

Abstract
The concept of orthology is widely used to relate genes across different species using comparative genomics, and it provides the basis for inferring gene function. Here we present the web accessible OrthoDB database that catalogs groups of orthologous genes in a hierarchical manner, at each radiation of the species phylogeny, from more general groups to more fine-grained delineations between closely related species. We used a COG-like and Inparanoid-like ortholog delineation procedure on the basis of all-against-all Smith-Waterman sequence comparisons to analyze 58 eukaryotic genomes, focusing on vertebrates, insects and fungi to facilitate further comparative studies. The database is freely available at http://cegg.unige.ch/orthodb
INTRODUCTION
Identification of orthologous genes is the cornerstone of comparative genomics, which is increasingly becoming an essential part of modern molecular biology. Functions of orthologous genes are often preserved through evolution, as by definition, orthologous genes descend by speciation from the common ancestor gene (1–3). Although the conservation of ortholog functions is not required or guaranteed, it is the most likely evolutionary scenario and provides a strong working hypothesis, particularly when the ortholog copy-number is preserved over a long period of time. Identification of orthologs is intricate as it assumes knowledge of ancestral state of the genes, and it requires knowledge of the complete gene repertoires. It is also complicated by gene duplication, fusion and exon shuffling, as well as pseudonization and loss, which make the problem particularly challenging with complex eukaryotic genomes. The fast growing number of available complete genomes facilitates a much better resolution of the gene genealogies, while at the same time greatly increasing the computational challenges.
There are two main approaches to delineate orthologous genes: (i) from reconciliation of gene trees with the species phylogeny and (ii) from classification of all-against-all sequence comparisons of complete genomes. The phylogeny approach takes advantage of well-studied evolution of conserved cores of globular proteins using quantitative models of amino acid substitutions (4–6). The notable examples of the tree-based approach to delineate orthologous genes are HOVERGEN (7) and TreeFam (8). The expert curation of the phylogenetic trees and the underlying multiple sequence alignments is both, advantageous, providing better accuracy and disadvantageous, limiting the comprehensiveness, homogeneity of quality and expandability to new species. Although given the appropriate data phylogenetic methods are likely to give more accurate models of ancestral sequences and therefore to yield more accurate orthology prediction, their applicability to current genomic data is hindered by several factors, most importantly: (i) they require substantially more computational resources, (ii) the reconciliation of gene and species trees relies on poorly quantified models of gene duplication and loss, and (iii) they are sensitive to completeness of predicted genes as the evolutionary models are designed for only well-conserved globular cores of proteins and missing data (gaps) render the approach inapplicable. The tree-based approaches also require the knowledge of the species phylogeny, and although the consensus on animal phylogeny seems to be close, it is still constantly challenged. The alternative approach of clustering orthologous genes on the basis of their whole-length similarity around Best-Reciprocal-Hits (BRHs, also known as SymBets, bi-directional BeTs and best–best hits, denoting sequences most similar to each other in between-genome comparisons) was first introduced by the database of Clusters of Orthologous Groups (COGs) (9). Triggered by the earlier availability of much smaller and simpler bacterial genomes the database has quickly gained wide recognition and was later extrapolated to eukaryotic genomes (KOGs) (9). The identification of BRHs is widely adopted currently in the field of comparative genomics for its simplicity and feasibility of application to large-scale data. In terms of phylogenetic trees, BRHs could be interpreted as genes from different species with the shortest connecting path over the distance-based tree. The simplest application of this approach using BLAST (10) for interspecies comparisons suffers from inaccuracies of sequence distance estimates and ignores many gene duplications after the speciation that are, in fact, co-orthologs that are difficult to differentiate functionally. However, using these genes as anchors of orthologous groups in different species, additional co-orthologs can be identified as genes that are more similar to them in intra-genome comparisons than to any other gene in the other genomes, as popularized by the pairwise Inparanoid approach (11). There are also a few alternative clustering heuristics with varying compromises between specificity and selectivity (12) that focus on the growing number of available eukaryotic genomes such as the probabilistic approach of OrthoMCL (13) and the vertebrate-centric Ensembl-Compara (14).
Another important feature of orthology and paralogy classification, which is currently underappreciated, is that it is relative to a particular ancestor, as orthology of genes is defined by their descent from a common ancestor gene by speciation (1–3). Therefore, the more distantly related species are considered the more general (inclusive) orthologous groups become, because all lineage-specific duplications since this last common ancestor should be considered as co-orthologs. Inversely, orthologous groups become more fine-grained (more 1 : 1 relations) when closely related species are considered, as there was less time for gene duplications to occur. The concept of hierarchical orthologous groups has already prompted development of Levels of Orthology From Trees (LOFT) (15) tool to interpret the gene-trees in the context of species tree, COrrelation COefficient-based CLustering (COCO-CL) (16) methodology to refine clusters of homologous genes, and PHOG approach (17) to resolve orthology at each taxonomy node using explicit modeling of the ancestral sequences and relying on PHOG-BLAST (18) profile–profile comparisons.
Aiming to fuel comparative genomic studies we focused here on the most represented eukaryotic phyla, namely, we analyzed 23 fungi, 19 insect (plus one crustacean) and 15 vertebrate species with complete proteomes available (Table 1). For this analysis, we employed our own implementation of COG-like and Inparanoid-like ortholog identification procedures from all-against-all sequence comparisons across multiple species (19–22), and here we explicitly delineate the hierarchy of the orthologous groups, consistently applying the procedure to the sets of species with varying levels of relatedness according to the species tree (Figure 1).
Table 1.
Sets of covered complete proteomes
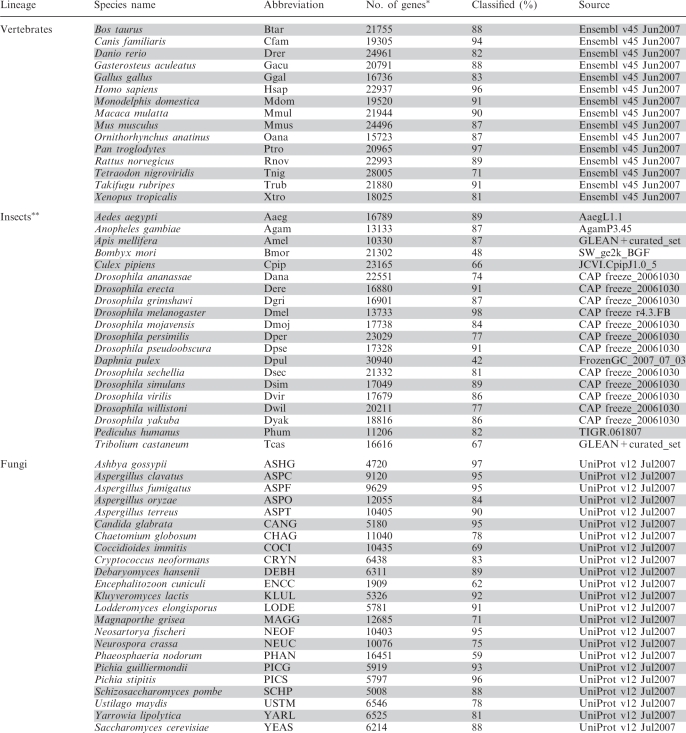 |
*Only the longest transcript per gene was considered.
**Including one crustacean.
Figure 1.

Example screenshot of the OrthoDB web interface (http://cegg.unige.ch/orthodb). The left panel enumerates the modes to query and browse the database by: (1) a keyword, (2) a user specified phylogenetic gene copy-number profile, (3) a common phylogenetic profile, or (4) sequence homology search; the middle panel is reserved for displaying results; and the right panel accommodates help and query history messages.
METHODS
Orthology delineation
Groups of orthologous genes were automatically identified using a strategy employed previously (19–22) that is based on all-against-all protein sequence comparisons using the Smith-Waterman algorithm as implemented in ParAlign (23) with default parameters, followed by clustering of best reciprocal hits from highest scoring ones to 10−6 e-value cutoff for triangulating BRH or 10−10 cutoff for unsupported BRH, and requiring a sequence alignment overlap of at least 30 amino acids across all members of a group. Furthermore, the orthologous groups were expanded by genes that are more similar to each other within a proteome than to any gene in any of the other species, and by very similar copies that share over 97% sequence identity, which were identified initially using CD-Hit (24). We considered only the longest transcript per gene or the most common as specified in UniProt (25). The outlined procedure was first applied to all species considered, and then to each subset of species according to the radiation of the phylogenetic tree.
Phylogeny reconstruction
To guide computation of the ortholog hierarchy we produced the multiple alignment of concatenated single-copy orthologs, using well-aligned regions extracted with Gblocks (26) from individually aligned orthologous sequences using Muscle (27). This was used to compute the phylogenetic trees using the maximum-likelihood method as implemented in PHYML (28), employing the JTT model, a gamma correction with four discrete classes, and an estimated alpha parameter and proportion of invariable sites.
DATABASE CONTENT
Overview statistics
As detailed in Table 1 we analyzed 23 complete proteomes of fungal species, 19 insects and 15 vertebrates at different levels of the species phylogeny. Overall, this effort spans 870 737 genes, 82% of which have been classified into 10 876 orthologous groups in fungi, 19 835 in insects and 23 940 in vertebrates, providing the first systematic classification of the wealth of data that will provide the basis for further comparative evolutionary analyses.
WEB INTERFACE
The database is freely accessible from http://cegg.unige.ch/orthodb
Hierarchy of the orthologous groups
Orthology and paralogy classification is relative to the set of species considered [namely, to the particular ancestor (1–3)] and is more general (inclusive) for distantly related species, and more fine-grained (specific) for closely related species. We therefore delineated orthologous groups at each radiation node of the species phylogeny. To clearly show the hierarchy level of the classifications and to allow easy navigation along the hierarchy we display the interactive species tree (Figure 1). The default level for an initial user query is set to fungi, arthropods or vertebrates and the level can be adjusted afterwards by selecting a radiation of interest on the phylogeny. Each result page provides a precompiled Bookmarklet, a snippet of JavaScript code that can be easily bookmarked in the user browser, to allow direct query to a particular phylogeny level.
Stable identifiers
We assigned short identifiers using Noid utility to the generated orthologous groups that we will maintain unique across subsequent updates of the database to allow stable references to the data.
Querying by keywords
The database is searchable by the relevant identifiers used for the proteins or orthologous groups, as well as by keywords associated with the protein annotation in UniProt (25) or Ensembl (14). Currently the search is implemented as mySQL full-text index and the query is interpreted in Boolean mode that allows use of ‘+’ and ‘−’operators to indicate that a word is required to be present or absent, respectively, for a match to occur; parentheses used to group words into subexpressions; ‘*’ serves as the wildcard operator; and a phrase is matched literally if it is enclosed within quotes (e.g. ‘cytochrome c’). The results always refer to the relevant orthologous groups, not separate genes.
Filtering by phylogenic profile
Another feature of the database interface is filtering orthologous groups by a phylogenic profile. This can be done by activating the set of selectors next to the phylogenetic tree and specifying the ortholog copy-number requirements in the species of interest, where ‘?’ notation stands for no restriction (‘any number’) and ‘0’, ‘1’, ‘>1’ are self explanatory. The ‘Filter’ button in the ‘Specify copy-number profile’ section will execute the corresponding query. In addition, we provide a set of precompiled queries for phylogenic profiles of common interest (via the selection list) that are more complicated to express, e.g. ‘all but one’ type: all single-copy orthologs but allowing for a loss or run-away in one of the species, or multigene orthologs in all but one species, etc. This allows viewing of the gene clusters that have undergone expansions or losses in the specific lineages, which is informative in the evolutionary context (29). These queries, as well as text search, are performed with respect to the selected speciation root, marked by red on the phylogenetic tree.
Query by sequence homology
Not all protein identifiers are widely known, particularly for automatically annotated genomes, and functional annotations for many genes are still anticipated. We therefore provide data querying by sample sequences, e.g. a user submitted sequence is matched using Blast against the collected proteomes, and the top five matches from distinct proteomes are shown to the user and used to retrieve the associated orthologous groups, ranking by the number of hits to each group. Please note that if a sequence of an as yet unanalyzed species is used, the query will return the best matching ortholog cluster, however, this may not be sufficient to assume orthology.
Export of data
All results or particular groups can be retrieved as tab-delimited text or as Fasta formatted protein sequences with annotation of the orthologous group.
FUTURE PERSPECTIVES
All current approaches to identify orthologous groups of genes have different deficiencies and there are ways to improve their sensitivity and specificity. The implemented infrastructure in principal does not depend on the particular choice of the method, although our own implementation of a COG-like and Inparanoid-like ortholog identification procedure seems to produce reliable results according to extensive checks in the frame of our previous research projects. We plan also to test other available orthology delineation procedures.
ACKNOWLEDGEMENTS
We thank Thomas Junier for technical support, Dr Stefan Wyder, Dr Ivo Pedruzzi and Robert M Waterhouse for feedback and quality checks. We would like to acknowledge UniProt, Ensembl, Vectorbase, Flybase, the AAA fly genomes initiative, genome analysis consortiums and sequencing centers Agencourt, Baylor, BGI, Broad Institute, JCVI and JGI for the data. Foundation Giorgi-Cavaglieri and Swiss National Science Foundation are acknowledged for funding (SNF 3100A0-112588 to EMZ). Funding to pay the Open Access publication charges for this article was provided by SNF 3100A0-112588.
Conflict of interest statement. None declared.
REFERENCES
- 1.Fitch WM. Distinguishing homologous from analogous proteins. Syst. Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- 2.Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 3.Sonnhammer EL, Koonin EV. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002;18:619–620. doi: 10.1016/s0168-9525(02)02793-2. [DOI] [PubMed] [Google Scholar]
- 4.Dayhoff MO. The origin and evolution of protein superfamilies. Fed. Proc. 1976;35:2132–2138. [PubMed] [Google Scholar]
- 5.Jones CH, Tatti KM, Moran C.P., Jr Effects of amino acid substitutions in the −10 binding region of sigma E from Bacillus subtilis. J. Bacteriol. 1992;174:6815–6821. doi: 10.1128/jb.174.21.6815-6821.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duret L, Mouchiroud D, Gouy M. HOVERGEN: a database of homologous vertebrate genes. Nucleic Acids Res. 1994;22:2360–2365. doi: 10.1093/nar/22.12.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, Coghlan A, Ruan J, Coin LJ, Heriche JK, Osmotherly L, Li R, Liu T, Zhang Z, et al. TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 2006;34:D572–D580. doi: 10.1093/nar/gkj118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Brien KP, Remm M, Sonnhammer EL. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005;33:476–480. doi: 10.1093/nar/gki107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen F, Mackey AJ, Vermunt JK, Roos DS. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PLoS ONE. 2007;2:e383. doi: 10.1371/journal.pone.0000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen F, Mackey AJ, Stoeckert C.J., Jr, Roos DS. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;34:D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Heijden RT, Snel B, van Noort V, Huynen MA. Orthology prediction at scalable resolution by phylogenetic tree analysis. BMC Bioinformatics. 2007;8:83. doi: 10.1186/1471-2105-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jothi R, Zotenko E, Tasneem A, Przytycka TM. COCO-CL: hierarchical clustering of homology relations based on evolutionary correlations. Bioinformatics. 2006;22:779–788. doi: 10.1093/bioinformatics/btl009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merkeev IV, Novichkov PS, Mironov AA. PHOG: a database of supergenomes built from proteome complements. BMC Evol. Biol. 2006;6:52. doi: 10.1186/1471-2148-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merkeev IV, Mironov AA. PHOG-BLAST–a new generation tool for fast similarity search of protein families. BMC Evol. Biol. 2006;6:51. doi: 10.1186/1471-2148-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waterhouse RM, Kriventseva EV, Meister S, Xi Z, Alvarez KS, Bartholomay LC, Barillas-Mury C, Bian G, Blandin S, et al. Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes. Science. 2007;316:1738–1743. doi: 10.1126/science.1139862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zdobnov EM, Bork P. Quantification of insect genome divergence. Trends Genet. 2007;23:16–20. doi: 10.1016/j.tig.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Zdobnov EM, von Mering C, Letunic I, Torrents D, Suyama M, Copley RR, Christophides GK, Thomasova D, Holt RA, et al. Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster. Science. 2002;298:149–159. doi: 10.1126/science.1077061. [DOI] [PubMed] [Google Scholar]
- 22.Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443:931–949. doi: 10.1038/nature05260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saebo PE, Andersen SM, Myrseth J, Laerdahl JK, Rognes T. PARALIGN: rapid and sensitive sequence similarity searches powered by parallel computing technology. Nucleic Acids Res. 2005;33:535–539. doi: 10.1093/nar/gki423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 25.Consortium. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2007;35:D193–D197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 27.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guindon S, Lethiec F, Duroux P, Gascuel O. PHYML Online–a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005;33:W557–559. doi: 10.1093/nar/gki352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wyder S, Kriventseva EV, Schröder R, Kadowaki T, Zdobnov EM. Quantification of ortholog losses in insects and vertebrates. Genome Biol. 2007 doi: 10.1186/gb-2007-8-11-r242. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]