cancer cell lines have recently been shown to possess elevated levels of cholesterol-rich lipid rafts [71], which may make these neoplastic cells less susceptible to killing by ACPs. Finally, cancer cells have a greater cell surface area than normal cells due to the presence of higher numbers of microvilli [72, 73], which are minute projections of the cell membrane, that may allow cancer cells to bind increased numbers of ACP molecules. The microvilli are also more irregular in size and shape, which may be due to changes in structural proteins [73, 74]. These irregularities could potentially affect receptor accessibility, cell adhesion, and other communications between the cancer cell and its environment, and could also play a role in the binding of ACPs selectively to the cancer cells. Therefore, differences in cell membrane composition, fluidity, and surface area between cells from different types of cancer may account for the variability in killing efficacy observed when a given ACP is tested against different types of cancer cells [67, 75].
Glycosylation of membrane-associated glycoproteins and glycolipids undergo changes when a cell becomes cancerous, which could play a role in determining the susceptibility of a given cancer cell to the cytotoxic activity of ACPs. Therefore, it would be useful to understand these important changes at the molecular level. These changes in glycoproteins are most often due to activation of certain glycosyltransferases, which catalyze biosynthesis of glycoproteins. Less often, the changes can be attributed to over-expression of glycosidases, which catalyze degradation processes [76].
Glycosylation is the most common post-translational modification of proteins. The first main type of glycosylation is N-linked. N-linked glycosylation occurs at the consensus sequence Asn-Xaa-Ser/Thr on a protein, Xaa being any amino acid except proline. The N-linkage occurs between N-acetylglucosamine (GlcNAc) and the asparagine residue; there are various subgroups of N-glycans, but all share a common core, the di-N-acetyl-chitobiose trimannosyl core (see Figure 2A) [77].
Figure 2.

(A) N-glycan di-N-acetyl-chitobiose trimannosyl core; (B) bisecting GlcNAc via addition of GlcNAc by GnT-III.
The bisecting GlcNAc found in various glycoproteins (see Figure 2B) and the enzyme that catalyzes its attachment, N-acetylglucosaminyltransferase III (GnT-III) may play a role in cancer. Over-expression of GnT-III has been shown to cause an increase in bisecting GlcNAc residues on surface glycoproteins or other specific proteins. This increase may affect a number of cell processes affecting cancer progression; some examples are: altered sensitivity to proteolysis, enhanced adhesion properties aiding in metastasis, resistance to natural killer cells of the immune system, impairment of signaling pathways, and sorting of proteins [76]. It has also been reported that cancerous cells have larger N-glycans, based on increased β1,6GlcNAc branching of the trimannosyl core. GlcNAc-IV, the glycosyltransferase responsible for this branching, activity is increased in cancerous cells and may be responsible for increased malignancy by altering regulation of growth and adhesion properties [78].
The second major type of glycosylation is O-linked, which is very common in mucins (large, heavily glycosylated proteins; transmembrane or secreted on mucosal surfaces or in saliva). This linkage occurs between the hemi-acetal function of N-acetylactosamine (GalNAc) and the −OH group of a serine or threonine residue. While composed of varying monosaccharide units, all O-glycans are made up of 3 structural units. The core consists of GalNAc linked to the peptide and 1 or 2 other monosaccharides. The backbone is made of galactose (Gal) and GalNAc sequences. Finally, the periphery (end of O-glycan) is made up of various monosaccharides.
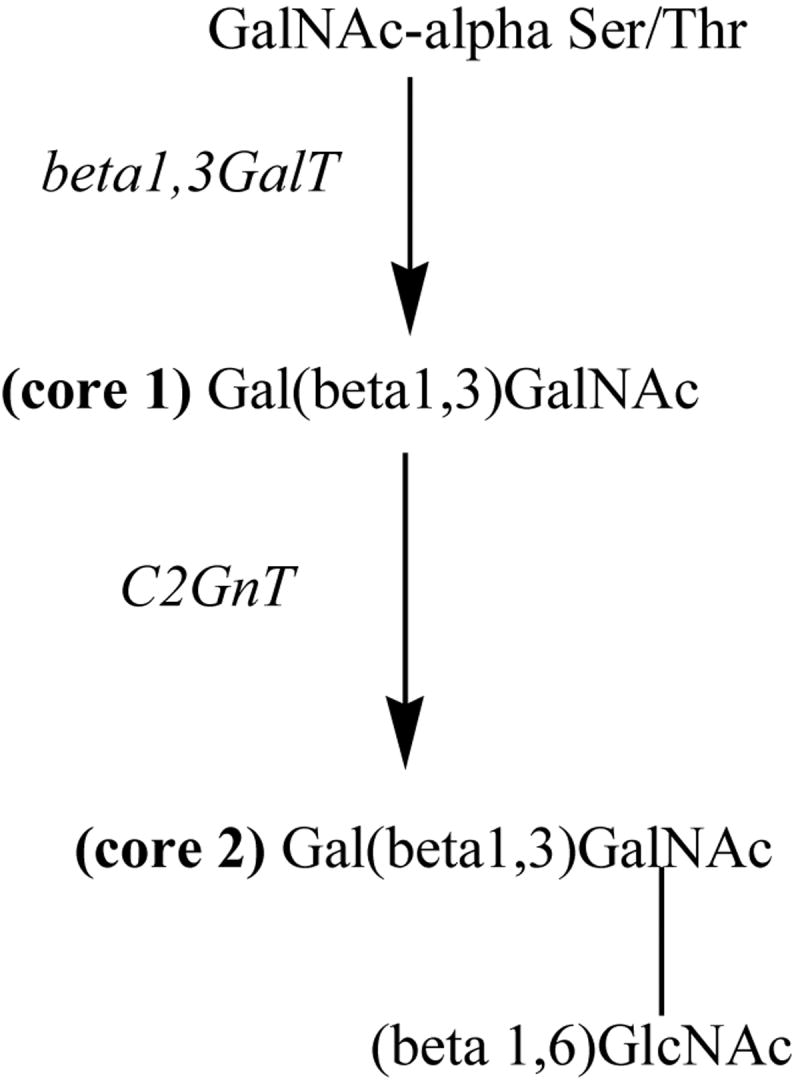
Changes in glycosylation of mucins in the mammary gland are often seen in breast cancer patients; one particular mucin, MUC1 in mammary tissue has been studied in detail. MUC1 is a major component of luminal epithelial cells of the mammary gland and is up-regulated during pregnancy, lactation, and in breast cancer tissue. The extracellular portion of the transmembrane form of MUC1 contains 25-125 tandem repeats, which are rich in serine and threonine residues, making them good candidates for O-linked glycosylation [79]. In normal MUC1, O-glycan structure is based on what is known as core 2. Core 1 consists of the GalNAc that has been added to a Ser or Thr residue plus galactose added via a β1,3 linkage. Core 2 results when a GlcNac is added (via β1,6 linkage) to the GalNAc of core 1 (see Figure 3). O-linked glycans are added sequentially as the protein moves through the Golgi apparatus and each addition is catalyzed by a specific glycosyltransferase. Cancer-associated MUC1 proteins show a higher overall level of glycosylation, have shorter glycans based on core 1 structure, and feature a high level of sialic acid [80]. Sialic acid is a terminal sugar and therefore is often found at the ends of the truncated core 1 based glycans of cancer cells. This difference in glycosylation may be due to over-expression of certain glycosyltransferases that catalyze formation of these core 1-based glycans. The shorter glycans also may promote higher glycosylation along the peptide chain as it moves through the Golgi, as they will not cause as much steric hindrance as the larger chains would. It has also been shown that the sequences flanking a certain Ser/Thr residue may also influence its likelihood of being glycosylated; higher glycosylation of nearby residues may therefore lead to even more glycosylation [80, 81].
Figure 3.
MUC1 O-glycan cores: non-cancerous (core 1 based) versus cancerous cells (core 2 based).
While the exact molecular mechanism by which altered glycosylation occurs as a cell becomes malignant is still unclear, such changes could play an important role in the selectivity of ACPs. It is known that glycosylation alters the secondary structure and dynamics of a membrane-associated peptide or protein. Moreover, differential branching and sialic acid content of N-linked glycans associated with transmembrane glycoproteins are believed to contribute to the net negative charge that is carried by the cell membrane of many cancer cells [82]. Interestingly, glycosylation has also been shown to increase the potency of the AMP drosocin [83]. It is therefore likely that the binding affinity of cationic AMPs (with and without glycosylation) for cancer cells and the subsequent permeabilization of membranes depend at least in part on altered glycosylation of cancer cell membrane proteins. Future investigations on the effect of ACP glycosylation on cytotoxic activity might yield more effective anticancer compounds.
The general membrane-targeted method of cell lysis shows potential for synergy with current cancer treatments and ACPs have, in fact, shown additional killing of cancer cells when tested in concert with antineoplastic drug treatments. AMPs with cytotoxic activity against cancer cells also show potential for overcoming common problems with multiple-drug resistant (MDR) proteins since AMPs should not select for resistant cancer cells. MDR proteins cause problems for current treatments because they give a cancerous cell the ability to resist treatment by simply pumping antineoplastic drugs out of the cell where they can do no harm. An AMP, however, will kill the cell simply by membrane disruption and avoid this resistance mechanism. Other beneficial traits of AMPs as anticancer agents include their wide range of activity, their ability to kill cancer cells quickly, their ability to destroy primary tumors as well as prevent metastasis, and the fact that they do not harm vital organs [84].
2. α-Helical Anticancer Peptides
2.1 BMAP-27 and BMAP-28
BMAP-27 (amino acid sequence: GRFKRFRKKFKKLFKKLSPVIPLLHL) and BMAP-28 (GGLRSLGRKILRAWKKYGPIIVPIIRI) are bovine cathelicidin-derived AMPs with sequences of 27 and 28 amino acid residues, respectively, that were originally deduced from their cDNAs [85]. The cationic NH2-terminal portion (residues 1 to18) of these peptides is predicted to form an amphipathic α-helix that is followed by a hydrophobic tail (residues 19 to 27 or 28) at the COOH-terminus. Interestingly, a truncated analogue of BMAP-28 composed of only the 18 NH2-terminal amino acid residues, as well as a modified form of full-length BMAP-28 in which highly hydrophobic amino acids in the COOH-terminal region were replaced with more hydrophilic amino acids, both showed a dramatic reduction in membrane-permeabilizing activity. This finding indicated that the hydrophobic tail is important for BMAP peptides to mediate their cytotoxic effect. The cytotoxic activity of BMAP-27 and -28 against neoplastic cells was revealed by the observation that freshly isolated human leukemia cells and various human leukemia cell lines exposed to 1.5-6 μM concentrations of the BMAP peptides exhibited membrane permeabilization and an influx of Ca++, followed by DNA fragmentation characteristic of apoptosis [86]. CEM-CCRF human T leukemia cells with a vinblastine-resistant phenotype were also susceptible to killing by BMAP-27 and -28. A subsequent study demonstrated that BMAP-28 treatment of U937 and K562 human leukemia cell lines caused a rapid reduction in mitochondrial membrane potential in situ that was related to a BMAP-28-induced opening of the mitochondrial permeability transition pore, which resulted in cytochrome c release and the initiation of cell death by apoptosis [87]. Although non-proliferating lymphocytes are not affected by the BMAP peptides at concentrations that are cytotoxic for human leukemia cells, at these concentrations both BMAP-27 and -28 are cytotoxic for activated human lymphocytes [86]. Low micromolar concentrations of BMAP-27 and -28 are therefore only active against cells that are actively proliferating. However, significant lysis of normal human neutrophils and erythrocytes occurs following exposure to higher concentrations (30 μM and greater) of the BMAP peptides [85]. The relatively narrow dosage range for BMAP-27- and BMAP-28-mediated killing of cancer cells without deleterious hemolytic activity, combined with the predicted inability of BMAP peptides to effectively kill slowly proliferating or dormant neoplastic cells, place severe restraints on the development of BMAP-27 and -28 as therapeutic anticancer agents.
2.2 Cecropin A and B
Cecropins are a class of antimicrobial peptides that were first described in insects, including the giant silk moth Hyalophora cecropia [88], but were later also found to be present in mammals [89]. Cecropins derived from insect sources consist of 34-39 amino acid residues [90, 91]. The best studied of these antimicrobial peptides are cecropin A (KWKLFKKIEKVGQNIRDGIIKAGPAVAVVGQATQIAK) and cecropin B (KWKVFKKIEKMGRNIRNGIVKAGPAIAVLGEAKAL), both of which assume a secondary structure that is characterized by the presence of two α helices [92, 93]. The NH2-terminal α-helix of cecropin A and cecropin B is highly amphipathic while the COOH-terminal α-helix is hydrophobic. Patch-clamp analysis of the effect of cecropin B on the Ags human stomach carcinoma cell line revealed that peptide treatment caused short outward currents that were consistent with the formation of transient channel-like pores [94]. In contrast, the cecropin B3 analogue, which consists of two hydrophobic α helices, failed to induce pore formation. Interestingly, the cecropin B1 analogue, which possesses two amphipathic α helices, shows potent cytotoxic activity against several human leukemia cell lines at peptide concentrations that do not lyse normal fibroblasts or erythrocytes [95]. In fact, cecropin B1 is a more potent cytolytic agent than cecropin B against HL-60 human promyelocytic leukemia cells [96]. Collectively, these findings indicate that the amphipathic NH2-terminal α-helix of cecropin B, which is believed to interact with anionic membrane components via its basic amino acid residues, is also able to mediate cytotoxic activity against cancer cells, whereas the hydrophobic COOH-terminal α-helix is dispensable in this regard. Nevertheless, the hydrophobic COOH-terminal α-helix may promote membrane insertion by the peptide, causing positive curvature strain on the membrane that creates an ion-permeable toroidal pore [93].
Cecropin A and B are able to lyse different types of human cancer cells at peptide concentrations that are not harmful to normal eukaryotic cells [91, 97, 98]. Cecropin B may have potential for use in the treatment of human cancers since this ACP exhibits in vivo antitumor activity in mice bearing ascitic colon adenocarcinoma cells, as well as in vitro cytotoxic activity against multidrug-resistant human breast and ovarian cancer cell lines [97]. Interestingly, the combination of cecropin A and the conventional chemotherapeutic agents 5-fluorouracil and cytarabine, at certain doses, shows a synergistic cytotoxic effect on CCRF-SB human lymphoblastic leukemia cells [98]. Conventional antineoplastic drugs might therefore be used in combination with ACPs such as cecropin A in order to lower the dosage of the drug that is required to have a therapeutic effect and thereby reduce chemotherapy-induced side effects. However, therapeutic use of cecropins or other ACPs will require repeated administration in order to maintain systemic levels of the peptide that are equivalent to the high concentrations required for ACPs to kill human cancer cells in culture or in mice. One attractive alternative is to transfer genes encoding ACPs directly into cancer cells or to the vicinity of solid tumors. In this regard, the introduction of expression constructs containing the cecropin A gene into EJ human bladder carcinoma cells prevented or reduced the growth of the tumor cells in immune-deficient mice [99]. Gene therapy using ACPs such as cecropin A may therefore one day be possible for the treatment of human cancers.
2.3 LL-37/hCAP-18
LL-37/hCAP-18 (human cationic AMP of 18 kDa) is the only human cathelicidin-derived AMP that has been identified to date [100]. This cathelicidin is initially synthesized as a preproprotein (hCAP-18) that is subsequently processed to its active form (LL-37) by proteinase 3-mediated extracellular cleavage [101]. hCAP-18 is expressed in a variety of cell types, including neutrophils [102] and squamous epithelial cells [103]. LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES) is a 4.5 kDa AMP that has antimicrobial activity at low micromolar concentrations against a variety of Gram-positive and Gram-negative bacteria. Unlike other AMPs, LL-37 is toxic to eukaryotic cells at a slightly higher concentration (25-30mM, which is 3-5 times its MIC value). LL-37 has been found to have additional defensive roles such as regulating the inflammatory response and chemo-attracting cells of the adaptive immune system to wound or infections sites, stimulation of angiogenesis and chemotaxis of neutrophils, monocytes and T-cells. This peptide is found throughout the body: in epithelial cells of testis, respiratory tract and gastrointestinal, in leukocytes and in skin. In addition, LL-37 has been expressed in E. coli and purified for biophysical studies [104]. CD and NMR studies have shown that LL-37 is unstructured in pure water but forms helical oligomers in solution either at a higher peptide concentration or in the presence of ions [105]. This property of the peptide is believed to be important as the oligomer can escape the proteolytic degradation [106]. Solid-state NMR studies have shown that LL-37 also assumes an α-helical conformation in a membrane environment and its oligomeric nature depends on the membrane composition. These studies revealed that the α-helical structure is comprised of both cationic and hydrophobic faces that orient in a parallel manner with lipid membranes, suggesting a carpet-like rather than channel-forming mechanism of cytotoxicity [105]. In addition, solid-state NMR and DSC experiments have shown that LL-37 is highly sensitive to the membrane composition, upon binding to membranes changes the head group conformation of phospholipids, induces positive curvature strain on lipid bilayers, and significantly disorders the hydrophobic core of the membrane [107]. More details on the structural and functional properties of this intriguing molecule can be found in a recent review article [20, 21].
A COOH-terminal fragment of hCAP-18 consisting of amino acid residues 109-135 (hCAP18109-135), which corresponds to amino acid residues 6-32 of LL-37, was recently found to induce apoptosis in a human oral squamous carcinoma cell line via a mechanism involving mitochondrial depolarization without any detectable activation of caspase-3 [108]. This finding is in line with a report that the core cytotoxic activity of LL-37 resides within a 13 amino acid residue fragment of the COOH-terminal region corresponding to amino acid residues 17-29, designated LL-37(17-29) [109]. Although LL-37(17-29) is equally cytotoxic to drug-sensitive and drug-resistant variants of the KB human squamous cancer cell line, LL-37(17-29) also kills untransformed human endothelial cells. Native LL-37 is similarly cytotoxic to human peripheral blood lymphocytes [110] and has significant hemolytic activity [106]. LL-37(17-29) and native LL-37 therefore lack the selectivity for cancer cells required of an ACP. In contrast, a tumoricidal concentration of hCAP18109-135 did not have any cytotoxic effect on either human gingival fibroblasts or the HaCaT human keratinocyte cell line [108]. The apparent selective cytotoxic activity of hCAP18109-135 against human cancer cells indicates that hCAP18109-135 warrants further investigation as a potential therapeutic ACP.
2.4 Magainins and Other Amphibian-derived Anticancer Peptides
The skin secretions of amphibians are a rich source of AMPs, several of which (aurein 1.2, citropin 1.1, gaegurins, magainins and analog peptides of magainins) have been reported to be selectively cytotoxic for human cancer cells [111-116]. The best studied of these ACPs are the magainins, which are AMPs isolated from the skin of the African clawed frog Xenopus laevis [117]. Magainins are comprised of 21 to 27 amino acid residues that create an α-helical secondary structure characterized by separate cationic and hydrophobic faces. Magainin 2 (GIGKFLHSAKKFGKAFVGEIMNS) and its more potent synthetic analogues (magainins A, B, and G) cause the rapid lysis of both hematopoietic and solid tumor cell lines at concentrations that are 5-10 fold lower than magainin concentrations that are lytic for normal human peripheral blood lymphocytes or neutrophils [115, 118]. Magainin 2 also shows selective cytotoxic activity against several human bladder cell lines with an average IC50 of approximately 200 μM [119]. Fluorescence spectrophotometric measurement of cancer cell membrane potential following exposure to magainin 2 or synthetic magainin analogues indicates that magainins lyse tumor cells by forming ion-conducting α-helical channels in the cancer cell membrane [115]. However, there is some evidence that, at lower concentrations, magainins destabilize the membrane bilayer via the carpet model [120]. A more recent study shows that a magainin 2 derivative is able to permeabilize and cross the cell membrane of HeLa human cervical carcinoma cells via an energy- and receptor-independent mechanism [121]. Magainins that gain access to the cytosolic compartment of cancer cells may trigger the mitochondrial pathway of apoptosis since magainins have been shown to form channels in the membranes of isolated rat liver mitochondria [122]. Interestingly, 10-50 nM concentrations of magainin 1 have recently been shown to induce apoptosis in HL-60 human promyelocytic leukemia cells via a mechanism that involves cytochrome c release from mitochondria and an increase in proteosome activity [116]. At the present time it is not clear whether magainins kill human cancer cells primarily through membrane lysis and/or apoptosis. A large number of recent biophysical studies focused on the mechanism of membrane-disruption by magainins (e.g., magainin 2 [38], PGLa [40-43], MSI-78 (commercially known as pexiganan) [32, 35, 36], MSI-594 [35], MSI-843 [39] and other synthetic variants) on model membranes. These peptides are unstructured in solution, form a α-helical structure in membranes, bind to the membrane with the helical axis parallel to the membrane surface and with a high affinity for negatively charged membranes, form oligomers, induce positive curvature strain, and disrupt membranes via carpet-toroidal-pore mechanism, which lead to fragmentation of membranes into bicelles and micelles after a sufficient time (>1 month). Some of these peptides like magainin2 [121], PGLa [43] and MSI-78 [35, 36] form antiparallel α-helical dimers in membranes.
A number of synthetic magainin analogues exhibit superior cytotoxic activity against neoplastic cells in comparison to native magainins [115, 124-126]. Superior selectivity for human cancer cells is exhibited by magainin G whereas magainin B is the most potent of the synthetic magainin analogues in terms of cytotoxicity [115]. Magainins A and G, which were designed to have increased α-helical potential and decreased hemolytic activity in comparison to native magainin 1 and 2, inhibit the growth of human small cell lung cancer cell lines, including drug-resistant tumor cell variants, with an average IC50 of approximately 9 μM [124]. In contrast, twice the concentration of magainin A or G is needed to inhibit the growth of normal human fibroblast cell lines. Magainin A and G also enhance the effectiveness of the chemotherapeutic agents DDP and VP-16, suggesting that these synthetic magainin analogues might be used in combination with conventional anticancer drugs in order to reduce chemotherapy-induced side effects. Two additional synthetic magainin analogues (MSI-136, comprised of L-amino acids and MSI-238, comprised of D-amino acids) designed to have an enhanced amphipathic α-helical structure, were found to have superior in vitro cytotoxic activity against human lung carcinoma cells in comparison to native magainin 2 and to increase survival of ovarian teratoma-bearing mice [125]. All-D-amino acid MSI-238 was more effective in vivo than all-L-amino acid MSI-136 or magainin 2, most likely because of decreased susceptibility to proteolytic degradation. An additional synthetic all-D-amino acid magainin analogue designated MSI-511 showed selective killing human melanoma cells in vitro at concentrations that do not harm normal melanocytes [126]. Moreover, intratumoral injection of MSI-511 completely eradicated human melanoma cells grown as subcutaneous xenografts in immune-deficient mice, providing evidence that locoregional therapy with protease-resistant magainin peptides may be useful for the treatment of malignant melanoma in humans.
Gaegurins are a class of six related AMPs that have been isolated from the skin of a Korean frog Rana rugosa [127]. The gaegurins assume a random-coil conformation in aqueous solution but adopt an amphipathic α-helical structure in membrane environments, allowing these AMPs to mediate cytolysis by the barrel stave and/or carpet mechanism [128]. More recent studies have revealed that gaegurin 5 (FLGALFKVASKVLPSVKCAITKKC) and gaegurin 6 (FLPLLAGLAANFLPTIICFISYKC) have selective cytotoxic activity against neoplastic cells [113, 114]. Gaegurin 5 and 6 are each comprised of 24 amino acid residues. Gaegurin 5 and two synthetic peptide analogues are able to selectively kill a range of human tumor cell types, including HCT116 colon and MCF-7 breast carcinoma cells, while showing only minimal hemolytic activity [113]. Gaegurin 6 and a synthetic peptide analogue (PTP7) have a similar broad spectrum of cytotoxic activity against human cancer cells with no detectable cytotoxicity against peripheral blood mononuclear cells and minimal hemolytic activity [114]. In addition, gaegurin 6 and PTP7 are active against a multidrug-resistant variant of the MCF-7 breast cancer cell line. Gaegurin 6- and PTP7-mediated cytotoxicity may involve apoptosis since DNA fragmentation was detected in MCF-7 breast cancer cells that were exposed to these ACPs.
Aurein 1.2 (GLFDIIKKIAESF) is a small cationic AMP comprised of 13 amino acid residues that has been isolated from the Australian bell frog Litoria raniformis [111]. NMR studies indicate that aurein 1.2 adopts an α-helical structure in solution. Aurein 1.2 is moderately cytotoxic for 57 of 60 human tumor cell lines but does not lyse erythrocytes at concentrations as high as 100 μg/ml, which is sufficient to kill most human cancer cells. Citropin 1.1 (GLFDVIKKVASVIGGL) is a relatively small AMP isolated from the tree frog Litoria citropa. This AMP is able to kill a wide range of human hematopoietic and non-hematopoietic tumor cell lines at concentrations that do not cause significant lysis of red blood cells [112]. Citropin 1.1, which is comprised of 16 amino acid residues, has an α-helical structure characterized by well defined hydrophobic and hydrophilic regions. The relatively small size of aurein 1.2 and citropin 1.1 implies that these ACPs mediate their cytotoxic effect via the carpet mechanism since ACPs need to be comprised of at least 20 amino acid residues in order to span eukaryotic cell membranes and cause cytolysis by the barrel-stave mechanism [129, 130]. However, it is important to note that peptides comprised of fewer than 20 amino acid residues may dimerize end-on in order to effect complete penetration of biological membranes [131].
2.5 Melittin
Melittin (GIGAVLKVLTTGLPALISWIKRKRQQ), an alkaline polypeptide comprised of 26 amino acid residues, is the major component of European honeybee (Apis mellifera) venom [132]. The NH2-terminal region of melittin is largely hydrophobic whereas the region at the COOH-terminus contains positively-charged amino acid residues and is hydrophilic [133]. Melittin forms channels in lipid bilayers [134, 135] and is lytic for both cancer cells and normal, healthy cells, including erythrocytes [133, 135, 136]. Nevertheless, murine L1210 leukemia cells have been reported to be several-fold more sensitive to melittin-mediated cytotoxicity than are normal mouse splenocytes or bone-marrow cells [137]. At low membrane concentrations, melittin assumes an α-helical structure that lies parallel to the bilayer plane [138]. However, recent studies indicate that monomeric melittin has little effect on a membrane mimetic bilayer structure whereas a dimeric form of melittin causes destabilization of membrane mimetic bilayers formed from dioleoylphosphatidylcholine [139]. Self association of amphipathic α-helical monomers of melittin is therefore suggested to be responsible for the large perturbations in membrane integrity that result in cellular lysis. Melittin is believed to cause damage to cell membranes via the barrel-stave mechanism [140]. However, melittin has additional effects on cancer cells. Melittin counter-selects for ras-over expressing cancer cells through a mechanism that involves the hyper-activation of phospholipase A2, an influx of Ca++, and the subsequent destruction of the transformed cells [141, 142]. Furthermore, melittin-mediated cytolysis of U937 human monocytic leukemia cells is associated with the transient activation of endogenous phospholipase D, which has been suggested to participate in an uncharacterized signal transduction pathway involved in the permeabilization of cancer cell membranes by this ACP [143].
Because of the relative lack of selectivity displayed by melittin for cancer cells, efforts have been focused on targeting melittin to neoplastic cells and/or tumor vasculature. One strategy is based on the use of matrix metalloproteinase-2, which is over expressed by human cancer cells and tumor-associated endothelium [144], to selectively cleave a melittin-avidin conjugate, thereby restoring the lytic function of melittin at the site of matrix metalloprotease-2 expression [145]. DU 145 prostate carcinoma cells and SKOV3 ovarian carcinoma cells that exhibit high levels of matrix metalloproteinase-2 activity are killed by the melittin-avidin conjugate whereas normal mouse L-cell fibroblasts that possess little matrix metalloprotease-2 activity are unaffected by the melittin-avidin conjugate. In addition, intratumoral administration of the melittin-avidin conjugate causes a significant reduction in the growth of B16 murine melanoma cells in syngeneic mice. Alternatively, tumor-specific antibodies can be used to target melittin to tumor cells. In this regard, administration of an immunoconjugate containing a melittin-like peptide improved the survival of immune-deficient mice bearing subcutaneous human prostate carcinoma xenografts [146]. Melittin-based gene therapy of human cancers is also a possibility, as demonstrated by the adenovirus-mediated transfer of the melittin gene under the control of the α-fetoprotein promoter to BEL-7402 human hepatocellular carcinoma cells, which resulted in a dramatic inhibition of the in vitro and in vivo (in immune-deficient mice) growth of the hepatocellular carcinoma cells [147]. Intratumoral injection of the adenovirus-melittin construct also caused subcutaneous BEL-7402 tumors in immune-deficient mice to shrink and eventually disappear. Clearly, the targeted delivery of melittin and other ACPs to tumor sites has exciting possibilities as a therapeutic approach.
3. β-Sheet Anticancer Peptides
3.1 Defensins
Defensins are a group of closely-related, Cys-Arg-rich, cationic AMPs comprised of 29 to 45 amino acid residues [148]. The six conserved Cys residues that are a characteristic feature of defensins form three intramolecular disulfide bridges between the NH2-terminal and COOH-terminal regions of the peptide, creating a cyclic, triple-stranded, amphiphilic β-sheet structure, making up the characteristic “defensin-like” fold and spatially separated hydrophobic and hydrophilic regions [149, 150]. Although defensins have been isolated from many different species, α- and β-defensins of human origin remain the best studied [10, 22, 151]. The disulfide connectivities in α-defensins are Cys1-Cys6, Cys2-Cys4 and Cys3-Cys5 (the number indicates the location of the Cys residue in the amino acid sequence from the N-terminus), while in β-defensin are Cys1-Cys5, Cys2-Cys4 and Cys3-Cys6.
Human neutrophil peptides (HNPs)-1 (ACYCRIPACIAGERRYGTCIYQGRLWAFCC), HNP-2 (CYCRIPACIAGERRYGTCIYQGRLWAFCC), and HNP-3 (DCYCRIPACIAGERRYGTCIYQGRLWAFCC) are α-defensins that were originally purified from the azurophilic granules of neutrophils [152] and were later found to have a cytotoxic effect on several different types of human and mouse tumor cells, including Raji human B-lymphoma cells, human oral squamous carcinoma cells, and MOT mouse teratocarcinoma cells [153-155]. Higher concentrations (> 25 μg/ml) of HNP-1, -2, and -3 suppress DNA synthesis in renal cell carcinoma lines, as well as reducing cancer cell viability [156]. Rabbit macrophage-associated defensins that are homologues of HNP-1 and -2 are also able to lyse murine tumor cells [157]. Tumor cell killing by HNP-1, -2, and -3 involves a membrane binding event, most likely mediated by electrostatic interactions, followed by rapid collapse of the membrane potential and loss of membrane integrity [154, 158]. Membrane permeabilization by HNPs has been attributed to the channel-forming ability of these peptides because HNP-1 has been shown to form voltage-dependent, ion-permeable channels in planar phospholipid bilayer membranes [159]. HNP-mediated cytotoxicity may also involve DNA damage since single strand DNA breaks were detected in HNP-treated target cells, although nucleosome-sized fragments of DNA that are characteristic of apoptosis were not observed [160]. In addition to their cytotoxic activity, HNP-1 and -3 may be able to interfere with neovascularization during tumor development because these α-defensins inhibit the α5β1 integrin-dependent migration and adhesion of endothelial cells to fibronectin in response to vascular endothelial growth factor (VEGF) [161]. In addition, HNP-1 and -3 inhibited VEGF-induced proliferation of endothelial cells via the induction of apoptosis. Unfortunately, the clinical utility of human α-defensins is limited by the fact that HNPs are not tumor-selective, causing the lysis of normal human leukocytes, epithelial cells, and fibroblasts [153, 162]. In addition, serum strongly inhibits HNP-mediated cytotoxicity [154], which poses an obstacle to the systemic administration of these human α -defensins.
3.2 Lactoferricin
Lactoferricin is a cationic AMP produced by acid-pepsin hydrolysis of mammalian lactoferrin [163], an iron-binding protein that is present in the secretory granules of neutrophils and in secreted fluids that include milk and saliva [164-166]. Bovine lactoferricin (LfcinB) isolated from cow’s milk consists of 25 amino acid residues (FKCRRWQWRMKKLGAPSITCVRRAF), including two Cys residues that create a disulfide bond linking the highly positively-charged NH2-terminal region and the COOH-terminal region of the peptide [167]. In aqueous solution, LfcinB assumes a twisted β-sheet configuration with the basic amino acid residues arranged on one face and most of the hydrophobic amino acid residues arranged on the other face [168]. LfcinB exhibits in vitro cytotoxic activity against many different types of mouse and human cancer cell lines, including leukemia cells, fibrosarcoma cells, various carcinomas, and neuroblastoma cells [169-172], at concentrations that do not substantially affect the viability of normal fibroblasts, lymphocytes, epithelial cells, endothelial cells, or erythrocytes [171, 173]. The cytotoxic activity of LfcinB against cancer cells is very much dependent on the amphipathic structure and high net positive charge of the peptide since cytotoxic activity is increased in LfcinB derivatives with clear cationic and hydrophobic sectors, while a glutamic acid-containing homologue of murine lactoferricin lacks the ability to kill cancer cells [174-176]. LfcinB binds to cancer cell membranes, causing membrane integrity to be lost due to the formation of transmembrane pores that allow the peptide to enter the cytoplasmic compartment of the cancer cell and co-localize with negatively-charged mitochondria [172, 177]. Although mouse fibrosarcoma cells and human neuroblastoma cells exposed to LfcinB die primarily via necrosis caused by a cell membrane lytic effect [172], LfcinB kills human leukemia and breast carcinoma cells by a process that involves the sequential generation of reactive oxygen species, loss of mitochondrial transmembrane potential, and activation of the caspase cascade culminating in cell death by apoptosis [171, 178]. Whether LfcinB-treated cancer cells die by necrosis or apoptosis may ultimately be determined by the degree of irreparable LfcinB-mediated damage to the cytoplasmic membrane relative to mitochondrial membrane damage caused by internalized LfcinB. Although the cytotoxic activity of LfcinB is reduced in the presence of high concentrations of serum [178], systemic or intratumoral administration of LfcinB is nonetheless able to inhibit the in vivo growth and/or metastasis of several different tumor types in mice [170, 172, 178].
Recently, LfcinB has been shown to suppress both basic fibroblast growth factor (bFGF)- and VEGF-driven proliferation and migration of human endothelial cells in vitro, as well as interfering with bFGF- and VEGF-induced angiogenesis in subcutaneous Matrigel plugs implanted in mice, by competing with bFGF and VEGF for growth factor receptor-associated heparan sulfate proteoglycans on the endothelial cell surface [179]. The antiangiogenic activity of LfcinB is dependent on the primary structure of the peptide since a scrambled peptide comprised of the same amino acid residues failed to effectively compete with bFGF or VEGF for heparin-like binding sites on endothelial cells. These findings are consistent with an earlier report that subcutaneous administration of LfcinB inhibited blood vessel development in B16-BL6 melanoma tumors implanted in syngeneic mice [178]. However, the relative contributions of the cytotoxic and antiangiogenic effects of LfcinB to the inhibition of tumor progression in vivo are not known at the present time.
3.3 Tachyplesin I
Hemocytes of the horseshoe crab (Tachypleus tridentatus) are the source of the cationic AMP tachyplesin I [180], which consists of 17 amino acid residues (KWCFRVCYRGICYRRCR) arranged in two anti-parallel β-sheets that are held in place by two disulfide bonds [181]. This configuration allows all six of the basic amino acid residues (Arg, Lys) of tachyplesin I to be exposed on the surface of the peptide, resulting in an amphipathic structure. While it is believed that a disulfide bond provides stability to the structure and also against proteolytic degradation, a recent showed that tachyplesin I without Cys residues retained the antimicrobial activity of the wild-type peptide, but may not be stable in serum [182]. Tachyplesin I has a unique method of killing cancer cells. In a recent study, tachyplesin I was shown to bind to hyaluronan on hyaluronan-over expressing human TSU prostate carcinoma cells, as well as to the C1q component of complement in human serum, leading to activation of the classical complement pathway and complement-mediated lysis of tachyplesin I-coated cancer cells [183]. The C1q-binding activity of tachyplesin I was dependent on the secondary structure of the peptide since denatured tachyplesin I bound significantly less C1q. Interactions between tachyplesin I and hyaluronan are believed to contribute to the selective killing of cancer cells by tachyplesin I because many tumor cells tend to express hyaluronan at levels that are much higher than those found on normal tissues [184]. Hyaluronan is also present in large amounts on the surface of endothelial cells involved in neovascularization [185], such as occurs during tumor development, suggesting that tachyplesin I might cause complement-mediated destruction of tumor-associated vasculature, as well as neoplastic cells. Interestingly, heat-inactivation of serum complement only partially protected tachyplesin I-treated TSU prostate cancer cells from lysis, suggesting that tachyplesin I has additional effects on cancer cells that can lead to cell death. In this regard, RGD-targeting of synthetic tachyplesin I to integrins on the surface of TSU prostate cancer cells and endothelial cells in vitro results in the inhibition of cell growth due to cell membrane damage and the induction of caspase-dependent apoptosis [186]. Administration of RGD-tachyplesin I also inhibits the in vivo growth of B16 melanoma cells in mice. It is possible that the overall positive charge of tachyplesin I allows internalized RGD-tachyplesin I to target and disrupt negatively-charged mitochondrial membranes in the cancer cells, thereby triggering the intrinsic pathway of apoptosis. However, at the present time it is not clear whether the hyaluronan-binding activity of tachyplesin I contributes to the cytotoxic effect of RGD-tachyplesin I on cancer cells. Although RGD-tachyplesin I-treated mice did not experience any obvious side-effects, it is important to note that, at higher concentrations, tachyplesin I interacts with neutral lipids in the plasma membrane of erythrocytes, resulting in membrane permeabilization and hemolysis [187].
Tachyplesin I also inhibits the growth of cancer cells through a non-cytolytic mechanism. Tachyplesin I treatment of SMMC-7721 human hepatoma cell and BGC-823 human gastric adenocarcinoma cell cultures leads to a decrease in the proliferative capacity of these cancer cells [188, 189]. Reduced growth of SMMC-7721 cells in the presence of tachyplesin I was associated with the reversal of malignant morphological and ultrastructural features, decreased expression of tumor-associated antigens (α-fetoprotein, proliferating cell nuclear antigen), modulation of differentiation-associated enzyme ((-glutamyltransferase, tyrosine aminotransferase) expression, decreased expression of the c-myc oncogene, and increased expression of the tumor suppressor gene p21WAF1/CIP1 [188]. Tachyplesin I treatment of BGC-823 cells also altered the morphology and ultrastructure of these cancer cells [190], as well as inhibiting their in vitro proliferation, decreasing c-myc, c-erbB-2 and mtp53 oncogene expression, and increasing p16 tumor suppressor gene expression [189]. Taken together, these findings indicate that tachyplesin I is capable of inducing tumor cell differentiation, thereby reversing the malignant phenotype.
4. Linear Anticancer Peptides
PR-39 is a Pro-Arg-rich member of the cathelicidin family that was originally isolated from porcine small intestine [191] and, later, from porcine neutrophils [192]. This linear AMP is comprised of 39 amino acid residues (RRRPRPPYLPRPRPPPFFPPRLPPRIPPGFPPRFPPRFP) and lacks the secondary structure that is characteristic of other ACPs. The mechanism by which PR-39 exerts anticancer activity is also unique among ACPs. Treatment of human hepatocellular carcinoma cell lines with PR-39 results in the induction of syndecan-1 expression [193]. Similar results were obtained when HLF hepatocellular carcinoma cells were transfected with the PR-39 gene. PR-39 might therefore be able to prevent or reduce the invasion and metastasis of tumor cells because heightened syndecan-1 expression by human myeloma and hepatocellular carcinoma cells has been associated with a reduction in the invasive activity of these cancer cells [193, 194]. Suppressed motile activity and altered actin structure in PR-39-transfected hepatocellular carcinoma cells might also contribute to the PR-39-mediated inhibition of cancer cell invasion [193]. In addition, ras-transformed cells transfected with the PR-39 gene were suppressed in their ability to proliferate both in vitro and in vivo [195].
PR-39 binds to eukaryotic cells in a receptor-dependent manner and subsequently rapidly gains access to the cytosolic compartment, without permeabilizing the plasma membrane [196]. Once inside the cell, SH3-binding motifs allow PR-39 to complex with multiple SH3-containing cytoplasmic proteins, including the signaling adaptor protein p130Cas and the p85α regulatory subunit of phosphatidylinositol 3-kinase [195, 196]. Recent studies on variants of the PR-39 peptide have shown that the SH3-binding and syndecan induction activities of PR-39 are dependent on the presence of NH2-terminal arginine residues [197]. The anticancer activity of PR-39 is most likely a consequence of the peptide interacting with and affecting the activity of SH3-bearing proteins that are involved in key cellular signaling processes. For example, endothelial cells treated with PR-39 show altered localization of p130Cas while phosphatidylinositol 3-kinase activity in ras-transformed NIH3T3 cells is suppressed as a consequence of PR-39 binding to p85α [195, 197].
5. Hybrid and Synthetic Anticancer Peptides
5.1 Cecropin A-based Hybrid Anticancer Peptides
In recent years, a number of attempts have been made to improve upon naturally occurring ACPs by creating hybrid ACPs that incorporate the best qualities of individual ACPs. Research on hybrid ACPs has been largely focused on combining the NH2-terminal positively-charged α-helical region of cecropin A with the NH2-terminal α-helical hydrophobic region of either melittin (CA-ME) or magainin 2 (CA-MA) [198-200]. Both CA(1-8)-ME(1-12) and CA(1-8)-MA(1-12) exhibit cytolytic activity against human small cell lung cancer cell lines, although at the highest peptide concentrations CA(1-8)-ME(1-12) has modest hemolytic activity whereas CA(1-8)-MA(1-12) has little or no lytic effect on erythrocytes [198]. Studies on CA(1-8)-ME(1-12) and CA(1-8)-MA(1-12) analogues containing amino acid substitutions suggested that flexibility in the hinge region of these hybrid peptides is an important factor in their cytolytic activity [199]. In this regard, a comparison between the three-dimensional structures of CA(1-8)-ME(1-12) and CA(1-8)-MA(1-12) revealed that the central hinge region (Gly-Ile-Gly) dictates whether these hybrid peptides have sufficient conformational flexibility to allow their cationic NH2-terminal α-helix to interact with and align in a parallel fashion with the cell membrane whilst their hydrophobic COOH-terminal α-helix inserts into and spans the cell membrane, thereby triggering cytolysis [200]. Thus, omission of the Gly-Ile-Gly hinge region resulted in a dramatic reduction in the anticancer and hemolytic activity of CA(1-8)-ME(1-12) and CA(1-8)-MA(1-12). The α-helical content and amphipathic properties of these hybrid peptides were also found to be important determinants of hemolytic activity since, in comparison to CA(1-8)-MA(1-12), hemolysis-inducing CA(1-8)-ME(1-12) has a longer and more stable α-helix and is a more hydrophobic molecule. P18 is an α-helical ACP that was designed from CA(1-8)-MA(1-12) [201]. This hybrid ACP is selectively cytotoxic for human cancer cells, including Jurkat T leukemia cells, K562 chronic myeloid leukemia cells, and MDA-MB-361 breast carcinoma cells, without causing any hemolysis. Structure-function analysis showed that the COOH-terminal α-helical region of P18 is important for cytolytic activity against human cancer cells whereas the NH2-terminal α-helical region is not. The design of hybrid ACPs such as P18 and its analogues that exert potent cytotoxic activity against neoplastic cells without any appreciable hemolytic activity or cytotoxicity against healthy cells represents an important advance in the quest for new anticancer agents.
5.2 Diastereomeric and Other Synthetic Anticancer Peptides
Synthetic diastereomeric lytic peptides are of considerable interest as ACPs because of their ability to selectively permeabilize negatively-charged phospholipid membranes, including those of cancer cells, while resisting enzymatic degradation by serum proteins [202, 203]. D-K4R2L9 is a diastereomeric amphipathic peptide comprised of Leu, Lys and Arg residues, totaling 15 amino acid residues, with one third of its sequence comprised of D-amino acids [204]. The D-K4R2L9 peptide binds to and lyses B16-F10 mouse melanoma cells in culture at concentrations that do not harm normal 3T3 fibroblasts or erythrocytes. Moreover, intravenous administration of D-K4R2L9 was effective in preventing intravenous-injected D122 lung carcinoma cells from forming lung tumors in mice. A subsequent study revealed that it is possible to obtain similar selective in vitro cytotoxic activity against human prostate cancer cell lines using a modified 15-amino acid diastereomeric amphipathic peptide designated D-K6L9, which contains D-Lys and D-Leu residues in one third of its sequence [205]. Interestingly, an analogue of the D-K6L9 peptide that is composed entirely of L-amino acids (L-K6L9) had similar anticancer activity but was also lytic for normal fibroblast and erythrocytes. This highlights another important advantage of employing D-amino acids in the construction of synthetic ACPs. In addition, the D-K6L9 peptide showed synergistic cytotoxic activity with the conventional anticancer drug doxorubicin against both androgen-dependent and androgen-independent prostate cancer cells. Importantly, intratumoral injections of D-K6L9 caused 22RV1 prostate carcinoma xenografts in immune-deficient mice to decrease in size and, in some cases, completely disappear. In contrast, L-K6L9 lacked in vivo antitumor activity due to its complete inactivation by serum proteins. Recently, systemic administration of D-K6L9 was shown to inhibit the growth and metastatic spread of 22RV1 prostate carcinoma cells and MB-231 breast carcinoma cells in immune-deficient mice, as shown in Figure 4 [206]. The selective cytotoxicity of D-K6L9 for human cancer cells was due to the binding of cationic D-K6L9 to anionic surface-exposed PS, followed by cytoplasmic membrane depolarization and necrosis.
Figure 4.

Reduction of tumor size by a synthetic anticancer peptide, D-K6L9: male mice without (left) and with treatment using the peptide (right) [206].
Targeting or membrane-penetrating sequences have also been used to enhance the selectivity or activity, respectively, of synthetic ACPs. Targeting domains (cyclic CNGRC or double-cyclic RGD-4C) have been coupled to the pro-apoptotic peptide (KLAKLAK)2, which is comprised of 14 D-amino acid residues and kills by disrupting mitochondrial membranes following its uptake into the cytosolic compartment [207]. These targeted synthetic peptides were selectively cytotoxic for endothelial cells under angiogenic conditions, i.e. tumor vasculature, but did not harm endothelial cells under angiostatic conditions. As a result, systemic administration of targeted (KLAKLAK)2 peptides significantly prolonged the survival of immune-deficient mice bearing human breast carcinoma xenografts, as well as dramatically reducing tumor volume without any apparent treatment-related toxicity. In another approach, the (KLAKLAK)2 peptide has been fused to the PTD-5 protein transduction domain in order to promote rapid internalization of the peptide [208]. The DP-1 peptide created in this way triggers apoptosis in cultures of mouse fibrosarcoma cells and human head and neck tumor isolates. Moreover, intratumoral injections of DP-1 caused established MCA205 mouse fibrosarcoma tumors to regress and, in some cases, totally disappear without any apparent side effects. In a similar approach, a cell-penetrating domain consisting of seven D-arginine residues (r7) was coupled to the D-amino acid configuration synthetic mitochondrial membrane-disrupting peptide kla [209]. In vitro treatment with r7-kla resulted in rapid peptide uptake and the induction of apoptosis in different types of human cancer cells, including breast, colorectal, and prostate carcinoma cell lines. In addition, a single intratumoral injection of r7-kla triggered apoptosis, along with significant tumor tissue loss and extensive necrosis, in HT1080 human fibrosarcoma xenografts grown in immune-deficient mice. However, it is important to note that synthetic ACPs such as DP-1 and r7-kla that have been engineered for rapid cellular uptake will also need to be selectively targeted to tumor cells in order to avoid unacceptable toxicity following systemic administration.
6. Summary and outlook
Current cancer treatments have many harmful side effects, since commonly used antineoplastic drugs target all rapidly dividing cells, rather than solely cancerous cells. In contrast, certain AMPs seem to show specificity towards malignant cells. It is clear that a great deal of further study must be devoted to studying the exact mechanism of interaction of ACPs with cancer cells and which of the characteristics of a cancer cell make some AMPs preferentially target them. In particular, biophysical studies dealing with the structure, dynamics, topology, mechanism of membrane disruption, and the role of individual components of membranes using a variety of model membranes mimicking cancer and normal cells would provide insights into the function of an ACP at a very high-resolution. For example, recently developed solid-state NMR approaches and model membranes to mimick various types of cell membranes [20] can be utilized to solve the high-resolution structure and oritentation of ACPs and also possible interaction with glycosylated proteins. Since NMR structures of most of ACPs are known, it would be relatively easy to interpret the experimental data that lead to membrane disruption by ACPs. It would also be worthwhile to design glycosylated ACPs to determine if glycosylation increases their potency. In addition, cross-linking and/or the use of fluorinated-amino acids to increase the stability of oligomeric structures as recently shown for MSI-78 could be useful. Such studies would greatly facilitate the design of more potent and highly selective ACPs with a variety of modes of action against human cancer cells, as well as notable advantages over current drug-based cancer treatments.
Also, current anticancer drugs must be taken into the cell in order to be effective, which makes it possible for cancer cells develop resistance to these drugs by pumping them out of the cell. Again, ACPs may have an advantage here, as they act on the cell membrane without having to be taken into the cancer cell. Major requirements for clinical usage of ACPs in cancer treatment include a very high specificity (low cytolytic activity to normal mammalian cells and red blood cells) and killing ability, as well as stability in serum. Serum has often been shown to affect the potency of AMPs, including ACPs; for example, LDL (low density lipoprotein), a type of cholesterol, is a strong inhibitor of peptide-mediated cell lysis. However, certain naturally occurring ACPs are more stable in serum. Synthetic ACPs have been specifically designed for this purpose as well. In this regard, recent studies have shown that ACPs containing D-amino-acids are more stable against proteolytic degradation. ACPs that are foreign to the human body might also elicit treatment-neutralizing antibodies and/or potentially dangerous allergic responses by cancer patients. One approach to avoid generating deleterious anti-ACP immune responses is to use peptides that are of human origin or to co-administer foreign ACPs with immunosuppressive drugs. Alternatively, immunogenic ACPs could be encapsulated in liposomes engineered to deliver their cargo to tumor sites, thereby minimizing the opportunity to develop anti-ACP immunity. Cost is an additional obstacle that will need to be overcome before ACPs can be employed in cancer treatment. Although the cost of isolating naturally occurring ACPs or producing synthetic ACPs currently exceeds that of manufacturing conventional chemotherapeutic agents, massive investment by pharmaceutical companies in the development of ACPs as cancer therapeutics will inevitably result in economies of scale and innovative strategies for ACP isolation/synthesis. In the relatively near future, AMPs that have been optimized for anticancer activity may be an economically viable and therapeutically superior alternative to the current generation of chemotherapeutic drugs.
Acknowledgments
We thank Dr. Yechiel Shai for providing Figure 4, Angela Richardson (Dalhousie University) for assistance with artwork, and Amy Krukemeyer (University of Michigan) for preparing Figures 2 and 3. Dr. David Hoskin’s research (Dalhousie University) was funded by the Natural Sciences and Engineering Research Council of Canada, the Dairy Farmers of Canada, the Canadian Breast Cancer Foundation, and the Leukemia and Lymphoma Society of Canada. Research in Dr. A. Ramamoorthy’s lab (University of Michigan) is funded by the National Institutes of Health (AI054515), American Heart Association, and Eli Lilly Co.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Edwards BK, Brown ML, Wingo PA, Howe HL, Ward E, Ries LAG, Schrag D, Jamison PM, Jemal A, Wu XC, Friedman C, Harlan L, Warren J, Anderson RN, Pickle LW. Annual report to the nation on the status of cancer, 1975-2002, featuring population-based trends in cancer treatment. J Natl Cancer Inst. 2005;97:1407–1427. doi: 10.1093/jnci/dji289. [DOI] [PubMed] [Google Scholar]
- 3.Renan MJ. How many mutations are required for tumorigenesis? Implications from human cancer cells. Mol Carcinog. 1993;7:139–146. doi: 10.1002/mc.2940070303. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 5.Espinosa E, Zamora P, Feliu J, Gonzalez Baron M. Classification of anticancer drugs-a new system based on therapeutic targets. Cancer Treat Rev. 2003;29:515–523. doi: 10.1016/s0305-7372(03)00116-6. [DOI] [PubMed] [Google Scholar]
- 6.Cassidy J, Misset JL. Oxaliplatin-related side effects: characteristics and management. Semin Oncol. 2002;29(Suppl 15):11–20. doi: 10.1053/sonc.2002.35524. [DOI] [PubMed] [Google Scholar]
- 7.Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S. Doxorubidin-induced apoptosis:implications in cardiotoxicity. Mol Cell Biochem. 2002;234-235:119–124. [PubMed] [Google Scholar]
- 8.Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VH, Groom AC, Chambers AF. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat. 2003;82:199–206. doi: 10.1023/B:BREA.0000004377.12288.3c. [DOI] [PubMed] [Google Scholar]
- 9.Gatti L, Zunino F. Overview of tumor cell chemoresistance mechanisms. Methods Mol Med. 2005;111:127–148. doi: 10.1385/1-59259-889-7:127. [DOI] [PubMed] [Google Scholar]
- 10.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 11.Hancock RE, Diamond G. The role of cationic antimicrobial peptides in innate host defences. Trends in Microbiology. 2000;8:402–410. doi: 10.1016/s0966-842x(00)01823-0. [DOI] [PubMed] [Google Scholar]
- 12.Rosenfeld Y, Shai Y. Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis. BBA-Biomembranes. 2006;1758:1513–1522. doi: 10.1016/j.bbamem.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Devine DA, Hancock REW. Cationic peptides: Distribution and mechanism of resistance. Curr Pharm Design. 2002;8:703–714. doi: 10.2174/1381612023395501. [DOI] [PubMed] [Google Scholar]
- 14.Mor A. Peptide-based antibiotics: A potential answer to raging antimicrobial resistance. Drug dev Res. 2000;50:440–447. [Google Scholar]
- 15.Koczulla AR, Bals R. Antimicrobial peptides – current status and therapeutics potential. Drugs. 2003;63:389–406. doi: 10.2165/00003495-200363040-00005. [DOI] [PubMed] [Google Scholar]
- 16.Giuliani A, Pirri G, Nicoletto SF. Antimicrobial peptides: an overview of a promising class of therapeutics. Cen Eur J Biol. 2007;2:1–33. [Google Scholar]
- 17.Mader JS, Hoskin DW. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Exp Opin Investing Drugs. 2006;15:933–946. doi: 10.1517/13543784.15.8.933. [DOI] [PubMed] [Google Scholar]
- 18.Chan DI, Prenner EJ, Vogel HJ. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. BBA Biomembranes. 2006;1758:1184–1202. doi: 10.1016/j.bbamem.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Shai Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta. 1999;1462:55–70. doi: 10.1016/s0005-2736(99)00200-x. [DOI] [PubMed] [Google Scholar]
- 20.Dürr UHN, Sudheendra US, Ramamoorthy A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. BBA-Biomembranes. 2006;1758:1408–1425. doi: 10.1016/j.bbamem.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 21.Wildman KAH. Ph.D thesis on The mechanism of lipid bilayer disruption by the human antimicrobial peptides, LL-37. University of Michigan; 2003. [DOI] [PubMed] [Google Scholar]
- 22.Dhople V, Krukemeyer A, Ramamoorthy A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. BBA-Biomembranes. 2006;1758:1499–1512. doi: 10.1016/j.bbamem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Oren Z, Shai Y. Mode of action of linear amphipathic ãhelical antimicrobial peptides. Biopolymers. 1998;47:451–463. doi: 10.1002/(SICI)1097-0282(1998)47:6<451::AID-BIP4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 24.Matsuzaki K, Murase O, Fujii N, Miyajima K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry. 1996;35:11361–11368. doi: 10.1021/bi960016v. [DOI] [PubMed] [Google Scholar]
- 25.Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 26.Bechinger B, Lohner K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. BBA Biomembranes. 2006;1758:1529–1539. doi: 10.1016/j.bbamem.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Hallock KJ, Lee Dong-Kuk, Omnaas John, Mosberg HI, Ramamoorthy A. Membrane composition determines pardaxin’s mechanism of lipid bilayer disruption. Biophys J. 2002;83:1004–1013. doi: 10.1016/S0006-3495(02)75226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porcelli F, Buck B, Lee DK, Hallock KJ, Ramamoorthy A, Veglia G. Structure and Orientation of Pardaxin Determined by NMR Experiments in Model Membranes. J Biol Chem. 2004;279:46815–46823. doi: 10.1074/jbc.M405454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cafisco DS. Alamethicin: A peptide model for voltage gating and protein-membrane interaction. Annu Rev Biophys Biomol Struct. 1994;23:141–165. doi: 10.1146/annurev.bb.23.060194.001041. [DOI] [PubMed] [Google Scholar]
- 30.Dave PC, Billington E, Pan YL, Straus SK. Interaction of alamethicin with ether-linked phospholipid bilayers : Oriented circular dichroism, 31P solid-state NMR, and differential scanning calorimetry studies. Biophys J. 2005;89:2434–2442. doi: 10.1529/biophysj.105.067678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stella L, Burattini M, Mazzuca C, Palleschi A, Venanzi M, Coin I, Peggion C, Toniolo C, Pispisa B. Chemistry & Biodiversity. 2007;4:1299. doi: 10.1002/cbdv.200790111. [DOI] [PubMed] [Google Scholar]
- 32.Hallock KJ, Lee DK, Ramamoorthy A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys J. 2003;84:3052. doi: 10.1016/S0006-3495(03)70031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pkorny A, Almeida PFF. Kinetics of dye efflux and lipid flip-flop induced by delta-lysin in phosphatidylcholine vesicle and the mechanism of graded release by amphipathic, alpha-helical peptides. Biochemistry. 2004;43:8846–8857. doi: 10.1021/bi0497087. [DOI] [PubMed] [Google Scholar]
- 34.Miteva M, Anderson M, Karshikoff A, Otting G. Molecular electrophoration: a unifying concept for the description of membrane pore formation by antibacterial peptides, exemplified with NK-lysin. FEBS Lett. 1999;462:155–158. doi: 10.1016/s0014-5793(99)01520-3. [DOI] [PubMed] [Google Scholar]
- 35.Ramamoorthy A, Thennarasu S, Lee DK, Tan A, Maloy Lee. Solid-State NMR Investigation of the Membrane-Disrupting Mechanism of Antimicrobial Peptides MSI-78 and MSI-594 Derived From Magainin 2 and Melittin. Biophys J. 2006;91:206–216. doi: 10.1529/biophysj.105.073890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porcelli F, Buck-Koehntop B, Thennarasu S, Ramamoorthy A, Veglia G. Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers by NMR spectroscopy. Biochemistry. 2006;45:5793–5799. doi: 10.1021/bi0601813. [DOI] [PubMed] [Google Scholar]
- 37.Mecke A, Lee DK, Ramamoorthy A, Orr BG, Holl MMB. Membrane thinning due to antimicrobial peptide binding: An atomic force microscopy study of MSI-78 in lipid bilayers. Biophys J. 2005;99:4043–4050. doi: 10.1529/biophysj.105.062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bechinger B, Zasloff M, Opella SJ. Structure and orientation of the antibiotic peptide magainin in membranes by solid-state Nuclear Magnetic Resonance Spectroscopy. Prot Sci. 1993;2:2077–2084. doi: 10.1002/pro.5560021208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thennarasu S, Lee DK, Tan A, Kari UP, Ramamoorthy A. Antimicrobial Actvity and Membrane Selective Interactions of a Synthetic Lipopeptide MSI-843. Biochim Biophys Acta. 2005;1711:49–58. doi: 10.1016/j.bbamem.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bechinger B, Zasloff M, Opella SJ. Structure and dynamics of the antibiotic peptide PGLa in membranes by solution and solid-state Nuclear Magnetic Resonance Spectroscopy. Biophys J. 1998;74:981–987. doi: 10.1016/S0006-3495(98)74021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strandberg E, Wadhwani P, Tremouilhac P, Durr UHN, Ulrich AS. Solid-state NMR analysis of the PGLa peptide orientation in DMPC bilayers: Structural fidelity of 2H-labels versus high sensitivity of 19F-NMR. Biophys J. 2006;90:1676–1686. doi: 10.1529/biophysj.105.073858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tremouilhac P, Strandberg E, Wadhwani P, Ulrich AS. Conditions affecting the re-alignment of the antimicrobial peptide PGLa in membranes as monitored by solid-state 2H NMR. BBA Biomembranes. 2006;1758:1330–1342. doi: 10.1016/j.bbamem.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 43.Tremouilhac P, Strandberg E, Wadhwani P, Ulrich AS. Synergistic transmembrane alignment of the antimicrobial heterodimer PGLa/magainin. J Biol Chem. 2006;281:32089–32094. doi: 10.1074/jbc.M604759200. [DOI] [PubMed] [Google Scholar]
- 44.Thennarasu S, Lee DK, Poon A, Kawulka KE, Vederas JC, Ramamoorthy A. Membrane permeabilization, orientation, and antimicrobial mechanism of subtilosin A. Chem Phys Lipids. 2005;137:38–51. doi: 10.1016/j.chemphyslip.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Lu JX, Blazyk J, Lorigan GA. Exploring membrane selectivity of the antimicrobial peptide KIGAKI using solid-state NMR spectroscopy. BBA Biomembranes. 2006;1758:1303–1313. doi: 10.1016/j.bbamem.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 46.Ramamoorthy A, Thennarasu S, Tan A, Lee DK, Clayberger C, Krensky AM. Cell selectivity correlates with membrane interactions: a case study on the antimicrobial peptide G15 derived from granulysin. BBA-Biomembranes. 2006;1758:154–163. doi: 10.1016/j.bbamem.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 47.Boland MP, Separovic F. Membrane interactions of antimicrobial peptides from Australian tree frogs. BBA-Biomembranes. 2006;1758:1178–1183. doi: 10.1016/j.bbamem.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 48.Pan YL, Cheng JTJ, Hale J, Pan JH, Hancock REW, Straus SK. Characterization of the structure and membrane interaction of the antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys J. 2007;92:2854–2864. doi: 10.1529/biophysj.106.097238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chekmenev EY, Vollmar BS, Forseth KT, Manion MN, Jones SM, Wagner TJ, Endicott RM, Kyriss BP, Homem LM, Pate M, He J, Raines J, Gor’kov PL, Brey WW, Mitchell DJ, Auman AJ, Ellard-Ivey MJ, Blazyk J, Cotton M. Investigating molecular recognition and biologicl function at interfaces using piscidins, antimicrobial peptides from fish. BBA-Biomembranes. 2006;1758:1359–1372. doi: 10.1016/j.bbamem.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 50.Marcotte I, Wegener KL, Lam YH, Chia BCS, de Planque MRR, Bowie JH, Auger M, Separovic F. Interaction of antimicrobial peptides from Australian amphibians with lipid membranes. Chem Phys Lipids. 2003;122:107–120. doi: 10.1016/s0009-3084(02)00182-2. [DOI] [PubMed] [Google Scholar]
- 51.Mani R, Buffy JJ, Waring AJ, Lehrer RI, Hong M. Solid-state NMR investigation of the selective disruption of lipid membranes by protegrin-1. Biochemistry. 2004;43:13839–13848. doi: 10.1021/bi048650t. [DOI] [PubMed] [Google Scholar]
- 52.Murakami T, Niwa M, Tokunaga F, Miyata T, Iwanaga S. Direct virus inactivation of tachyplesin I and its isopeptides from horseshoe crab hemocytes. Chemotherapy. 1991;37:327–334. doi: 10.1159/000238875. [DOI] [PubMed] [Google Scholar]
- 53.Morimoto M, Mori H, Otake T, Ueba N, Kunita N, Niwa M, Murakami T, Iwanaga S. Inhibitory effect of tachyplesin I on the proliferation of human immunodeficiency virus in vitro. Chemotherapy. 1991;37:206–211. doi: 10.1159/000238855. [DOI] [PubMed] [Google Scholar]
- 54.Masuda M, Nakashima H, Ueda T, Naba H, Ikoma R, Otaka A, et al. A novel anti-HIV synthetic peptide T-22 ([Tyr5,12,Lys7]-polyphemusin II) Biochem Biophys Res Commun. 1992;189:845–850. doi: 10.1016/0006-291x(92)92280-b. [DOI] [PubMed] [Google Scholar]
- 55.Murakami T, Nakajima T, Koyanagi Y, Tachibana K, Fujii N, Tamamura H, Yoshida N, Waki M, Matsumoto A, Yoshie O, Kishimoto T, Yamamoto N, Nagasawa T. A small molecule CXCR4 inhibitor that blocks T cell line-tropic HIV-1 infection. J Exp Med. 1997;186:1389–1393. doi: 10.1084/jem.186.8.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andersen JH, Osbakk SA, Vorland LH, Traavik T, Gutteberg TJ. Lactoferrin and cyclic lactoferricin inhibit the entry of human cytomegalovirus into human fibroblasts. Antiviral Res. 2001;51:141–149. doi: 10.1016/s0166-3542(01)00146-2. [DOI] [PubMed] [Google Scholar]
- 57.Andersen JH, Jenssen H, Sandvik K, Gutteberg TJ. Anti-HSV activity of lactoferrin and lactoferricin is dependent on the presence of heparan sulfate at the cell surface. J Med Virol. 2004;74:262–271. doi: 10.1002/jmv.20171. [DOI] [PubMed] [Google Scholar]
- 58.Mistry N, Drobi P, Näslund J, Sunkari VG, Jenssen H, Evander M. The anti-papillomavirus activity of human and bovine lactoferricin. Antiviral Res. 2007;75:258–265. doi: 10.1016/j.antiviral.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 59.Utsugi T, Schroit AJ, Connor J, Buccana CD, Fidler IJ. Elevated expression of phosphatidylserine in the outer leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991;51:3062–3066. [PubMed] [Google Scholar]
- 60.Dobrzynska J, Szachowicz-Petelska B, Sulkowski S, Figaszewski Z. Changes in electric charge and phospholipids composition in human colorectal cancer cells. Mol Cell Biochem. 2005;276:113–119. doi: 10.1007/s11010-005-3557-3. [DOI] [PubMed] [Google Scholar]
- 61.Yoon WH, Park HD, Lim K, Hwang BD. Effect of O-glycosylated mucin on invation and metastasis of HM7 human colon cancer cells. Biochem Biophys Res Commun. 1996;222:694–699. doi: 10.1006/bbrc.1996.0806. [DOI] [PubMed] [Google Scholar]
- 62.Burdick MD, Harris A, Reid CJ, Iwamura T, Hollingsworth MA. Oligosaccharides expressed on MUC1 by pancreatic and colon tumor cell lines. J Biol Chem. 1997;272:24198–24202. doi: 10.1074/jbc.272.39.24198. [DOI] [PubMed] [Google Scholar]
- 63.Cruciani RA, Barker JL, Zasloff M, Chen H-C, Colamonici O. Antibiotic magainins exert cytolytic activity against transformed cell lines through channel formation. Proc Natl Acad Sci USA. 1991;88:3792–3796. doi: 10.1073/pnas.88.9.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zachowski A. Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem J. 1993;294(Pt 1):1–14. doi: 10.1042/bj2940001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kozlowska K, Nowak J, Kwiatkowski B, Cichorek M. ESR study of plasmatic membrane of the transplantable melanoma cells in relation to their biological properties. Exp Toxicol Pathol. 1999;51:89–92. doi: 10.1016/S0940-2993(99)80074-8. [DOI] [PubMed] [Google Scholar]
- 66.Sok M, Sentjurc M, Schara M. Membrane fluidity characteristics of human lung cancer. Cancer Lett. 1999;139:215–220. doi: 10.1016/s0304-3835(99)00044-0. [DOI] [PubMed] [Google Scholar]
- 67.Leuschner C, Hansel W. Membrane disrupting lytic peptides for cancer treatments. Curr Pharm Des. 2004;10:2299–2310. doi: 10.2174/1381612043383971. [DOI] [PubMed] [Google Scholar]
- 68.Simons K, Ikonen E. How cells handle cholesterol. Science. 2000;290:1721–1726. doi: 10.1126/science.290.5497.1721. [DOI] [PubMed] [Google Scholar]
- 69.Steiner H, Andreu D, Merrifield RB. Binding and action of cecropin and cecropin analogues: antibacterial peptides from insects. Biochim Biophys Acta. 1988;939:260–266. doi: 10.1016/0005-2736(88)90069-7. [DOI] [PubMed] [Google Scholar]
- 70.Silvestro L, Gupta K, Weiser JN, Axelson PH. The concentration-dependent membrane activity of cecropin A. Biochemistry. 1997;36:11452–11460. doi: 10.1021/bi9630826. [DOI] [PubMed] [Google Scholar]
- 71.Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168:1107–1118. doi: 10.2353/ajpath.2006.050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Domagala W, Koss LG. Surface configuration of human tumor cells obtained by fine needle aspiration biopsy. Scan Electron Microsc. 1980;1:101–108. [PubMed] [Google Scholar]
- 73.Chaudhary J, Munshi M. Scanning electron microscopic analysis of breast aspirates. Cytopathology. 1995;6:162–167. doi: 10.1111/j.1365-2303.1995.tb00469.x. [DOI] [PubMed] [Google Scholar]
- 74.Allred LE, Porter KR. Morphology of Normal and Transformed Cells. In: Hynes R, editor. Surfaces of Normal and Malignant Cells. New York, NY: John Wiley & Sons; 1979. pp. 21–61. [Google Scholar]
- 75.Mader JS, Salsman J, Conrad DM, Hoskin DW. Bovine lactoferricin selectively induces apoptosis in human leukemia and carcinoma cell lines. Mol Cancer Ther. 2005;4:612–624. doi: 10.1158/1535-7163.MCT-04-0077. [DOI] [PubMed] [Google Scholar]
- 76.Taniguchi N, Gao CX, Ihara Y, Miyoshi E, Yoshimura M, Sheng Y, Sultan AS, Ikeda Y. The Involvement of Bisecting N-Acetylglucosamine in Cancer. In: Aubery M, editor. Glycans in Cell Interaction and Recognition: Therapeutic Aspects. Japan: Harwood Academic Publishers; 2001. pp. 73–88. [Google Scholar]
- 77.Michalski JC. Structure and Analysis of Glycoprotein-Associated Oligosaccharides. In: Aubery M, editor. Glycans in Cell Interaction and Recognition: Therapeutic Aspects. Japan: Harwood Academic Publishers; 2001. pp. 17–35. [Google Scholar]
- 78.Dennis JW, Granovsky M. β1,6 N-Acetylglucosaminyltransferase V is a Determinant of Cancer Growth and Metastasis. In: Aubery M, editor. Glycans in Cell Interaction and Recognition: Therapeutic Aspects. Japan: Harwood Academic Publishers; 2001. pp. 89–104. [Google Scholar]
- 79.Taylor-Papadimitriou J, Burchell JM, Plunkett T, Graham R, Correa I, Miles D, Smith M. J Mammary Gland Biol Neoplasia. 2002;7:209–221. doi: 10.1023/a:1020360121451. [DOI] [PubMed] [Google Scholar]
- 80.Burchell JM, Mungul A, Taylor-Papadimitriou J. J Mammary Gland Biol Neoplasia. 2001;6:335–364. doi: 10.1023/a:1011331809881. [DOI] [PubMed] [Google Scholar]
- 81.Karsten U, von Mensdorff-Pouilly S, Goletz S. Tumor Biology. 2005;26:217–220. doi: 10.1159/000086956. [DOI] [PubMed] [Google Scholar]
- 82.Dennis JW. N-linked oligosaccharide processing and tumor cell biology. Semin Cancer Biol. 1991;2:411–420. [PubMed] [Google Scholar]
- 83.McManus AM, Otvos L, Hoffmann R, Craik DJ. Conformational studies by NMR of the antimicrobial peptide, drosocin, and its non-glycosylated derivatives: Effects of glycosylation on solution conformation. Biochemistry. 1999;38:705–714. doi: 10.1021/bi981956d. [DOI] [PubMed] [Google Scholar]
- 84.Papo N, Shai Y. Host defense peptides as new weapons in cancer treatment. Cell Mol Life Sci. 2005;62:784–790. doi: 10.1007/s00018-005-4560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Skerlavaj B, Gennaro R, Bagella L, Merluzzi L, Risso A, Zanetti M. Biological characterization of two novel cathelicidin-derived peptides and identification of structural requirements for their antimicrobial and cell lytic activities. J Biol Chem. 1996;271:28375–28381. doi: 10.1074/jbc.271.45.28375. [DOI] [PubMed] [Google Scholar]
- 86.Risso A, Zanetti M, Gennaro R. Cytotoxicity and apoptosis mediated by two peptides of innate immunity. Cell Immunol. 1998;189:107–115. doi: 10.1006/cimm.1998.1358. [DOI] [PubMed] [Google Scholar]
- 87.Risso A, Braidot E, Sordano MC, Vianello A, Macri F, Skerlavaj B, Zanetti M, Gennaro R, Bernardi P. BMAP-28, an antibiotic peptide of innate immunity, induces cell death through opening of the mitochondrial permeability transition pore. Mol Cell Biol. 2002;22:1926–1935. doi: 10.1128/MCB.22.6.1926-1935.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Steiner H, Bennich H, Boman HG, Engstrom A, Hultmark D. Sequence and specificity of 2 anti-bacterial proteins involved in insect immunity. Nature. 1981;292:246–248. doi: 10.1038/292246a0. [DOI] [PubMed] [Google Scholar]
- 89.Lee JY, Boman A, Sun CX, Andersson M, Jornvall H, Mutt V, Boman HG. Antibacterial peptides from pig intestine: isolation of a mammalian cecropin. Proc Natl Acad Sci USA. 1989;86:19159–19162. doi: 10.1073/pnas.86.23.9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bowman HG, Hultmark D. Cell-free immunity in insects. Annu Rev Microbiol. 1987;41:103–126. doi: 10.1146/annurev.mi.41.100187.000535. [DOI] [PubMed] [Google Scholar]
- 91.Chen HM, Wang W, Smith D, Chan SC. Effects of the anti-bacterial peptide cecropin B and its analogs, cecropins B-1 and B-2, on liposomes, bacteria, and cancer cells. Biochim Biophys Acta. 1997;1336:171–179. doi: 10.1016/s0304-4165(97)00024-x. [DOI] [PubMed] [Google Scholar]
- 92.Holak TA, Engstrom A, Kraulis PJ, Lindeberg G, Bennich H, Jones TA, Gronenborn AM, Clore GM. The solution conformation of the antibacterial peptide cecropin A: a nuclear magnetic resonance and dynamical simulated annealing study. Biochemistry. 1988;27:7620–7629. doi: 10.1021/bi00420a008. [DOI] [PubMed] [Google Scholar]
- 93.Hung SC, Wang W, Chan SI, Chen HM. Membrane lysis by the antibacterial peptides cecropin B1 and B3: a spin-label electron spin resonance study on phospholipids bilayers. Biophys J. 1999;77:3120–3133. doi: 10.1016/S0006-3495(99)77142-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ye J-S, Zheng X-J, Leung KW, Chen HM, Sheu F-S. Induction of transient ion channel-like pores in a cancer cell by antibiotic peptide. J Biochem. 2004;136:255–259. doi: 10.1093/jb/mvh114. [DOI] [PubMed] [Google Scholar]
- 95.Srisailam S, Kumar TKS, Arunkumar AI, Leung KW, Yu C, Chen HM. Crumpled structure of the custom hydrophobic lytic peptide cecropin B3. Eur J Biochem. 2001;268:4278–4284. doi: 10.1046/j.1432-1327.2001.02345.x. [DOI] [PubMed] [Google Scholar]
- 96.Chan SC, Yau WL, Wang W, Smith DK, Sheu F-S, Chen HM. Microscopic observations of the different morphological changes caused by anti-bacterial peptides on Klebsiella pneumoniae and HL-60 leukemia cells. J Peptide Sci. 1998;4:413–425. doi: 10.1002/(SICI)1099-1387(199811)4:7%3C413::AID-PSC160%3E3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 97.Moore AJ, Devine DA, Bibby MC. Preliminary experimental anticancer activity of cecropins. Pept Res. 1994;7:265–269. [PubMed] [Google Scholar]
- 98.Hui L, Leung K, Chen HM. The combined effects of antibacterial peptide cecropin A and anti-cancer agents on leukemia cells. Anticancer Res. 2002;22:2811–2816. [PubMed] [Google Scholar]
- 99.Winder D, Günzburg WH, Erfle V, Salmons B. Expression of antimicrobial peptides has an antitumour effect in human cells. Biochem Biophys Res Comm. 1998;242:608–612. doi: 10.1006/bbrc.1997.8014. [DOI] [PubMed] [Google Scholar]
- 100.Larrick JW, Michimasa H, Balint RF, Lee J, Zhong J, Wright SC. Human CAP18: a novel antimicrobial lipopolysaccharide-binding protein. Infect Immun. 1995;63:1291–1297. doi: 10.1128/iai.63.4.1291-1297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sørensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, Borregaard N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. 2001;97:3951–3959. doi: 10.1182/blood.v97.12.3951. [DOI] [PubMed] [Google Scholar]
- 102.Sørensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood. 1997;90:2796–2803. [PubMed] [Google Scholar]
- 103.Nilsson MF, Sandstedt B, Sørensen O, Weber G, Borregaard N, Ståhle-Bäckdahl M. The human cationic antimicrobial protein (hCAP18), a peptide antibiotic, is widely expressed in human squamous epithelia and co-localizes with interleukin 6. Infect Immun. 1999;67:2561–2566. doi: 10.1128/iai.67.5.2561-2566.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moon JY, Henzler-Wildman KA, Ramamoorthy A. Expression and purification of a recombinant LL-37 from Escherichia coli. BBA-Biomembranes. 2006;1758:1351–1358. doi: 10.1016/j.bbamem.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 105.Henzler-Wildman KA, Lee DK, Ramamoorthy A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry. 2003;42:6545–6558. doi: 10.1021/bi0273563. [DOI] [PubMed] [Google Scholar]
- 106.Oren Z, Lerman JC, Gudmundsson GH, Agerberth B, Shai Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-selective activity. Biochem J. 1999;341:501–513. [PMC free article] [PubMed] [Google Scholar]
- 107.Henzler-Wildman KA, Martinez GV, Brown MF, Ramamoorthy A. Perturbation of the Hydrophobic Core of Lipid Bilayers by the Human Antimicrobial Peptide LL-37. Biochemistry. 2004;43:8459–8469. doi: 10.1021/bi036284s. [DOI] [PubMed] [Google Scholar]
- 108.Okumura K, Itoh A, Isogai E, Hirose K, Hosokawa Y, Abiko Y, Shibata T, Hirata M, Isogai H. C-terminal domain of human CAP18 antimicrobial peptide induces apoptosis in oral squamous cell carcinoma SAS-H1 cells. Cancer Lett. 2004;212:185–194. doi: 10.1016/j.canlet.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 109.Li X, Li Y, Han H, Miller DW, Wang G. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J Am Chem Soc. 2006;128:5776–5785. doi: 10.1021/ja0584875. [DOI] [PubMed] [Google Scholar]
- 110.Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J Biol Chem. 1998;273:3718–3724. doi: 10.1074/jbc.273.6.3718. [DOI] [PubMed] [Google Scholar]
- 111.Rozek T, Wegener KL, Bowie JH, Olver IN, Carver JA, Wallace JC, Tyler MJ. The antibiotic and anticancer active aurein peptides from the Australian Bell Frogs Litoria aurea and Litoria raniformis. The solution structure of aurein 1.2. Eur J Biochem. 2000;267:5330–5341. doi: 10.1046/j.1432-1327.2000.01536.x. [DOI] [PubMed] [Google Scholar]
- 112.Doyle J, Brinkworth CS, Wegener KL, Carver JA, Llewellyn LE, Olver IN, Bowie JH, Wabnitz PA, Tyler MJ. nNOS inhibition, antimicrobial and anticancer activity of the amphibian skin peptide, citropin 1.1 and synthetic modifications. The solution structure of a modified citropin 1.1. Eur J Biochem. 2003;270:1141–1153. doi: 10.1046/j.1432-1033.2003.03462.x. [DOI] [PubMed] [Google Scholar]
- 113.Won H-S, Seo M-D, Jung SJ, Lee S-J, Kang S-J, Son W-S, Kim H-J, Park T-K, Park S-J, Lee B-J. Structural determinants for the membrane interaction of novel bioactive undecapeptides derived from gaegurin 5. J Med Chem. 2006;49:4886–4895. doi: 10.1021/jm050996u. [DOI] [PubMed] [Google Scholar]
- 114.Kim S, Kim SS, Bang Y-J, Kim S-J, Lee BJ. In vitro activities of native and designed peptide antibiotics against drug sensitive and resistant tumor cell lines. Peptides. 2003;24:945–953. doi: 10.1016/s0196-9781(03)00194-3. [DOI] [PubMed] [Google Scholar]
- 115.Cruciani RA, Barker JL, Zasloff M, Chen H-C, Colamonici O. Antibiotic magainins exert cytolytic activity against transformed cell lines through channel formation. Proc Natl Acad Sci USA. 1991;88:3792–3796. doi: 10.1073/pnas.88.9.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cruz-Chamoro L, Puertollano MA, Puertollano E, de Cienfuegos GA, de Pablo MA. In vitro biological activities of magainin 1 alone or in combination with nisin. Peptides. 2006;27:1201–1209. doi: 10.1016/j.peptides.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 117.Zasloff M. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial DNA sequence of a precursor. Proc Natl Acad Sci USA. 1987;84:5449–5453. doi: 10.1073/pnas.84.15.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jacob L, Zasloff M. Potential therapeutic applications of magainins and other antimicrobial agents of animal origin. Ciba Found Symp. 1994;186:197–216. doi: 10.1002/9780470514658.ch12. [DOI] [PubMed] [Google Scholar]
- 119.Lehmann J, Retz M, Sidhu SS, Suttmann H, Sell M, Paulsen F, Harder J, Unteregger G, Stöckle M. Antitumor activity of the antimicrobial peptide magainin II against bladder cancer cell lines. Eur Urol. 2006;50:141–147. doi: 10.1016/j.eururo.2005.12.043. [DOI] [PubMed] [Google Scholar]
- 120.Ludtke SJ, He K, Wu Y, Huang HW. Cooperative membrane insertion of magainin correlated with its cytolytic activity. Biochim Biophys Acta. 1994;1190:181–184. doi: 10.1016/0005-2736(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 121.Takeshima K, Chikushi A, Lee K-K, Yonehara S, Matsuzaki K. Translocation of analogues of the antimicrobial peptides magainin and buforin across human cell membranes. J Biol Chem. 2003;278:1310–1315. doi: 10.1074/jbc.M208762200. [DOI] [PubMed] [Google Scholar]
- 122.Westerhoff HV, Hendler RW, Zasloff M, Juretic D. Interactions between a new class of eukaryotic antimicrobial agents and isolated rat mitochondria. Biochim Biophys Acta. 1989;975:361–369. doi: 10.1016/s0005-2728(89)80344-5. [DOI] [PubMed] [Google Scholar]
- 123.Wakamatsu K, Takeda A, Tachi T, Matsizaki K. Dimer structure of magainin 2 bound to phospholipid vesicles. Biopolymers. 2002;64:314–327. doi: 10.1002/bip.10198. [DOI] [PubMed] [Google Scholar]
- 124.Ohsaki Y, Gazdar AF, Chen H-C, Johnson BE. Antitumor activity of magainin analogues against human lung cancer cell lines. Cancer Res. 1992;52:3534–3538. [PubMed] [Google Scholar]
- 125.Baker MA, Maloy WL, Zasloff M, Jacob LS. Anticancer efficacy of magainin 2 and analogue peptides. Cancer Res. 1993;53:3052–3057. [PubMed] [Google Scholar]
- 126.Soballe PW, Maloy WL, Myrgra ML, Jacob LS, Herlyn M. Experimental local therapy with lytic magainin peptides. Int J Cancer. 1995;60:280–284. doi: 10.1002/ijc.2910600225. [DOI] [PubMed] [Google Scholar]
- 127.Park JM, Jung JE, Lee BJ. Antimicrobial peptides from the skin of a Korean frog, Rana rugosa. Biochem Biophys Res Commun. 1994;205:948–954. doi: 10.1006/bbrc.1994.2757. [DOI] [PubMed] [Google Scholar]
- 128.Oren Z, Shai Y. Mode of action of linear amphipathic α-helical antimicrobial peptides. Biopolymers. 1998;47:451–463. doi: 10.1002/(SICI)1097-0282(1998)47:6<451::AID-BIP4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 129.Shai YC. Molecular recognition between membrane-spanning peptides. Trends Biochem Sci. 1995;20:460–464. doi: 10.1016/s0968-0004(00)89101-x. [DOI] [PubMed] [Google Scholar]
- 130.Epand RM, Shai YC, Segrest JP, Anantharamaiah GM. Mechanisms for the modulation of membrane bilayer properties by amphipathic helical peptides. Biopolymers. 1995;37:319–338. doi: 10.1002/bip.360370504. [DOI] [PubMed] [Google Scholar]
- 131.Andreu D, Ubach J, Boman A, Wåhlin B, Wade D, Merrifield RB, Boman HG. Shortened cecropin A-melittin hybrids. Significant size reduction retains potent antibiotic activity. FEBS Lett. 1992;296:190–194. doi: 10.1016/0014-5793(92)80377-s. [DOI] [PubMed] [Google Scholar]
- 132.Gauldie J, Hanson JM, Shipolini RA, Vernon CA. The structures of some peptides from bee venom. Eur J Biochem. 1978;83:405–410. doi: 10.1111/j.1432-1033.1978.tb12106.x. [DOI] [PubMed] [Google Scholar]
- 133.Habermann E, Jentsch J. Sequenzanalyse des Melittins aus den tryptischen und peptischen Spaltstucken. Hope-Seyler’s Z Physiol Chem. 1967;348:37–50. [PubMed] [Google Scholar]
- 134.Tosteson MT, Alvarez O, Hubbell W, Bieganski RM, Attenbach C, Caporales LH, Levy JJ, Nutt RF, Rosenblatt M, Tosteson DC. Primary structure of peptides and ion channels. Role of amino acids side chains in voltage gating melittin channels. Biophys J. 1990;58:1367–1375. doi: 10.1016/S0006-3495(90)82483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tosteson MT, Tosteson DC. The sting Melittin forms channels in lipid bilayers. Biophys J. 1981;36:109–116. doi: 10.1016/S0006-3495(81)84719-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tosteson MT, Holmes SJ, Razin M, Tosteson DC. Melittin lysis of red cells. J Membr Biol. 1985;87:35–44. doi: 10.1007/BF01870697. [DOI] [PubMed] [Google Scholar]
- 137.Killion JJ, Dunn JD. Differential cytolysis of murine spleen, bone-marrow and leukemia cells by melittin reveals differences in membrane topography. Biochem Biophys Res Commun. 1986;139:222–227. doi: 10.1016/s0006-291x(86)80102-4. [DOI] [PubMed] [Google Scholar]
- 138.Frey S, Tamm LK. Orientation of melittin in phospholipid bilayers: a polarized attenuated total reflection infrared study. Biophys J. 1991;60:922–930. doi: 10.1016/S0006-3495(91)82126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hristova K, Dempsey CE, White SH. Structure, location, and lipid perturbations of melittin at the membrane interface. Biophys J. 2001;80:801–811. doi: 10.1016/S0006-3495(01)76059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sui SF, Wu H, Guo Y, Chen KS. Conformational changes of melittin upon insertion into phospholipids monolayer and vesicles. J Biochem. 1994;116:482–487. doi: 10.1093/oxfordjournals.jbchem.a124550. [DOI] [PubMed] [Google Scholar]
- 141.Sharma SV. Melittin resistance: a counterselection for ras transformation. Oncogene. 1992;7:193–201. [PubMed] [Google Scholar]
- 142.Sharma SV. Melittin-induced hyperactivation of phospholipase A2 activity and calcium influx in ras-transformed cells. Oncogene. 1993;8:939–947. [PubMed] [Google Scholar]
- 143.Saini SS, Chopra AK, Peterson JW. Melittin activates endogenous phospholipase D during cytolysis of human monocytic leukemia cells. Toxicon. 1999;37:1605–1619. doi: 10.1016/s0041-0101(99)00110-5. [DOI] [PubMed] [Google Scholar]
- 144.Vihinen P, Kahari VM. Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer. 2002;10:155–167. doi: 10.1002/ijc.10329. [DOI] [PubMed] [Google Scholar]
- 145.Holle L, Song W, Holle F, Wei Y, Wagner T, Yu X. A matrix metalloproteinase 2 cleavable melittin/avidin conjugate specifically targets tumor cells in vitro and in vivo. Int J Oncol. 2003;22:93–98. [PubMed] [Google Scholar]
- 146.Russell PJ, Hewish D, Carter T, Sterling-Levis K, Ow K, Hattarki M, Doughty L, Guthrie R, Shapira D, Molloy PL, Werkmeister JA, Kortt AA. Cytotoxic properties of immunoconjugates containing melittin-like peptide 101 against prostate cancer: in vitro and in vivo studies. Cancer Immunol Immunother. 2004;53:411–421. doi: 10.1007/s00262-003-0457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ling C-Q, Li B, Zhang C, Zhu D-Z, Huang X-Q, Gu W, Li S-X. Inhibitory effect of recombinant adenovirus carrying melittin gene on hepatocellular carcinoma. Ann Oncol. 2005;16:109–115. doi: 10.1093/annonc/mdi019. [DOI] [PubMed] [Google Scholar]
- 148.Lehrer RI, Lichtenstein AK, Ganz T. Defensins: antimicrobial and cytotoxic peptides of mammalian cells. Annu Rev Immunol. 1993;11:105–128. doi: 10.1146/annurev.iy.11.040193.000541. [DOI] [PubMed] [Google Scholar]
- 149.Selsted ME, Harwig SSL. Determination of the disulfide array in the human defensin HNP-2: a covalently cyclized peptide. J Biol Chem. 1989;264:4003–4007. [PubMed] [Google Scholar]
- 150.Hill PC, Yee J, Selsted ME, Eisenberg D. Crystal structure of defensin HNP-3, an anphiphilic dimmer: mechanisms of membrane permeabilization. Science. 1991;251:1481–1485. doi: 10.1126/science.2006422. [DOI] [PubMed] [Google Scholar]
- 151.Martin E, Gantz T, Lehrer R. Defensins and other endogenous peptide antibiotics of vertebrates. J Leukoc Biol. 1995;58:128–136. doi: 10.1002/jlb.58.2.128. [DOI] [PubMed] [Google Scholar]
- 152.Ganz T, Selsted ME, Sklarek D, Harwig SSL, Daher K, Bainton DF, Lehrer RI. Defensins: natural peptide antibiotics of human neutrophils. J Clin Invest. 1985;76:1427–1435. doi: 10.1172/JCI112120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lichtenstein A, Ganz T, Selsted ME, Lehrer RI. In vitro tumor cell cytolysis mediated by peptide defensins of human and rabbit granulocytes. Blood. 1986;68:1407–1410. [PubMed] [Google Scholar]
- 154.Lichtenstein AK, Ganz T, Nguyen T-M, Selsted ME, Lehrer RI. Mechanism of target cytolysis by peptide defensins. Target cell metabolic activities, possibly involving endocytosis, are crucial for expression of cytotoxicity. J Immunol. 1988;140:2686–2694. [PubMed] [Google Scholar]
- 155.McKeown STW, Lundy FT, Nelson J, Lockhart D, Irwin CR, Cowan CG, Marley JJ. The cytotoxic effects of human neutrophil peptide-1 (HNP-1) and lactoferrin on oral squamous cell carcinoma (OSCC) in vitro. Oral Oncol. 2006;42:685–690. doi: 10.1016/j.oraloncology.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 156.Müller CA, Markovic-Lipkovski J, Klatt T, Gamper J, Schwarz G, Beck H, Deeg M, Kalbacher H, Widmann S, Wessels JT, Becker V, Müller GA, Flad T. Human α-defensins HNPs-1, -2, and-3 in renal cell carcinoma. Influences on tumor cell proliferation. Am J Pathol. 2002;160:1311–1324. doi: 10.1016/s0002-9440(10)62558-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sheu MJ-T, Baldwin WW, Brunson KW. Cytotoxicity of rabbit macrophage peptides MCP-1 and MCP-2 for mouse tumor cells. Antimicrob Agents Chemother. 1985;28:626–629. doi: 10.1128/aac.28.5.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lichtenstein A. Mechanism of mammalian cell lysis mediated by peptide defensins. Evidence for an initial alteration of the plasma membrane. J Clin Invest. 1991;88:93–100. doi: 10.1172/JCI115310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kagan BL, Selsted ME, Ganz T, Lehrer RI. Antimicrobial defensin peptides form voltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc Natl Acad Sci USA. 1990;87:210–214. doi: 10.1073/pnas.87.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gera JF, Lichtenstein A. Human neutrophil peptide defensins induce single strand DNA breaks in target cells. Cell Immunol. 1991;138:108–120. doi: 10.1016/0008-8749(91)90136-y. [DOI] [PubMed] [Google Scholar]
- 161.Chavakis T, Cines DB, Rhee J-S, Liang OD, Schubert U, Hammes H-P, Al-Roof Higazi A, Nawroth PP, Preissner KT, Bdeir K. Regulation of neovascularization by human neutrophil peptides (α-defensins): a link between inflammation and angiogenesis. FASEB J. 2004;18:1306–1308. doi: 10.1096/fj.03-1009fje. [DOI] [PubMed] [Google Scholar]
- 162.Nishimura M, Abiko Y, Kurashige Y, Takeshima M, Yamazaki M, Kusano K, Saitoh M, Nakashima K, Inoue T, Kaku T. Effect of defensin peptides on eukaryotic cells: primary epithelial cells, fibroblasts and squamous cell carcinoma cell lines. J Dermatol Sci. 2004;36:87–95. doi: 10.1016/j.jdermsci.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 163.Tomita M, Bellamy W, Takase M, Yamauchi K, Wakabayashi H, Kawase K. Potent antibacterial peptides generated by pepsin digestion of bovine lactoferrin. J Dairy Sci. 1991;74:4137–4142. doi: 10.3168/jds.S0022-0302(91)78608-6. [DOI] [PubMed] [Google Scholar]
- 164.Masson PL, Heremans JF, Schonne E. Lactoferrin, an iron-binding protein in neutrophilic leukocytes. J Exp Med. 1969;130:643–658. doi: 10.1084/jem.130.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Masson PL, Heremans JF. Lactoferrin in milk from different species. Comp Biochem Physiol B. 1971;39:119–129. doi: 10.1016/0305-0491(71)90258-6. [DOI] [PubMed] [Google Scholar]
- 166.Dipaola C, Mandel ID. Lactoferrin concentration in human parotid saliva as measured by an enzyme-linked immunosorbent assay (ELISA) J Dent Res. 1980;59:1463–1465. doi: 10.1177/00220345800590090101. [DOI] [PubMed] [Google Scholar]
- 167.Bellamy W, Takase M, Yamauchi K, Wakabayashi H, Kawase K, Tomita M. Identification of the bactericidal domain of lactoferrin. Biochim Biophys Acta. 1992;1121:130–136. doi: 10.1016/0167-4838(92)90346-f. [DOI] [PubMed] [Google Scholar]
- 168.Hwang PM, Zhou N, Shan X, Arrowsmith CH, Vogel HJ. Three-dimensional solution structure of lactoferricin B, an antimicrobial peptide derived from bovine lactoferrin. Biochemistry. 1998;37:4288–4298. doi: 10.1021/bi972323m. [DOI] [PubMed] [Google Scholar]
- 169.Yoo Y-C, Watanabe R, Koike Y, Mitobe M, Shimazaki K, Watanabe S, Azuma I. Apoptosis in human leukemic cells induced by lactoferricin, a bovine milk protein-derived peptide: involvement of reactive oxygen species. Biochem Biophys Res Commun. 1997;237:624–628. doi: 10.1006/bbrc.1997.7199. [DOI] [PubMed] [Google Scholar]
- 170.Eliassen LT, Berge G, Sveinbjørnsson B, Svendsen JS, Vorland LH, Rekdal Ø. Evidence for a direct antitumour mechanism of action of bovine lactoferricin. Anticancer Res. 2002;22:2703–2710. [PubMed] [Google Scholar]
- 172.Mader JS, Salsman J, Conrad DM, Hoskin DW. Bovine lactoferricin selectively induces apoptosis in human leukemia and carcinoma cell lines. Mol Cancer Ther. 2005;4:612–624. doi: 10.1158/1535-7163.MCT-04-0077. [DOI] [PubMed] [Google Scholar]
- 172.Eliassen LT, Berge G, Leknessund A, Wikman M, Lindin I, Løkke C, Ponthan F, Johnsen JI, Sveinbjørnsson B, Kogner P, Flæstad T, Rekdal Ø. The antimicrobial peptide, Lactoferricin B, is cytotoxic to neuroblastoma cells in vitro and inhibits xenograft growth in vivo. Int J Cancer. 2006;119:493–500. doi: 10.1002/ijc.21886. [DOI] [PubMed] [Google Scholar]
- 173.Furlong SJ, Mader JS, Hoskin DW. Lactoferricin-induced apoptosis in estrogen-nonresponsive MDA-MB-435 breast cancer cells is enhanced by C6 ceramide or tamoxifen. Oncol Rep. 2006;15:1385–1390. [PubMed] [Google Scholar]
- 174.Eliassen LT, Haug BE, Berge G, Rekdal Ø. Enhanced antitumour activity of 15-residue bovine lactoferricin derivatives containing bulky aromatic amino acids and lipophilic N-terminal modifications. J Pept Sci. 2003;9:510–517. doi: 10.1002/psc.472. [DOI] [PubMed] [Google Scholar]
- 175.Yang N, Strøm MB, Mekonnen SM, Svendsen JS, Rekdal Ø. The effects of shortening lactoferrin derived peptides against tumour cells, bacteria and normal human cells. J Pept Sci. 2004;10:37–46. doi: 10.1002/psc.470. [DOI] [PubMed] [Google Scholar]
- 176.Gifford JL, Hunter HN, Vogel HJ. Lactoferricin: a lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell Mol Life Sci. 2005;62:2588–2598. doi: 10.1007/s00018-005-5373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mader JS, Richardson A, Salsman J, Top D, de Antueno R, Duncan R, Hoskin DW. Bovine lactoferricin causes apoptosis in Jurkat T-leukemia cells by sequential permeabilization of the cell membrane and targeting of mitochondria. Exp Cell Res. 2007 doi: 10.1016/j.yexcr.2007.05.015. in press. [DOI] [PubMed] [Google Scholar]
- 178.Yoo Y-C, Watanabe S, Watanabe R, Hata K, Shimazaki K, Azuma I. Bovine lactoferrin and lactoferricin, a peptide derived from bovine lactoferrin, inhibit tumor metastasis in mice. Jpn J Cancer Res. 1997;88:184–190. doi: 10.1111/j.1349-7006.1997.tb00364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mader JS, Smyth D, Marshall JS, Hoskin DW. Bovine lactoferricin inhibits basic fibroblast growth factor- and vascular endothelial growth factor165-induced angiogenesis by competing for heparin-like binding sites on endothelial cells. Am J Pathol. 2006;169:1753–1766. doi: 10.2353/ajpath.2006.051229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Nakamura T, Furunaka H, Miyata T, Tokunaga F, Muta T, Iwanaga S, Niwa M, Takao T, Shimonishi Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus). Isolation and chemical structure. J Biol Chem. 1988;263:16709–16713. [PubMed] [Google Scholar]
- 181.Rao AG. Conformation and antimicrobial activity of linear derivatives of tachyplesin lacking disulfide bonds. Arch Biochem Biophys. 1999;361:127–134. doi: 10.1006/abbi.1998.0962. [DOI] [PubMed] [Google Scholar]
- 182.Ramamoorthy A, Thennarasu S, Tan A, Gottipati K, Sreekumar S, Heyl DL, An FYP, Shelburne CE. Deletion of all cycteines in Tachyplesin I abolishes hemolytic activity and retains antimicrobial activity and LPS selective binding. Biochemistry. 2006;45:6529–6540. doi: 10.1021/bi052629q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chen J, Xu X-M, Underhill CB, Yang S, Wang L, Chen Y, Hong S, Creswell K, Zhang L. Tachyplesin activates the classic complement pathway to kill tumor cells. Cancer Res. 2005;65:4614–4622. doi: 10.1158/0008-5472.CAN-04-2253. [DOI] [PubMed] [Google Scholar]
- 184.Adamia S, Maxwell CA, Pilarski LM. Hyaluronan and hyaluronan synthase: potential therapeutic targets in cancer. Curr Drug Targets Cardivasc Haematol Disord. 2005;5:3–14. doi: 10.2174/1568006053005056. [DOI] [PubMed] [Google Scholar]
- 185.Rooney P, Kumar S, Ponting J, Wang M. The role of hyaluronan in tumour neovascularization (review) Int J Cancer. 1995;60:632–636. doi: 10.1002/ijc.2910600511. [DOI] [PubMed] [Google Scholar]
- 186.Chen Y, Xu X, Hong S, Chen J, Liu N, Underhill CB, Creswell K, Zhang L. RGD-tachyplesin inhibits tumor growth. Cancer Res. 2001;61:2434–2438. [PubMed] [Google Scholar]
- 187.Katsu T, Nakao S, Iwanaga S. Mode of action of an anti-microbial peptide, tachyplesin I, on biomembranes. Biol Pharm Bull. 1993;16:178–181. doi: 10.1248/bpb.16.178. [DOI] [PubMed] [Google Scholar]
- 188.Ouyang G-L, Li Q-F, Peng X-X, Liu Q-R, Hong S-G. Effects of tachyplesin on proliferation and differentiation of human hepatocellular carcinoma SMMC-7721 cells. World J Gastroenterol. 2002;8:1053–1058. doi: 10.3748/wjg.v8.i6.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shi S-L, Wang Y-Y, Liang Y, Li Q-F. Effects of tachyplesin and n-sodium butyrate on proliferation and gene expression of human gastric adenocarcinoma cell line BGC-823. World J Gastroenterol. 2006;12:1694–1698. doi: 10.3748/wjg.v12.i11.1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li QF, Ou Yang GL, Li CY, Hong SG. Effects of tachyplesin on the morphology and ultrastructure of human gastric carcinoma cell line BGC-823. World J Gastroenterol. 2000;6:676–680. doi: 10.3748/wjg.v6.i5.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Agerberth B, Lee J-Y, Bergman T, Carlquist M, Bowman HG, Mutt V, Jornvall H. Amino acid sequence of PR-39: isolation from pig intestine of a new member of the family of proline-arginine-rich antibacterial peptides. Eur J Biochem. 1991;202:849–854. doi: 10.1111/j.1432-1033.1991.tb16442.x. [DOI] [PubMed] [Google Scholar]
- 192.Shi J, Ross CR, Chengappa MM, Sylte MJ, McVey DS, Blecha F. Antibacterial activity of a synthetic peptide (PR-26) derived from PR-39, a proline-arginine-rich neutrophil antimicrobial peptide. Antimicrob Agents Chem. 1996;40:115–121. doi: 10.1128/aac.40.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ohtake T, Fujimoto Y, Ikuta K, Saito H, Ohhira M, Ono M, Kohgo Y. Proline-rich antimicrobial peptide, PR-39 gene transduction altered invasive activity and actin structure in human hepatocellular carcinoma cells. Br J Cancer. 1999;81:393–403. doi: 10.1038/sj.bjc.6690707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Liebersbach BF, Sanderson RD. Expression of syndecan-1 inhibit cell invasion into type I collagens. J Biol Chem. 1994;269:20013–20019. [PubMed] [Google Scholar]
- 195.Tanaka K, Fujimoto Y, Suzuki M, Suzuki Y, Ohtake T, Saito H, Kohgo Y. PI3-kinase p85α is a target molecule of proline-rich antimicrobial peptide to suppress proliferation of ras-transformed cells. Jpn J Cancer Res. 2001;92:959–967. doi: 10.1111/j.1349-7006.2001.tb01187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chan YR, Gallo RL. PR-39, a syndecan-inducing antimicrobial peptide, binds and affects p130Cas. J Biol Chem. 1998;273:28978–28985. doi: 10.1074/jbc.273.44.28978. [DOI] [PubMed] [Google Scholar]
- 197.Chan YR, Zanetti M, Gennaro R, Gallo RL. Anti-microbial activity and cell binding are controlled by sequence determinants in the anti-microbial peptide PR-39. J Invest Dermatol. 2001;116:230–235. doi: 10.1046/j.1523-1747.2001.01231.x. [DOI] [PubMed] [Google Scholar]
- 198.Shin SY, Lee MK, Kim KL, Hahm K-S. Structure-antitumor and hemolytic activity relationships of synthetic peptides derived from cecropin A-magainin 2 and cecropin A-melittin hybrid peptides. J Pept Res. 1997;50:279–285. doi: 10.1111/j.1399-3011.1997.tb01469.x. [DOI] [PubMed] [Google Scholar]
- 199.Shin SY, Kang JH, Hahm K-S. Structure-antibacterial, antitumor and hemolytic activity relationships of cecropin A-magainin 2 and cecropin A-melittin hybrid peptides. J Pept Res. 1999;53:82–90. doi: 10.1111/j.1399-3011.1999.tb01620.x. [DOI] [PubMed] [Google Scholar]
- 200.Oh D, Shin SY, Kang JH, Hahm K-S, Kim KL, Kim Y. NMR structural characterization of cecropin A(1-8)-magainin 2 (1-12) and cecropin A (1-8)-melittin (1-12) hybrid peptides. J Pept Res. 1999;53:578–589. doi: 10.1034/j.1399-3011.1999.00067.x. [DOI] [PubMed] [Google Scholar]
- 201.Shin SY, Lee S-H, Yang S-T. Antibacterial, antitumor and hemolytic activities of α-helical antibiotic peptide, P18 and its analogues. J Pept Res. 2001;58:504–514. doi: 10.1034/j.1399-3011.2001.00934.x. [DOI] [PubMed] [Google Scholar]
- 202.Oren Z, Hong J, Shai Y. A repertoire of novel antibacterial diastereomeric peptides with selective cytolytic activity. J Biol Chem. 1997;272:14643–14649. doi: 10.1074/jbc.272.23.14643. [DOI] [PubMed] [Google Scholar]
- 203.Papo N, Shahar M, Eisenbach L, Shai Y. A novel lytic peptide composed of D,L-amino acids selectively kills cancer cells in culture and in mice. J Biol Chem. 2003;278:21018–21023. doi: 10.1074/jbc.M211204200. [DOI] [PubMed] [Google Scholar]
- 204.Papo N, Shai Y. New lytic peptides based on the D,L-amphipathic helix motif preferentially kill tumor cells compared to normal cells. Biochemistry. 2003;42:9346–9354. doi: 10.1021/bi027212o. [DOI] [PubMed] [Google Scholar]
- 205.Papo N, Braunstein A, Eshhar Z, Shai Y. Suppression of human prostate tumor growth in mice by a cytolytic D-, L-amino acid peptide: Membrane lysis, increased necrosis, and inhibition of prostate-specific antigen seretion. Cancer Res. 2004;64:5779–5786. doi: 10.1158/0008-5472.CAN-04-1438. [DOI] [PubMed] [Google Scholar]
- 206.Papo N, Seger D, Makovitzki A, Kalchenko V, Eshhar Z, Degani H, Shai Y. Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res. 2006;66:5371–5378. doi: 10.1158/0008-5472.CAN-05-4569. [DOI] [PubMed] [Google Scholar]
- 207.Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Del Rio G, Krajewski S, Lombardo CR, Rao R, Ruoslahti E, Bredesen DE, Pasqualini R. Anticancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5:1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- 208.Mai JC, Mi Z, Kim S-H, Ng B, Robbins PD. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001;61:7709–7712. [PubMed] [Google Scholar]
- 209.Law B, Quinti L, Choi Y, Weissleder R, Tung C-H. A mitochondrial targeted fusion peptide exhibits remarkable cytotoxicity. Mol Cancer Ther. 2006;5:1944–1949. doi: 10.1158/1535-7163.MCT-05-0509. [DOI] [PubMed] [Google Scholar]